

Abstract
We investigated the impact of poor maternal nutrition and metabolic health on the development of islets of the nonhuman primate (NHP). Interestingly, fetal offspring of high fat diet (HFD) fed animals had normal total islet and β cell mass; however, there was a significant reduction in α cell mass, and decreased expression of transcription factors involved in α cell differentiation. In juvenile animals all offspring maintained on a HFD during the postweaning period demonstrated increases in total islet mass, however, the control offspring displaying increased islet number, and HFD offspring displayed increased islet size. Finally, while control offspring had increases in α and β cells, the HFD offspring had increases only in β cell number. These studies indicate that consumption of a HFD diet during pregnancy in the NHP, independent of maternal metabolic health, causes long-term abnormalities in α cell plasticity that may contribute to chronic disease susceptibility.
Abbreviations: HFD, High-fat diet; CTR, Control.
Keywords: Obesity, Pregnancy, Pancreas, Development, High fat diet, Diabetes
1. Introduction
According to the National Health and Nutrition Examination Survey (NHANES), the incidence of childhood obesity in the United States has tripled in the last 25 years [1] and has become a worldwide epidemic. Since early childhood and adolescent obesity is a strong predictor of being obese or overweight as an adult [2,3], this rise in childhood obesity indicates that we have not reached the peak in the obesity health crisis that is plaguing our world today. Recent studies have demonstrated that children exposed to maternal obesity in utero are at increased risk for developing both obesity and metabolic syndrome [4–7]. If the incidence of obesity continues to rise, especially among women, we will likely see an increased incidence of childhood metabolic syndrome in future generations. This rise will lead to increased health burdens for the general population as the frequency of diabetes and cardiovascular disease increases. The mechanisms that lead to the association between maternal obesity and childhood obesity and/or metabolic syndrome need to be determined in order to treat and prevent this effect from perpetuating.
Using a nonhuman primate (NHP) model, our group has demonstrated that chronic consumption of a high-fat diet (HFD) during pregnancy, independent of the maternal metabolic phenotype, can lead to a broad range of complications in fetal development that may increase the susceptibility of the offspring to obesity and diabetes later in life. In the fetus, we have observed that a maternal HFD leads to increased liver triglycerides, a shift in genes involved in hepatic gluconeogenesis, and increased hepatic apoptosis in the fetal offspring, suggesting early signs of NAFLD [8,9]. We also observed placental insufficiency and inflammation in pregnant macaques fed a HFD. Importantly, the cytokines produced in the placenta are secreted specifically into the developing fetus, where we hypothesized that they initiate inflammatory responses in several developing organs [10]. Also, because placental insufficiency has been demonstrated to lead to reduced islet vascularization [11,12], it is important to determine how this will affect islet cell development and function.
The primary purpose of these studies was to determine the relative impact of early programming events (i.e., maternal diet) versus post-weaning dietary influence on pancreas development and long-term susceptibility to diabetes. To accomplish this, we examined the pancreata from fetuses of pregnant NHPs who consumed a control (CTR) or a HFD. We also measured post-weaning effects of HFD consumption on islet development and function in juvenile macaques that were born to CTR or HFD mothers and maintained on this diet or switched to the opposing diet after weaning, producing four post-weaning groups. These groups allowed us to determine the relative impact of maternal HFD versus the impact of the HFD during the post-weaning period on islet structural morphology and function.
Principally we chose to examine islet morphology, α cell and β cell mass, and changes in expression of genes involved in glucose metabolism and islet development. Previous animal model studies indicate that much of the dysregulation of glucose homeostasis in offspring associated with in utero obesity exposure is due to β cell dysfunction, or the suppression of β cell proliferation in response to this early life insult [13–16]. We chose to investigate these hypotheses in our well-characterized NHP model that has developmental similarities to humans, and in which we can control and manipulate their environment. Our aim was to determine which islet cells may be affected by maternal obesity and/or HFD consumption, and how these changes may lead to dysregulation of glucose homeostasis in the postnatal offspring. We proposed that any defects in islet function in the HFD offspring would be perpetuated by continued HFD exposure through the post-weaning period. Moreover, we determined whether a post-weaning dietary intervention to a standard control (CTR) diet could reverse the effects of HFD consumption during early development.
2. Materials and method
2.1. Animals
All animal procedures were in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Oregon National Primate Research Center (ONPRC) as previously described [8,17–20]. Briefly, age- and weight-matched adult female Japanese macaques were fed a CTR diet or HFD ad libitum. The CTR diet (Monkey Diet no. 5052, Lab Diet, Richmond, IN, USA) supplies 2.87 kcal/g with 26.8% energy from protein, 58.5% from carbohydrate, and 14.7% from fat and its major source of fat is soya bean oil. In contrast, the main sources of fat in the HFD (Custom Diet 5A1F, Test Diet, Richmond, IN, USA) are lard, animal fat, butter, and safflower oil, and this supplies 4.2 kcal/g with 16.7% energy from protein, 51.5% from carbohydrate, and 31.8% from fat. In depth characterization of the diets has been previously published [9]. As previously described, dams varied in their sensitivity to weight gain and insulin resistance in response to the HFD [8]. Mothers were fasted overnight and fetal offspring were obtained by c-section at gestational day 130 (G130) (early 3rd trimester) and immediately delivered to necropsy for tissue collection. Pancreas tissue was harvested from CTR and HFD fetuses and analyzed using immunohistochemisty (IHC) and quantitative RT-PCR (qPCR). Over a five year period, juvenile offspring were born naturally from CTR or HFD mothers who had been consuming the diet for 2–5 years, and these offspring were maintained with their mothers until weaning at approximately 8 months of age (249±4 days). Infant offspring began independent ingestion of the maternal diet by 4 months of age and were primarily consuming this diet, instead of nursing, by 6 months of age. At 8 months, the juveniles were weaned and ad libitum fed the same diet or switched to the opposing diet, for 5 months (143±3 days), thus generating four juvenile groups: CTR/CTR, CTR/HFD, HFD/CTR, & HFD/HFD. Groups were balanced for sex, maternal sensitivity to the diet, maternal age, and length of maternal consumption of the diets. The juvenile animals were necropsied at approximately 13 months of age (393±4 days).
2.2. Intravenous glucose tolerance test (IVGTT)
We performed IVGTTs at 90, 180 days, and 1 week prior to necropsy in the juvenile offspring. In the dams, we performed the IVGTTs at 123 days (±5 days) gestation and at approximately 130 days (±6 days) after delivery. Mothers were fasted overnight prior to a morning IVGTT, while offspring were fasted for 5 h and the IVGTT was performed in the afternoon. Animals were sedated with ketamine (10 mg/kg) and administered a glucose bolus (50% dextrose solution) at a dose of 0.6 g/kg via the saphenous vein. Baseline blood samples were obtained prior to the infusion, and 500 μL blood samples were taken at 1, 3, 5, 10, 20, 40, and 60 min after infusion via the femoral artery. Glucose was measured immediately using a OneTouch Ultra2 Blood Glucose Monitor (LifeScan, Milpitas, CA), and the remainder of the blood was kept in heparinized tubes on ice for insulin measurement. After IVGTT, samples were centrifuged, and plasma was stored with 5% aprotinin at −80 °C until assayed. Insulin was assayed in plasma by RIA (Catalog no. RI-13K; EMD Millipore, Billerica, MA). HOMA-IR was calculated as fasting serum insulin (μU/ml)×fasting plasma glucose (mmol/l)/22.5, as previously described [21,22]. Fasting glucagon was assayed in plasma by RIA (Catalog no. GL-32K; Millipore) and fasting serum c-peptide levels were assayed by ELISA (Catalog no. K6220; Dako, Carpinteria, CA). Glucose stimulated insulin secretion (GSIS) was measured as the insulin area under the curve (IAUC) minus basal fasting insulin levels, while total IAUC is a measure of insulin secretion from 0.
2.3. Intravenous insulin tolerance test (IVITT)
We performed IVITTs at 210 days on a subset of animals. After a 5 h morning fast, animals were sedated with telazol (5 mg/kg) and a baseline blood sample was collected and measured for glucose. Insulin was infused at 0.05 U/Kg and sequential blood samples were taken at 1, 3, 5, 10, and 15 min. Glucose was measured immediately using a OneTouch Ultra2 Blood Glucose Monitor (LifeScan, Milpitas, CA). The insulin sensitivity index (ISI) was calculated as the delta glycemia from baseline to 15 min (estimated using the linear regression from baseline to 15 min) divided by the initial glycemia (average of both baselines), as previously described [23].
2.4. RNA isolation and quantitative RT-PCR
Samples for qPCR were collected from both fetal and juvenile offspring. Total RNA was isolated from 5 head-to-tail grouped sections (Figure A.1) using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA was DNase treated and reverse transcribed. mRNA was measured by real time qPCR using a PE Applied Biosystems PRISM 7900 Sequences Detector System (Invitrogen Life Technologies, Carlsbad, CA). Premade qPCR gene expression assays directed against human sequences were purchased from Applied Biosystems (Table A.1). The amplification was performed as follows: 2 min at 50 °C, 10 min at 95 °C, then 45 cycles each at 95 °C for 15 s, and at 60 °C for 60 s. Standard curves on serial dilutions of the cDNA (1:10, 1:50, 1:100 1:500, 1:1000, and 1:10,000) were performed using cDNA from pooled pancreas samples. Standard curves were drawn on the basis of the log of the input RNA versus the critical threshold (CT) cycle, in which the fluorescence of the sample is greater than the threshold of the baseline fluorescence. The housekeeping gene β actin was assayed, and it was shown to be consistently expressed between juvenile groups (Figure 8F), while RNA Polymerase II was used to normalize real-time expression in the fetal groups. Therefore, all qPCR results are normalized to the appropriate housekeeping gene expression. It should be noted that we have also normalized to mean β cell area, α cell area, or islet area, depending on the cell types that the gene of interest is expressed. This methodology has been previously described when whole pancreas mRNA is used [24].
Figure 8.
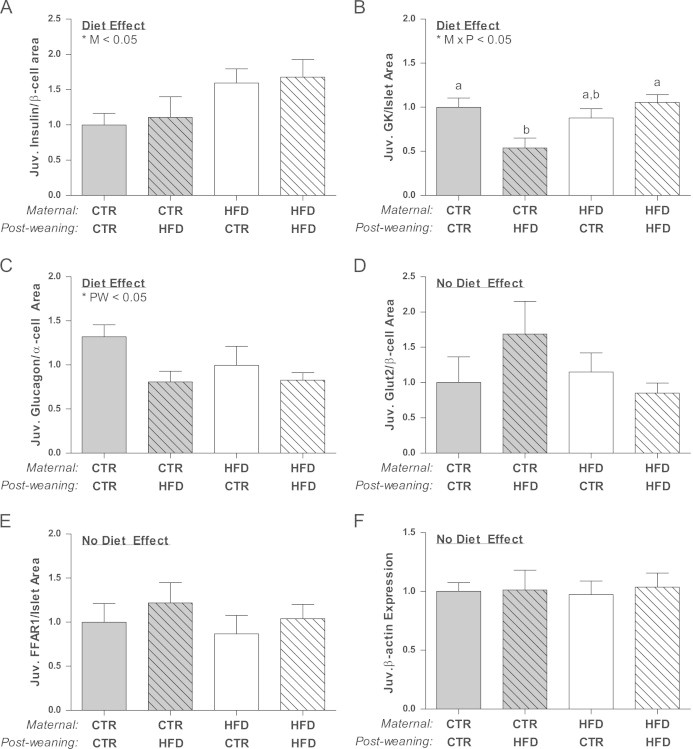
Insulin mRNA/β-cell area is significantly affected by the maternal diet (M; p<0.05), but not different between juvenile groups (A). Glucokinase mRNA/islet area is significantly down-regulated in the CTR/HFD animals and affected by an interaction between the maternal and post-weaning diet (M×PW; p<0.05) (B). Glucagon mRNA/α cell area is affected by the post-weaning diet (PW; p<0.05), but not significantly different between groups (C). Glut2/β cell area is not significantly affected (D). FFAR1 mRNA/islet area is not significantly affected (E).Two-way ANOVA with Bonferroni's post-hoc analysis; bars labeled with different letters “a” or “b” indicate a significant difference (p<0.05) from each other, while bars with the same letter are not significantly different from each other. Samples were excluded if values were more than two standard deviations away from the mean. CTR/CTR, n=12; CTR/HFD, n=6; HFD/CTR, n=9; HFD/HFD, n=12.
2.5. Immunohistochemistry
Tissue was collected from a subset of the fetal offspring (CTR=7, HFD=8) for immunohistochemistry (IHC). Preliminary analysis found that 75% of islet mass was located in the tail portion of the fetal pancreas (not shown). Therefore tissue for the fetal offspring was collected from randomly selected sections within the tail region of the pancreas. Samples for juvenile IHC were collected during the first three years of this study. Pancreas tissue was collected from 5 head to tail areas (Figure A.1) and cut into 5 μm paraffin embedded sections. Sections were deparaffinized in xylene and then washed with 100%, 95%, 85%, and 70% graded ethanol for 5 min each. Microwaving in citrate buffer was used for antigen retrieval. Standard immunohistochemical methods were used. Briefly, sections were washed in KPBS, and then blocked in 2% donkey serum in 0.4% triton X/KPBS. Antibodies against insulin (#20056, 1:2000, Immmunostar, Hudson, WI) and glucagon (#4031-01F, 1:10,000, Linco, St. Charles, MO) were used, which have been previously described for immunohistochemical identification of β cells and α cells, respectively [25,26]. Primary antibodies were diluted in 2% donkey serum in 0.4% triton X/KPBS and applied to tissue sections where they were incubated overnight. Tissues were then washed in KPBS and secondary antibodies were applied for 1 h at room temperature at a dilution of 1:200 (Jackson ImmunoResearch Laboratories, Inc, West Grove, PA). The specificity of the antibodies were determined by omitting the primary and using only the secondary. These tests did not produce staining.
2.6. Quantitative analysis
Images were acquired with a MarianasTM imaging workstation (Intelligent Imaging Innovations, Denver, CO), using a 20× objective and the stereology module to montage images covering whole sections. Areas occupied by β and α cells were measured based on intensity segmentation of each fluorescence channel using Slidebook 5.0. The β and α cells fractions were calculated by dividing the area of the whole section. Total islet area (insulin and glucagon stained area/DAPI stained area), islet mass (total islet area×pancreas weight), β and α cell mass [(total insulin or glucagon stained area/DAPI stained area)×pancreas weight], and islet density (total islet number/DAPI area) were calculated as previously reported [27]. Islets were classified based on size:stained cell clusters with an area greater than 1000 μm2 or diameter approximately 35 μm were arbitrarily defined as large islets, while any clusters smaller than this were classified as small islets. In juvenile offspring, individual α and β cell size and number were quantified in a manner similar to that previously described [28–30]. Briefly, stereologically sampled 40× images from the tail region of each pancreata were systematically randomly selected and 3–5 whole islets within randomly generated stereology counting frames were quantified from each animal. Individual α and β cell area was measured based on glucagon and insulin staining and the number of nuclei were quantified in that area. Individual islet cell size was determined by the number of nuclei divided by the stained area for each islet (i.e., number of nuclei in glucagon stained area/glucagon stained area). All islets stereologically sampled for each diet group were included in statistical analyses.
2.7. Data analysis
For all analyses not specifically described above, data was compiled and tested for normality with SPSS Statistics 19 software (Armonk, New York). Data were tested for maternal or post-weaning diet effect using two-way ANOVA, then Bonferroni post-hoc analysis to test difference between individual groups. In order to determine a sex effect, a three-way ANOVA was performed on all data for sex, maternal diet and post-weaning diet. Significance was reported as p<0.05. All graphs were made with Prism software (GraphPad Software, Inc., La Jolla, CA).
3. Results
3.1. Alpha cell mass is decreased in HFD fetal offspring
Overall, the physiological phenotype in the HFD fetus was minimally affected by the maternal phenotype or diet. HFD fetal weight and plasma glucagon was significantly reduced in only the offspring of HFD mothers who were resistant to weight gain (Table A.2, Figure A.2A). Furthermore, maternal HFD consumption did not lead to significant differences in pancreas weight, blood glucose (Table A.2), or circulating insulin levels in the fetus (Table A.2; Figure A.2B). However, serum c-peptide was significantly decreased in HFD fetuses (Table A.2; Figure A.2C). Analysis of islet morphology demonstrated no significant difference in total islet mass between the fetal groups (Figure 1A), although HFD offspring had a significant decrease in the mass of small islets (Figure A.3A). Representative images are presented in Figure 1B and C. There was no significant effect of the HFD on islet density or diameter (Figs. A.3B and A.3C). In addition, an analysis of specific cell types within the islets demonstrated a significant decrease in α cell mass in the HFD offspring, regardless of maternal metabolic phenotype (Figure 1D). While glucagon mRNA expression was significantly decreased in the pancreas of HFD fetuses (Figure 1E) when this expression was normalized to α cell mass the difference was lost (Figure 1F). On the contrary, there was no significant difference in β cell mass (Figure 1G). Insulin gene expression was slightly suppressed in the HFD offspring (Figure 1H), and normalizing to β cell area did indicate that there was a significant decline in insulin transcription on a per cell basis (Figure 1I). The effect on islet cell mass led to a significant increase in the β/α cell ratio in HFD offspring (Figure 2A), suggesting altered paracrine interactions.
Figure 1.
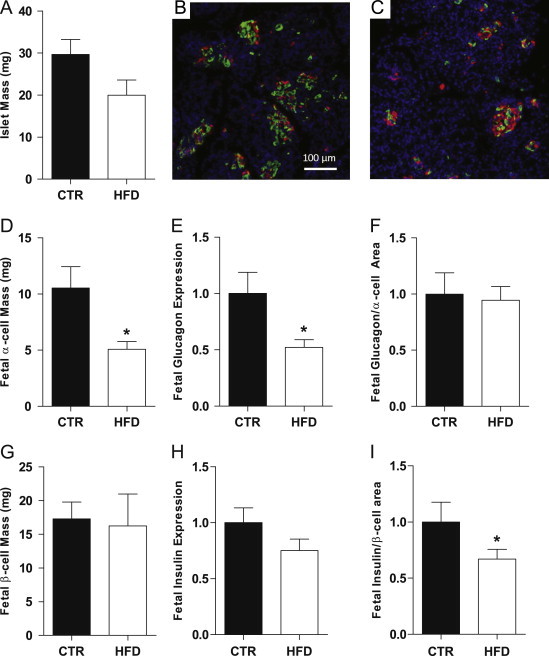
Islet mass was not significantly different between fetal groups (A) Representative images of CTR (B) and HFD (C) IHC, glucagon green and insulin red. α cell mass was significantly decreased in the HFD fetal pancreas (D). Glucagon expression was suppressed in HFD fetuses (E). When normalized to α cell area this effect was no longer significant (F). β cell mass was not affected in the HFD fetuses (G). Insulin fetal mRNA expression was unchanged (H) but suppressed in the HFD fetuses when normalized to β cell area (I). *p<0.05 versus CTR (Student's t-test). IHC: CTR, n=7; HFD, n=8; real-time: CTR n=11; HFD, n=17.
Figure 2.
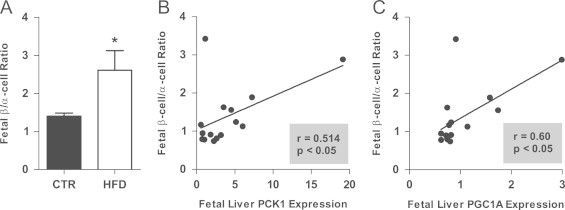
β/α cell ratio was increased in the HFD fetal pancreas (A). *p<0.05 versus CTR (Student's t-test). CTR, n=7; HFD, n=8. Hepatic expression of PCK1 (B) and PGC1A (C) significantly correlates with β/α cell ratio in the fetus.
While circulating, fetal insulin was not significantly affected in the fetal HFD group, c-peptide levels were reduced, which may indicate altered clearance by the liver. Since we have previously demonstrated an increase in gluconeogenic gene expression in the fetal HFD liver [8], we chose to investigate the relationship between islet cell mass and the expression of these genes. The β/α cell ratio significantly correlated with the expression of phosphoenolpyruvatecarboxykinase 1 (PCK1) and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC1A) (Figure 2B and C).
There are three primary mechanisms that may contribute to altered islet morphology: changes in neogenesis, proliferation and/or apoptosis. An analysis of the colocalization of CK7, a ductal marker, with insulin or glucagon indicated that there were no differences in islet neogenesis (data not shown). Similarly, we were unable to detect any significant differences in islet cell proliferation, as assessed by Ki67 staining (data not shown). Though apoptosis was not directly measured, TUNEL-positive cells were difficult to detect in either group. Thus, the mechanism leading to an altered β to α cell ratio in HFD offspring remains unclear.
3.2. Transcription factor expression in fetal pancreas
Due to the decrease demonstrated in α cell mass, we analyzed transcription factor expression by qPCR. Both FoxA2 and Irx2, which have been demonstrated to colocalize with glucagon expressing cells [31,32], were significantly suppressed in the HFD offspring (Figure 3A and B). Arx, which, in the rodent, is required for α cell differentiation [33], was unchanged at this stage (Table A.3). Pax6 has been demonstrated to regulate endocrine cell differentiation and proglucagon gene expression by modulating NeuroD1/Beta2 expression [34,35]. Analysis of expression of both Pax6 and NeuroD1 demonstrated no significant effect of the maternal HFD (Table A.3). PDX1, a transcriptional regulator of early pancreatic development, islet cell neogenesis, and insulin gene transcription [36], was not affected in the HFD fetal pancreas (Table A.3). Likewise, NKX6.1, which is more specific to expansion and maintenance of β cells at this stage of development [37], was unchanged (Table A.3).
Figure 3.
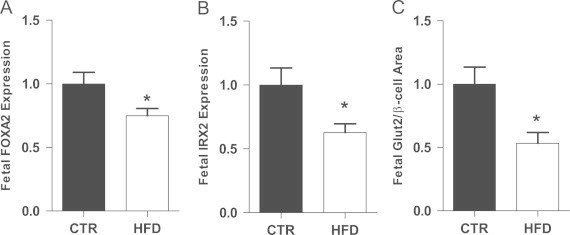
FOXA2 and IRX2 expression were down-regulated in the HFD fetal offspring (A and B). GLUT2 gene expression was suppressed the HFD fetuses when normalized to β cell area (C). *p<0.05 versus CTR (Student's t-test). CTR, n=11; HFD, n=17.
3.3. Expression of genes involved in glucose stimulated insulin secretion (GSIS) in the fetal offspring
Since we were unable to directly test GSIS in fetal offspring, we analyzed gene expression of proteins involved in this pathway, in order to determine if GSIS may be affected by the β/α cell ratio. The two initial factors in the glucose stimulated insulin secretion cascade are the facilitated glucose transporter 2 (Glut2) that allows glucose to enter the cell and glucokinase (GK), the rate-limiting enzyme in glycolysis [38–40]. Glut2 mRNA expression, which is expressed in β cells in macaques [41], when normalized to β cell area, was decreased in the HFD animals (Figure 3C). Since, GK is expressed in both α cells and β cells [42], we normalized GK expression to islet area. GK was not significantly affected in the HFD offspring (Tables A.3A and A.3B). The ATP sensitive K+ channel present in both α and β cells is critical for regulating insulin and glucagon secretion. This channel is composed of a sulfonylurea receptor (SUR1) and an inwardly rectifying potassium channel Kir6.2 (encoded by KCNJ11), both of which are necessary for the channel to function [43]. SUR1 and KCNJ11 expression, when normalized to islet area, were not significantly affected by the HFD (Tables A.3A and A.3B). Further analysis of real-time results demonstrated that mRNA expression patterns from the fetal groups were not dependent on maternal sensitivity to the HFD.
3.4. Physiology of infant offspring of CTR and HFD dams prior to weaning
To determine if the abnormalities in islet development have implications on early postnatal pancreas function, we measured glucose homeostasis and insulin resistance in the post-natal offspring. While there was no significant difference in weight or body fat at post-natal day 90 (P90), the insulin response to an IVGTT was significantly suppressed in the HFD offspring (Table A.4). P90 males were heavier and had elevated circulating glucose compared to females (Table A.4). However, there was no significant effect of diet on glucose or body weight at this stage. By postnatal day 180 (P180), HFD offspring had approximately a 10% increase in body weight and the sex effect was no longer demonstrable (Table A.5). While still very lean (less than 3% body fat), the HFD animals had a 50% increase in body fat (Table A.5). By P180, HFD offspring insulin secretion in response to an IVGTT was similar to control levels (Table A.5). At 210 days (P210), we measured glucose response to an insulin infusion, which demonstrated that animals on the HFD had become insulin resistant as indicated by a 30% decrease in their insulin sensitivity index (Table A.5).
3.5. Metabolic characteristics of juvenile offspring after weaning
At weaning, age matched offspring from both CTR and HFD mothers were either maintained on the same diet as their mothers or weaned to the opposing diet, thereby producing four post-natal groups: CTR/CTR, CTR/HFD, HFD/CTR, and HFD/HFD. During the post-weaning period, both CTR/HFD and HFD/HFD animals gain significantly more weight than CTR/CTR animals(data not shown). However, by 13 months, only the HFD/HFD animals were significantly heavier than CTR/CTR animals (Figure 4A). Maternal diet, did not significantly affect final weight, rather the post-weaning diet was the most significant predictive factor of final weight. Interestingly, for the HFD/HFD and HFD/CTR groups, males had a higher (approximately 10% higher) final weight than females; in contrast, for the CTR/CTR and CTR/HFD, the final weights were similar for males and females (Figure A.4A). Overall body fat (as determined by DEXA) was not significantly different between groups (Table A.6A). However, retroperitoneal white adipose tissue (RWAT) mass was elevated in both post-wean fed HFD groups (Figure 4B; Table A.6B), and subscapular caliper measures, an indicator of subcutaneous fat mass, were also significantly greater in these groups (not shown). It should be recognized that, as a whole, these animals are very lean at 13 months, with less than 3% mean body fat (Table A.6A). Consistent with the sex dependent differences in adiposity, fasting glucose was significantly elevated specifically in the males that had been exposed to the maternal HFD (HFD/HFD and HFD/CTR), but was not significantly different between the female diet groups (Figure A.4B, Table A.6B). In contrast, fasting insulin, GSIS, and HOMA-IR was similarly increased in both sexes in response to the post-weaning HFD exposure (Figure 4C and D, Table A.6B); however, this was only significant in the HFD/HFD group. There was no significant difference in basal glucagon levels between the different groups (Table A.6A).
Figure 4.
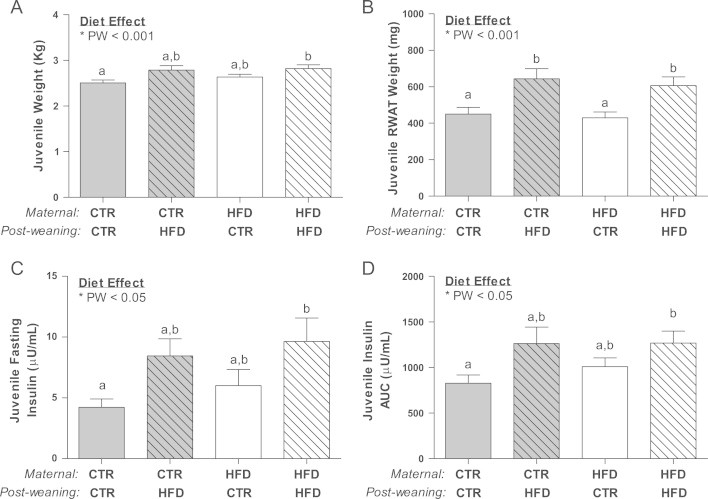
HFD/HFD juvenile animals were significantly heavier than the CTR/CTR, while CTR/HFD displayed a similar trend (A) and RWAT was significantly elevated in the CTR/HFD and HFD/HFD juveniles (B), which was due to a significant effect of the post-weaned diet (PW; p<0.01). HFD/HFD fasting (C) insulin and insulin area under the curve (D) were significantly elevated when compared to CTR/CTR and significantly affected by the post-weaned diet (PW; p<0.05). Weight, RWAT weight, fasting insulin and insulin AUC were significantly affected by the post-weaning diet. Two-way ANOVA with Bonferroni's post-hoc analysis; bars labeled with different letters “a” or “b” indicate a significant difference (p<0.05) from each other, while bars with the same letter are not significantly different from each other. CTR/CTR, n=22; CTR/HFD, n=9; HFD/CTR, n=14; HFD/HFD, n=21.
3.6. Altered islet morphology in high-fat diet juveniles
Stereological analysis of pancreatic islets was performed to determine the relative contribution of islet mass, size, density, and islet composition upon the physiological effects noted in the juveniles. Importantly, pancreas weight was specifically lower in the CTR/HFD group (Figure 5A). Islet mass was increased by 30% due to the post-weaning diet compared to CTR/CTR, while the HFD/CTR was not significantly different from CTR/CTR (Figure 5B). Interestingly, the increase in islet mass in the HFD/HFD group was due to an increase in the average islet diameter (Figure 5C), due to a maternal diet effect on both the HFD/CTR and HFD/HFD animals. However, the increase in the CTR/HFD was due to an increase in islet density (Figure 5D). Further analysis of islets by size demonstrated an increase in large islet mass in the HFD/HFD group and an increase in small islet density in the CTR/HFD group (Figs. A.5A and A.5C). In order to determine which cell type contributed to the increase in islet mass, we quantified total α and β cell area separately. Surprisingly, while the CTR/HFD group displayed a near doubling of both β cell and α cell area (Figure 6A and B), only β cell area was significantly increased in the HFD/HFD (Figure 6A). This leads to a normal β/α cell ratio in the CTR/HFD group, but an increase in the β/α cell ratio in HFD/HFD animals (Figure 6C). Since an increase in β cell or α cell area could be a result of hyperplasia or hypertrophy, we also determined β cell and α cell size and number. While β cell size did not change in any of the groups (Figure 7A), β cell number was increased in both the CTR/HFD and HFD/HFD groups (Figure 7B); however, the increase in the CTR/HFD was significantly greater than the HFD/HFD group. In contrast, α cell size was increased in both the CTR/HFD and HFD/HFD groups (Figure 7C), even though only the CTR/HFD group had an increase in total area occupied by α cells (Figure 6B). The HFD/HFD group also had a significant decrease in α cell number (Figure 7D). This would indicate that the increase in β cell area in the CTR/HFD and HFD/HFD groups is due to hyperplasia, while the increased α cell area in the CTR/HFD group is due to hypertrophy.
Figure 5.
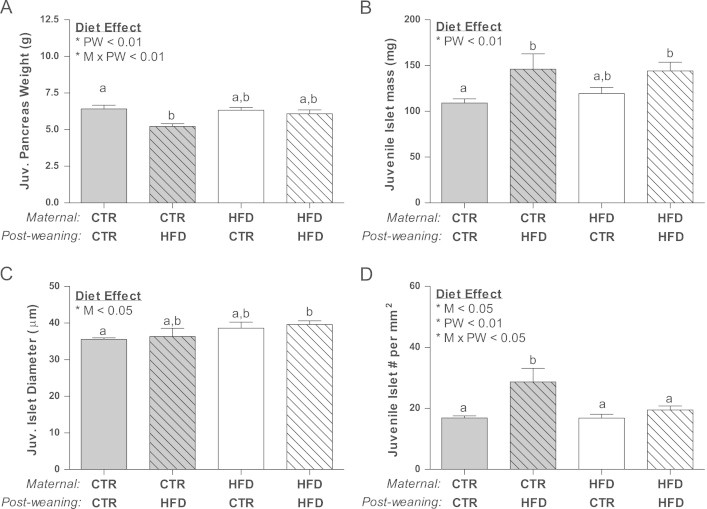
Pancreas weight was decreased in the juvenile CTR/HFD group due to post-weaning (PW<0.01) and an interacting diet effect (MxPW; p<0.05) (A). Both CTR/HFD and HFD/HFD juveniles had a 30% increase in islet mass as compared to CTR/CTR animals, which is significantly affected by the post-weaning diet (PW; p<0.01) (B). Mean islet diameter was significantly elevated only in the HFD/HFD offspring, due to a maternal diet effect (M; p<0.05) (C) while islet density was only significantly elevated in CTR/HFD and displayed a maternal (M; p<0.05), post-weaning (PW; p<0.01) and interacting diet effect (MxPW; p<0.05) (D).Two-way ANOVA with Bonferroni's post-hoc analysis; bars labeled with different letters “a” or “b” indicate a significant difference (p<0.05) from each other, while bars with the same letter are not significantly different from each other. CTR/CTR, n=7, CTR/HFD, n=6; HFD/CTR, n=7; HFD/HFD, n=7.
Figure 6.
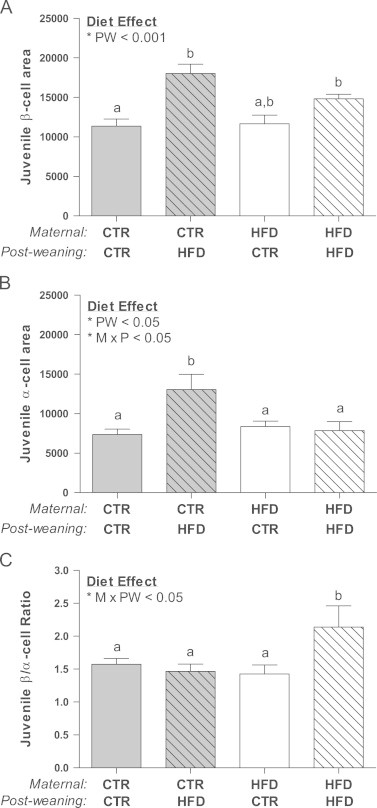
β cell area was increased in both the CTR/HFD the HFD/HFD juveniles, demonstrating a post-weaning diet effect (PW; p<0.001) (A). Only the CTR/HFD juveniles had significantly increased α cell area due to a post-weaning (PW; p<0.05) and interacting diet effect (M×PW; p<0.05) (B). HFD/HFD offspring had nearly a 50% increase in β/α cell ratio due to an interaction between the maternal and post-weaning diet (M×PW; p<0.05).Two-way ANOVA with Bonferroni's post-hoc analysis; bars labeled with different letters “a” or “b” indicate a significant difference (p<0.05) from each other, while bars with the same letter are not significantly different from each other. CTR/CTR, n=7; CTR/HFD, n=6; HFD/CTR, n=7; HFD/HFD, n=7.
Figure 7.
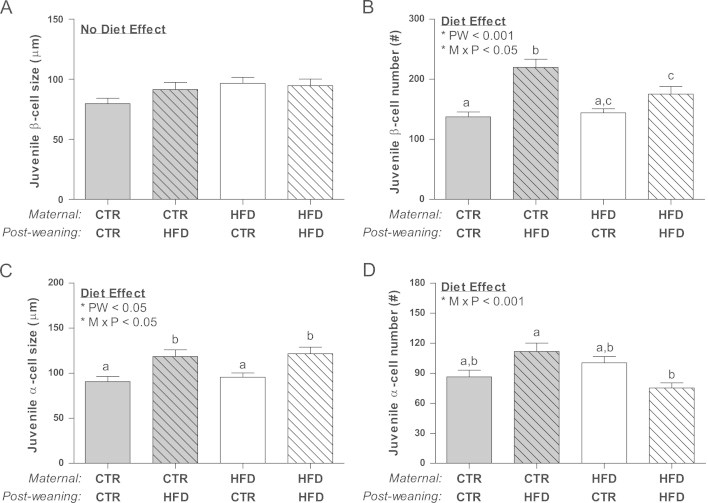
There was no change in size of β cells from all juvenile groups (A) while β cells from both juvenile post-wean fed HFD groups displayed significant hyperplasia and had a significant post-weaning (PW; p<0.01) and interacting diet effect (M×PW; p<0.05) (B). α cell area hypertrophy was significant in both CTR/HFD and HFD/HFD offspring due to a post-weaning (PW; p<0.05) and interacting diet (M×PW;p<0.05) effect (C). Finally, while CTR/HFD α-cells tended to be hyperplasic, HFD/HFD displayed a decreased number compared to the CTR/HFD animals. This response is due to an interacting diet effect (M×PW; p<0.05) (D).Two-way ANOVA with Bonferroni's post-hoc analysis; bars labeled with different letters “a” or “b” or “c” indicate a significant difference (p<0.05) from each other, while bars with the same letter are not significantly different from each other. CTR/CTR, n=30; CTR/HFD, n=35; HFD/CTR, n=30; HFD/HFD, n=25.
3.7. Differential islet gene expression of high-fat diet juveniles
In order to determine which factors may be responsible for the increase in insulin secretion observed in offspring exposed to the HFD, we analyzed the mRNA expression in the whole pancreas of various genes involved in insulin production and secretion. Absolute insulin mRNA expression was significantly elevated only in HFD/HFD pancreas, when compared to the CTR/CTR group (Sup. Figure 6A). When insulin mRNA levels were normalized to the β cell area (insulin producing cells), the effect in the HFD/HFD group was lost, but there was a significant overall maternal diet effect (Figure 8A). This indicates that some of the increase in the HFD groups is dependent on the increase in β cell area, but that maternal HFD diet causes an increase in cellular expression. Consistent with the lack of difference in circulating glucagon levels, glucagon mRNA was not significantly different between the groups (Sup. Figure 6B) even after normalizing to the α cell area (Figure 8C). The two key genes involved in glucose stimulated insulin secretion are the glucose transporter Glut2 that transports glucose into the cell and glucokinase (GK), the rate-limiting enzyme in glycolysis [38–40]. In preliminarily studies using immunohistochemistry, we demonstrated that the primary glucose transporter in β cells is Glut2 (Sup. Figure 7). Interestingly, Glut2 expression (Sup. Figure 6C) was significantly increased specifically in the CTR/HFD, while GK expression was specifically elevated in HFD/HFD group (Sup. Figure 6D). However, when normalized to β cell area, the differences in Glut2 mRNA were completely eliminated (Figure 8D), suggesting that the change in Glut2 is dependent on changes in β cell area. In contrast, when GK expression was normalized to islet area, the effect in the HFD/HFD group was lost, but a decrease in expression in the CTR/HFD group was unmasked (Figure 8B). This suggests that cellular expression of GK in the islet is normal in HFD/HFD animals, but lower in the CTR/HFD group.
Juveniles on the HFD are exposed to 4 times more fatty acids than the CTR animals [9], which can augment insulin secretion through activation of the G-protein coupled receptor GPR40/FFAR1 (free fatty acid receptor 1) [44,45]. Offspring on the post-weaning HFD had significantly increased FFAR1 gene expression (Sup. Figure 6E). Since FFAR1 has been confirmed in α cells as well as β cells, FFAR1 expression was normalized to islet area. There was no significant effect on normalized FFAR1 expression (Figure 8E), indicating that the up-regulation was likely secondary to the increase in islet mass, not individual cell gene expression.
4. Discussion
In our current study we demonstrated a significant decrease in α cell mass in the HFD fetus, resulting in near doubling of the β to α cell ratio in these animals. The alternation in the islet cell ratio may lead to a significant dysregulation of fetal islet function because of the diminished paracrine regulation of β cells by α cell secretory products. The correlation between gluconeogenic genes in the liver and the increase in this ratio indicates that α cell mass may be adapting to the increased glucose production by the liver of fetal HFD offspring. In addition, the concomitant decrease in c-peptide, with no change in circulating insulin levels, indicates that HFD animals are secreting less insulin, but may have a parallel reduction in insulin clearance. This adjustment may allow the fetuses to maintain glucose homeostasis. However, while the changes in islet cells may protect the fetus during development, this may be a mal-adaptive response that will ultimately lead to significant physiological consequences that are not measureable at the fetal stage.
In order to understand what factors may be leading to the differences in α cell mass, we analyzed the transcription factors involved in islet cell differentiation. The differences in transcription factor expression may be indicative of the timing of α cell differentiation in the offspring of CTR versus HFD mothers. Based on the reduction in FoxA2 and Irx2 expression, along with the lack of effect on other transcription factors analyzed, we suggest that the HFD offspring may be lagging in terminal differentiation of α cells, which results in a decline in α cell mass and total glucagon expression at G130. While the decreased α cell mass in the HFD fetal pancreas may be due to reduced neogenesis, proliferation or increased apoptosis, we have not been able to detect changes at this stage and further studies are needed to determine the mechanism of change.
In contrast, while there have been several studies demonstrating changes in β cell development and function in response to maternal overnutrition, undernutrition, and obesity in rodent and sheep models, they have either not investigated abnormalities in the α cell, or have reported no changes in this cell type [15,46,47]. However, the effect on α cell mass in the NHP offspring may represent a novel, unexplored avenue that clarifies the islet defects that may occur prior to childhood obesity onset. Furthermore, because of the essential nature of islet paracrine regulation for normal endocrine function, the change in β/α cell ratio may lead to islet cell dysfunction [48,49]. Our results indicate that while glucose metabolism is normal, insulin secretory response is suppressed. While Glut2 expression was decreased, GK, the rate-limiting enzyme in glycolysis, was unchanged. In addition, expression of genes involved in the terminal portion of the GSIS pathway (KCNJ11 and SUR1) was also unchanged, indicating that glucose metabolism may be unaffected. Yet, since insulin gene transcription is decreased, this will decrease the reserved supply of insulin in the β cell, thereby limiting the availability of the readily releasable insulin pool, which was demonstrated by the decreased c-peptide levels. In the studies from disassociated islets, β cells have a poor insulin secretory response, despite normal glucose metabolism [50,51], indicating that there is a basal level of glucagon or other α cell secretagogues that are required for efficient insulin secretion. The decreased α cell mass demonstrated in these animals elucidates a possible mechanism by which decreased α cell paracrine regulation of β cells results in decreased insulin secretion. However, studies from isolated fetal islets are essential to determine how the GSIS mechanism may be biochemically altered.
In order to determine if changes in islet composition and gene expression might lead to lasting physiological effects, we examined the physiology of these animals after birth until 13 months of age. Infant offspring consume solely maternal milk during the first three months of life. Since previous studies from our lab have shown no significant difference in the percentage of fatty acids in milk from dams on the CTR versus HFD [9], it is likely that they will not be exposed to the specific metabolic pressure of the HFD until after P90. In fact, at P90, we demonstrated decreased insulin secretion in the HFD offspring, which is reflective of the high insulin content of the mother's milk [9], and may be the perpetuation of the insulin secretory deficit demonstrated in the HFD fetus. By 7 months of age, animals are principally consuming the available diet and the HFD infants were heavier, had increased body fat, and were insulin resistant. However, at this time, there was no significant difference in fasting insulin or insulin response to an IVGTT. This indicates that while HFD exposure during the early post-natal period may lead to whole body insulin resistance, there were no measureable effects that could be specifically attributed to islets.
It was essential to follow these animals after weaning to determine if these adaptations are beneficial or harmful to the post-weaned juvenile animal. By examining the juvenile animals, it is clear that exposure to a HFD during the postnatal period can have a similar impact on overall adiposity and glucose homeostasis, irrespective of whether the animal was born to a mother on a CTR or HFD. However, these studies point to some fundamental differences in the underlying mechanisms leading to the impaired endocrine pancreas function between CTR and HFD offspring. First, it should be noted that HFD/HFD and CTR/HFD offspring displayed a higher average body weight, which was only significant in the HFD/HFD group, although the increase is only approximately 10%. Furthermore, while these animals have increased adiposity, overall the animals are very lean; with less than 3% mean body fat. In spite of this modest phenotype, the HFD/HFD offspring have significantly increased fasting insulin and GSIS. There is also a modest sex difference in the metabolic effects. Body weight in females is not significantly elevated, but CTR/HFD and HFD/HFD are insulin resistant at this stage. However, males are heavier and the effect of the HFD is more evident in this sex. Glucose levels are significantly elevated in both the HFD/CTR and the HFD/HFD males, indicating that the metabolic effect of maternal HFD is more robust in these animals. The sex effect that we have demonstrated is consistent with the reports in rodent models in which males tend to be more susceptible to fetal HFD-induced metabolic effects [52–55]. Importantly, these effects were independent of maternal obesity or insulin resistance.
Islet mass is similarly increased in both the CTR/HFD and HFD/HFD offspring. However, the underlying factors that contribute to this increase are different. Animals exposed to the maternal HFD have larger islets, while the post-weaning HFD alone leads to increased number of islets. Both effects result in increased islet mass, yet the morphological discrepancies are likely to have differential long-term outcomes in these animals. By increasing small islet density, CTR/HFD animals will likely be able to combat HFD effects for longer. Since islet maturation in the macaque continues through puberty, the smaller islets may be able to continue to expand in response to metabolic pressure and allow for increased insulin secretion in response to chronic, long-term HFD exposure. To the contrary, larger islets indicate that the HFD/HFD islets have already started expanding in size in response to chronic HFD. The HFD/HFD juveniles will likely progress through the stages of insulin resistance leading to diabetes more rapidly by reaching the threshold of islet expansion at a faster pace than the CTR/HFD juveniles. Future studies will investigate whether there are changes in islet cell neogenesis, proliferation or apoptosis in the juvenile animals.
We demonstrated that the CTR/HFD animals have increased α cell and β cell mass, while the HFD/HFD offspring only have increased β cell mass. In contrast, the HFD/CTR α cell and β cell mass are normalized to control levels, indicating that islet cells are highly plastic during the peri-weaning period. Consistent with the decreased α cell mass in the fetal HFD offspring, the lack of α cell expansion in the HFD/HFD juveniles indicates that there is likely a programmed suppression of α cell expansion that is a result of chronic HFD throughout the peri-natal period. This results is nearly a 40% increase in the β/α cell ratio in HFD/HFD offspring. While the CTR/HFD juveniles do have larger islets, the ratio of β cells to α cells is maintained at normal levels, thus expectedly maintaining paracrine function of these islets. Healthy, paracrine regulation of islet cells is essential for long-term glucose homeostasis [56,57], indicating that the relative contribution of α cells and β cells to islet composition will ultimately predict islet efficiency. It is not known whether the increase in β/α cell ratio in HFD/HFD juveniles is adaptive or maladaptive at this stage. Therefore, future studies in these offspring should be performed in order to determine the long-term effect that the differential ratio will have on glucose homeostasis.
We also established that the increase in β cell mass in both CTR/HFD and HFD/HFD offspring is due to β cell hyperplasia. Since there were no measurable differences in fetal β cell mass from HFD mothers, this increase is likely due to the post-natal diet. This effect is similar to a rodent model in which post-natal HFD exposure leads to increased β cell mass [58]. However, the effects of maternal versus post-weaning HFD exposure on α cell development are novel when compared to other model species. In rodents and sheep models, α cell area tends to be elevated or unaffected by maternal obesity or high calorie diet consumption [46,47]. The HFD/HFD juveniles have increased α cell size, but display decreased α cell number, suggesting that they are unable to yield more α cells in response to chronic HFD pressure. In contrast, the CTR/HFD offspring retain α cell plasticity, demonstrably by increasing cell density when hypertrophy is not sufficient to balance augmented β cell mass. These differences indicate that maternal HFD exposure limits the ability of α cells to replicate in response to chronic post-natal HFD pressure.
Ultimately, both CTR/HFD and HFD/HFD juveniles are hyperinsulinemic, but analysis of gene expression in the pancreas provides evidence that the mechanism underlying the elevation in insulin is maternally programmed. However, total gene expression of the pancreas may be biased by the islet cell mass or density when isolated from whole pancreas tissue. Also, gene expression from isolated islets is biased toward large islets, because isolation protocols generally score islets as high-quality if they have a minimum specified diameter above at least 100 μM [59]. Therefore, in order to obtain gene expression that is representative of islet content, we normalized gene expression to the area of tissue in which the gene is expressed (i.e., α cell area, β cell area, or islet area). After normalization, the key differences observed were an up-regulation of insulin expression, which is programmed by the maternal HFD, and a down-regulation in GK in the CTR/HFD juveniles. Previous studies have shown that GK is the rate-limiting step in the initial phases of GSIS [60,61]. Decreased GK expression in the CTR/HFD animals could cause these islets to be less responsive to glucose, while the β cells of HFD/HFD juveniles are primed to respond to glucose. It is unclear that what causes the increased secretion demonstrated in CTR/HFD offspring. FFAR1 expression analysis indicates that expression of this receptor rises with increasing islet area. Fatty acid potentiation of GSIS is an additional factor that may contribute to the increased insulin secretion measured in both post-weaned HFD juvenile groups. However, future in-vivo and gene expression studies are essential to determine the role of both glucose and FFAs that play in modulation of GSIS in all diet groups.
In conclusion, upon review of the effects of HFD on these animals, it is evident that HFD consumption during pregnancy leads to decreased α cell plasticity in the juvenile Japanese macaque, when chronically exposed to the HFD after weaning. When animals are returned to a control diet, most factors are ameliorated, indicating that this is a dynamic period of islet development. However, maternally programmed metabolic effects are still evident to some extent in these animals, since not all factors are normalized to control levels. Post-natal HFD exposure appears to exacerbate the effects of early HFD programming. Uniquely, β cells are hyper-responsive in the HFD/HFD juveniles (fasting insulin levels are doubled, but β cell mass is increased only by 30%).This hyper-responsiveness is indicative of early insulin resistance and the ultimate progression toward diabetes [62–64].
While there is a rise in β cell to α cell ratio in HFD/HFD juveniles, apparently, these animals are still capable of managing the current metabolic insult. In HFD/HFD animals, α cells hyper-secrete glucagon. It is likely that by suppressing α cell mass, glucagon levels are maintained at control levels. It is unclear if this imbalance in the islet cell ratio prepares the animal to deal with future metabolic pressure, or if they will reach a peak at which islets are no longer able to withstand the metabolic insult. We anticipate that, by following the HFD/HFD through the adolescent period, these animals will ultimately reach the plateau of adaptation and start to decline into early metabolic syndrome. Eventually the CTR/HFD and HFD/HFD are likely to develop similar metabolic complications because adaptation to this HFD is unsustainable. However, these studies suggest that the mechanisms in which the disease progresses are different between the post-weaned HFD groups and, ultimately, the timing of disease progression will be accelerated in the HFD/HFD animals
Conflict of interest
None declared.
Acknowledgments
This work was funded by the NIH Grants DK-060685 (to K.L. Grove), DK-079194 (to K.L. Grove), and P51 OD011092 (to K.L. Grove and ONPRC). The authors would like to acknowledge Anne Evans, Lindsay Bader, Sandra Joachim, Christopher Osman, Rene Lindsley, Elizabeth Hoyt, Jeanette Valleau and the Division of Comparative Medicine and Veterinary staff at ONPRC for their technical assistance with these studies. The authors also acknowledge Francis Pau and the RIA Core, and Anda Cornea and the Imaging Core at ONPRC.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at 10.1016/j.molmet.2012.11.001.
Appendix A. Supplementary materials
Supplementary material
References
- 1.Ogden C.L., Carroll M.D., Curtin L.R., Lamb M.M., Flegal K.M. Prevalence of high body mass index in US children and adolescents, 2007–2008. Journal of the American Medical Association. 2010;303(3):242–249. doi: 10.1001/jama.2009.2012. [DOI] [PubMed] [Google Scholar]
- 2.Guo S.S., Wu W., Chumlea W.C., Roche A.F. Predicting overweight and obesity in adulthood from body mass index values in childhood and adolescence. American Journal of Clinical Nutrition. 2002;76(3):653–658. doi: 10.1093/ajcn/76.3.653. [DOI] [PubMed] [Google Scholar]
- 3.Magarey A.M., Daniels L.A., Boulton T.J., Cockington R.A. Predicting obesity in early adulthood from childhood and parental obesity. International Journal of Obesity and Related Metabolic Disorders. 2003;27(4):505–513. doi: 10.1038/sj.ijo.0802251. [DOI] [PubMed] [Google Scholar]
- 4.Whitaker R.C. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004;114(1):e29–e36. doi: 10.1542/peds.114.1.e29. [DOI] [PubMed] [Google Scholar]
- 5.Koupil I., Toivanen P. Social and early-life determinants of overweight and obesity in 18-year-old Swedish men. International Journal of Obesity. 2008;32(1):73–81. doi: 10.1038/sj.ijo.0803681. [DOI] [PubMed] [Google Scholar]
- 6.Forsén T., Eriksson J.G., Tuomilehto J., Teramo K., Osmond C., Barker D.J. Mother's weight in pregnancy and coronary heart disease in a cohort of Finnish men: follow up study. British Medical Journal. 1997;315(7112):837–840. doi: 10.1136/bmj.315.7112.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boney C.M., Verma A., Tucker R., Vohr B.R. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115(3):e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 8.McCurdy C.E., Bishop J.M., Williams S.M., Grayson B.E., Smith M.S., Friedman J.E. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. Journal of Clinical Investigation. 2009;119(2):323–335. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grant W.F. Maternal high-fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS One. 2011;6(2):e17261. doi: 10.1371/journal.pone.0017261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frias A.E., Morgan T.K., Evans A.E., Rasanen J., Oh K.Y., Thornburg K.L. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology. 2011;152(6):2456–2464. doi: 10.1210/en.2010-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin-Gronert M.S., Ozanne S.E. Experimental IUGR and later diabetes. Journal of Internal Medicine. 2007;261(5):437–452. doi: 10.1111/j.1365-2796.2007.01800.x. [DOI] [PubMed] [Google Scholar]
- 12.Fahy A. Microbiology. University of Arizona; Tucson: 2009. Vascularization of the pancreas and islets of langerhans in intrauterine growth restricted fetuses and lambs. p. 17. [Google Scholar]
- 13.Van Assche F.A., De Prins F., Aerts L., Verjans M. The endocrine pancreas in small-for-dates infants. British Journal of Obstetrics and Gynaecology. 1977;84(10):751–753. doi: 10.1111/j.1471-0528.1977.tb12486.x. [DOI] [PubMed] [Google Scholar]
- 14.Hales C.N., Barker D.J., Clark P.M., Cox L.J., Fall C., Osmond C. Fetal and infant growth and impaired glucose tolerance at age 64. British Medical Journal. 1991;303(6809):1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Long N.M., Hein S.M., Ma Y., Nathanielsz P.W., Ford S.P. Maternal obesity in ewes results in reduced fetal pancreatic beta-cell numbers in late gestation and decreased circulating insulin concentration at term. Domestic Animal Endocrinology. 2011;40(1):30–39. doi: 10.1016/j.domaniend.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertin E., Gangnerau M.N., Bellon G., Bailbé D., Arbelot De Vacqueur A., Portha B. Development of beta-cell mass in fetuses of rats deprived of protein and/or energy in last trimester of pregnancy. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2002;283(3):R623–R630. doi: 10.1152/ajpregu.00037.2002. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan E.L., Grayson B., Takahashi D., Robertson N., Maier A., Bethea C.L. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. Journal of Neuroscience. 2010;30(10):3826–3830. doi: 10.1523/JNEUROSCI.5560-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grayson B.E., Levasseur P.R., Williams S.M., Smith M.S., Marks D.L., Grove K.L. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology. 2010;151(4):1622–1632. doi: 10.1210/en.2009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grant W.F., Gillingham M.B., Batra A.K., Fewkes N.M., Comstock S.M., Takahashi D. Maternal high-fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS One. 2011;6(2):e17261. doi: 10.1371/journal.pone.0017261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frias A.E., Morgan T.K., Evans A.E., Rasanen J., Oh K.Y., Thornburg K.L. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology. 2011;152(6):2456–2464. doi: 10.1210/en.2010-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews D.R., Hosker J.P., Rudenski A.S., Naylor B.A., Treacher D.F., Turner R.C. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 22.Bonora E., Targer G., Alberiche M., Bonadonna R.C., Saggiani F., Zenere M.B. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care. 2000;23(1):57–63. doi: 10.2337/diacare.23.1.57. [DOI] [PubMed] [Google Scholar]
- 23.Grulet H., Durlach V., Hecart A.C., Gross A., Leutenegger M. Study of the rate of early glucose disappearance following insulin injection: insulin sensitivity index. Diabetes Research and Clinical Practice. 1993;20(3):201–207. doi: 10.1016/0168-8227(93)90079-k. [DOI] [PubMed] [Google Scholar]
- 24.Gatford K.L., Mohammad S.N., Harland M.L., De Blasio M.J., Fowden A.L., Robinson J.S. Impaired beta-cell function and inadequate compensatory increases in beta-cell mass after intrauterine growth restriction in sheep. Endocrinology. 2008;149(10):5118–5127. doi: 10.1210/en.2008-0233. [DOI] [PubMed] [Google Scholar]
- 25.Prado C.L., Pugh-Bernard A.E., Elghazi L., Sosa-Pineda B., Sussel L. Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):2924–2929. doi: 10.1073/pnas.0308604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elghazi L., Cras-Méneur C., Czernichow P., Scharfmann R. Role for FGFR2IIIb-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(6):3884–3889. doi: 10.1073/pnas.062321799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchida T., Nakamura T., Hashimoto N., Matsuda T., Kotani K., Sakaue H. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nature Medicine. 2005;11(2):175–182. doi: 10.1038/nm1187. [DOI] [PubMed] [Google Scholar]
- 28.Montanya E., Tellez N. Pancreatic remodeling: beta-cell apoptosis, proliferation and neogenesis, and the measurement of beta-cell mass and of individual beta-cell size. Methods in Molecular Biology. 2009;560:137–158. doi: 10.1007/978-1-59745-448-3_11. [DOI] [PubMed] [Google Scholar]
- 29.Béringue F., Blondeau B., Castellotti M.C., Bréant B., Czernichow P., Polak M. Endocrine pancreas development in growth-retarded human fetuses. Diabetes. 2002;51(2):385–391. doi: 10.2337/diabetes.51.2.385. [DOI] [PubMed] [Google Scholar]
- 30.Weibel E.R., Kistler G.S., Scherle W.F. Practical stereological methods for morphometric cytology. Journal of Cell Biology. 1966;30(1):23–38. doi: 10.1083/jcb.30.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee C.S., Sund N.J., Behr R., Herrera P.L., Kaestner K.H. Foxa2 is required for the differentiation of pancreatic alpha-cells. Developmental Biology. 2005;278(2):484–495. doi: 10.1016/j.ydbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 32.Petri A., Ahnfelt-Rønne J., Frederiksen K.S., Edwards D.G., Madsen D., Serup P. The effect of neurogenin3 deficiency on pancreatic gene expression in embryonic mice. Journal of Molecular Endocrinology. 2006;37(2):301–316. doi: 10.1677/jme.1.02096. [DOI] [PubMed] [Google Scholar]
- 33.Collombat P., Mansouri A., Hecksher-Sorensen J., Serup P., Krull J., Gradwohl G. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes & Development. 2003;17(20):2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gosmain Y., Marthinet E., Cheyssac C., Guérardel A., Mamin A., Katz L.S. Pax6 controls the expression of critical genes involved in pancreatic {alpha} cell differentiation and function. Journal of Biological Chemistry. 2010;285(43):33381–33393. doi: 10.1074/jbc.M110.147215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.St-Onge L., Sosa-Pineda B., Chowdhury K., Mansouri A., Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387(6631):406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- 36.Jonsson J., Carlsson L., Edlund T., Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 37.Rivas-Carrillo J.D., Okitsu T., Tanaka N., Kobayashi N. Pancreas development and beta-cell differentiation of embryonic stem cells. Current medicinal chemistry. 2007;14(14):1573–1578. doi: 10.2174/092986707780831096. [DOI] [PubMed] [Google Scholar]
- 38.Matschinsky F.M. Banting lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45(2):223–241. doi: 10.2337/diab.45.2.223. [DOI] [PubMed] [Google Scholar]
- 39.Iynedjian P.B. Mammalian glucokinase and its gene. Biochemical Journal. 1993;293(Pt 1):1–13. doi: 10.1042/bj2930001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newgard C.B., McGarry J.D. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annual Review of Biochemistry. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 41.Kramer J., Moeller E.L., Hachey A., Mansfield K.G., Wachtman L.M. Differential expression of GLUT2 in pancreatic islets and kidneys of new and old World nonhuman primates. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2009;296(3):R786–R793. doi: 10.1152/ajpregu.90694.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heimberg H., De Vos A., Moens K., Quartier E., Bouwens L., Pipeleers D. The glucose sensor protein glucokinase is expressed in glucagon-producing alpha-cells. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(14):7036–7041. doi: 10.1073/pnas.93.14.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moritz W., Leech C.A., Ferrer J., Habener J.F. Regulated expression of adenosine triphosphate-sensitive potassium channel subunits in pancreatic beta-cells. Endocrinology. 2001;142(1):129–138. doi: 10.1210/endo.142.1.7885. [DOI] [PubMed] [Google Scholar]
- 44.Nolan C.J., Madiraju M.S., Delghingaro-Augusto V., Peyot M.L., Prentki M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes. 2006;55(Suppl. 2):S16–S23. doi: 10.2337/db06-s003. [DOI] [PubMed] [Google Scholar]
- 45.Briscoe C.P., Tadayyon M., Andrews J.L., Benson W.G., Chambers J.K., Eilert M.M. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. Journal of Biological Chemistry. 2003;278(13):11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 46.Ford S.P., Zhang L., Zhu M., Miller M.M., Smith D.T., Hess B.W. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2009;297(3):R835–R843. doi: 10.1152/ajpregu.00072.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerf M.E., Williams K., Nkomo X.I., Muller C.J., Du Toit D.F., Louw J. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2005;288(5):R1122–R1128. doi: 10.1152/ajpregu.00335.2004. [DOI] [PubMed] [Google Scholar]
- 48.Franklin I., Gromada J., Gjinovci A., Theander S., Wolheim C.B. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes. 2005;54(6):1808–1815. doi: 10.2337/diabetes.54.6.1808. [DOI] [PubMed] [Google Scholar]
- 49.Leung Y.M., Ahmed I., Sheu L., Gao X., Hara M., Tsushima R.G. Insulin regulates islet alpha-cell function by reducing KATP channel sensitivity to adenosine 5'-triphosphate inhibition. Endocrinology. 2006;147(5):2155–2162. doi: 10.1210/en.2005-1249. [DOI] [PubMed] [Google Scholar]
- 50.Huypens P., Ling Z., Pipeleers D., Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia. 2000;43(8):1012–1019. doi: 10.1007/s001250051484. [DOI] [PubMed] [Google Scholar]
- 51.Holz G.G.T., Kuhtreiber W.M., Habener J.F. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37) Nature. 1993;361(6410):362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gómez-Pérez Y., Gianotti M., Lladó I., Proenza A.M. Sex-dependent effects of high-fat-diet feeding on rat pancreas oxidative stress. Pancreas. 2011;40(5):682–688. doi: 10.1097/MPA.0b013e31821f2645. [DOI] [PubMed] [Google Scholar]
- 53.Medrikova D., Jilkova Z.M., Bardova K., Janovska P., Rossmeisl M., Kopecky J. Sex differences during the course of diet-induced obesity in mice: adipose tissue expandability and glycemic control. International Journal of Obesity. 2012;36(2):262–272. doi: 10.1038/ijo.2011.87. [DOI] [PubMed] [Google Scholar]
- 54.Estrany M.E., Proenza A.M., Lladó I., Gianotti M. Isocaloric intake of a high-fat diet modifies adiposity and lipid handling in a sex dependent manner in rats. Lipids in Health and Disease. 2011;10:52. doi: 10.1186/1476-511X-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunn G.A., Bale T.L. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology. 2009;150(11):4999–5009. doi: 10.1210/en.2009-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cabrera O., Berman D.M., Kenyon N.S., Ricordi C., Berggren P.O., Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2334–2339. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker J.N., Ramracheya R., Zhang Q., Johnson P.R., Braun M., Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes, Obesity & Metabolism. 2011;13(Suppl 1):95–105. doi: 10.1111/j.1463-1326.2011.01450.x. [DOI] [PubMed] [Google Scholar]
- 58.Matveyenko A.V. Differential effects of prenatal and postnatal nutritional environment on beta-cell mass development and turnover in male and female rats. Endocrinology. 2010;151(12):5647–5656. doi: 10.1210/en.2010-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsumoto S., Qualley S.A., Goel S., Hagman D.K., Sweet I.R., Poitout V. Effect of the two-layer (University of Wisconsin solution-perfluorochemical plus O2) method of pancreas preservation on human islet isolation, as assessed by the Edmonton isolation protocol. Transplantation. 2002;74(10):1414–1419. doi: 10.1097/00007890-200211270-00013. [DOI] [PubMed] [Google Scholar]
- 60.De Vos A., Heimberg H., Quartier E., Huypens P., Bouwens L., Pipeleers D. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. Journal of Clinical Investigation. 1995;96(5):2489–2495. doi: 10.1172/JCI118308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang Y., Najafi H., Matschinsky F.M. Glucose regulates glucokinase activity in cultured islets from rat pancreas. Journal of Biological Chemistry. 1990;265(28):16863–16866. [PubMed] [Google Scholar]
- 62.Ahlgren U., Jonsson J., Jonsson L., Simu K., Edlund H. Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes & Development. 1998;12(12):1763–1768. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weir G.C., Marselli L., Marchetti P., Katsuta H., Jung M.H., Bonner-Weir S. Towards better understanding of the contributions of overwork and glucotoxicity to the beta-cell inadequacy of type 2 diabetes. Diabetes, Obesity & Metabolism. 2009;11(Suppl. 4):82–90. doi: 10.1111/j.1463-1326.2009.01113.x. [DOI] [PubMed] [Google Scholar]
- 64.Weir G.C., Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(Suppl. 3):S16–S21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material