

Abstract
Hypothalamic Prolyl carboxypeptidase (PRCP) plays a role in the regulation of energy metabolism by inactivating hypothalamic α-melanocyte stimulating hormone (α-MSH) levels and thus affecting melanocortin signaling. Alpha-MSH production is highly regulated both at transcriptional and posttranslational levels. Here we show that fasting induces a hypothalamic-specific up-regulation of Prcp mRNA and protein levels. Since fasting is characterized by elevated circulating ghrelin levels, we tested the effect of peripheral and central administration of ghrelin, and found that ghrelin increases hypothalamic Prcp mRNA expression. No changes in Prcp mRNA levels were detected in ghrelin knockout mice compared to their controls. Finally, ghrelin effect on PRCP expression was ghrelin receptor-mediated. Altogether our data show that ghrelin is a key regulator of hypothalamic PRCP expression, and up-regulation of PRCP by ghrelin may be an additional mechanism to decrease melanocortin signaling.
Abbreviations: (POMC), Proopiomelanocortin; (α-MSH), α-Melanocyte stimulating hormone; (PRCP), Prolyl carboxypeptidase; (Prcpgt/gt), Prcp-ablated mice; (DMH), Dorsomedial nucleus; (LH), Lateral hypothalamus; (ARC), Arcuate nucleus; (VMH), Ventromedial nucleus; (CTX), Cortex; (NPY), Neuropeptide Y; (AgRP), Agouti related peptide; (Hcrt), Hypocretin; (MCH), Melanin concentrating hormone; (GHS-R), Growth hormone secretagogue receptor
Keywords: Fasting, Ghrelin, Prolyl carboxypeptidase, Alpha-melanocyte stimulating hormone, Hypothalamus
1. Introduction
Prolyl carboxypeptidase (PRCP), a small glycosylated protein and a member of serine proteases, catalyzes the cleavage of one amino acid at the c-terminal of substrates containing a proline at the penultime amino acid position [1–3]. PRCP is expressed in the hypothalamus in key nuclei involved in the regulation of energy metabolism [4–6]. We have shown that alpha-melanocyte stimulating hormone (α-MSH) is a substrate of PRCP [4]. By the removal of the last amino acid, α-MSH is unable to activate melanocortin 4 receptor (MC4R)-expressing neurons in the paraventricular nucleus of the hypothalamus (PVN). In support of its role in α-MSH inactivation, Prcp-ablated mice (Prcpgt/gt) show increased α-MSH levels in the hypothalamus and lower body weight, body length, food intake with a concomitant increase in energy expenditure compared to wild type controls [4–6]. This increase in energy expenditure was, at least in part, due to an increase of circulating thyroid hormones due to an up-regulation of TRH gene expression in the PVN [5]. In addition, Prcpgt/gt mice are protected from diet-induced obesity having a significant decrease in body weight gain and fat mass and improved metabolic parameters, such as glucose tolerance, insulin sensitivity and liver metabolism compared to diet-induced obese (DIO) wild type mice [6].
PRCP is widely distributed in the brain. For example, PRCP is expressed in the cerebral cortex, hippocampus, brain stem and the hypothalamus. Specifically, in the mediobasal hypothalamus PRCP is mostly expressed in the dorsomedial nucleus (DMH), in the lateral hypothalamus (LH) and moderately in the arcuate nucleus (ARC) [4]. In the ARC, PRCP is not localized in α-MSH-expressing neurons, while in the LH, PRCP is expressed in subpopulations of melanin concentrating (MCH)- and hypocretin/orexin (Hcrt)-containing neurons [4]. Neurons in all these nuclei are known to project to the PVN where they could release PRCP, which would then inactivate α-MSH at the synaptic levels influencing body energy metabolism [4]. In addition, in the DMH PRCP may cleave α-MSH intracellularly after ligand-mediated receptor endocytosis since MC4R-containing neurons are also expressed in this nucleus [7]. All of these anatomical and functional data demonstrate that PRCP action is important for controlling α-MSH concentration in the hypothalamus, and thus, to regulate whole body energy metabolism, representing a potential therapeutic target for obesity treatment [8].
To date, the regulation of PRCP in the hypothalamus is undetermined. Thus, our study was undertaken to assess whether PRCP expression is regulated in different metabolic states.
2. Materials and methods
2.1. Animals
All animal studies were approved by Yale University Institutional Animal Care and Use Committee. Male mice on a C57Bl6 background (3–5 months old) were used in all of these studies [4–6,9]. Animals were housed in a temperature-controlled environment (25 °C) with a 12 h light/12 h dark cycle, and had a free access to standard chow diet (Harlan Teklad#2018). Fasted animals were food deprived overnight (about 16 h). For intraperitoneal (i.p) ghrelin injection, animals received a single dose of either ghrelin (10 nmol/mouse; PolyPeptide Laboratories, INC, Torrance, CA; cat.# SC1356) or the equivalent volume of vehicle (saline) just before the beginning of the dark phase. Food was removed and animals sacrificed after three hours from the injection. For central administration (icv) of ghrelin, mice were individually housed for a week. Then, a cannula was implanted into the lateral ventricle as described elsewhere [9], and animals were allowed to recover for another week. Mice were then icv injected with either vehicle (saline) or ghrelin (2 μg) at the beginning of the dark phase. Animals were sacrificed after one hour from the administration. Brains were collected for either in situ hybridization or Western Blot analysis.
2.2. Western blot analysis
The hypothalamic regions including the ARC, DMH, VMH and LH, as well as cortex were dissected from 4 months old C57BL/6 mice and lysed by modified RIPA buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton-X-100, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride) supplemented with protease inhibitor cocktail (Roche, Cat# 11 836 170 001) on ice for 30 minutes followed by centrifugation at 14,000 rpm for 15 min. Protein concentrations were determined using the BCA kit (Thermo scientific, Cat# 23228 and 1859078). 20 μg of proteins were resolved on 8% SDS-PAGE and transferred to PVDF membrane (Millipore, Cat# IPVH 15150). Membranes were blocked with 5% dry milk in TBS (50 mM Tris–HCl, pH 7.5, 150 mM NaCl) for 1 h and incubated with anti-PRCP antibody (Santa Cruz Biotechnology, Cat# sc-49272) overnight at 4 °C. After three washes with TBST (TBS including 0.05% Tween 20), membranes were incubated with anti-rabbit IgG conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Cat# sc-2004) for 1 h and washed three times with TBST. Immunoreactive bands were visualized using the ECL kit (Thermo scientific, Cat# 32016). Membranes were stripped using stripping buffer (Thermo scientific, Cat# 21059) and reused to detect β-actin (Sigma, Cat# A5441).
2.3. Radioactive in situ hybridization
Five hundred bp of S35-labeled sense and antisense riboprobes specific to mice PRCP were generated, and purified using G-50 columns (supplement Figure 1). Cryostat sections (20 μm; Leica's CM 1850, Wetzlar, Germany) were collected at a 200 μm interval and stored at −80 °C. In situ hybridization was performed as previously reported [4,5] with an overnight hybridization at 52 °C. Radioactive signal on sections was visualized by a phosphorimager (STORM 860 II phosphorimager, GE Health Care, USA) and sections were then processed for emulsion autoradiography.
2.4. Double fluorescence in situ hybridization (dFISH)
We performed a double FISH to label PRCP and ghrelin receptor mRNAs. To this end, antisense riboprobes directed against PRCP (biotinylated PRCP) and ghrelin receptor (digoxygenin-ghrelin receptor) were simultaneously hybridized overnight at 52 °C, as described above. After hybridization and washes, sections were developed for ghrelin receptor, using a peroxidase-conjugated anti-DIG antibody (1:1000 in TNB; 0.1 M Tris–HCl, pH 7.4, 0.05% Triton-X 100, 5 M NaCl, 2 mg/ml BSA in RNase-free water) for 1.5 h followed by incubation for 30 min in tyramide coupled with Alexa-594 (1:200 dilution in working solution, Invitrogen, Carlsbad, CA). Sections were then incubated in 0.3% H2O2 for 15 min. To detect biotinylated PRCP probes, sections were incubated with a peroxidase-conjugated anti-biotin antibody (1:1000 dilution in TNB; Vector Laboratories, Burlingame, CA) for 1.5 h, then incubated in Alexa 488-coupled tyramide (1:200 dilution; Invitrogen, Carlsbad, CA) for 30 min. TNT washing (3×10 min; 0.1 M Tris–HCl, pH 7.4, 0.15 M NaCl, 0.05% Triton-X-100) was performed between each steps. Sections were then coverslipped with aquamount for microscopic examination.
2.5. Statistical analysis
Radioactive in situ hybridization signal after emulsion autoradiography was analyzed using the NIH ImageJ program. In every section, both hemispheres of each area were analyzed for silver grain density. A background density outside of the brain section was used to normalize the data. The values of each group were compared to the value of either fed or saline group (considered to be 100). For analysis of Western Blot data, band density was normalized to β-actin, and then compared to value of fed or vehicle-injected groups (considered to 100). All data are expressed as means±SE and were compared by Student's t test, and P<0.05 was considered significant.
3. Results
3.1. Fasting increases hypothalamic PRCP mRNA and protein levels
Brain sections from fed and fasted C57Bl/6 mice were used to assess Prcp mRNA expression levels by radioactive in situ hybridization. Prcp mRNA expression in the hypothalamus was significantly increased by fasting (Figure 1A–I and suppl Figure 2). Specifically, increase in Prcp gene expression was observed in the ARC (100.0±14.0 in fed vs. 469.0±135.4 in fasting, n=6 and 4, respectively, p=0.0048; Figure 1A–B, I), in the VMH (100.0±21.7 in fed vs. 279.5±99.8 in fasting, n=6 and 3, respectively, p=0.0427; Figure 1A–B. I), in the DMH (100.0±9.8 in fed vs. 359.5±77.7 in fasting, n=6 and 4, respectively, p=0.0032; Figure 1C–D, I), and in the LH (100.0±18.4 in fed vs. 279.0±43.6 in fasting, n=6 and 4, respectively, p=0.0026; Figure 1E–F, I). On the other hand, no difference in Prcp mRNA expression was observed in the cortex after fasting (100.0±18.4 in fed vs. 156.8±28.0 in fasting, n=6 and 4, respectively, p=0.1126; Figure 1G–I).
Figure 1.
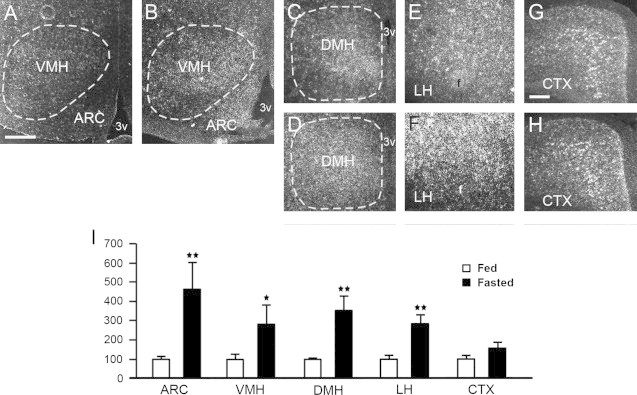
Representative micrographs of several hypothalamic areas including the arcuate nucleus (ARC), ventromedial nucleus (VMH), dorsomedial nucleus (DMH), lateral hypothalamus (LH), as well as the cerebral cortex (CTX) showing silver grains from in situ hybridization of Prcp mRNA in fed (panels A, C, E, G) and fasted mice (panels B, D, F, H). Prcp gene expression in both hemispheres of each area was analyzed using a NIH imageJ program, and relative density was compared between fed and overnight fasted animals. 3v=third ventricle; f=fornix. *p<0.05; **p<0.01. Bar scales in panels A (for A–F) and G (for G and H) represent 200 μm.
We also determined PRCP protein levels by Western blot analysis in the brain of fed and fasted animals. To this end, we extracted total proteins from micro-punched hypothalamic areas including the ARC, VMH, DMH, LH, as well as from the cortex (Figure 2). Similar to its mRNA levels, PRCP protein expression was increased in different areas of the hypothalamus of fasted mice compared to fed animals. Specifically, statistical significance was observed in the ARC (100.0±4.0 in fed vs. 127.0±7.0 in fasting, n=3 for each, p=0.029), VMH (100.0±1.5 in fed vs. 123.9±1.1 in fasting, n=3 for each, p=0.0002) and in the DMH (100.0±1.7 in fed vs. 118.2±1.3 in fasted mice, n=3 for each, p=0.0045). Although increased, no significant differences were found in the LH of fasted versus fed mice (100.0±6.7 in fed vs. 124.8±7.6 in fasted, n=3 for each, p=0.0704). PRCP expression in the cortex was not affected by fasting (100.0±2.0 in fed vs. 101.3±1.2 in fasting, n=3 for each, p=0.6022).
Figure 2.
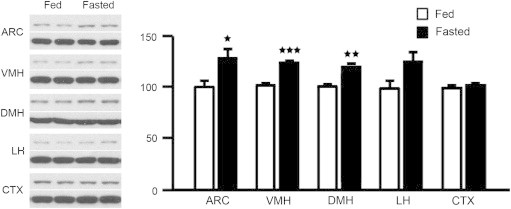
Representative Western blot images and density results of PRCP protein levels in several hypothalamic areas including the ARC, VMH, DMH, LH, and CTX of fed and fasted mice. The density of PRCP protein (upper band) was first normalized to β-actin (lower band) using a NIH imageJ program, and then compared between fed and overnight fasted groups. *p<0.05; **p<0.01; ***p<0.001.
3.2. Ghrelin effect on PRCP mRNA expression in the brain
We next assessed whether ghrelin might be a key metabolic signal for the induction of PRCP expression. We first determined the role of circulating ghrelin on Prcp gene expression in brain. Animals were i.p. injected with either saline (vehicle) or ghrelin just before the beginning of the dark cycle. Three hours later, animals were sacrificed and hypothalamic Prcp mRNA levels were measured by in situ hybridization. Similar to fasting, increased Prcp mRNA expression was observed in different hypothalamic nuclei after peripheral ghrelin administration (Figure 3A–I and suppl Figure 3). A statistically significant induction of PRCP gene expression was detected in the ARC (100.0±14.5 in ip saline- vs. 184.8±17.3 in ip ghrelin-treated mice, n=4 and 3, respectively, p=0.0129; Figure 3A–B, I), VMH (100.0±12.9 in ip saline- vs. 182.7±11.9 in ip ghrelin-treated mice, n=4 and 3, respectively, p=0.0061; Figure 3A–B, I), and DMH (100.0±7.1 in ip saline- vs. 291.2±49.9 in ip ghrelin-treated mice, n=4 and 3, respectively, p=0.0065; Figure 3C–D,I), while in the LH (100.0±21.0 in ip saline- vs. 142.1±7.1 in ip ghrelin-treated mice, n=4 and 3, respectively, p=0.1604; Figure 3E–F, I) and in the cortex (100.0±14.6 in ip saline- vs. 144.9±7.7 in ip ghrelin-treated mice, n=4 for each, p=0.0532; Figure 3G–I), Prcp mRNA levels did not show significant differences.
Figure 3.
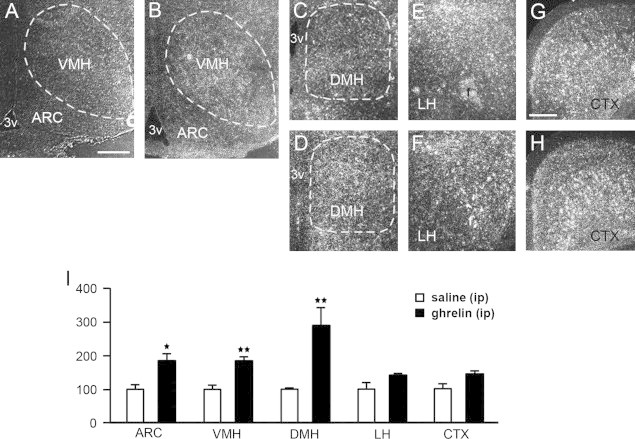
Representative micrographs of several hypothalamic areas including ARC, VMH, DMH, LH, and CTX showing silver grains from in situ hybridization of Prcp mRNA in ip saline- (panels A, C, E, G) and ip ghrelin-treated mice (panels B, D, F, H). Prcp mRNA expression in both hemispheres of each area was analyzed using a NIH imageJ program, and relative density was compared between ip saline- and ip ghrelin-injected groups. 3v=third ventricle; f=fornix. *p<0.05; **p<0.01. Bar scales in panels A (for A–F) and G (for G and H) represent 200 μm.
Next, to determine whether the increased Prcp mRNA level in the brain was due to the direct action of ghrelin, a single dose of ghrelin was injected intracerebroventricularly (Figure 4A–I and suppl Figure 4). A statistically significant induction of Prcp gene expression following central administration of ghrelin was observed in the ARC (100.0±9.1 in icv saline vs. 141.1±8.6 in icv ghrelin-treated mice, n=3 for each, p=0.03; Figure 4A–B, I), VMH (100.0±6.5 in icv saline vs. 138.6±6.3 in icv ghrelin-treated mice, n=4 and 3, respectively, p=0.009; Figure 4A–B, I) and DMH (100.0±9.7 in icv saline vs. 134.5±5.2 in icv ghrelin-treated mice, n=3 for each, p=0.035; Figure 4C–D, I), while no significant difference was detected in the LH (100.0±12.3 in saline vs. 106.3±10.4 in icv ghrelin-treated mice, n=4 and 3, respectively, p=0.725; Figure 4E–F, I) and cortex (100.0±12.3 in saline vs. 108.8±8.3 in icv ghrelin-treated mice, n=4 and 3, respectively, p=0.609; Figure 4G–I).
Figure 4.
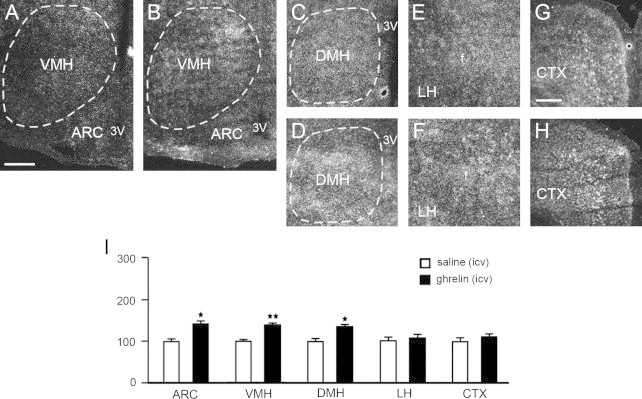
Representative micrographs of several hypothalamic areas including ARC, VMH, DMH, LH, and CTX showing silver grains from in situ hybridization of Prcp mRNA in icv saline- (panels A, C, E, G) and icv ghrelin-injected mice (panels B, D, F, H). Prcp mRNA expression in both hemispheres of each brain area, was analyzed using a NIH imageJ program, and relative density was compared between icv saline- and icv ghrelin-injected groups. 3v: third ventricle; f: fornix. *p<0.05; **p<0.01. Bar scales in panels A (for A–F) and G (for G and H) represent 200 μm.
To further test the role of ghrelin in PRCP regulation, we assessed Prcp gene expression in fed and fasted ghrelin knockout mice (Ghr−/−; [10]; Figure 5A–I and suppl Figure 5). No differences in Prcp gene expression in the hypothalami of fed and fasted Ghr−/− mice was found in all the hypothalamic areas examined (ARC: 100.0±7.2 in fed vs. 80.0±11.7 fasted mice, p=0.1689, Figure 5A–B, I; VMH: 100.0±11.1 in fed vs. 67.3±11.8 fasted, p=0.1129, Figure 5A–B, I; DMH: 100.0±9.5 in fed vs. 78.0±16.7 fasted mice, p=0.2527, Figure 5C–D, I; LH: 100.0±6.8 in fed vs. 79.6±6.9 fasted mice, p=0.1040, Figure 5E–F, I; CTX: 100.0±15.4 in fed vs. 77.4±10.9 fasted mice, p=0.3703, Figure 5G–I; n=6 for fed and 4 for fasted mice).
Figure 5.
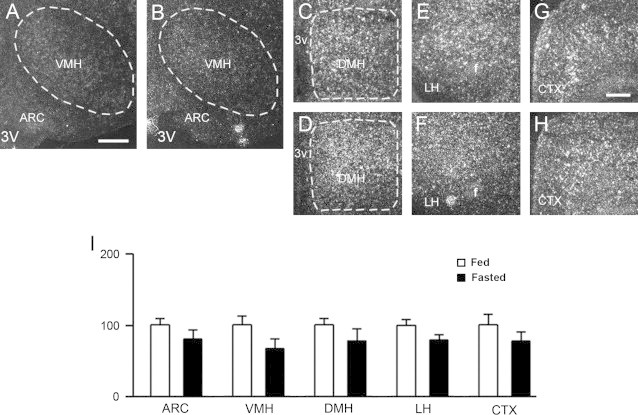
Representative micrographs of several hypothalamic areas including ARC, VMH, DMH, LH, and CTX showing silver grains from in situ hybridization of Prcp mRNA in fed (panels A, C, E, G) and fasted ghrelin knockout mice (panels B, D, F, H). Prcp mRNA expression in both hemispheres of each brain area, was analyzed using a NIH imageJ program, and relative density was compared between fed and overnight fasted ghrelin knockout animals. 3v: third ventricle; f: fornix. *p<0.05; **p<0.01. Bar scales in panels A (for A–F) and G (for G and H) represent 200 μm.
3.3. Ghrelin action on PRCP expression is mediated by ghrelin receptors
Considering the effect of ghrelin on Prcp mRNA levels, we then assessed whether ghrelin action on central Prcp expression was mediated by ghrelin receptors (Ghs-r). By double fluorescent in situ hybridization, we first assessed whether Prcp and ghrelin receptors are co-expressed in the same neurons (Figure 6). A vast majority of Prcp-expressing cells in the hypothalamus were found to express ghrelin receptor, suggesting that ghrelin action on Prcp gene expression may be mediated by its receptor. In one animal, we analyzed the colocalization of Prcp and Ghs-r mRNAs. We found that in the ARC out of 119 Prcp mRNA-expressing neurons, 96 cells were expressing Ghs-r mRNA (80.7%), in the VMH out of 156 Prcp mRNA-expressing neurons, 137 cells were expressing Ghs-r mRNA (87.8%), in the DMH out of 61 Prcp mRNA-expressing neurons, 51 cells were expressing Ghs-r mRNA (83.6%); in the LH out of 131 Prcp mRNA-expressing neurons, 101 cells were expressing Ghs-r mRNA (77.0%) and in CTX out of 164 Prcp mRNA-expressing neurons, 143 cells were expressing Ghs-r mRNA (87.2%). To further determine whether ghrelin-induced increase of Prcp expression is mediated by Ghs-r, we measured PRCP protein levels in the hypothalamus of ip saline- (vehicle) or ghrelin-injected Ghs-r knockout mice (Ghsr−/−; 9). While hypothalamic PRCP protein level was increased by ghrelin administration in wild type control mice (100.0±0.1 in saline vs. 112.9±1.0 ghrelin-treated mice, p=0.006; Figure 7), ghrelin treatment to Ghsr−/−mice did not alter hypothalamic PRCP protein levels compared to saline-treated Ghsr−/− controls (100.5±3.3 in saline vs. 97.3±1.9 ghrelin-treated mice, p=0.42; Figure 7).
Figure 6.
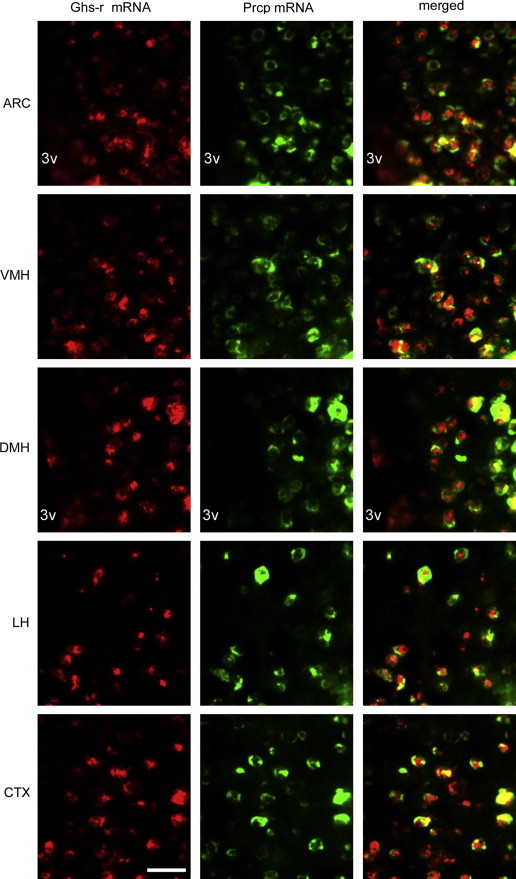
Representative photographs showing colocalization of ghrelin receptors (Ghs-r) and Prcp mRNA in the hypothalamic ARC, VMH, DMH, LH and the CTX, by double fluorescent in situ hybridization. Red and green represent Ghs-r- and Prcp-expressing cells, respectively. Merged images show co-labeled cells producing both Ghs-r and Prcp. 3v: third ventricle. Bar scale represents 30 μm for all panels. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Figure 7.
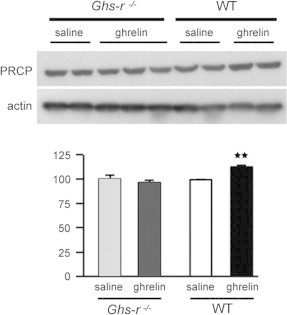
Representative Western blot images and density results of PRCP protein levels in the whole hypothalamus of wild type and Ghsr−/− mice after ip ghrelin administration. The density of PRCP protein levels (upper bands) was first normalized to β-actin (lower bands) using a NIH imageJ program, and then relative density was compared between saline and ip ghrelin-injected wild type and Ghsr−/− animals. **p<0.01.
4. Discussion
Anatomical and functional data have shown that PRCP, a member of the serine protease family [1–3], plays a role in metabolism regulation by degrading central α-MSH and thus affecting melanocortin signaling [4–6]. In support of these, mice in which Prcp gene was deleted showed reduced food intake, body weight and fat mass and increased energy expenditure [5]. Furthermore, when exposed to high fat diet, they showed resistance to diet-induced obesity compared to their wild type controls [4,6]. Considering the role of PRCP in metabolism regulation through the control of the central melanocortin system, it is conceivable that its expression and activity levels may be under the control of peripheral metabolic signals including hormones. To test this, we first analyzed Prcp expression both at mRNA and protein levels in several hypothalamic nuclei in fed and fasting conditions. Hypothalamic Prcp mRNA and protein levels were found up-regulated by fasting. However, in the cortex, fasting did not induce changes in Prcp mRNA and protein levels. Since ghrelin is a key orexigenic hormone [11], we determined the potential role of ghrelin in regulating hypothalamic Prcp gene expression. We found that both peripheral and central administration of ghrelin promoted an increase in hypothalamic Prcp expression. In support of this, fasting did not induce the increase of Prcp expression in ghrelin knockout (Ghr−/−) mice. To further assess whether ghrelin effect is mediated by its receptor (GHS-R), we first determine the pattern of expression of Ghs-r and Prcp mRNAs and found that they indeed co-localize in the hypothalamus. We then determined PRCP protein levels in the hypothalamus of vehicle- and ghrelin-treated Ghsr−/− mice and found that ghrelin did not affect PRCP levels compared to vehicle-treated Ghsr−/− controls. Altogether our data suggest that ghrelin is an important regulator of PRCP and, thus, α-MSH degradation.
Arcuate nucleus levels of α-MSH are tightly regulated and they change according to the metabolic state of the organism [12]. For example, during fasting, a reduction of Pomc mRNA has been shown [13,14]. This decrease is mainly due to the regulation of Pomc gene by the peripheral adipose-derived hormone, leptin [14]. Leptin-deficient mice (ob/ob) indeed also have reduced levels of Pomc mRNA in the hypothalamus, and leptin replacement induces an increase in Pomc mRNA [13–16]. Furthermore, α-MSH, is generated by extensive posttranslational processes that involve several enzymes such as the prohormone convertases 1 and 2 (PC1 and PC2), carboxypeptidase E (CPE) and peptidyl a-amidating monooxygenase (PAM). These posttranslational processes have been shown regulated by metabolic signals. For example, PC1 and PC2 are lower during fasting because of diminishing leptin levels [17] suggesting that leptin-dependent stimulation of α-MSH involves the activation of Pomc transcription as well as the increase of PC1 and PC2 expression, which together lead to the increased production of α-MSH. However, little is known about the regulation of α-MSH degradation. PRCP is expressed in key hypothalamic areas, including the DMH, ARC and LH [4]. Interestingly, the DMH and LH do not express POMC, and within the ARC, PRCP is not localized in α-MSH-expressing neurons [4]. This pattern of expression suggests that PRCP may function at the synaptic levels, inactivating α-MSH once released. Indeed, all of these areas project to the target region of α-MSH projections, the PVN, where α-MSH terminals innervate MC4R-expressing neurons [18]. Thus, PRCP may control the output and efficacy of the melanocortin system. In support of this, Shinyama and collaborators [19] have shown that α-MSH degradation is independent of MC4R.
Fasting and both peripheral and central administration of ghrelin promoted Prcp gene expression in the ARC, VMH, and DMH. Given that arcuate POMC neurons do not express ghrelin receptor or PRCP [20], and that ghrelin activate Neuropeptide Y/ Agouti related peptide (NPY/AgRP)-expressing neurons, it is conceivable that PRCP is expressed in the arcuate NPY/AgRP neurons. These orexigenic neurons are known to project to the MC4R-expressing neurons in the PVN where they will control melanocortin signaling by releasing both AgRP, the endogenous reverse agonist of MC4R, and PRCP which will inactivate MC4R endogenous agonist, α-MSH.
Interestingly, areas such as the LH, although showed a significant change in PRCP expression after fasting, did not show a significant increase in PRCP expression after either central or peripheral ghrelin administration. PRCP is expressed in subpopulations of hypocretin/orexin (Hcrt)- and melanin concentrating hormone (MCH)-expressing neurons of the LH [4] and ghrelin receptors are expressed in the LH [21,22]. However, the inability of ghrelin to induce significant changes in PRCP expression may be due to a selective expression of GHS-R in this region. In support of this, Toshinai and collaborators [23] found that ghrelin induces c-fos activation in Hcrt neurons, but not in MCH neurons of the LH, suggesting that MCH neurons may not express GHS-R and therefore, ghrelin may be able to regulate PRCP only in a selected subpopulation of PRCP-expressing neurons. In our study we analyzed PRCP expression in the entire LH, which may preclude us from finding changes in selective neuronal subpopulations. It is also possible that other metabolic signals affected by fasting may be responsible for Prcp regulation in the LH. Similarly, despite the colocalization of Prcp and Ghs-r in the cortex, no effect on Prcp expression was found after ghrelin treatment. However, fasting also failed to alter Prcp levels in the cortex suggesting that fasting-induced changes in Prcp expression in the brain are region-specific.
In conclusion, our data provide evidence for a dynamic relationship between central PRCP and metabolic signals. Prcp is an inducible obesity gene in the hypothalamus, and ghrelin is necessary for fasting-induced PRCP expression in the hypothalamus.
Conflict of interest
None declared.
Acknowledgment
This work was supported by the NIH grant DK 084065 (to S.D.).
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.molmet.2013.01.002.
Appendix A. Supporting information
Supplemental Figure 1.
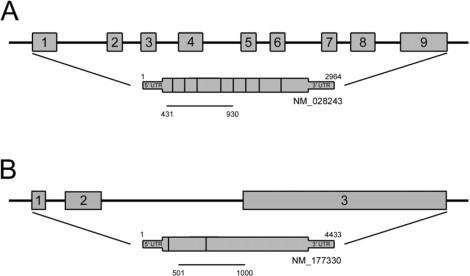
Cloning of cDNAs specific to mice PRCP (A) and ghrelin receptor (B). A 500 bp of mice Prcp probe was obtained from NCBI gene database (gene accession number: NM_028243, mRNA is from combination of nine exons), and represents Prcp specific, conserved domain between vertebrates. A 500 bp of ghrelin receptor was also obtained from NCBI gene database (accession number: NM_177330, mRNA is from combination of three exons), and represents conserved area between mRNAs of ghrelin receptor variant.
Supplemental Figure 2.
Supplemental Figure 3.
Supplemental Figure 4.
Supplemental Figure 5.
References
- 1.Abeywickrema P.D., Patel S.B., Byrne N.J., Diehl R.E., Hall D.L., Ford R.E. Expression, purification and crystallization of human prolylcarboxypeptidase. Acta Crystallographica Section F: Structural Biology and Crystallization Communications. 2010;66:702–705. doi: 10.1107/S1744309110014041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soisson S.M., Patel S.B., Abeywickrema P.D., Byrne N.J., Diehl R.E., Hall D.L. Structural definition and substrate specificity of the S28 protease family: the crystal structure of human prolylcarboxypeptidase. BMC Structural Biology. 2010;10:16. doi: 10.1186/1472-6807-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumamoto K., Stewart T.A., Johnson A.R., Erdos E.G. Prolylcarboxypeptidase (angiotensinase C) in human lung and cultured cells. The Journal of Clinical Investigation. 1981;67:210–215. doi: 10.1172/JCI110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallingford N., Perroud B., Gao Q., Coppola A., Gyenqesi E., Liu Z.W. Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. The Journal of Clinical Investigation. 2009;119:2291–2303. doi: 10.1172/JCI37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeong J.K., Szabo G., Diano S. Prolyl carboxypeptidase regulates energy expenditure and the thyroid axis. Endocrinology. 2012;153:683–689. doi: 10.1210/en.2011-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeong J.K., Szabo G., Raso G.M., Meli R., Diano S. Deletion of prolyl carboxypeptidase attenuates the metabolic effects of diet-induced obesity. American Journal of Physiology Endocrinology and Metabolism. 2012;302:E1502–E1510. doi: 10.1152/ajpendo.00544.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu H., Kishi T., Roseberry A.G., Cai X., Lee C.E., Montez J.M. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou C., Garcia-Calvo M., Pinto S., Lombardo M., Feng Z., Bender K. Design and synthesis of prolylcarboxypeptidase (PrCP) inhibitors to validate PrCP as a potential target for obesity. Journal of Medicinal Chemistry. 2010;53:7251–7263. doi: 10.1021/jm101013m. [DOI] [PubMed] [Google Scholar]
- 9.Andrews Z.B., Liu Z.W., Wallingford N., Erion D.M., Borok E., Friedman J.M. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;14:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diano S., Farr S.A., Benoit S.C., McNay E.C., da Silva I., Horvath B. Ghrelin controls hippocampal spine synapse density and memory performance. Nature Neuroscience. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 11.Tschöp M., Smiley D.L., Heiman M.L. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 12.Spiegelman B.M., Flier J.S. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz M.W., Seeley R.J., Woods S.C., Weigle D.S., Campfield L.A., Burn P. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 14.Mizuno T.M., Kleopoulos S.P., Bergen H.T., Roberts J.L., Priest C.A., Mobbs C.V. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- 15.Bergendahl M., Wiemann J.N., Clifton D.K., Huhtaniemi I., Steiner R.A. Short-term starvation decreases POMC mRNA but does not alter GnRH mRNA in the brain of adult male rats. Neuroendocrinology. 1992;56:913–920. doi: 10.1159/000126324. [DOI] [PubMed] [Google Scholar]
- 16.Korner J., Chua S.C., Jr, Williams J.A., Leibel R.L., Wardlaw S.L. Regulation of hypothalamic proopiomelanocortin by leptin in lean and obese rats. Neuroendocrinology. 1999;70:377–383. doi: 10.1159/000054499. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez V.C., Goldstein J., Stuart R.C., Hovanesian V., Huo L., Munzberg H. Regulation of hypothalamic prohormone convertases 1 and 2 and effects on processing of prothyrotropin-releasing hormone. The Journal of Clinical Investigation. 2004;114:357–369. doi: 10.1172/JCI21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim M.S., Rossi M., Abusnana S., Sunter D., Morgan D.G., Small C.J. Hypothalamic localization of the feeding effect of agouti-related peptide and alpha-melanocyte-stimulating hormone. Diabetes. 2000;49:177–182. doi: 10.2337/diabetes.49.2.177. [DOI] [PubMed] [Google Scholar]
- 19.Shinyama H., Masuzaki H., Fang H., Flier J.S. Regulation of melanocortin-4 receptor signaling: agonist-mediated desensitization and internalization. Endocrinology. 2003;144:1301–1314. doi: 10.1210/en.2002-220931. [DOI] [PubMed] [Google Scholar]
- 20.Willesen M.G., Kristensen P., Romer J. Co-localization of groth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology. 1999;70:306–316. doi: 10.1159/000054491. [DOI] [PubMed] [Google Scholar]
- 21.Michell V., Bouret S., Beauvillain J.C., Schilling A., Perret M., Kordon C. Comparative distribution of mRNA encoding the groth hormone secretagogue-receptor (GHS-R) in Microcebus murinus (Primate, lemurian) and rat forebrain and pituitary. The Journal of Comparative Neurology. 2001;429:469–489. doi: 10.1002/1096-9861(20010115)429:3<469::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 22.Olszewski P.K., Li D., Grace M.K., Billington C.J., Kotz C.M., Levine A.S. Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides. 2003;24:597–602. doi: 10.1016/s0196-9781(03)00105-0. [DOI] [PubMed] [Google Scholar]
- 23.Toshinai K., Date Y., Murakami N., Shimada M., Mondal M.S., Shimbara T. Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology. 2003;144:1506–1512. doi: 10.1210/en.2002-220788. [DOI] [PubMed] [Google Scholar]