

Abstract
Superoxide dismutase (EC-SOD) controls the level of superoxide in the extracellular space by catalyzing the dismutation of superoxide into hydrogen peroxide and molecular oxygen. In addition, the enzyme reacts with hydrogen peroxide in a peroxidase reaction which is known to disrupt enzymatic activity. Here, we show that the peroxidase reaction supports a site-specific bond cleavage. Analyses by peptide mapping and mass spectrometry shows that oxidation of Pro112 supports the cleavage of the Pro112–His113 peptide bond. Substitution of Ala for Pro112 did not inhibit fragmentation, indicating that the oxidative fragmentation at this position is dictated by spatial organization and not by side-chain specificity. The major part of EC-SOD inhibited by the peroxidase reaction was not fragmented but found to encompass oxidations of histidine residues involved in the coordination of copper (His98 and His163). These oxidations are likely to support the dissociation of copper from the active site and thus loss of enzymatic activity. Homologous modifications have also been described for the intracellular isozyme, Cu/Zn-SOD, reflecting the almost identical structures of the active site within these enzymes. We speculate that the inactivation of EC-SOD by peroxidase activity plays a role in regulating SOD activity in vivo, as even low levels of superoxide will allow for the peroxidase reaction to occur.
Abbreviations: DDC, diethyldithiocarbamate; DMPO, 5,5-dimethyl-pyrroline N-oxide; EC-SOD, extracellular superoxide dismutase; FA, formic acid; MALDI, matrix assisted laser desorption/ionization; TFA, trifluoroacetic acid
Keywords: EC-SOD, Peroxidase activity, Inhibition, Oxidation
Graphical abstract
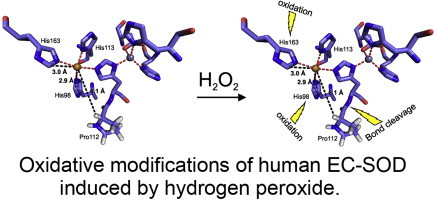
Oxidative modifications of human EC-SOD induced by hydrogen peroxide.
Highlights
► Hydrogen peroxide inhibits EC-SOD by oxidation of amino acid residues in spatial proximity to the active site copper. ► The coordination sphere of copper is disrupted by the oxidation of His98 or His163. ► Protein fragmentation is mediated by cleavage of the Pro112-His113 peptide bond. ► Fragmentation is induced by hydrogen abstraction on the carbon of Pro112.
Introduction
Extracellular superoxide dismutase (EC-SOD) is an extracellular antioxidant that catalyzes the dismutation of superoxide (O2•−) into hydrogen peroxide (H2O2) and molecular oxygen (O2). The protein is highly expressed in the lung and the vasculature where it is immobilized by the binding to a number of ligands in the extracellular space [1–4]. Hence, EC-SOD plays an important role in diseases where reactive oxygen species are known to be involved, including hypertension, atherosclerosis, and pulmonary fibrosis [5]. Recently, EC-SOD has also been shown to be involved in bacterial clearance [6,7], suggesting a role not only in protection against superoxide-mediated macromolecular damage [3,4,8,9] but also as a component involved in modulating the immune response to bacterial infection.
The amino acid sequence representing the central part of EC-SOD is homologous to the sequence of the intercellular counterpart, Cu/Zn-SOD [10]. Structural analyses show that Cu/Zn-SOD and the central part of EC-SOD shares the same fold albeit with some minor differences in loop structures [11,12]. The amino acid residues involved in coordination of the catalytic active copper atom and the zinc atom are conserved and share nearly identical spatial orientations, supporting the finding that the enzymatic properties of these enzymes are comparable. Apart from the ability to dismutate the superoxide radical, Hodgson and Fridovich showed that Cu/Zn-SOD also displayed peroxidase activity and found that this reaction inhibited the enzyme [13]. A large body of literature has described this activity in great detail suggesting the formation of a copper-bound hydroxyl radical (OH•) that mediates the loss of SOD activity by subsequent oxidations [14]. By using HPLC and electrochemical detection, Uchida and Kawakishi showed that His118 (numbering according to bovine protein) was oxidized to 2-oxo-histidine in Cu/Zn-SOD inactivated by H2O2 [15]. A more recent study using LC–MS/MS revealed that His44, His46, as well as His118 were oxidized after exposure to H2O2 [16]. These histidine residues are all known to coordinate copper [11] and oxidation is thus likely to destroy enzymatic activity by disrupting the coordination sphere. In addition, studies have shown that Pro60 can be oxidized leading to fragmentation of the Pro60–His61 peptide bond [16,17]. However, although several mechanisms for oxidative fragmentation at the prolyl bond have been proposed [18–20], it is not clear how oxidation at Pro60 mediates fragmentation of Cu/Zn-SOD.
The peroxidase activity and subsequent inhibition of enzymatic activity has also been shown for EC-SOD [21]. Interestingly, these authors showed that the presence of uric acid at physiological relevant concentrations could prevent inhibition in vitro. Moreover, when using an in vivo model of atherosclerosis, they were able to show that EC-SOD was partially inhibited by H2O2 during oxidative stress and that the activity was restored after increasing the level of urate in the circulation [21]. In line with these results, inhibition of EC-SOD activity has also been suggested to play a role in high-volume hypertension [22] and in persistent pulmonary hypertension of the newborn [23]. Collectively, these studies show that the inhibition of EC-SOD by H2O2 is relevant to disease, and that inhibition may allow for the development and progression of diseases involving reactive oxygen species.
Here we describe the mechanism of H2O2-induced EC-SOD inhibition and show that the modifications are similar to those described for Cu/Zn-SOD including histidine oxidation and site-specific fragmentation. Our data underscore that EC-SOD is subject to product inhibition and that the presence of EC-SOD subunits in tissue extracts and extracellular fluids may not necessarily correlate with protein activity.
Materials and methods
Proteins and reagents
Human EC-SOD was purified from aorta tissue or from cell culture supernatants by heparin-affinity chromatography and anion exchange chromatography as previously described [24]. Standard chemicals including diethylene triamine pentaacetic acid (DTPA) and α-cyano-4-hydroxysinnamic acid were obtained from Sigma. The MALDI matrix 2,5-dihyroxyacetophenone (DHAP) and standards for calibration of the mass spectrometer were obtained from Bruker Daltonics. The spin trap 5,5-dimethyl-pyrroline N-oxide (DMPO) was purchased from Enzo Life Sciences and diethyldithiocarbamate (DDC) and hydrogen peroxide (30%) were from Merck. Sequence grade porcine trypsin and bovine chymotrypsin were obtained from Promega and xanthine oxidase and PNGaseF were from Roche.
Expression of recombinant P112A human EC-SOD
The sequence encoding full-length EC-SOD with an optimized Kozak sequence was previously established in the pIRES vector [25]. The Pro112Ala substitution was introduced by PCR using the Quick change site-directed mutagenesis kit provided by Stratagene and the sequence of the obtained expression plasmid was verified by sequencing. HEK293 cells were stably transfected as previously described and protein expression conducted in serum-free medium [25]. The expressed P112A EC-SOD was active as evaluated by using the cytochrome C assay (see below) and activity staining [24] indicating that the protein was folded correctly.
Exposure of purified EC-SOD to hydrogen peroxide
Samples containing purified EC-SOD were prepared in PBS containing 0.1 mM DTPA and increasing amounts of H2O2 as indicated. In order to evaluate the role of the copper ion within the EC-SOD subunit, DDC was added to the samples and allowed to incubate 5 min prior to the addition of H2O2. When indicated, 1 mM DMPO was added to the reaction mixture to allow for detection of any protein-centered radicals generated. The reaction mixtures were incubated for 1 h at 37 °C and processed for further analysis (see below).
SDS-PAGE and protein visualization
Prior to electrophoresis, H2O2 was removed by reverse-phase chromatography using Poros50 R1 micro-columns as previously described [26]. Proteins were separated by polyacrylamide gel electrophoresis using uniform 10% polyacrylamide gels and the glycine/2-amino-2-methyl-1,3-propanediol-HCl buffer system [27]. Samples were analyzed under reducing conditions by boiling in the presence of 0.5% (w/v) SDS and 50 mM dithiothreitol prior to electrophoresis. Separated proteins were subsequently visualized by silver staining.
SOD activity assay
The activity of EC-SOD exposed to H2O2 was evaluated using the cytochrome C assay [28] modified for use in a 96-well plate format. In brief, samples containing EC-SOD and H2O2 were diluted in 50 mM NaHCO3, 0.1 mM EDTA, pH 10 containing 0.1 mM xanthine and cytochrome C and 100 μl added to wells of a microtiter plate. As negative control, wells received 100 μl reaction mixtures without EC-SOD. To initiate the reaction, 100 μl of 50 mM NaHCO3, 0.1 mM EDTA, pH 10 containing cytochrome C and xanthine oxidase was added and the absorbance at 550 nm measured between 0 and 2 min with intervals of 20 s using an EnSpire 2300 multimode plate reader (Perkin Elmer). The SOD activity was evaluated as ΔAbs/min and the relative activity determined by defining the activity of the sample containing no H2O2 as 100%.
Peptide maps of EC-SOD exposed to H2O2
EC-SOD exposed to H2O2 was desalted into 50 mM Tris–HCl, pH 8.0 containing 6 M guanidinium hydrochloride, 10 mM 8-hydroxyquinoline and 30 mM DTT by using Zeba spin desalting columns (Thermo Scientific). The material was incubated at 37 °C for 30 min and subsequently added 60 mM iodoacetamide to block free cysteine residues and incubated for another 30 min at 22 °C in the dark. The material was acidified and recovered by desalting using a Poros50 R1 reverse-phase micro-column, lyophilized, and subsequently added porcine trypsin in 50 mM ammonium bicarbonate. The digestion was allowed to proceed at 37 °C over night. The generated peptides were separated by UPLC reverse-phase chromatography by using a BEH300 C18 column (2.1 mm×15 cm; 1.7 μm) operated by an Aquity UPLC system (Waters). The column was developed at a flow rate of 300 μl min−1 using a 1% B min−1 linear gradient of solvent B (90% acetonitrile, 0.08% (v/v) trifluoroacetic (TFA)) in solvent A (0.1% (v/v) TFA). Fractions were collected manually and analyzed by MALDI mass spectrometry.
Mass spectrometric analyses
Samples containing EC-SOD were acidified by the addition of TFA and H2O2 was removed by desalting using Poros50 R1 reverse phase micro-columns. The recovered protein was lyophilized and resuspended in 0.1% TFA and mixed thoroughly with DHAP matrix solution prepared in 20 mM diammonium hydrogen citrate, 75% (v/v) ethanol. The mass spectra were acquired using a Bruker Autoflex III instrument operated in linear mode and calibrated in the mass range from 5000 Da to 17,500 Da using Protein calibration standard I (Bruker Daltronics). The analysis of tryptic peptides recovered by off-line reverse phase chromatography was performed using α-cyano-4-hydroxycinnamic acid prepared in 70% (v/v) acetonitrile, 0.1% (v/v) TFA acid. The instrument was operated in reflector mode and calibrated in the mass range from 1000 Da to 3200 Da using Peptide calibration standard (Bruker Daltronics). When appropriate, MALDI spectra were evaluated by using the GPMAW software (Lighthouse data).
For analysis of material by SDS-PAGE and in-gel digestion, desalted protein was solubilized in PBS containing 0.1% SDS and boiled for 3 min. The material was allowed to cool and added PNGaseF and incubated at 37 °C for 2 h. The cleaved material was subjected to SDS-PAGE as described above and protein bands were excised and subjected to in-gel digestion using sequence grade trypsin or chymotrypsin as indicated [29]. The material was lyophilized and resuspended in 0.1% TFA for analysis by MALDI-MS/MS as described above or in 0.1% formic acid (FA) for analysis by LC–MS/MS by using an Ultimate 3000 system (Thermo Scientific) connected to a MircoTOF-Q II mass spectrometer (Bruker Daltronics). The peptides were recovered using an Acclaim PepMap100 column (75 μm ID, 15 cm length) and eluted by a linear gradient of solvent B (90% acetonitrile, 0.08% (v/v) FA) in solvent A (0.1% (v/v) FA) from 5% to 50% with a flow rate of 200 nl min−1. The obtained mass spectra were analyzed using the Mascot 2.3.02 and the SwissProt database as reference. Search parameters were set to allow for one missed cleavage, propionamide (C) as a fixed modification, and oxidation of methionine, histidine, and tryptophan as variable modifications. Due to PNGaseF treatment, the substitution of Asp for Asn was also included as a variable modification. Peptide mass tolerance offset and fragment ion tolerance offset were set to 20 ppm and 0.8 Da, respectively.
Results and discussion
EC-SOD is inhibited and fragmented by H2O2
The impact of H2O2 on the enzymatic activity of EC-SOD was evaluated by titrating a fixed concentration of EC-SOD with increasing levels of H2O2 and the residual activity was subsequently determined using the cytochrome C assay [28]. The activity of the protein was reduced dose dependently and showed a 50% reduction in the presence of 0.8 mM H2O2 and complete inhibition was obtained in the presence of 2 mM H2O2 (Fig. 1A). To investigate the impact of oxidative stress on protein structure, we further subjected the samples to analysis by SDS-PAGE under reducing conditions. The analyses revealed that upon increasing amounts of H2O2 the EC-SOD subunits migrated the same (Fig. 1B). However, three bands with apparent masses of 20, 15, and 13 kDa could be detected by extended silver staining in samples exposed to >0.2 mM H2O2, suggesting that a minor fraction of the protein was fragmented. Fragmentation of EC-SOD induced by oxidative stress has previously not been reported, but incubation of Cu/Zn-SOD at conditions allowing for glycation was found to induce fragmentation [17]. As estimated by protein staining (Fig. 1A), it is clear that only a minor part of EC-SOD is degraded and that EC-SOD subunits with and without activity migrate the same, implying that protein visualization by staining or western blotting may not correlate directly to protein activity.
Fig. 1.
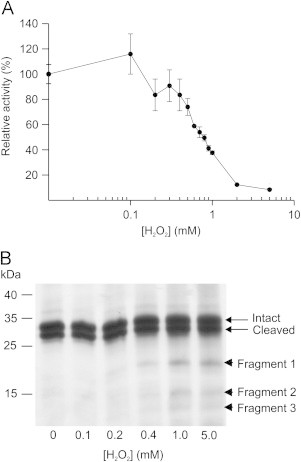
EC-SOD is sensitive to H2O2 exposure. (A) The activity of EC-SOD was monitored by using the cytochrome C/xanthine oxidase assay. The activity was inhibited by H2O2 (37 °C, 1 h) in a dose dependent manner producing an IC50-value of 0.8 mM and complete inhibition at ∼2 mM. Data points represent the mean and error bars the standard deviation of triplicates. (B) The structural integrity of EC-SOD exposed to the indicated concentrations of H2O2 was evaluated by reduced SDS-PAGE and followed by silver staining. Apart from the intact and cleaved subunit, three fragments can be detected at increasing H2O2 concentrations as indicated on the right. These analyses show that EC-SOD is inhibited by H2O2 and that the major part of inhibited EC-SOD maintains structural integrity albeit fragmentation of the protein can be detected.
Peptide map of EC-SOD subjected to oxidative stress
As the exposure of EC-SOD to H2O2 produces three well defined protein fragments, this suggests that the site of fragmentation is specific. To characterize the fragmentation, we subjected EC-SOD to tryptic digestion and separated the generated peptides by reverse-phase chromatography. When EC-SOD was subjected to increasing amounts of H2O2, the intensity of a single chromatographic peak was found to be significantly reduced (Fig. 2). Mass spectrometric analysis of this peak identified an ion of m/z 4491.1 corresponding to the tryptic peptide Ala94–Arg134 modified by S-carbamidomethylation at Cys107 (m/zcalc. 4491.1). No concomitant increase in absorption from peptides representing fragments could be appreciated. To evaluate the involvement of the copper ion in the fragmentation by H2O2, we added DDC to the reaction mixture in order to chelate the catalytic copper ion of EC-SOD [30]. The analysis in the absence of coordinated copper showed that the intensity of the peak representing Ala94–Arg134 remained unchanged compared to the holo-enzyme. This indicates that the presence of copper within the active site of EC-SOD is required to support the fragmentation of the protein, likely by the formation of a copper-bound hydroxyl radical as detected in Cu/Zn-SOD [13,14].
Fig. 2.
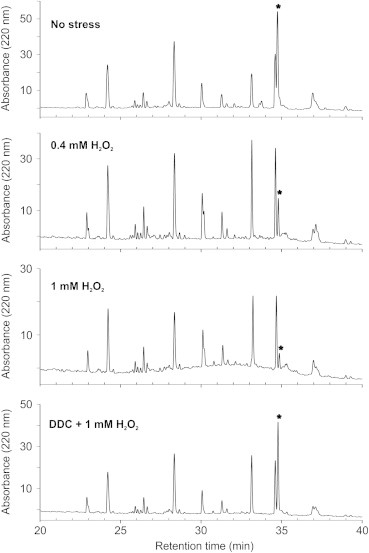
Tryptic peptide maps of EC-SOD exposed to H2O2. EC-SOD and H2O2 were incubated at the indicated conditions and protein subsequently digested using trypsin. The digests were separated by reverse-phase UPLC and the peptides were detected by absorbance at 220 nm. The peptide maps showed that the absorption of the S-carbamidomethylated peptide Ala94–Arg143 (⁎) was reduced by increasing amounts of H2O2 indicating that this peptide is modified by oxidation. No significant increase in concomitant absorption could be detected in the maps. The presence of DDC inhibits the modification of the peptide showing that copper is central for the process.
Mass spectrometric characterization of EC-SOD fragmentation induced by H2O2
To further characterize the fragmentation induced by H2O2, we subjected exposed material to analysis by linear mode MALDI mass spectrometry. In the absence of stressor, the mass spectrum showed the presence of the intact (m/zcalc. 26,485.0)1 and cleaved (m/zcalc. 24,885.1) EC-SOD subunit containing a sialylated complex-type glycan on Asn89 [31]. In addition, masses representing the corresponding subunits lacking one sialic acid on the glycan structure were evident (Δm/z 291; Fig. 3). Moreover, the double charged species could be observed. When material subjected to 1 mM H2O2 was analyzed, the intensity of ions representing the intact and cleaved subunits were significantly reduced and was only observed just above background level. However, one intense peak of m/z 10,232.9 was observed, indicating fragmentation of the EC-SOD subunits (Fig. 3). This peak does not show any indication of sialic acid loss, suggesting that the fragment does not encompass Asn89. When EC-SOD was incubated with DDC prior to H2O2 exposure, the resulting mass spectrum showed the ions representing the intact and cleaved subunits as detected in the sample without stress (Fig. 3). This finding is in line with the peptide map analysis showing that the fragmentation of EC-SOD by H2O2 is mediated by the copper ion. A corresponding conclusion has likewise been established from the study of H2O2-induced Cu/Zn-SOD fragmentation [14]. The formation of a copper-bound hydroxyl radical is likely to mediate the subsequent formation of protein-centered radicals. With the aim to trap such radicals, we subjected protein to H2O2 in the presence of the spin trap DMPO, which is known to quench radical formation [32]. Mass spectrometric analysis showed that no complexes between EC-SOD and DMPO could be detected (Fig. 3). However, two fragment ions of m/z 10,233.1 and m/z 11,833.5 was observed. In addition, two minor peaks with low resolution were observed representing an ion of m/z∼14,657 and an associated ion corresponding to loss of sialic acid indicating that this fragment includes the glycosylated Asn89 residue (Fig. 3). These data show that trapping of protein-centered radicals could not be facilitated by the presence of DMPO. The inability of DMPO to quench the fragmentation has previously been shown for Cu/Zn-SOD subjected to H2O2 [14]. To determine the nature of the observed fragments, the obtained m/z values was searched against the EC-SOD sequence by using the GPMAW software allowing a mass offset of 200 ppm. The fragments of m/z 10,233.1 and m/z 11,833.5 are likely to represent His113–Glu209 (m/zcalc.10,234.4) and His113–Ala222 (m/zcalc.11,834.3), respectively. Despite the low resolution, the ion of m/z∼14,657 is likely to represent the glycosylated Trp1–Pro112 fragment (m/zcalc.14,669.8). These data indicate that the Pro112–His peptide bond in EC-SOD is subject to cleavage induced by the peroxidase reaction. The homologous peptide bond in Cu/Zn-SOD was also identified as the site of fragmentation by amino acid sequence analysis [17]. Radical-mediated cleavage on the C-terminal side of a prolyl peptide bond has previously been described, and suggests the formation of a 2-pyrrolidone structure in the final product representing a mass reduction of the terminal proline residue corresponding to 30 Da [19,20]. Due to the low resolution of the N-terminal protein fragment obtained by mass spectrometry, it was not possible to address the presence of a 2-pyrrolidone structure.
Fig. 3.
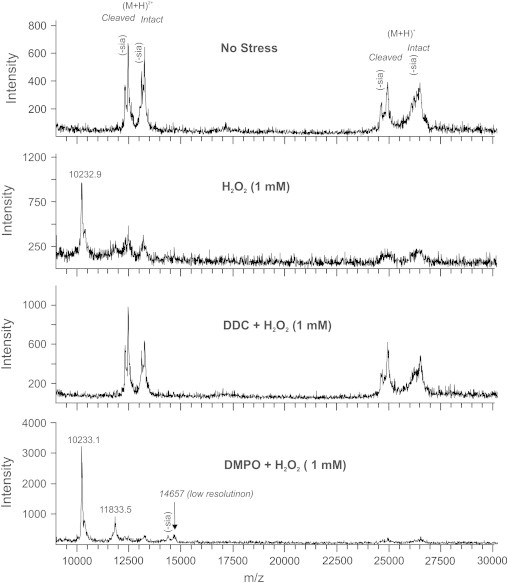
Mass spectrometric analysis of fragmentation. EC-SOD subjected to the indicated conditions was analyzed by MALDI-MS using 2,5-DHAP as matrix. In the absence of H2O2 the intact and cleaved subunits could be detected with indication of sialic acid loss in the carbohydrate moiety (-sia). Both the single and double charged ions could be detected. In the presence of 1 mM H2O2 an ion of m/z 10,232.9 was detected indicating protein fragmentation. No fragmentation could be observed in the presence of DDC showing the involvement of copper in the fragmentation process. The presence of DMPO did not inhibit fragmentation and fragment ions of m/z 10,233.1 and 11,833.5 could be detected. Moreover, an ion of low resolution with the indication of sialic acid loss could be detected (m/z∼14,657). These analyses suggest that the fragmentation locates to the Gly111–Pro112–His113 segment.
The EC-SOD fragments 1–3 identified by SDS-PAGE analysis (Fig. 1B) are likely to represent the ions corresponding to m/z 14,657, 11,833.5, and 10,233.1, respectively. To further characterize the peptide bond cleaved by H2O2, we subjected fragments 1–3 to in-gel digestion using trypsin. Analysis of the resulting peptides by mass spectrometry showed that fragment 1 encompassed peptides representing the N-terminal region of EC-SOD, and that peptides generated from fragments 2 and 3 were encompassed by the C-terminal region (Supplementary Fig. S1) corroborating the findings by mass spectrometry. Close inspection of the spectra obtained, identified one ion in fragment 1 (m/z 1957.1) and one ion in both fragments 2 and 3 (m/z 2470.2) that did not correlate with theoretical masses of tryptic peptides generated from EC-SOD. To further evaluate the nature of these ions, we subjected them to MALDI-MS/MS analysis. The fragment ions of m/z 1957.1 provided evidence that this ion represented Ala94–Gly111 with Cys107 modified by propionamide (introduced on reduced cysteine residues during electrophoresis in acrylamide gel) (m/zcalc.1956.9) (Fig. 4A) and the ion detected in fragments 2 and 3 was identified to represent un-modified His113–Arg134 (m/zcalc. 2470.2) (Fig. 4B). However, the tryptic peptide representing the N-terminal fragment did not encompass a C-terminal Pro112 residue although the analysis by linear mode mass spectrometry indicated the presence of this residue in the protein fragment. The apparent lack of this residue in the analyzed peptide cannot be explained, but may suggest that it is unstable at conditions used for in-gel digestion or MALDI mass spectrometry.
Fig. 4.
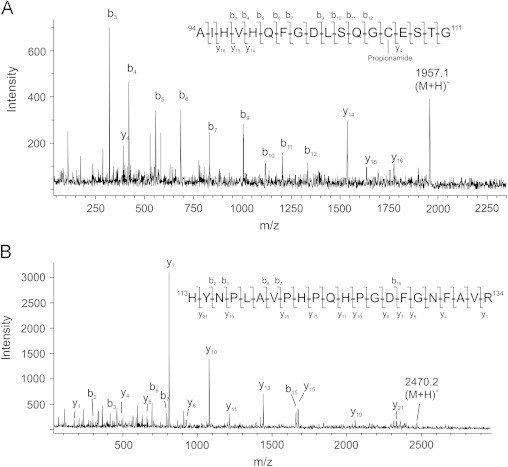
In-gel digestion and MALDI-MS/MS analysis of EC-SOD fragments. (A) MALDI MS analysis of fragment 1 showed at peptide of m/z 1957.1 that did not correspond to any theoretical mass of tryptic peptides. MALDI-MS/MS analysis showed that this ion corresponded to the Ala94–Gly111 peptide modified by propionamide at Cys107. (B) MALDI-MS analysis of fragments 2 or 3 produced an ion of m/z 2470.2 representing the peptide His113–Arg134 as determined by MALDI-MS/MS analysis. The parent ions are indicated by (M+H)+ and the detected fragment ions are indicated in the spectra and in the amino acid sequence of the peptides.
Substitution of Ala for Pro112 does not inhibit fragmentation
To investigate the importance of the proline residue in fragmentation, we generated recombinant EC-SOD substituting Ala for Pro112 (P112A). Purified P112A EC-SOD was subjected to H2O2 exposure and the products analyzed by SDS-PAGE (Supplementary Fig. S2). The analysis showed that P112A EC-SOD was fragmented generating products corresponding to fragments 1 and 2 of EC-SOD (Fig. 1B). The lack of fragment 3 correlates with the absence of cleaved subunits in recombinant EC-SOD protein expressed in HEK293 cell lines [25,26]. In line with this observation, it has previously been noticed that equine Cu/Zu-SOD – encompassing an equivalent Ala–His bond – was fragmented by H2O2 exposure [17]. Analysis by in-gel digestion and tandem mass spectrometry showed that fragment 1 contained the tryptic peptide corresponding to Ala94–Gly111 (m/z 1956.0) (Fig. 5A), however, this value differs by 1 mass unit to the expected size (m/zcalc.1956.9). Close investigation of the y- and b-ion series, showed that the mass of all b-ions correlated with the expected values, whereas the y-ions were determined to be one mass unit lower than expected. This finding suggests that the structure of the C-terminal residue is altered. Fragment 2 produced a tryptic peptide of m/z 2540.2 which was determined to represent Ala112–Arg134 by MALDI-MS/MS analysis (m/zcalc. 2541.3) (Fig. 5B). The determined m/z values representing the parent ion and all identified b-ions were found to be reduced by 1 mass unit, suggesting that the N-terminal Ala residue was modified as a result of peptide bond cleavage. Several authors have described a sequence of reactions initiated by radical-mediated hydrogen abstraction on the Cα-carbon of alanine resulting in the cleavage of the peptide bond on the N-terminal side of alanine [33,34]. The disruption of the peptide bond on the N-terminal side of alanine generates modified termini in both fragments supporting the loss of one mass unit (pseudoGly and pseudoAla) (Fig. 5C). These data show that the radical-mediated bond cleavage is likely to be initiated by hydrogen abstraction on the α carbon supporting the establishment of a carbon-centered radical. Based on amino acid side chain structure, this will facilitate the cleavage on the N-terminal side (Ala112) or the C-terminal side (Pro112).
Fig. 5.
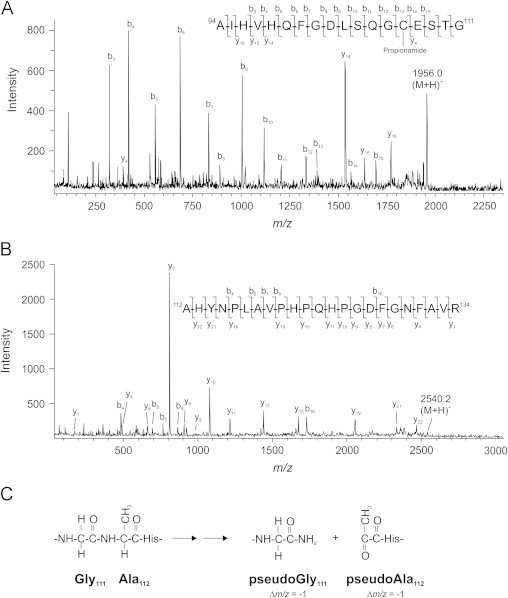
In-gel digestion and MALDI-MS/MS analysis of P112A EC-SOD fragments. (A) In-gel digestion of fragment 1 of P112A EC-SOD generated an ion of m/z 1956.0, which produced b-ions corresponding to the Ala94–Gly111 peptide modified by propionamide at Cys107 when subjected to MALDI-MS/MS analysis. However, the detected y-ions were one mass unit below the calculated values. (B) In gel digestion of fragment 2 of P112A EC-SOD produced an ion of m/z 2540.2, which generated y-ions representing the Ala112–Arg134 peptide when analyzed by MALDI-MS/MS. The b-ions detected were all one mass unit below the calculated value. (C) The reduced mass of parent and fragment ions can be explained by the radical-mediated cleavage of the Gly111–Ala112 peptide bond generating new C- and N-termini reduced with one mass unit. The parent ions are indicated by (M+H)+ and the detected fragment ions are indicated in the spectra and in the amino acid sequence of the peptides.
Oxidation of chelating histidine residues disrupt activity
Although there is a significant loss of SOD activity when EC-SOD is subjected to increasing concentrations of H2O2 (Fig. 1A), it is clear from the SDS-PAGE analysis, that the majority of the protein remains intact (Fig. 1B). It is thus likely that the loss of activity is accounted for by oxidation of amino acid residues central to maintaining the activity of the protein. Therefore, we analyzed the status of these histidine residues in EC-SOD exposed to 2 mM H2O2 by ESI-MS/MS. Subunits were separated by electrophoresis and subjected to in-gel digestion using both trypsin and chymotrypsin to allow for the analysis of the histidine residues involved in copper (His96/98/113/163) and zinc coordination (His113/His121/His124) [10]. Prior to electrophoresis, the protein was deglycosylated by using endoglycosidase F to remove the glycan structure on Asn89 converting Asn to Asp in the enzymatic process. This analysis showed that His98 and His163 were subject to oxidation (Table 1). The data presented here shows that the peptides containing non-oxidized histidine residues could be observed as well (Table 1), suggesting that oxidation of one residue supports the destruction of protein integrity by dissociation of copper from the active site [13,17,35]. This suggestion is supported by the finding that protein fragmentation of EC-SOD induced by oxidation of Pro112 or Ala112 did not show any oxidation of His98 when the N-terminal fragment analyzed by in-gel digestion (Figs. 4A and 5A). Homologous residues were also found to be oxidized in Cu/Zn-SOD exposed to H2O2 [15,16].
Table 1.
LC–MS/MS analysis of peptides containing histidine residues involved in chelation of zinc and copper.
| Peptidea | Calculated mass |
−H2O2 |
+H2O2 |
||
|---|---|---|---|---|---|
| Observed massb | Modification | Observed mass | Modification | ||
| A159VVVH163AGEDDLGR171 (T) | 1336.67 | 1336.65 (99) | None | 1336.65 (99) | None |
| 1352.64 (51) | His163(OX) | ||||
| A80LEGFPTEPDSSSRAIH96VH98QF100 (C)c | 2324.12 | 2324.08 (55) | None | 2324.09 (55) | None |
| 2340.09 (30) | His98(OX) | ||||
| S104QGCESTGPH113YNPL117 (C) | 1559.67 | 1559.64 (50) | None | 1559.65 (57) | None |
| A118VPH121PQH124PGDFGNF128 (C) | 1518.70 | 1518.68 (34) | None | 1518.68 (41) | None |
Peptides were generated by using trypsin (T) or chymotrypsin (C).
Observed masses represent the molecular mass of detected and deconvoluted ions. The MASCOT score is given in parenthesis.
The sequence is given with an Asn89→Asp substitution due to the conversion of Asn to Asp upon deglycosylation using PNGaseF.
Conclusions
We have shown that the mechanism of H2O2-induced EC-SOD inhibition is similar to that of Cu/Zn-SOD, including oxidation of proline and histidine residues proximal to the active site copper atom. The molecular structure of the oxidized histidine residues detected in EC-SOD was not determined, but is likely to be represented by 2-oxo-histidine as shown for oxidized Cu/Zn-SOD [15]. The mechanism involved in the formation of 2-oxo-histidine may involve the addition of the copper-bound hydroxyl radical to the immidazole ring of histidine at position C2 or C5 followed by dehydration to support the formation of 2-oxo-histidine [36]. An alternative pathway of oxidation is initiated by electron transfer from the immidazole ring of histidine to the hydroxyl radical generating a carbon-centered radical [37]. The addition of oxygen subsequently generates a highly reactive peroxyl radical which decomposes to form 2-oxo-histidine. Structural evaluation clearly shows that His98 and His163 are within 3 Å of the copper atom supporting oxidation [12] (Fig. 6). However, the hydrogen atom on the α-carbon of Pro112 is located 6.1 Å away from the copper atom and it is this highly unlikely that oxidation is supported by the copper-bound hydroxyl radical, but more likely indirectly by, e.g., a histidyl radical or histidine peroxide established at His98. The analysis of the fragmented peptide (Fig. 4A) shows that His98 was not oxidized, indicating hydrogen abstraction and subsequently bond cleavage is supported by a histidyl-radical. The finding that P112A EC-SOD also was fragmented, indicates that oxidation at this site is less dependent on residue structure and likely supported by abstraction of the hydrogen atom on the α-carbon. As site-specific fragmentation only accounts for a minor part of inhibition, this suggests that the oxidation of Pro112 is kinetically less favorable, in line with the notion that an oxidizing intermediate participates in the oxidation.
Fig. 6.
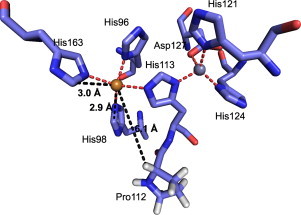
Structural representation of H2O2-induced modifications of EC-SOD. The structural region of EC-SOD involved in the coordination of the copper and zinc ions is shown. The residues susceptible to oxidation (His98 and His163) and central to fragmentation (Pro112) are shown in sticks. The distances between copper and C2 of His98 and His163 and the hydrogen atom at the α-carbon of Pro112 are indicated.
We have shown that the peroxidase reaction of EC-SOD induce oxidations similar to those found in Cu/Zn-SOD, accounting for the loss of enzymatic activity. Although the concentrations of oxidant used to study these modifications are in the millimolar scale, the inhibition of EC-SOD induced by peroxidase activity appears to play a role in disease [21–23]. This may be accounted for by the establishment of a local and high concentration of H2O2, as observed in the process of wound repair [38]. Moreover, it could be speculated that since the peroxidase reaction is supported by the reaction of H2O2 with cuprous EC-SOD, superoxide and H2O2 competes for the same substrate. Hence, if the level of superoxide is relatively low, the peroxidase reaction will dominate and hence regulate dismutase activity by substrate inhibition and thereby reduce the level of H2O2 in the extracellular space.
Acknowledgments
Dr. Rune T. Kidmose is gratefully acknowledged for the preparation of Fig. 6. This work was supported by The Lundbeck Foundation (S.V.P.).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Mature EC-SOD contains an Ala/Thr polymorphism at position 40 (SNP: rs2536512) which corresponds to a mass difference of 30 Da. The calculated mass is given using Ala40, reduced cysteine residues, and a sialylated, biantennary, and core fucosylated complex type glycan structure at Asn89 [31].
Appendix A. Supplementary materials
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2012.12.004.
Appendix A. Supplementary materials
Fig. S1.
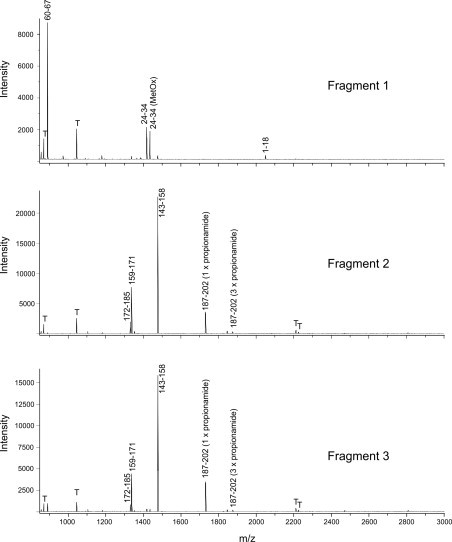
Fragments 1–3 were subjected to in-gel digestion and the resulting peptides analyzed by MALDI-MS using α-cyano-4-hydroxysinnamic acid as matrix. The ions representing tryptic peptides of EC-SOD are shown by amino acid numbering and peptides encompassing cysteine residues modified by propionamide (+71 Da) induced by electrophoresis are indicated. The ions representing peptides originating from porcine trypsin are labeled (T).
Fig. S2.
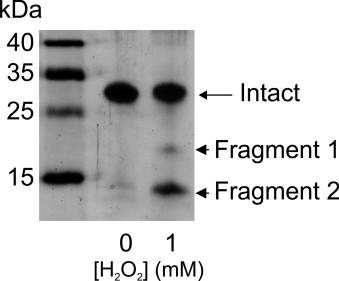
Purified P112A EC-SOD was exposed to 1 mM H2O2 and subsequently separated by electrophoresis and protein visualized by silver staining. P112A EC-SOD receiving buffer only was included as control (0 mM H2O2). The position of the intact subunit and generated fragments are indicated on the right and a molecular weight marker shown on the left.
References
- 1.Sandstrom J., Carlsson L., Marklund S.L., Edlund T. The heparin-binding domain of extracellular superoxide dismutase C and formation of variants with reduced heparin affinity. Journal of Biological Chemistry. 1992;267:18205–18209. [PubMed] [Google Scholar]
- 2.Adachi T., Kodera T., Ohta H., Hayashi K., Hirano K. The heparin binding site of human extracellular-superoxide dismutase. Archives of Biochemistry and Biophysics. 1992;297:155–161. doi: 10.1016/0003-9861(92)90654-f. [DOI] [PubMed] [Google Scholar]
- 3.Petersen S.V., Oury T., Oestergaard L., Valnickova Z., Wegrzyn J., Thogersen I.B., Jacobsen C., Bowler R.P., Fattman C.L., Crapo J.D., Enghild J.J. Extracellular superoxide dismutase (EC-SOD) binds to type I collagen and protects against oxidative fragmentation. Journal of Biological Chemistry. 2004;279:13705–13710. doi: 10.1074/jbc.M310217200. [DOI] [PubMed] [Google Scholar]
- 4.Gao F., Koenitzer J.R., Tobolewski J.M., Jiang D., Liang J., Noble P.W., Oury T.D. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. Journal of Biological Chemistry. 2008;283:6058–6066. doi: 10.1074/jbc.M709273200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nozik-Grayck E., Suliman H.B., Piantadosi C.A. Extracellular superoxide dismutase. International Journal of Biochemistry and Cell Biology. 2005;37:2466–2471. doi: 10.1016/j.biocel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Manni M.L., Tomai L.P., Norris C.A., Thomas L.M., Kelley E.E., Salter R.D., Crapo J.D., Chang L.Y., Watkins S.C., Piganelli J.D., Oury T.D. Extracellular superoxide dismutase in macrophages augments bacterial killing by promoting phagocytosis. American Journal of Pathology. 2011;178:2752–2759. doi: 10.1016/j.ajpath.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Break T.J., Jun S., Indramohan M., Carr K.D., Sieve A.N., Dory L., Berg R.E. Extracellular superoxide dismutase inhibits innate immune responses and clearance of an intracellular bacterial infection. Journal of Immunology. 2012;188:3342–3350. doi: 10.4049/jimmunol.1102341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kliment C.R., Oury T.D. Extracellular superoxide dismutase protects cardiovascular syndecan-1 from oxidative shedding. Free Radical Biology and Medicine. 2011;50:1075–1080. doi: 10.1016/j.freeradbiomed.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kliment C.R., Tobolewski J.M., Manni M.L., Tan R.J., Enghild J., Oury T.D. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxidants and Redox Signaling. 2008;10:261–268. doi: 10.1089/ars.2007.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hjalmarsson K., Marklund S.L., Engstrom A., Edlund T. Isolation and sequence of complementary DNA encoding human extracellular superoxide dismutase. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:6340–6344. doi: 10.1073/pnas.84.18.6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tainer J.A., Getzoff E.D., Beem K.M., Richardson J.S., Richardson D.C. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. Journal of Molecular Biology. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 12.Antonyuk S.V., Strange R.W., Marklund S.L., Hasnain S.S. The structure of human extracellular copper–zinc superoxide dismutase at 1.7 Å resolution: insights into heparin and collagen binding. Journal of Molecular Biology. 2009;388:310–326. doi: 10.1016/j.jmb.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 13.Hodgson E.K., Fridovich I. The interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide: inactivation of the enzyme. Biochemistry. 1975;14:5294–5299. doi: 10.1021/bi00695a010. [DOI] [PubMed] [Google Scholar]
- 14.Jewett S.L., Rocklin A.M., Ghanevati M., Abel J.M., Marach J.A. A new look at a time-worn system: oxidation of CuZn-SOD by H2O2. Free Radical Biology and Medicine. 1999;26:905–918. doi: 10.1016/s0891-5849(98)00274-3. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K., Kawakishi S. Identification of oxidized histidine generated at the active site of Cu,Zn-superoxide dismutase exposed to H2O2. Selective generation of 2-oxo-histidine at the histidine 118. Journal of Biological Chemistry. 1994;269:2405–2410. [PubMed] [Google Scholar]
- 16.Kurahashi T., Miyazaki A., Suwan S., Isobe M. Extensive investigations on oxidized amino acid residues in H(2)O(2)-treated Cu,Zn-SOd protein with LC-ESI-Q-TOF-MS, MS/MS for the determination of the copper-binding site. Journal of the American Chemical Society. 2001;123:9268–9278. doi: 10.1021/ja015953r. [DOI] [PubMed] [Google Scholar]
- 17.Ookawara T., Kawamura N., Kitagawa Y., Taniguchi N. Site-specific and random fragmentation of Cu,Zn-superoxide dismutase by glycation reaction. Implication of reactive oxygen species. Journal of Biological Chemistry. 1992;267:18505–18510. [PubMed] [Google Scholar]
- 18.Wolff S.P., Garner A., Dean R.T. Free-radicals, lipids and protein-degradation. Trends in Biochemical Sciences. 1986;11:27–31. [Google Scholar]
- 19.Uchida K., Kato Y., Kawakishi S. A novel mechanism for oxidative cleavage of prolyl peptides induced by the hydroxyl radical. Biochemical and Biophysical Research Communications. 1990;169:265–271. doi: 10.1016/0006-291x(90)91463-3. [DOI] [PubMed] [Google Scholar]
- 20.Kato Y., Uchida K., Kawakishi S. Oxidative fragmentation of collagen and prolyl peptide by Cu(II)/H2O2. Conversion of proline residue to 2-pyrrolidone. Journal of Biological Chemistry. 1992;267:23646–23651. [PubMed] [Google Scholar]
- 21.Hink H.U., Santanam N., Dikalov S., McCann L., Nguyen A.D., Parthasarathy S., Harrison D.G., Fukai T. Peroxidase properties of extracellular superoxide dismutase: role of uric acid in modulating in vivo activity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:1402–1408. doi: 10.1161/01.atv.0000027524.86752.02. [DOI] [PubMed] [Google Scholar]
- 22.Jung O., Marklund S.L., Xia N., Busse R., Brandes R.P. Inactivation of extracellular superoxide dismutase contributes to the development of high-volume hypertension. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:470–477. doi: 10.1161/01.ATV.0000254823.15843.1f. [DOI] [PubMed] [Google Scholar]
- 23.Wedgwood S., Lakshminrusimha S., Fukai T., Russell J.A., Schumacker P.T., Steinhorn R.H. Hydrogen peroxide regulates extracellular superoxide dismutase activity and expression in neonatal pulmonary hypertension. Antioxidants and Redox Signaling. 2011;15:1497–1506. doi: 10.1089/ars.2010.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oury T.D., Crapo J.D., Valnickova Z., Enghild J.J. Human extracellular superoxide dismutase is a tetramer composed of two disulphide-linked dimers: a simplified, high-yield purification of extracellular superoxide dismutase. Biochemical Journal. 1996;317:51–57. doi: 10.1042/bj3170051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottfredsen R.H., Tran S.M., Larsen U.G., Madsen P., Nielsen M.S., Enghild J.J., Petersen S.V. The C-terminal proteolytic processing of extracellular superoxide dismutase is redox regulated. Free Radical Biology and Medicine. 2012;52:191–197. doi: 10.1016/j.freeradbiomed.2011.10.443. [DOI] [PubMed] [Google Scholar]
- 26.Petersen S.V., Kristensen T., Petersen J.S., Ramsgaard L., Oury T.D., Crapo J.D., Nielsen N.C., Enghild J.J. The folding of human aEC-sod and iEC-sod is an intracellular event. Journal of Biological Chemistry. 2008;283:15031–15036. doi: 10.1074/jbc.M801548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bury A.F. Analysis of protein and peptide mixtures. Evaluation of three sodium dodecyl sulfate-polyacrylamide gel electrophoresis buffer systems. Journal of Chromatography. 1981;213:491–500. [Google Scholar]
- 28.McCord J.M., Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) Journal of Biological Chemistry. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 29.Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 30.Misra H.P. Reaction of copper–zinc superoxide dismutase with diethyldithiocarbamate. Journal of Biological Chemistry. 1979;254:11623–11628. [PubMed] [Google Scholar]
- 31.Stromqvist M., Holgersson J., Samuelsson B. Glycosylation of extracellular superoxide dismutase studied by high-performance liquid chromatography and mass spectrometry. Journal of Chromatography. 1991;548:293–301. doi: 10.1016/s0021-9673(01)88611-8. [DOI] [PubMed] [Google Scholar]
- 32.Deterding L.J., Ramirez D.C., Dubin J.R., Mason R.P., Tomer K.B. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. Journal of Biological Chemistry. 2004;279:11600–11607. doi: 10.1074/jbc.M310704200. [DOI] [PubMed] [Google Scholar]
- 33.Chen H.Y., Jang S., Jinn T.R., Chang J.Y., Lu H.F., Li F.Y. Oxygen radical-mediated oxidation reactions of an alanine peptide motif—density functional theory and transition state theory study. Chemistry Central Journal. 2012;6 doi: 10.1186/1752-153X-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan P.E., Pattison D.I., Davies M.J. Quantification of hydroxyl radical-derived oxidation products in peptides containing glycine, alanine, valine, and proline. Free Radical Biology and Medicine. 2012;52:328–339. doi: 10.1016/j.freeradbiomed.2011.10.448. [DOI] [PubMed] [Google Scholar]
- 35.Salo D.C., Pacifici R.E., Lin S.W., Giulivi C., Davies K.J. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. Journal of Biological Chemistry. 1990;265:11919–11927. [PubMed] [Google Scholar]
- 36.Schoneich C. Mechanisms of metal-catalyzed oxidation of histidine to 2-oxo-histidine in peptides and proteins. Journal of Pharmaceutical and Biomedical Analysis. 2000;21:1093–1097. doi: 10.1016/s0731-7085(99)00182-x. [DOI] [PubMed] [Google Scholar]
- 37.Gunther M.R., Peters J.A., Sivaneri M.K. Histidinyl radical formation in the self-peroxidation reaction of bovine copper–zinc superoxide dismutase. Journal of Biological Chemistry. 2002;277:9160–9166. doi: 10.1074/jbc.M107342200. [DOI] [PubMed] [Google Scholar]
- 38.Niethammer P., Grabher C., Look A.T., Mitchison T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]