

Abstract
Acute anemia increases the risk for perioperative morbidity and mortality in critically ill patients who experience blood loss and fluid resuscitation (hemodilution). Animal models of acute anemia suggest that neuronal nitric oxide synthase (nNOS)-derived nitric oxide (NO) is adaptive and protects against anemia-induced mortality. During acute anemia, we have observed a small but consistent increase in methemoglobin (MetHb) levels that is inversely proportional to the acute reduction in Hb observed during hemodilution in animals and humans. We hypothesize that this increase in MetHb may be a biomarker of anemia-induced tissue hypoxia. The increase in MetHb may occur by at least two mechanisms: (1) direct hemoglobin oxidation by increased nNOS-derived NO within the perivascular tissue and (2) by increased deoxyhemoglobin (DeoxyHb) nitrite reductase activity within the vascular compartment. Both mechanisms reflect a potential increase in NO signaling from the tissue and vascular compartments during anemia. These responses are thought to be adaptive; as deletion of nNOS results in increased mortality in a model of acute anemia. Finally, it is possible that prolonged activation of these mechanisms may lead to maladaptive changes in redox signaling. We hypothesize, increased MetHb in the vascular compartment during acute anemia may reflect activation of adaptive mechanisms which augment NO signaling. Understanding the link between anemia, MetHb and its treatments (transfusion of stored blood) may help us to develop novel treatment strategies to reduce the risk of anemia-induced morbidity and mortality.
Abbreviations: oxyhemoglobin, OxyHb; deoxyhemoglobin, DeoxyHb; methemoglobin, MetHb; neuronal nitric oxide synthase, nNOS
Keywords: Anemia, Methemoglobin, Nitrite reductase
Graphical abstract

Highlights
► Anemia is a risk for increased morbidity and mortality in surgical patients. ► Neuronal nitric oxide synthase (nNOS)-derived nitric oxide (NO) is adaptive in animal models of anemia. ► Decreasing hemoglobin levels leads to progressive tissue hypoxia and an associated increase in methemoglobin (MetHb) levels in peripheral blood. ► The increase in MetHb levels may be a biomarker of anemic tissue hypoxia. ► The mechanism for increased MetHb during anemia may include direct oxidation of Hb by NO and increased deoxyhemoglobin (DeoxyHb) nitrite reductase activity. ► Tissue nNOS and vascular DeoxyHb nitrite reductase may act cooperatively to generate increased levels of biologically active NO.
Introduction: what is methemoglobin?
Methemoglobin (MetHb) is a form of oxidized hemoglobin (Hb) normally maintained as a very small proportion of total hemoglobin (<1%), primarily by the action of red blood cell (RBC) MetHb reductase activity. Clinically significant methemoglobinemia can occur due to metabolic deficiencies in MetHb reductase and due to acquired exposure to oxidizing medications [1]. Until recently, the primary clinical significance of acute increases in MetHb levels was related to its inability to carry oxygen, leading to tissue hypoxia when MetHb levels increased to represent a significant portion of total Hb. However, we have recently proposed the novel hypothesis that small increases in MetHb levels may actually reflect an adaptive increased nitric oxide (NO) signaling during acute anemia [2,3].
What is the problem with anemia?
Acute and chronic anemia are well defined independent predictors of increased mortality in patients with systemic disease [4]. Acute blood loss and fluid resuscitation (hemodilution) are also associated with mortality in surgical patients in a manner that is proportional to the degree of Hb loss [4,5]. Currently, no treatment strategy has been shown to reduce this risk. While transfusion of stored allogeneic red blood cells (RBC) is utilized to restore optimal oxygen carrying capacity; collective data from randomized clinical trials has not demonstrated any survival benefit of RBC transfusion in critically ill or surgical patients [6–10]. In addition, new analysis suggests that stored RBCs may also be associated with increased mortality [11]. In search for an understanding of the mechanism of acute anemia-induced mortality, we have assessed the potentially adaptive role of nNOS-derived NO during acute anemia [12–14]. Our recent studies have shown that neuronal nitric oxide synthase (nNOS) knockout mice suffer increased mortality during acute anemia, supporting an adaptive role for nNOS in this setting [14]. In addition, we have proposed that the proportional but small increase in MetHb observed in acutely anemic animals and humans [2,3] may provide a means of assessing the degree of anemia-induced tissue hypoxia in the clinic. In this hypothesis article we propose a mechanism to link these experimental observations and propose that nNOS-derived NO and/or nitrite is important in MetHb formation which in turn may represent a novel indicator for anemia-dependent toxicity.
Anemia disrupts oxygen homeostasis and lead to tissue hypoxia
Experimental studies demonstrate that while global oxygen consumption is maintained during anemia, regional tissue hypoxia occurs and oxygen homeostasis is jeopardized. For example, during acute hemodilution (Hb∼50), overall brain oxygen consumption is maintained (to match constant O2 demand). This is achieved by many factors including a proportional increase in cerebral blood flow which compensates for the reduction in blood oxygen carrying capacity (in attempts to sustain O2 supply) and which would explain the mechanism for acute increases in the proportion of oxygen extracted by the brain as demonstrated in animal models [15]. In addition and as illustrated in Fig. 1, a rightward shift in the oxyhemoglobin dissociation curve (i.e. decreasing oxygen affinity due to increased 2,3 DPG) would facilitate increased oxygen extraction [16,17]. Whereas this phenomenon has been demonstrated in chronic anemia, this has never been demonstrated in the microcirculation of acutely anemic mammals. Finally, the contributing effects of increased microvascular blood flow, increased functional capillary density and/or a reduction in overall cerebral metabolism, are all mechanisms that may maintain cerebral oxygen homeostasis during acute anemia [18].
Fig. 1.
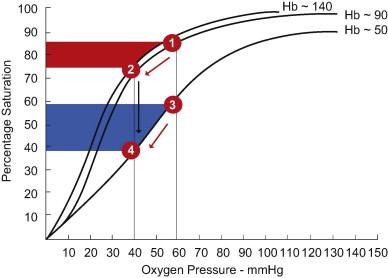
The impact of acute and chronic anemia on the oxyhemoglobin (OxyHb) dissociation curve. With an acute reduction in hemoglobin (from 140 through to 50 g/L), without any shift in the curve, the measured reduction in tissue oxygen tension (from 60 to 40 mmHg) would result in movement along the existing OxyHb dissociating curve (Hb∼140) from point 1 to 2. This would increase oxygen off-loading (extraction) as indicated by the red shaded area. During chronic anemia, a moderate reduction in Hb∼90 g/L does not result in a shift in the curve. However, with more severe anemia (Hb∼50), there is a significant rightward shift in the OxyHb dissociation curve and a decrease in Hb affinity for Oxygen. This effect would allow for a greater proportional off-loading of oxygen (blue shaded area) represented by a drop in OxyHb saturation from 3 to 4. The maximal effect of both acute and chronic anemia would be reflected by movement from point 1 to 4, thus optimizing oxygen unloading and maximally increased DeoxyHb levels. Modified from Rodman et al. [17]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Importantly however, compensated oxygen supply is not sufficient to fully meet the tissue demand during anemia as brain microvascular PO2 decreases proportionally with decreased Hb [14]. This reduction in local tissue PO2 results in-part from sustained tissue metabolic activity which facilitates increased dissociation of oxygen from hemoglobin (i.e. increased DeoxyHb) according to the well-defined oxyhemoglobin dissociation curve [16,17]. Recent assessment of the oxygen dependence of oxidative phosphorylation and the ability of mitochondria to act as oxygen sensors, suggest that tissue oxygen demand regulates physiologic responses such as the increase in cardiac output and regional tissue blood flow observed during anemia [19]. Evidence of increased expression of hypoxic cellular mediators including HIF and nNOS, suggests that the measured drop in tissue PO2 is biologically relevant and sensed by the brain parenchymal cell [13,14]. The importance of these adaptive responses is emphasized by the finding that mice lacking nNOS are susceptible to anemia induced mortality [14]. Thus, nNOS derived NO support survival during anemia.
Anemia, NO signaling and increased MetHb
The importance of nNOS to survival during anemia supports the paradigm that tissue derived NO signaling is important to maintain overall oxygen homeostasis during anemia. This effect is not observed with eNOS or iNOS indicating specificity regarding the mechanism and/ or compartmentalization of NO-formation in protecting against anemia [14]. One possibility is that nNOS activity primes or loads the vascular compartment with stable NO-metabolites, which are subsequently used to maintain vital tissue perfusion. Evidence of an nNOS mediated tissue-to-intravascular NO gradient are supported by three independent findings: Firstly, nNOS-derived NO is required for perivascular (endothelial) HIF stabilization in the anemic brain [14] which may serve to optimize homeostatic mechanisms during anemia, such as glucose transport [20]; Secondly, anemia results in a proportional increase in the oxidation of hemoglobin (Hb) to methemoglobin (MetHb) in a manner [2,3] that is partially dependent on nNOS (Fig. 2). In these experiments, progressive systematic normovolemic hemodilution was performed in wildtype (nNOS replete) and nNOS deficient mice (nNOS−/−), as previously reported [14]. Comparison of MetHb levels demonstrate an nNOS dependent increase in MetHb as total Hb levels decrease to levels near 60 g/L. Below this Hb level, there is a more marked increase in MetHb that is independent of nNOS (Fig. 2, possible reflecting increased DeoxHb nitrite reductase activity). During anemia, nNOS may be particularly well suited to provide a source of tissue NO as its activity requires a relatively high tissue PO2, which is the case during anemia, in contrast to hypoxia [21]. Finally, changes in vascular tone during anemia appear to be mediated by factors that are extrinsic to the resistance artery [14]. Thus, influences from the local environment on both the adventitial and/or luminal aspect of the microvasculature may regulate vascular tone and local blood flow during anemia. This constellation of findings led us to focus on the importance of NO-hemoglobin interactions in anemia.
Fig. 2.
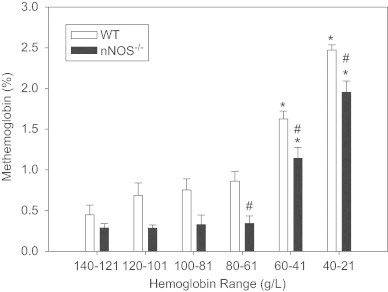
In acutely anemic mice, there is a progressive increase in MetHb levels as the total hemoglobin (Hb) decreases. This effect is attenuated in nNOS-deficient mice, demonstrating a degree of nNOS dependence to this process. However, at lower Hb levels, nNOS independent factors accentuate the increased in MetHb in both strains; possibly due to an increased nitrite reductase activity of DeoxyHb. These experiments were performed as per the hemodilution protocol outlined in Tsui et al. PNAS 2011 (ANOVA, group and hemoglobin effect, p<0.001 for both; post-hoc Tukeys test, ⁎p<0.05 from baseline, #p<0.05 between groups).
Herein we build upon the seminal findings that acute anemia causes a small (1–2%) but proportional and significant increase in MetHb in animal and human studies [2,3]. Potential mechanisms for increased methemoglobin include: (1) direct oxidation of Hb to MetHb by NO or peroxides [22,23]; (2) production of MetHb as an intermediate molecule in the biosynthesis of SNO-Hb [24]; and (3) as a product of deoxyhemoglobin (DeoxyHb) nitrite reductase activity [25]. Recently, a new mechanism has been described whereby MetHb can modulate NO signaling in the vascular compartment [26]. Straub et al provide evidence that regulation of MetHb levels by MetHb reductase (also known as cytochrome b5 reductase 3) can facilitate NO signaling at the level of the junction between endothelium and vascular smooth muscle (VSM). An increased level of MetHb (Fe3+ state), reduces NO binding and allows NO to readily diffuse to neighboring cells and activate the NO signaling pathways in the VSM. Thus, multiple paradigms exist for redox mediated changes in Hb can influence NO signaling within the vasculature. Regardless of the mechanism, we hypothesize that increased intraerythrocytic MetHb is an intravascular marker which reflects inadequate tissue oxygen delivery during acute anemia and reflects an associated increase in NO signaling, the goal of which is to improve tissue blood flow and maintain oxygen homeostasis during anemia.
Within the paradigm hypothesized above, methemoglobin represents an intriguing player, particularly as putative biomarker to indicate anemia. The degree of methemoglobin formation in this setting is still relatively low and unlikely to compromise oxygen carrying capacity (since methemoglobin does not bind oxygen). However, methemoglobin formation can also be viewed as a gain of toxic function, due to its potential to promote oxidative stress by reacting with peroxides (hydrogen peroxide, lipid hydroperoxides) and forming ferrylhemoglobin, a process illustrated by acellular hemoglobin transfusion dependent toxicity [27]. Red blood cells (RBC) are naturally endowed with a MetHb reductase system that maintains intraerythrocytic levels of MetHb at relatively low (<0.5%) levels. Since anemia usually occurs in the setting of a background inflammatory (and likely oxidative) stress it is possible that the ability of RBC to limit MetHb dependent oxidative stress is compromised. Thus, initially MetHb may represent a by-product of the RBC reflecting processes aimed at promoting NO-bioavailability and maintaining oxygen homeostasis. However, prolonged anemia (together with addition of stored RBCs which is pro-inflammatory, see below), may lead to an environment in which MetHb is driving the pathogenesis via oxidative stress.
The observed increased in tissue nNOS-derived NO could serve to link the cellular requirement for oxygen to the vascular compartment by an NO gradient from tissue to the vascular space. nNOS (vs. eNOS) is particularly suited to this function as the dependence of its activity on substrate oxygen, favors the production of large amounts of NO at relatively preserved high tissue pO2 values seen in anemia [21]. We hypothesize that nNOS-derived NO could serve to preload the vascular compartment with stable NO metabolites (e.g. nitrite). In addition, the ongoing tissue demand for oxygen and increased oxygen extraction is known to increase the proportion of DeoxyHb from ∼30% to ∼50% in the brain microvasculature. We posit that this metabolically driven increase in the proportion of DeoxyHb would facilitate production of vascular NO by DeoxyHb dependent nitrite reduction. The kinetics of this reaction may be favored by an increased in DeoxyHb and an overall reduction in total hemoglobin (i.e. NO binding sequestering capacity). Thus, by two coordinated mechanisms increases in tissue and vascular NO production may maintain oxygen homeostasis during acute anemia.
Nitrite reductase activity and NO-scavenging balance
Together with oxygen carrying capacity, red cells also control blood flow, thereby ensuring oxygen delivery meets local metabolic demands. Blood flow is controlled by mechanism(s) that couple hemoglobin deoxygenation with the stimulation of NO-dependent vasodilation. The proposed mechanisms have been discussed previously [28] with nitrite-reduction being one. The latter occurs by DeoxyHb mediated reduction of nitrite, intermediate formation of reactive nitrogen species, and ultimate formation of NO and metHb [29]. The initial rate of this reaction displays a bell shaped dependence on oxygen fractional saturation, increasing with desaturation from 100% to ∼50%, and then decreasing thereafter [30]. Thus over the range of oxygen fractional saturations observed in the cerebral circulation during anemia, without overt hypoxia, nitrite reduction by DeoxyHb, is predicted to be graded and proportional to the degree of desaturation. Importantly however, in order for any NO-generated by RBC to elicit a signaling effect in the vasculature, the rapid scavenging of NO- by oxy- or deoxyhemoglobin has to be overcome and underscores the overall paradigm that the balance between RBC-dependent NO-scavenging and formation will control function, with this balance being shifted to the latter during anemia (Fig. 3).
Fig. 3.
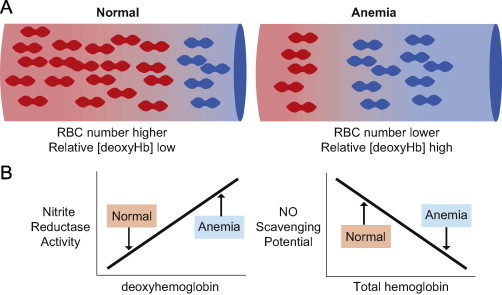
Hypothesis: anemia shifts the balance between nitrite reduction and NO-scavenging to the former. Relative to the normal situation, anemia is characterized by fewer RBC and more deoxyhemoglobin (Panel A). DeoxyHb can reduce nitrite to stimulate NO-dependent signaling (nitrite reductase activity). In contrast ferrous (oxy- or deoxy-) hemoglobin rapidly scavenges, and inhibits NO-signaling (NO-scavenging potential). Panel B shows hypothesized changes in these parameters during anemia. Specifically, a higher deoxyhemoglobin concentration coupled with decreased NO-scavenging (due to less hemoglobin) is proposed to promote NO-signaling and represent an adaptive response to maintain tissue perfusion during acute anemia.
Effect of RBC aging on nitrite reductase activity and increased mortality
The unresolved clinical paradox, that anemia and its treatment namely stored red blood cell transfusion are both associated with increased mortality may be partially explained by the effect of storage on RBC reactions with NO. Recently published data has demonstrated that stored human RBCs have an increase in NO scavenging [31,32]. In addition, we posit that due to storage dependent increased oxygen affinity, which in turn will limit deoxygenation, NO-formation from nitrite reduction will also be compromised [32]. These mechanisms may also help explain why blood transfusion of old stored blood has not improved survival. Indeed, transfusion of stored blood may actually decrease survival. Thus, further exploration of the hypothesis that old blood may short circuit the ‘normal’ balance between RBC dependent NO-formation vs. scavenging towards the latter and thereby impede regulatory mechanisms for increasing NO and local tissue blood flow during anemia is required.
Transfusion of HBOCs may increase mortality by impeding NO signaling as reflected by enhanced MetHb production
An additional link between the NO, hemoglobin and MetHb can be made by assessing the relationship between hemoglobin based oxygen carrier therapy, increased MetHb and mortality. In both experimental and clinical settings, transfusion of HBOCs results in a disproportionate increase in MetHb, likely due to the limited MetHb reductase activity in the plasma compartment. Experimental models show a constant rate of MetHb production over time in vivo. Increased rates of NO scavenging compared to erythrocytic hemoglobin, together with increased peroxide dependent oxidation of acellular hemoglobin (due to lower peroxide scavenging potential in the extracellular environment) most likely underscores MetHb formation in this setting. Thus, production of MetHb no longer reflects adaptive biological processes and NO is globally sequestered in the vascular compartment. This may explain the consistent finding that HBOC's can increase blood oxygen content and prevent transfusion but uniformly increase myocardial injury and mortality [33].
Conclusion
Acute anemia provides a unique environment within which to assess the importance of tissue and RBC based NO signaling mechanisms which attempt to maintain oxygen homeostasis and promote survival. The complexity of the interaction between tissue and vascular NO production in the setting of acutely reduced RBC Hb is complex and requires assessment in vivo in intact vascular beds. Based on experimental and clinical studies, we have proposed a potential mechanism by which anemia-induced tissue hypoxia may activate nNOS to generate biologically relevant tissue NO. The associated increase in tissue oxygen extraction, facilitated by the imbalance in oxygen supply and demand, may provide a vital link between the tissue and intravascular responses. By increasing the proportion of DeoxyHb during acute anemia, the kinetics of DeoxyHb nitrite reductase may be favored, leading to a further increase in local tissue NO signaling. Both mechanisms may be associated with an increase in MetHb levels providing a strong rationale for continued assessment of the hypothesis that increased MetHb is an important biomarker of anemia-induced tissue hypoxia.
Acknowledgments
Gregory M.T. Hare has received peer reviewed funds from the Canadian Anesthesiologists' Society and the Society of Cardiovascular Anesthesiologists to support this research. Support for his academic time is provided by the Department of Anesthesia, St. Michael's Hospital; a peer reviewed merit award from the Department of Anesthesia, University of Toronto and non-peer reviewed Salary Support from Johnson & Johnson Medical Companies as Co-Director of Center of Excellence for Patient Blood Management at St. Michael's Hospital. RPP acknowledges support from NIH RO1 HL095468 and HL092624.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Gregory M.T. Hare, Email: hareg@smh.ca.
Rakesh P. Patel, Email: rakeshp@uab.edu.
References
- 1.Ash-Bernal R., Wise R., Wright S.M. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004;83:265–273. doi: 10.1097/01.md.0000141096.00377.3f. [DOI] [PubMed] [Google Scholar]
- 2.Hare G.M., Mu A., Romaschin A. Plasma methemoglobin as a potential biomarker of anemic stress in humans. Canadian Journal of Anaesthesia. 2012;59:348–356. doi: 10.1007/s12630-011-9663-7. [DOI] [PubMed] [Google Scholar]
- 3.Tsui A.K., Dattani N.D., Marsden P.A. Reassessing the risk of hemodilutional anemia: some new pieces to an old puzzle. Canadian Journal of Anaesthesia. 2010;57:779–791. doi: 10.1007/s12630-010-9329-x. [DOI] [PubMed] [Google Scholar]
- 4.Shander A., Javidroozi M., Ozawa S., Hare G.M. What is really dangerous: anaemia or transfusion? British Journal of Anaesthesia. 2011;107(1):i41–i59. doi: 10.1093/bja/aer350. [DOI] [PubMed] [Google Scholar]
- 5.Carson J.L., Duff A., Poses R.M. Effect of anaemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348:1055–1060. doi: 10.1016/S0140-6736(96)04330-9. [DOI] [PubMed] [Google Scholar]
- 6.Carson J.L., Terrin M.L., Noveck H. Liberal or restrictive transfusion in high-risk patients after hip surgery. New England Journal of Medicine. 2011;365:2453–2462. doi: 10.1056/NEJMoa1012452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carson J.L., Carless P.A., Hebert P.C. Transfusion thresholds and other strategies for guiding allogeneic red blood cell transfusion. Cochrane Database of Systematic Reviews. 2012;4:CD002042. doi: 10.1002/14651858.CD002042.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajjar L.A., Vincent J.L., Galas F.R. Transfusion requirements after cardiac surgery: the TRACS randomized controlled trial. Journal of the American Medical Association. 2010;304:1559–1567. doi: 10.1001/jama.2010.1446. [DOI] [PubMed] [Google Scholar]
- 9.Hebert P.C., Wells G., Blajchman M.A. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. New England Journal of Medicine. 1999;340:409–417. doi: 10.1056/NEJM199902113400601. [DOI] [PubMed] [Google Scholar]
- 10.Lacroix J., Hebert P.C., Hutchison J.S. Transfusion strategies for patients in pediatric intensive care units. New England Journal of Medicine. 2007;356:1609–1619. doi: 10.1056/NEJMoa066240. [DOI] [PubMed] [Google Scholar]
- 11.Wang D., Sun J., Solomon S.B., Klein H.G., Natanson C. Transfusion of older stored blood and risk of death: a meta-analysis. Transfusion. 2012;52:1184–1195. doi: 10.1111/j.1537-2995.2011.03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hare G.M., Mazer C.D., Mak W. Hemodilutional anemia is associated with increased cerebral neuronal nitric oxide synthase gene expression. Journal of Applied Physiology. 2003;94:2058–2067. doi: 10.1152/japplphysiol.00931.2002. [DOI] [PubMed] [Google Scholar]
- 13.McLaren A.T., Marsden P.A., Mazer C.D. Increased expression of HIF-1{alpha}, nNOS, and VEGF in the cerebral cortex of anemic rats. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2007;292:R403–R414. doi: 10.1152/ajpregu.00403.2006. [DOI] [PubMed] [Google Scholar]
- 14.Tsui A.K., Marsden P.A., Mazer C.D. Priming of hypoxia-inducible factor by neuronal nitric oxide synthase is essential for adaptive responses to severe anemia. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17544–17549. doi: 10.1073/pnas.1114026108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Bommel J., Trouwborst A., Schwarte L., Siegemund M., Ince C., Henny C. Intestinal and cerebral oxygenation during severe isovolemic hemodilution and subsequent hyperoxic ventilation in a pig model. Anesthesiology. 2002;97:660–670. doi: 10.1097/00000542-200209000-00021. [DOI] [PubMed] [Google Scholar]
- 16.Sladen R.N. The oxyhemoglobin dissociation curve. International Anesthesiology Clinics. 1981;19:39–70. doi: 10.1097/00004311-198119030-00006. [DOI] [PubMed] [Google Scholar]
- 17.Rodman T., Close H.P., Purcell M.K. The oxyhemoglobin dissociation curve in anemia. Annals of Internal Medicine. 1960;52(295–309):295–309. doi: 10.7326/0003-4819-52-2-295. [DOI] [PubMed] [Google Scholar]
- 18.Hare G.M., Tsui A.K., McLaren A.T., Ragoonanan T.E., Yu J., Mazer C.D. Anemia and cerebral outcomes: many questions, fewer answers. Anesthesia and Analgesia. 2008;107:1356–1370. doi: 10.1213/ane.0b013e318184cfe9. [DOI] [PubMed] [Google Scholar]
- 19.Wilson D.F., Harrison D.K., Vinogradov S.A. Oxygen, pH, and mitochondrial oxidative phosphorylation. Journal of Applied Physiology. 2012;113(12):1838–1845. doi: 10.1152/japplphysiol.01160.2012. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y., Lei L., Liu D. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17478–17483. doi: 10.1073/pnas.1209281109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Semenza G.L. New insights into nNOS regulation of vascular homeostasis. Journal of Clinical Investigation. 2005;115:2976–2978. doi: 10.1172/JCI26792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel R.P., Hogg N., Kim-Shapiro D.B. The potential role of the red blood cell in nitrite-dependent regulation of blood flow. Cardiovascular Research. 2011;89:507–515. doi: 10.1093/cvr/cvq323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alayash A.I., Patel R.P., Cashon R.E. Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxidants and Redox Signaling. 2001;3:313–327. doi: 10.1089/152308601300185250. [DOI] [PubMed] [Google Scholar]
- 24.Singel D.J., Stamler J.S. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annual Review of Physiology. 2005;67(99–145):99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 25.Gladwin M.T., Raat N.J., Shiva S. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. American Journal of Physiology Heart and Circulatory Physiology. 2006;291:H2026–H2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 26.Straub A.C., Lohman A.W., Billaud M. Endothelial cell expression of haemoglobin alpha regulates nitric oxide signalling. Nature. 2012;491:473–477. doi: 10.1038/nature11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buehler P.W., D'Agnillo F., Schaer D.J. Hemoglobin-based oxygen carriers: From mechanisms of toxicity and clearance to rational drug design. Trends in Molecular Medicine. 2010;16:447–457. doi: 10.1016/j.molmed.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Owusu B.Y., Stapley R., Patel R.P. Nitric oxide formation versus scavenging: the red blood cell balancing act. Journal of Physiology. 2012;590:4993–5000. doi: 10.1113/jphysiol.2012.234906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu S., Grubina R., Huang J. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nature Chemical Biology. 2007;3:785–794. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- 30.Crawford J.H., Isbell T.S., Huang Z. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donadee C., Raat N.J., Kanias T. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465–476. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stapley R., Owusu B.Y., Brandon A. Erythrocyte storage increases rates of NO and nitrite scavenging: implications for transfusion-related toxicity. Biochemical Journal. 2012;446:499–508. doi: 10.1042/BJ20120675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natanson C., Kern S.J., Lurie P., Banks S.M., Wolfe S.M. Cell-free hemoglobin-based blood substitutes and risk of myocardial infarction and death: a meta-analysis. Journal of the American Medical Association. 2008;299:2304–2312. doi: 10.1001/jama.299.19.jrv80007. [DOI] [PMC free article] [PubMed] [Google Scholar]