

Abstract
Excessive production of unsaturated aldehydes from oxidized lipoproteins and membrane lipids is a characteristic feature of cardiovascular disease. Our previous studies show that unsaturated lipid peroxidation-derived aldehydes such as 4-hydroxy-trans-2-nonenal (HNE) promote autophagy in rat aortic smooth muscle cells (RASMC). In this study, we examined the mechanism by which HNE induces autophagy. Exposure of RASMC to HNE led to the modification of several proteins, most of which were identified by mass spectrometry and confocal microscopy to be localized to the endoplasmic reticulum (ER). HNE stimulated the phosphorylation of PKR-like ER kinase and eukaryotic initiation factor 2α and increased heme oxygenase-1 (HO-1) abundance. HNE treatment also increased LC3-II formation and the phosphorylation of JNK and p38. Pharmacological inhibition of JNK, but not p38, prevented HNE-induced HO-1 expression and LC3-II formation. Inhibition of JNK increased cell death in HNE-treated cells. Pretreatment with the chemical chaperone phenylbutryic acid prevented LC3-II formation as well as JNK phosphorylation and HO-1 induction. Taken together, these data suggest that autophagic responses triggered by unsaturated aldehydes could be attributed, in part, to ER stress, which stimulates autophagy by a JNK-dependent mechanism and promotes cell survival during oxidative stress.
Abbreviations: RASMC, rat aortic smooth muscle cells; ER, endoplasmic reticulum; UPR, unfolded protein response; Grp, glucose regulated protein; PDI, protein disulfide isomerase; PERK, PKR-like ER kinase; IRE, inositol requiring enzyme; LC3, microtubule-associated protein 1 light chain 3; HNE, 4-hydroxy-trans-2-nonenal; JNK, c-jun N-terminal kinase; eIF2α, elongation initiation factor 2 α; DTT, dithiothreitol; DNPH, 2,4-dinitrophenylhydrazine; ATF6, activating transcription factor 6; ECL, enhanced chemiluminescence; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; NP-40, noniodet P40; SDS, sodium dodecyl sulfate; PVDF, polyvinylidene fluoride; DMEM, Delbucco's Eagle Modified Medium; HRP, horseradish peroxidase; DMSO, dimethylsulfoxide; FBS, fetal bovine serum
Keywords: Oxidative stress, Autophagy, 4-hydroxynonenal, JNK, Unfolded protein response, Smooth muscle cells
Graphical abstract
Highlights
► The lipid peroxidation product 4-hydroxynonenal (HNE) modifies endoplasmic reticulum (ER) proteins in vascular smooth muscle cells. ► HNE activates autophagy. ► PKR-like ER kinase (PERK) and the stress kinase JNK are activated by HNE. ► Pharmacological inhibition of JNK or ER stress prevents autophagy in vascular smooth muscle cells.
Introduction
Polyunsaturated fatty acids esterified in lipoproteins or membrane phospholipids are highly susceptible to oxidation. Alternating single and double bonds in these lipids efficiently stabilize alkene radicals generated from electron abstraction by reactive species such as transition metals. In the presence of oxygen, the alkyl radicals are readily converted into peroxyl radicals that undergo a series of autocatalytic chain reactions leading to the generation of other radical species or hydroperoxides. If unquenched these reactions lead to extensive fragmentation of fatty acids, generating a variety of stable molecules, particularly saturated and unsaturated carbonyls [1,2]. High levels of lipid peroxidation-derived carbonyls have been detected in biological tissues under a variety of etiologically unrelated disease states and toxicological conditions associated with oxidative stress [3–7].
The carbonyls generated by lipid peroxidation have high chemical and biological activity. Some carbonyls, such as the α, β unsaturated aldehydes, react avidly with nucleophilic groups found in glutathione, proteins, phospholipids, and nucleic acids [8]. Chemical reactions of unsaturated aldehydes with nucleophiles incite a range of effects. At low concentrations, oxidized lipids have the capacity to promote a protected cellular phenotype [9,10], whereas at high concentrations they disrupt cell structure and function leading to either necrotic or apoptotic cell death. Recent investigations suggest that the carbonyls derived from lipid peroxidation are critical determinants of oxidative stress, inflammation, and cell death occurring in several pathologies, particularly cardiovascular disease [11,12]. Exposure to lipid oxidation products such as 4-hydroxy-trans-2-nonenal (HNE), 4-oxo-trans-2-nonenal (ONE), and acrolein results in glutathione depletion, changes in ion homeostasis, and stimulation of stress signaling [1,8,13]. Our previous studies show that lipid peroxidation-derived aldehydes stimulate autophagy, which removes aldehyde-modified proteins [14].
Autophagy is initiated in cells in response to conditions associated with nutrient deprivation and/or oxidative stress [15–17]. While excessive autophagy triggers cell death, removal of damaged organelles by autophagy or recycling of cell constituents promotes survival under conditions of nutrient deprivation [15]. However, the mechanisms by which HNE stimulates autophagy remain unknown. Hence, to determine which proteins are modified by HNE and how they stimulate autophagy, we examined the subcellular localization of protein-HNE adducts and the signaling pathways they activate. Our results show that exposure to HNE results in the formation of protein adducts that accumulate in the endoplasmic reticulum (ER) and that HNE triggers components of the unfolded protein response (UPR) that stimulate autophagy by a JNK-dependent mechanism.
Experimental
Materials
HNE was synthesized as described previously [18]. All materials and chemicals were obtained from Sigma (St. Louis, MO) unless otherwise stated. Electrophoresis supplies were purchased from Bio-Rad (Hercules, CA). Primary antibodies against microtubule-associated protein 1 light chain 3 (LC3) were obtained from MBL International (Woburn, MA, U.S.A.). Primary antibodies against phospho-PKR-like ER kinase (PERK), PERK, and glucose regulated protein 78 (Grp78) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-elongation initiation factor 2α (eIF2α), -eIF2α, -phospho-c-jun N-terminal kinase (JNK), -JNK, -phospho-p38, and –p38 antibodies as well as the horseradish peroxidase (HRP)-linked secondary goat-anti-rabbit or goat-anti-mouse antibodies were from Cell Signaling (Danvers, MA). The anti-CHOP antibody was purchased from Affinity Bioreagents (Rockford, IL). The antibodies against KDEL and heme oxygenase-1 (HO-1) were obtained from Stressgen (Ann Arbor, MI). Polyclonal antibodies against protein-HNE were raised and tested using previously published protocols [14,19]. Fluorescently labeled secondary antibodies (Alexa Fluor 488 anti-rabbit-IgG, Texas-red anti-mouse-IgG) and the Slow Fade® Gold anti-fade reagent were purchased from Molecular Probes (Invitrogen, Carlsbad, CA). Phenylbutric acid (PBA) was from Pfaltz & Bauer (Waterbury, CT). ECL® reagent was purchased from GE Healthcare (Amersham Biosciences, Pittsburgh, PA). The Oxyblot kit was obtained from Chemicon International (Temecula, CA).
Cell culture experiments
Primary rat aortic smooth muscle cells (RASMC) were isolated from Sprague-Dawley rats and cultured in Dulbecco's Modified Eagle's Medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin under standard cell culture conditions. For HNE exposure, cells grown to 90% confluence were washed with Hank's balanced salt solution (HBSS, pH 7.4: 20 mM HEPES, 135 mM NaCl, 5.4 mM KCl, 1.0 mM MgCl2, 2.0 mM CaCl2, 2.0 mM NaH2PO4, 5.5 mM glucose) and exposed to HNE in 1 ml of HBSS. Cells were then treated with HNE, tunicamycin, or thapsigargin for the indicated times. For inhibiting ER stress and JNK, cells were pretreated with the chemical chaperone PBA (10 mM for 16 h) or the JNK inhibitor (SP600125; 25 μM for 30 min) prior to HNE treatment. After this pre-incubation, the cells were treated with HNE in HBSS in the absence or presence of PBA or JNK inhibitor. After treatment, the cells were washed with HBSS and then scraped with a rubber policeman in lysis buffer containing 25 mM HEPES, pH 7.0, 1 mM EDTA, 1 mM EGTA, 1.0% NP-40, 0.1% SDS, 1:100 mammalian protease inhibitor cocktail (Sigma) and 1:50 phosphatase inhibitor cocktail (Pierce, Rockland, IL). Cell suspensions were lysed further by sonication, and the homogenates were centrifuged at 14,000×g for 15 min at 4 °C. Total protein concentration was measured by a modified Lowry method [20].
Protein identification
For identification of oxidatively-modified proteins, proteins were separated by 2D electrophoresis and protein carbonyls were derivatized “in-stripe” using 2,4-dinitrophenylhydrazine (DNPH) [21]. Briefly, protein from HNE-treated or control RASMC was loaded on 3–10 NL IPG strips (Bio-Rad) for isoelectric focusing. The proteins were then separated by SDS-PAGE, electroblotted to PVDF membranes, and probed with anti-DNP antibodies. Proteins displaying oxidative modifications were excised from parallel silver-stained gels, trypsinized, and subjected to MALDI-TOF MS for protein identification by peptide mass fingerprinting using the NCBI database.
Confocal immunocytochemistry
Cells grown on cover slips were treated for 30 min with 25 μM HNE as described above. After treatment, the cells were washed three times with ice-cold phosphate-buffered saline (PBS; Invitrogen, Carlsbad, CA) and fixed for 10 min at room temperature with 4% PFA/PBS (pH 7.4). Following fixation, the cells were permeabilized by treatment with 0.1% Triton X-100/PBS at room temperature for 3 min and incubated for 15 min in 1% BSA/PBS. Primary antibody incubations (1:250 and 1:1000 dilutions for anti-KDEL and -HNE antibodies, respectively) were performed in PBS at 4 °C overnight. The cells were then incubated with the fluorescently labeled secondary antibodies (1:500 dilution) for 1 h at room temperature in the dark. Cells were mounted with Slow Fade® Gold antifade reagent, and fluorescence was visualized by confocal microscopy (Olympus Bx51W1).
Immunoblot analyses
For Western blots, 20–50 μg protein was loaded on 5%, 7.5%, 12%, or 15% SDS polyacrylamide gels for protein separation. The proteins were transferred to PVDF membranes. PVDF membranes were then blocked in 5% blotting milk (made in Tris-buffered saline/0.3% Tween 20). Proteins or post-translational protein modifications were detected using the appropriate primary antibodies (anti-DNP, 1:150 dilution; anti-phospho-PERK, PERK, P-eIF2α, eIF2α, CHOP, and LC3 at 1:1000 dilution; anti-Grp78, 1:500 dilution; anti-actin, 1:2000 dilution; anti-HO-1, 1:5000 dilution), HRP-linked secondary antibodies (1:2000 dilution), and ECL plus detection reagents. Bands were visualized and quantified using a Typhoon 9400 Variable Mode Imager (Amersham) and ImageQuant TL software.
Measurements of cell viability
Cell viability was estimated by measuring lactate dehydrogenase (LDH) release into the medium. For this, cells were treated with the indicated amount of HNE for 30 min in HBSS. The HNE-containing medium was then removed, and fresh DMEM containing 0.1% FBS was added for 16 h. The medium was then collected, and cell lysates were prepared by rinsing culture plates with ice-cold PBS and incubating on ice for 5 min with 100 μl of PBS, pH 7.4, containing 0.1% Triton X-100. Then, 5 μl of cell lysate or 200 μl of the culture medium were incubated with NADH (300 μM) and pyruvate (10 mM). Conversion of NADH into NAD was followed by the change in absorbance at 340 nm for 4 min at 37 °C. Relative LDH release was expressed as the percentage LDH activity in the supernatant of cultured cells (medium) compared with total LDH activity present per well (medium+cell lysate).
Results
HNE modifies ER proteins
Our previous studies show that RASMC treated with HNE accumulate protein-HNE adducts and that the formation and accumulation of these adducts stimulate autophagy. However, neither the identity of the proteins modified nor their subcellular location is known. These are important to know to understand how autophagy could be activated by HNE. To identify proteins modified by HNE, we used a two-dimensional (2D) electrophoresis technique [21] followed by mass spectrometry. For this, RASMC were treated with HNE (50 μM) for 30 min, and the cells were lysed and separated by isoelectric focusing. The proteins were then derivatized with 2,4-dinitrophenylhydrazine (DNP), separated in the second dimension by SDS-PAGE, and assessed for DNP modification by Western blotting with anti-DNP antibodies. Shown in Fig. 1A are specific regions of the 2D immunoblots from control and HNE-treated cells that showed the highest immunoreactivity with anti-DNP antibodies. Corresponding proteins on parallel silver-stained gels were excised, digested with trypsin, and the resultant peptides were analyzed by MALDI-TOF/MS. Results from peptide mass fingerprinting showed that several proteins found in the endoplasmic reticulum (ER) were carbonylated after HNE exposure (Table 1). These proteins included protein disulfide isomerase (PDI; 1), glucose regulated protein 58 (Grp58; 2), and Grp78 (5), all of which are chaperones critical for protein folding in the ER. The ER calcium binding protein reticulocalbin (6), the cytoskeletal protein vimentin (3), and heat shock protein 60 (HSP60; 4) were also found to be modified.
Fig. 1.
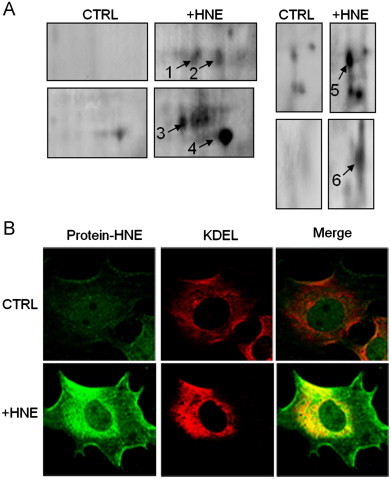
HNE modifies proteins in the endoplasmic reticulum in rat aortic smooth muscle cells (RASMC). Immunological detection of protein modifications induced by HNE: (A) 2D electrophoretic analysis of oxidatively modified proteins after treatment with HNE (50 μM) for 30 min. Proteins (1–6) identified by MS are summarized in Table 1. (B) Confocal fluorescence microscopy showing the subcellular localization of protein-HNE adducts in untreated (CTRL) and HNE-treated (+HNE) cells. Fixed cells were immunostained with anti-protein-HNE (green) and anti-KDEL (red) antibodies. Merged images (yellow) demonstrate the colocalization of protein-HNE adducts with the ER marker sequence, KDEL (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Identification of carbonylated proteins in rat aortic smooth muscle cells exposed to HNE.
| # | Protein | Accession gi number | MW (kDa) | pI | Coverage (%) | Function |
|---|---|---|---|---|---|---|
| 1 | PDI | 1352384 | 57.0 | 5.88 | 37 | ER chaperone/foldase |
| 2 | Grp58 | 8393322 | 57.0 | 5.88 | 22 | ER chaperone/foldase |
| 3 | Vimentin | 14389299 | 53.8 | 5.06 | 69 | Cytoskeletal protein |
| 4 | HSP 60 | 1334284 | 58.1 | 5.35 | 32 | Mitochondrial chaperone |
| 5 | Grp78 | 38303969 | 72.4 | 5.07 | 45 | ER chaperone |
| 6 | Reticulocalbin | 34856626 | 38.1 | 4.67 | 35 | ER calcium binding protein |
RASMC were exposed to 50 μM HNE for 30 min. Proteins in the cell lysates were subjected to 2D electrophoresis and analysis for protein carbonyls. Immunoreactive proteins were excised from parallel 2D silver-stained gels for MALDI-TOF/MS. Proteins were identified by peptide mass fingerprinting using the NCBI database and Mascot software (www.matrixscience.com). Rows in bold indicate proteins localized to the endoplasmic reticulum.
To confirm that the carbonylation of ER proteins observed by 2D Oxyblot analysis was due to direct protein-HNE modification, we immuno-labeled untreated and HNE-treated RASMC with anti-KDEL and anti-protein-HNE antibodies and analyzed their subcellular localization by confocal microscopy. Resident ER proteins commonly have a KDEL sequence at their carboxy terminus which functions as a retention motif and is recognized by the KDEL receptor on the ER membrane [22]; hence, the anti-KDEL antibody was used as a marker of the ER. Shown in Fig. 1B are representative fluorescence images of cells left either untreated (upper panel) or treated with HNE (lower panel) that were labeled with antibodies against protein-HNE adducts (green) and KDEL (red). The merged images (yellow) are shown at the far right. Control cells show light and diffuse HNE staining that does not localize strongly with the ER. Cells treated with HNE, however, show dense perinuclear staining that colocalizes primarily with the anti-KDEL signal. These data are consistent with mass spectrometric analysis, indicating that most of the HNE-modified proteins are located in the ER.
Activation of ER stress responses by HNE
To determine whether the formation of protein-HNE adducts in the ER triggers UPR, we examined changes in the PKR-like ER kinase (PERK) pathway. The interaction of Grp78 with PERK and inositol requiring enzyme-1 (IRE-1) has been proposed to regulate the ER stress response [23]. Accordingly, we examined whether HNE treatment results in the activation of PERK and its substrate for phosphorylation, eIF2α. As shown in Fig. 2A–i and ii, treatment of the cells with HNE for 30 min increased PERK and eIF2α phosphorylation to levels similar to that induced by tunicamycin treatment, which was used as a positive control. Interestingly, the anti-phospho-PERK antibody detected only one band at ∼250 kDa. Reprobing the same blot with the anti-PERK antibody showed a band at ∼125 kDa, and this band was decreased after HNE exposure when P-PERK immunoreactivity was increased (Supplementary Fig. 1). This high molecular mass form of phosphorylated PERK was also observed in cells treated with the ER stress inducers thapsigargin and tunicamycin. These data suggest that HNE treatment promotes the phosphorylation of PERK and that PERK activation in RASMC results in the formation of an SDS-resistant, irreducible dimer.
Fig. 2.
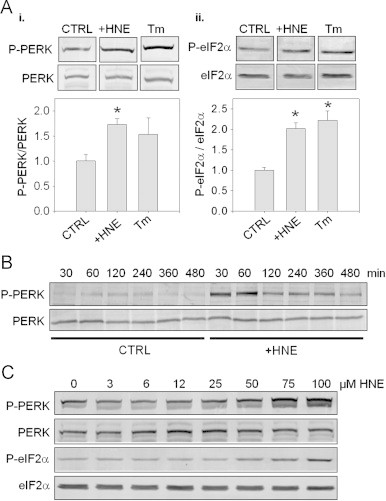
HNE activates the PERK pathway of the unfolded protein response. Detection of ER stress pathways by immunoblotting: (A) Western blot analysis of PERK (panel i) and eIF2α (panel ii) phosphorylation in lysates obtained from RASMC incubated in the absence (CTRL) or presence of 50 μM HNE (+HNE) or 10 μg/ml tunicamycin (+Tm) for 30 min. The extent of phosphorylation was calculated by normalizing the band intensities of the phospho-proteins to their respective loading controls. The values were then normalized to control groups (CTRL). Group data are mean±SEM. *p<0.02 vs. CTRL; n=3 per group. (B) Time course of PERK phosphorylation: cells were incubated in the absence (CTRL) or presence (+HNE) of 50 μM HNE for 30 min. The cells were then either lysed immediately or the medium was replaced and incubated for the indicated times in fresh culture medium. Lysates of cells were then used to assess PERK phosphorylation and expression. (C) Concentration-dependence of PERK and eIF2α phosphorylation by HNE: cells were treated with HNE (0–100 μM) for 30 min, and the cell lysates were used to analyze PERK and eIF2α phosphorylation.
To examine the temporal activation of PERK, RASMC were treated with HNE for 30 min. The HNE-containing medium was then removed and replaced with culture medium, and the cells were incubated for an additional 7.5 h. On completion of the treatment protocol, the cells were harvested at the indicated times, and phosphorylation of PERK in the lysates was analyzed by Western blotting. As shown in Fig. 2B, cells treated with HNE showed maximum PERK phosphorylation at 30 min, which declined to near control levels 480 min after treatment. It should be pointed out that the relative intensity of PERK phosphorylation in control cells appeared to be dependent on cell passage; however, in all passages used, HNE consistently increased PERK phosphorylation relative to that in untreated cells.
To examine the concentration-dependence of activation of the PERK-eIF2α pathway by HNE, RASMC were exposed to 0–100 μM HNE for 30 min. The cells were then lysed, and the phosphorylation of PERK and eIF2α were assessed by Western blotting. As shown in Fig. 2C, phosphorylation of PERK and eIF2α were observed after treatment with 50–100 μM HNE. This is in accordance with our previous studies showing that protein-HNE adducts form at these concentrations under identical conditions [14] and supports the notion that modification of proteins by HNE promotes ER stress.
In addition to the PERK pathway, ER stress could also result in the activation of inositol requiring enzyme 1 (IRE1), which directs the alarm phase of the UPR by stimulating alternative splicing of XBP-1 mRNA. Although RASMC treated with tunicamycin showed evidence of XBP-1 splicing and therefore IRE-1 activation, HNE did not affect the XBP-1 transcript (Suppl. Fig. 2), suggesting that under the conditions tested HNE does not activate the IRE1-XBP-1 pathway.
HNE activates JNK and p38
Previous studies show that HNE activates the stress kinases JNK and p38 [24,25]; however, the mechanisms remain unknown. Because these kinases are activated in response to ER stress by both the IRE1-TRAF and PERK pathways [26–28], we examined whether activation of the PERK pathway by HNE was accompanied by JNK activation. As shown in Fig. 3, exposure of RASMC to HNE led to a marked increase in the phosphorylation of both JNK and p38 (Fig. 3A). A significant increase in the levels of phospho-p38 was observed within 5 min of the addition of HNE to the medium, whereas, at least 30 min were required for an increase in JNK phosphorylation. Significantly, HNE treatment did not result in the induction of CHOP or Grp78 (Fig. 3A and C), although robust activation of CHOP and Grp78 was observed in tunicamycin- or thapsigargin-treated cells (Fig. 3B and C). Interestingly, we noticed that, at the 480 min timepoint, HNE-treated cells contained a lower molecular weight form of Grp78 (Fig. 3A); separate experiments confirmed the presence of this band (Fig. 3C), which may be due to degradation of oxidatively modified Grp78. Collectively, these data suggest that HNE activates the stress kinases JNK and p38, without increasing overall abundance of CHOP or native Grp78.
Fig. 3.
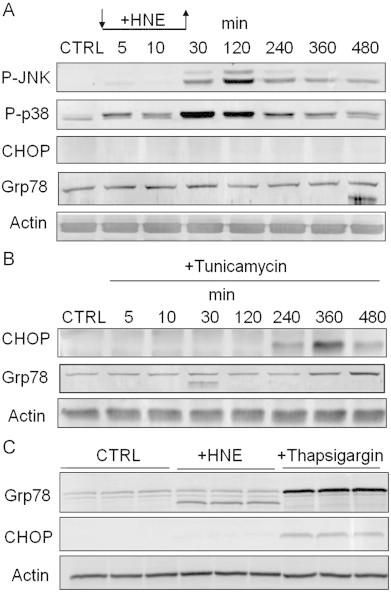
HNE activates stress kinases in smooth muscle cells. Immunoblot analysis of HNE- or ER stress inducer-treated cells: (A) Western blot analysis of stress kinase phosphorylation and the expression of the ER stress-related proteins CHOP and Grp78. Cells were incubated in the absence (CTRL) or presence of 50 μM HNE (+HNE), and the cells were harvested after 5, 10, or 30 min. For incubation at longer time points (120–480 min), HNE-containing medium was removed after 30 min, and the cells were incubated in fresh culture medium for the indicated times. (B) Cells were incubated in culture medium alone (CTRL) or in medium supplemented with 10 μg/ml tunicamycin for the indicated times. The expression of CHOP, Grp78, and actin was analyzed by Western blotting. (C) Cells were incubated in culture medium alone (CTRL) or in medium supplemented with 1 μg/ml thapsigargin for 8 h. The expression of CHOP, Grp78, and actin was analyzed by Western blotting. Results are representative of at least 3 separate experiments.
HNE-induced JNK phosphorylation, LC3 conversion, and HO-1 induction are dependent on ER stress
Because ER stress has been shown previously to regulate autophagy [29] and stress kinase activation [26,28], we pretreated RASMC with the chemical chaperone phenylbutyric acid (PBA), which facilitates protein folding and ameliorates ER stress [30]. RASMC pretreated with PBA (10 mM, 16 h) were exposed to HNE for 30 min, followed by incubation in the culture medium without or with PBA for an additional 1.5 h. The cells were then lysed and probed for JNK phosphorylation by Western blotting. As shown in Fig. 4A, pre-incubation with PBA decreased HNE-induced JNK phosphorylation by ∼40%. Interestingly, control cells treated with PBA also showed slightly lower levels of JNK phosphorylation, suggesting that even under basal conditions, JNK activation is, in part, regulated by ER-regulated pathways. In addition to preventing JNK activation, PBA also prevented HNE-induced LC3-II formation (Fig. 4B) and HO-1 induction (Fig. 4C). Taken together, these data support the notion that the activation of JNK by HNE is, in part, mediated by ER stress and that an increase in the protein folding capacity of the ER diminishes JNK activation and the stimulation of downstream events that lead to LC3-II conversion and HO-1 induction.
Fig. 4.
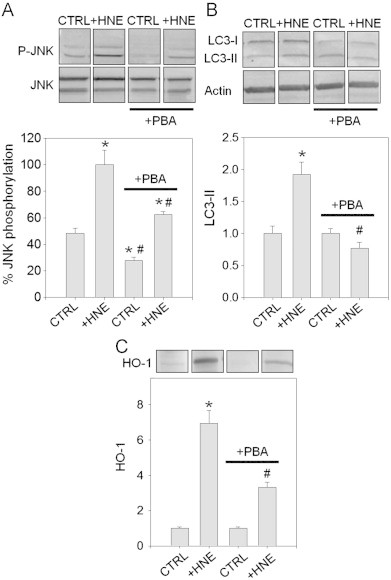
The chemical chaperone phenylbutyric acid (PBA) prevents HNE-induced JNK phosphorylation, LC3 conversion, and HO-1 induction in RASMCs. RASMCs were cultured in the absence or presence of 10 mM PBA (+PBA) for 16 h. The cells were then exposed to 50 μM HNE (for analysis of JNK phosphorylation and HO-1 induction) or 25 μM HNE (for LC3 conversion) in HBSS for 30 min in the absence or presence of PBA. After 30 min, the HBSS was replaced with culture medium without or with PBA, and the cells were incubated for 1.5, 2.5, or 7.5 h for analysis of JNK phosphorylation, LC3 conversion, or HO-1 induction, respectively. (A) JNK phosphorylation by HNE, as examined by Western blotting. For quantification, the band intensities of phospho-JNK (P-JNK) were normalized to total JNK in untreated (CTRL) or HNE-treated cells (+HNE) without or with PBA treatment. JNK phosphorylation after HNE treatment without PBA treatment=100%; *p<0.05 vs. CTRL without PBA, #p<0.01 vs. +HNE without PBA; n=3 per group. (B) LC3 conversion analyzed by Western blotting and normalized to actin expression. Data are mean±SEM normalized to controls. *p<0.005 vs. CTRL without PBA, #p<0.05 vs. +HNE group without PBA; n=5 per group. (C) HO-1 analyzed by Western blotting: band intensities of HO-1 were normalized to control groups. Data are mean±SEM. *p<0.002 vs. CTRL without PBA, #p<0.02 vs.+HNE without PBA; n=3 per group.
HNE-induced JNK activation, LC3-II conversion, and HO-1 induction are dependent on ER stress
Because JNK has been shown previously to regulate autophagy [31,32], we next determined whether JNK is required for the induction of autophagy by HNE. For this, we pretreated cells with the JNK inhibitor SP600125 and then with HNE for 30 min in the absence or presence of the inhibitor. The HNE-containing medium was then removed and replaced with culture medium for an additional 3.5 h. Following cell lysis, LC3-II formation was measured by Western blotting as a marker of autophagy. In agreement with previous findings [14], HNE significantly increased LC3-II formation (Fig. 5A). This increase in LC3-II was abrogated by the JNK inhibitor.
Fig. 5.
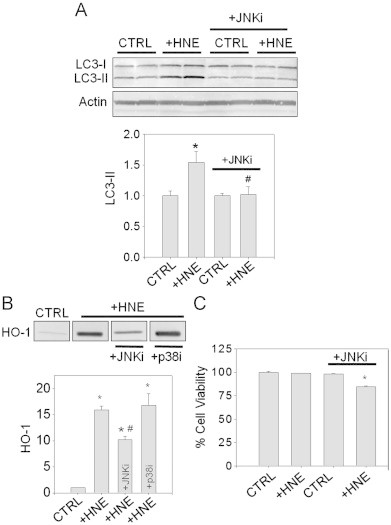
Autophagy and cellular protective responses to HNE are dependent on JNK. (A) JNK-dependent conversion of LC3: cells were incubated without (CTRL) or with HNE (25 μM; +HNE) for 30 min in the absence or presence of the JNK inhibitor SP600125 (+JNKi). The culture medium was then replaced with DMEM containing vehicle (DMSO) or JNKi, and cells were incubated further for 2.5 h before harvest. Lysates were used to analyze LC3 conversion and expression of actin by Western blotting. Band intensities of LC3-II were normalized to the band intensities of the loading control actin. Group data are mean±SEM, normalized to the controls. *p<0.03 vs. CTRL; #p<0.05 vs. +HNE group without JNKi; n=4 per group. (B) JNK-dependent HO-1 induction: Cells were treated with 50 μM HNE (+HNE) in the absence or presence of the JNK inhibitor SP600125 (+JNKi) or the p38 inhibitor SB203580 (+p38i). After 30 min of exposure, the medium was replaced with DMEM containing either inhibitor for 7.5 h. The cells were then lysed and equal amounts of protein were loaded for SDS-PAGE and Western blot analysis. Band intensities were normalized to the CTRL group. Data are mean±SEM. *p<0.001 vs. CTRL; #p<0.002 vs. +HNE group; n=3 per group. (C) JNK-dependent cell survival: RASMC were treated with 50 μM HNE (+HNE) in HBSS in the absence or presence of SP600125 (+JNKi). Control cells (CTRL) received vehicle (DMSO). After 30 min, this medium was removed and the cells were incubated in fresh medium in the absence or presence of the JNKi for 16 h. Cell viability was determined by lactate dehydrogenase assay. Data are mean±SEM as a percent of control. *p<0.0001 vs. all other groups; n=3 per group.
To determine whether JNK also plays a role in the induction of HO-1, RASMC were pre-incubated with the JNK inhibitor SP600125 or the p38 inhibitor SB203580 followed by treatment with HNE (50 μM) for 30 min. The HNE-containing medium was then removed and replaced with culture medium in the absence or presence of each inhibitor for an additional 7.5 h. As shown in Fig. 5B, treatment with SP600125, but not SB203580, prevented the increase in HO-1 in HNE-treated cells. These results demonstrate that JNK activation is required for HO-1 induction in HNE-treated cells.
To determine whether JNK activation is also required for cell survival following HNE treatment, RASMC were pretreated with vehicle or SP600125 for 30 min followed by treatment with 50 μM HNE for 30 min in the absence or presence of SP600125. The HNE-containing medium was then removed, and the cells were incubated for an additional 16 h in culture medium containing either the vehicle or SP600125. Cell viability was determined by LDH assay. As shown in Fig. 5C, cells treated with HNE or SP600125 alone did not show apparent cell death; however, cells treated with both SP600125 and HNE showed ∼15% cell death. Taken together, these data support the notion that, in HNE-treated cells, JNK activation is required for cell survival as well as for the induction of cytoprotective responses, i.e., stimulation of autophagy and the induction of HO-1 (Fig. 6).
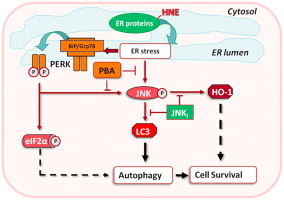
Fig. 6.
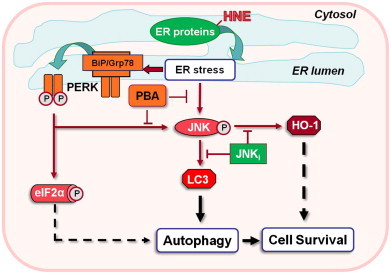
Proposed model of pro-survival pathways activated by HNE. HNE modification of ER proteins results in the activation of the adaptive arm of the unfolded protein response. This HNE-mediated response is characterized by the phosphorylation of PERK, eIF2α, and JNK, activation of autophagy, and the induction of HO-1. HNE-stimulated conversion of LC3 (an essential step in autophagosome formation and marker of autophagy) and HO-1 induction is shown to be mediated by ER stress via JNK activation.
Discussion
Our previous study shows that oxidized lipids stimulate autophagy, which promotes the removal of HNE-modified proteins [14]. In that study, we found that formation of protein-HNE adducts in RASMC was accompanied by the ultrastructural changes and LC3-II formation characteristic of autophagy. Moreover, the removal of protein-HNE adducts was accelerated by the autophagy stimulator rapamycin and decreased when autophagy was inhibited by 3-methyladenine. The goal of the current study was to determine the mechanism by which oxidized lipids stimulate autophagy. We found that HNE preferentially modifies chaperone proteins in the ER that regulate the initiation of UPR signaling and are required for protein folding. Exposure of the cells to HNE led to the selective activation of the PERK pathway, the upregulation of the cytoprotective protein HO-1, and the activation of stress kinases. Using chemical and molecular approaches, we found that induction of ER stress was obligatory for HNE-induced autophagy, with JNK activation being an essential downstream effector for LC3 conversion and maintenance of cell viability under conditions of excessive protein-HNE accumulation.
The mechanisms by which cells respond to proteins modified by lipid peroxidation products are not well understood. It is known that oxidation of unsaturated fatty acids generates a wide range of reactive carbonyls that trigger stress signaling and cause inflammation [1,24,25]. A key feature of the cytotoxicity caused by unsaturated carbonyls such as HNE is the formation of covalently modified proteins at nucleophilic amino acid side chains [1,8]. Proteins modified by HNE and related carbonyls have been detected in diseased tissues such as atherosclerotic plaques [33,34], cerebral lesions of Alzheimer's and Parkinson patients [4,6], and the ischemic myocardium [5]. Modification of proteins by products of lipid peroxidation and other electrophiles is likely to be harmful, not only because it disrupts protein function, but also because it leads to the accumulation of cross-linked proteins [35], which can inhibit the proteasome [36] and induce tissue dysfunction, inflammation, or apoptotic cell death [11,37]. The activation of signaling pathways by oxidized lipids appears to be particularly important for regulating these responses [24,25]. However, it is important to note that not all electrophiles will promote identical responses and that target specificity is likely important to the biological response (for review, see [38]).
The UPR is comprised of several signaling pathways that are responsive to oxidized lipids [39,40]. This response is triggered by protein misfolding and appears to have two primary roles in the cell: to re-establish proper protein folding or to trigger cell death. It prevents the accumulation of unfolded or misfolded proteins by transiently suppressing protein synthesis and increasing the ability of the ER to fold proteins; however, if protein folding and degradation pathways are completely overwhelmed, it coordinates signaling events to induce cell death. In mammalian cells the UPR is comprised of three distinct pathways—IRE1, PERK, and ATF6—that are thought to be activated following their dissociation from Grp78 [26]. However, not all inducers of ER stress trigger an identical UPR. Homocysteine, for instance, induces Grp78 and eIF2α in ARPE-10 cells [41]; but, in the same cells, arsenite does not induce Grp78, yet activates eIF2α [42]. Thapsigargin and dithiothreitol (DTT) trigger PERK-dependent JNK phosphorylation, but no such response was observed with tunicamycin [28]. Similarly in AR42J cells, IRE1α dissociation from Grp78 was more robust with DTT than with thapsigargin, although PERK was equally responsive to both agents [30]. Hence, it appears that UPR is a graded and cell-specific response tuned to particular metabolic and environmental conditions.
In HNE-treated RASMC we found significant increases in PERK and eIF2α phosphorylation as well as changes in JNK phosphorylation and HO-1 expression (Figs. 2 and 3). However, we found no evidence for robust stimulation of IRE1 or ATF6 as measured by XBP-1 splicing (Suppl. Fig. 2) or Grp78 expression (Fig. 3). The selective activation of the PERK pathway in HNE-treated cells therefore appears to be an early adaptive response that stimulates autophagy to counter oxidative insults rather than a full-blown response which could lead to apoptosis. This differs from HNE-induced responses in endothelial cells, where, in addition to PERK, HNE increases XBP-1 splicing and induces Grp78 [43]. That the ER stress response in SMCs is adaptive rather than deleterious is further supported by the fact that HNE treatment did not result in appreciable cell death or the upregulation of the death effector, CHOP, but induced the cytoprotective protein HO-1. These observations suggest that, in RASMC, the accumulation of HNE-modified proteins activates only a subset of the UPR pathways normally activated by misfolded proteins.
Our observations that HNE induces ER stress are consistent with previous work showing stimulation of UPR components in endothelial cells by oxidized phospholipids [39], HNE [40], and acrolein [44]. Reactive carbonyls such as HNE could trigger ER stress by several mechanisms. They could inhibit proteolysis [36,45], which would result in the accumulation of damaged proteins. Because a large fraction of proteins are incorrectly folded, even under basal conditions [46], inhibition of proteolysis results in accumulation of misfolded proteins and triggers UPR. Also, ER stress could be triggered by HNE by inducing cell-wide changes in calcium or reactive oxygen species production, which could secondarily affect protein folding pathways. Alternatively, HNE could interfere with protein folding by modifying ER proteins directly, thereby rendering them incapable of assisting in protein folding. Indeed, our proteomic analysis suggests that HNE modifies PDI, Grp58, and Grp78, which are key participants in protein folding. Previous studies have shown that modification by HNE prevents the catalytic activity of PDI [47]. Hence, modification of ER proteins by HNE could interfere with their folding capacity, leading to an increase in the accumulation of misfolded proteins or proteins that tend to aggregate.
That ER proteins are susceptible to modification by HNE is also supported by the localization of the protein-HNE adducts. By confocal microscopy, the majority of HNE-modified proteins appeared to colocalize with the ER sequence retention marker, KDEL (Fig. 1B). Reasons for the high vulnerability of ER proteins to modification by HNE are unclear, but may be related to the unique redox state of the ER, which maintains a highly oxidizing environment. In the cytosol, the ratio of reduced to oxidized glutathione is >50:1, whereas in the ER this ratio is 1:1 to 3:1 [48]. As a result, unsaturated carbonyls such as HNE appearing in the ER cannot likely be detoxified by glutathione. Moreover, aldehyde-metabolizing enzymes such as glutathione-S-transferase and aldehyde reductases and dehydrogenases (as well as their cofactors NAD+ and NADPH) may not be as abundant in the ER as they are in the cytosol. As a result, it is likely that proteins in the ER are more vulnerable to modification by HNE and related electrophiles.
Experiments using the chemical chaperone PBA support the concept that ER stress regulates autophagy. These interventions decreased HNE-induced LC3-II formation and prevented HO-1 induction in HNE-treated cells. From our analysis of ER pathways associated with the UPR, it appears that PERK activation is required for autophagy in RASMC in response to HNE. Several previous studies show that PERK activates autophagy [49–51]. Of interest was the degree to which HNE activated PERK and its shift to an apparent high molecular mass when phosphorylated. As shown in Fig. 2, HNE was just as robust as tunicamycin at inducing PERK phosphorylation. With HNE, tunicamycin, and thapsigargin treatment, PERK formed an apparent dimer that was not dissociated by reducing SDS-PAGE (e.g., see Suppl. Fig. 1). Reasons for this non-characteristic phosphorylated form of PERK remain unknown.
Our results also demonstrate that JNK activation is required for autophagy in HNE-treated cells. Pharmacological inhibition of JNK prevented LC3-II formation and decreased HO-1 expression in response to HNE treatment. Furthermore, treatment with the JNK inhibitor led to a small, but significant increase in cell death in HNE-treated cells. Inhibition of ER stress pharmacologically prevented JNK activation, LC3-II formation, and HO-1 induction, suggesting that activation of JNK by HNE is a response downstream of ER stress that is responsible for the induction of autophagy. Previous studies also suggest that ER stress activates JNK and that JNK regulates autophagy. The mechanisms by which ER stress leads to JNK activation appear to depend on the experimental condition and cell type. In fibroblasts, activation of JNK by ER stress was attenuated by deletion of IRE1α [31], but JNK activation by ER stress inducers was also impaired in PERK-null fibroblasts [28]. Because PERK, but not IRE1α, was activated by HNE, it appears that JNK activation during HNE-induced ER stress is mediated by PERK. PERK has been shown to be required for autophagy [51]; yet, in some cell types, IRE1α appears to be the predominant ER stress pathway that regulates autophagy [31,52]. Further studies are required to distinguish between the cytoprotective and death-effector roles of JNK and how it stimulates autophagy when the pathways of protein folding and degradation are compromised. In addition, it could be surmised that ER stress- and JNK-mediated autophagy or mitophagy impact other common targets of electrophiles such as mitochondria. Of note, smooth muscle cells in diseased vessels are characterized by increased levels of protein-HNE adducts [19,34,53], ER stress [54,55], and autophagosomes [32,56]. Relating these changes to disease progression and discerning molecular and therapeutic interventions are exciting prospects for future studies.
Acknowledgments
This work was supported in part by the NIH (National Institutes of Health) grant RR24489.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary Information
Supplementary data associated with this article can be found in the online version at doi:S0021-9290(13)00003-1.
Appendix A. Supplementary materials
Supplementary material
Reference
- 1.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology and Medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Porter N.A. Chemistry of lipid peroxidation. Methods in Enzymology. 1984;105:273–282. doi: 10.1016/s0076-6879(84)05035-7. [DOI] [PubMed] [Google Scholar]
- 3.Benedetti A., Fulceri R., Ferrali M., Ciccoli L., Esterbauer H., Comporti M. Detection of carbonyl functions in phospholipids of liver microsomes in CCl4- and BrCCl3-poisoned rats. Biochimica Biophysica Acta. 1982;712:628–638. doi: 10.1016/0005-2760(82)90292-2. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield D.A., Reed T., Perluigi M., De M.C., Coccia R., Cini C. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neuroscience Letters. 2006;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Eaton P., Li J.M., Hearse D.J., Shattock M.J. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. American Journal of Physiology. 1999;276:H935–H943. doi: 10.1152/ajpheart.1999.276.3.H935. [DOI] [PubMed] [Google Scholar]
- 6.Yoritaka A., Hattori N., Uchida K., Tanaka M., Stadtman E.R., Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proceedings of the National Academy of Sciences USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srivastava S., Chandrasekar B., Bhatnagar A., Prabhu S.D. Lipid peroxidation-derived aldehydes and oxidative stress in the failing heart: role of aldose reductase. American Journal of Physiology—Heart and Circulatory Physiology. 2002;283:H2612–H2619. doi: 10.1152/ajpheart.00592.2002. [DOI] [PubMed] [Google Scholar]
- 8.Lopachin R.M., Gavin T., Petersen D.R., Barber D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chemical Research in Toxicology. 2009 doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z.H., Saito Y., Yoshida Y., Sekine A., Noguchi N., Niki E. 4-Hydroxynonenal induces adaptive response and enhances PC12 cell tolerance primarily through induction of thioredoxin reductase 1 via activation of Nrf2. Journal of Biological Chemistry. 2005;280:41921–41927. doi: 10.1074/jbc.M508556200. [DOI] [PubMed] [Google Scholar]
- 10.Oh J.Y., Giles N., Landar A., Darley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochemical Journal. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glass C.K., Witztum J.L. Atherosclerosis—the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez J., Ballinger S.W., Darley-Usmar V.M., Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circulation Research. 2006;99:924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 13.Doorn J.A., Petersen D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chemico-Biological Interactions. 2003;143-144:93–100. doi: 10.1016/s0009-2797(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 14.Hill B.G., Haberzettl P., Ahmed Y., Srivastava S., Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochemical Journal. 2008;410:525–534. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 15.Shintani T., Klionsky D.J. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinet W., De Meyer G.R. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circulation Research. 2009;104:304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- 17.Higdon A.N., Benavides G.A., Chacko B.K., Ouyang X., Johnson M.S., Landar A. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. American Journal of Physiology—Heart and Circulatory Physiology. 2012;302:H1394–H1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava S., Chandra A., Wang L.F., Seifert W.E., Jr., DaGue B.B., Ansari N.H. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. Journal of Biological Chemistry. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava S., Ramana K.V., Tammali R., Srivastava S.K., Bhatnagar A. Contribution of aldose reductase to diabetic hyperproliferation of vascular smooth muscle cells. Diabetes. 2006;55:901–910. doi: 10.2337/diabetes.55.04.06.db05-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dulley J.R., Grieve P.A. A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Analytical Biochemistry. 1975;64:136–141. doi: 10.1016/0003-2697(75)90415-7. [DOI] [PubMed] [Google Scholar]
- 21.Reinheckel T., Korn S., Mohring S., Augustin W., Halangk W., Schild L. Adaptation of protein carbonyl detection to the requirements of proteome analysis demonstrated for hypoxia/reoxygenation in isolated rat liver mitochondria. Archives of Biochemistry and Biophysics. 2000;376:59–65. doi: 10.1006/abbi.1999.1680. [DOI] [PubMed] [Google Scholar]
- 22.Munro S., Pelham H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 23.Bertolotti A., Zhang Y., Hendershot L.M., Harding H.P., Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 24.Uchida K., Shiraishi M., Naito Y., Torii Y., Nakamura Y., Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. Journal of Biological Chemistry. 1999;274:2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 25.Usatyuk P.V., Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. Journal of Biological Chemistry. 2004;279:11789–11797. doi: 10.1074/jbc.M311184200. [DOI] [PubMed] [Google Scholar]
- 26.Xu C., Bailly-Maitre B., Reed J.C. Endoplasmic reticulum stress: cell life and death decisions. Journal of Clinical Investigation. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang K., Kaufman R.J. Signaling the unfolded protein response from the endoplasmic reticulum. Journal of Biological Chemistry. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 28.Liang S.H., Zhang W., McGrath B.C., Zhang P., Cavener D.R. PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochemical Journal. 2006;393:201–209. doi: 10.1042/BJ20050374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yorimitsu T., Klionsky D.J. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy. 2007;3:160–162. doi: 10.4161/auto.3653. [DOI] [PubMed] [Google Scholar]
- 30.Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R.O. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogata M., Hino S., Saito A., Morikawa K., Kondo S., Kanemoto S. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and Cellular Biology. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia G., Cheng G., Gangahar D.M., Agrawal D.K. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunology and Cell Biology. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 33.Gaut J.P., Heinecke J.W. Mechanisms for oxidizing low-density lipoprotein. Insights from patterns of oxidation products in the artery wall and from mouse models of atherosclerosis. Trends in Cardiovascular Medicine. 2001;11:103–112. doi: 10.1016/s1050-1738(01)00101-3. [DOI] [PubMed] [Google Scholar]
- 34.Salomon R.G., Kaur K., Podrez E., Hoff H.F., Krushinsky A.V., Sayre L.M. HNE-derived 2-pentylpyrroles are generated during oxidation of LDL, are more prevalent in blood plasma from patients with renal disease or atherosclerosis, and are present in atherosclerotic plaques. Chemical Research in Toxicology. 2000;13:557–564. doi: 10.1021/tx000007u. [DOI] [PubMed] [Google Scholar]
- 35.Cohn J.A., Tsai L., Friguet B., Szweda L.I. Chemical characterization of a protein-4-hydroxy-2-nonenal cross-link: immunochemical detection in mitochondria exposed to oxidative stress. Archives of Biochemistry and Biophysics. 1996;328:158–164. doi: 10.1006/abbi.1996.0156. [DOI] [PubMed] [Google Scholar]
- 36.Friguet B., Szweda L.I. Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Letters. 1997;405:21–25. doi: 10.1016/s0014-5793(97)00148-8. [DOI] [PubMed] [Google Scholar]
- 37.Uchida K. A lipid-derived endogenous inducer of COX-2: a bridge between inflammation and oxidative stress. Molecules and Cells. 2008;25:347–351. [PubMed] [Google Scholar]
- 38.Higdon A., Diers A.R., Oh J.Y., Landar A., Darley-Usmar V.M. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochemical Journal. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gargalovic P.S., Imura M., Zhang B., Gharavi N.M., Clark M.J., Pagnon J. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proceedings of the National Academy of Sciences USA. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanson M., Auge N., Vindis C., Muller C., Bando Y., Thiers J.C. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circulation Research. 2009;104:328–336. doi: 10.1161/CIRCRESAHA.108.183749. [DOI] [PubMed] [Google Scholar]
- 41.Roybal C.N., Yang S., Sun C.W., Hurtado D., Vander Jagt D.L., Townes T.M. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. Journal of Biological Chemistry. 2004;279:14844–14852. doi: 10.1074/jbc.M312948200. [DOI] [PubMed] [Google Scholar]
- 42.Roybal C.N., Hunsaker L.A., Barbash O., Vander Jagt D.L., Abcouwer S.F. The oxidative stressor arsenite activates vascular endothelial growth factor mRNA transcription by an ATF4-dependent mechanism. Journal of Biological Chemistry. 2005;280:20331–20339. doi: 10.1074/jbc.M411275200. [DOI] [PubMed] [Google Scholar]
- 43.Vladykovskaya E., Sithu S.D., Haberzettl P., Wickramasinghe N.S., Merchant M.L., Hill B.G. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. Journal of Biological Chemistry. 2012;287:11398–11409. doi: 10.1074/jbc.M111.320416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haberzettl P., Vladykovskaya E., Srivastava S., Bhatnagar A. Role of endoplasmic reticulum stress in acrolein-induced endothelial activation. Toxicology and Applied Pharmacology. 2009;234:14–24. doi: 10.1016/j.taap.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farout L., Mary J., Vinh J., Szweda L.I., Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Archives of Biochemistry and Biophysics. 2006 doi: 10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Schubert U., Anton L.C., Gibbs J., Norbury C.C., Yewdell J.W., Bennink J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 47.Carbone D.L., Doorn J.A., Kiebler Z., Petersen D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chemical Research Toxicology. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 48.Hwang C., Sinskey A.J., Lodish H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 49.Park M.A., Yacoub A., Sarkar D., Emdad L., Rahmani M., Spiegel S. PERK-dependent regulation of MDA-7/IL-24-induced autophagy in primary human glioma cells. Autophagy. 2008;4:513–515. doi: 10.4161/auto.5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park M.A., Curiel D.T., Koumenis C., Graf M., Chen C.S., Fisher P.B. PERK-dependent regulation of HSP70 expression and the regulation of autophagy. Autophagy. 2008;4:364–367. doi: 10.4161/auto.5593. [DOI] [PubMed] [Google Scholar]
- 51.Kouroku Y., Fujita E., Tanida I., Ueno T., Isoai A., Kumagai H. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell death and Differentiation. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 52.Ding W.X., Ni H.M., Gao W., Yoshimori T., Stolz D.B., Ron D. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. American Journal of Pathology. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jurgens G., Chen Q., Esterbauer H., Mair S., Ledinski G., Dinges H.P. Immunostaining of human autopsy aortas with antibodies to modified apolipoprotein B and apoprotein(a) Arteriosclerosis, Thrombosis and Vascular Biology. 1993;13:1689–1699. doi: 10.1161/01.atv.13.11.1689. [DOI] [PubMed] [Google Scholar]
- 54.Werstuck G.H., Khan M.I., Femia G., Kim A.J., Tedesco V., Trigatti B. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes. 2006;55:93–101. [PubMed] [Google Scholar]
- 55.Zhou J., Lhotak S., Hilditch B.A., Austin R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 56.Martinet W., De B.M., Schrijvers D.M., De Meyer G.R., Herman A.G., Kockx M.M. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arteriosclerosis, Thrombosis and Vascular Biology. 2004;24:2296–2301. doi: 10.1161/01.ATV.0000146266.65820.a1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material