

Abstract
Glioma survival is dismal, in part, due to an imbalance in antioxidant expression and activity. Peroxisome proliferator-activated receptor (PPAR) agonists have antineoplastic properties which present new redox-dependent targets for glioma anticancer therapies. Herein, we demonstrate that treatment of primary cultures of normal rat astrocytes with PPAR agonists increased the expression of catalase mRNA protein, and enzymatic activity. In contrast, these same agonists had no effect on catalase expression and activity in malignant rat glioma cells. The increase in steady-state catalase mRNA observed in normal rat astrocytes was due, in part, to de novo mRNA synthesis as opposed to increased catalase mRNA stability. Moreover, pioglitazone-mediated induction of catalase activity in normal rat astrocytes was completely blocked by transfection with a PPARγ-dominant negative plasmid. These data suggest that defects in PPAR-mediated signaling and gene expression may represent a block to normal catalase expression and induction in malignant glioma. The ability of PPAR agonists to differentially increase catalase expression and activity in normal astrocytes but not glioma cells suggests that these compounds might represent novel adjuvant therapeutic agents for the treatment of gliomas.
Abbreviations: 9cRA, 9 cis retinoic acid; ActD, actinomycin D; BCNU, 1,3-bis (2-chloroethyl)-1-nitrosourea; CNS, central nervous system; DCF, dichlorofluorescein; GPx, glutathione peroxidase; GSH, glutathione; Pio, Pioglitazone; PPAR, Peroxisome proliferator-activated receptor; PPARγ-d.n, PPARγ-dominant negative; PPRE, PPAR-response elements; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; Rosi, Rosiglitazone; RXR, retinoid X receptor; shRNA, short hairpin RNA; SOD, superoxide dismutase; TZDs, thiazolidinediones
Keywords: Catalase, Primary rat astrocytes, C6 glioma cells, PPAR agonists, PPARγ-dominant negative, Transfection
Graphical Abstract
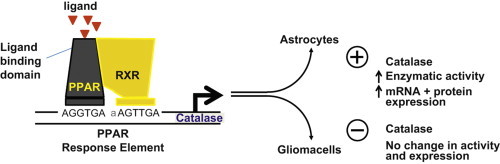
Highlights
► PPAR agonists increase catalase expression and activity. ► Differential upregulation of catalase in astrocytes but not glioma cells. ► PPAR agonists induce de novo transcription of catalase mRNA. ► PPARγ-dominant negative plasmid blocks agonist induced catalase enzymatic activity. ► Implications for protecting astrocytes during anti-glioma therapy.
Introduction
Novel therapeutic approaches that selectively protect normal brain cells and/or sensitize glioma cells to anti-cancer therapies are urgently needed. Roughly 70,000 new cases of primary brain tumors will be diagnosed in the US, the majority being grade 4 astrocytoma (glioblastoma) [1]. Despite multimodality therapy including surgery, radiation therapy and chemotherapy with temozolomide, median survival remains only approximately 15 months [2] and the 5-year survival rate is 1% [3]. This outcome reflects both glioma cell resistance to therapy and the risk of radiation-induced normal brain injury, which limits the total dose that can be safely administered to the tumor [4]. Recent data suggest that progressive cognitive impairment occurs in ∼50% of brain tumor patients who are long-term survivors after treatment with partial or whole-brain irradiation [5,6]. This negative prognosis is, in part, due to radiation-induced oxidative tissue injury.
The central nervous system (CNS) is inherently susceptible to oxidative stress. This is evidenced by the CNS being: (1) highly active in oxidative metabolism, leading to a relatively high rate of reactive oxygen species (ROS) production [7]; (2) relatively low in the specific activity of the key antioxidant enzymes superoxide dismutase (SOD), catalase and glutathione peroxidase (GPx) [8] in oligodendrocytes, neurons and endothelial cells [9,10]; and (3) rich in readily oxidizable polyunsaturated fatty acids (PUFAs) such as docosahexaenoic acid and eicosapentaenoic acid [11].
These issues suggest that a lipid signaling-based therapeutic strategy may be beneficial in treating CNS cancers. PUFAs and oxidized lipids are ligands for peroxisome proliferator-activated receptors (PPARs) [12,13], which are members of the nuclear hormone receptor (NHR) superfamily. PPARs are ligand-activated transcription factors that heterodimerize with the retinoid X receptor (RXR), bind to PPAR-response elements (PPRE), and thereby regulate gene expression [14]. Three isotypes of PPARs have been identified and designated α, β/δ, and γ [15]. PPARα is activated by fibrates, a class of cholesterol-lowering drugs used in the treatment of dyslipidemia, and functions by enhancing β-oxidation in the liver [16]. PPARγ agonists, such as thiazolidinediones (TZDs), are best known for controlling hyperglycemia and treating type 2 diabetes mellitus, in part, through the regulation of adipocyte differentiation and lipid storage/metabolism [17]. PPARβ/δ is expressed in tissues controlling lipid metabolism and acts as a lipid sensor, thus representing a molecular target for treating metabolic syndrome [18]. Data supports the concept that PPARγ mediates anti-proliferative and pro-apoptotic activities in tumor cells [19].
Studies have shown that activation of PPARγ has antineoplastic effects through redox-dependent mechanisms. The PPARγ agonists Rosiglitazone (Rosi) and Pioglitazone (Pio) decreased cell viability in both human (U87 and A172) and rat (C6) malignant glioma cells whereas normal rat astrocytes were not affected [19,20]. This cytotoxic effect was spared by N-acetylcysteine suggesting reactive oxygen species (ROS) are playing an integral role. PPARγ agonists decreased glutathione (GSH) levels in C6 rat glioma cells and increased dichlorofluorescein (DCF) fluorescence in both the primary rat astrocytes and C6 rat glioma cells [20]. Moreover, Pio treatment significantly reduced tumor volume in a rat glioma model by decreasing proliferation, suppressing MMP9 induction, and inhibiting tumor cell invasion [19].
The cytotoxic effect of PPARγ agonists on glioma cells is partially mediated by enhanced redox reactions. Yet, normal rat astrocytes, which are the most prevalent cell type in the CNS, comprising more than 50% of brain volume and outnumbering neurons approximately 9:1 [21], appear to be resistant to PPARγ-mediated cytotoxicity [22]. Catalase gene expression and enzymatic activity was the only antioxidant enzyme significantly increased by PUFAs [23]. In light of this, it is hypothesized that catalase is responsible for the protective effects following PPAR agonist treatment of primary cultures of normal rat astrocytes. To investigate this putative role of PPARs in catalase regulation, we treated primary cultures of normal rat astrocytes and C6 rat glioma cells with PPAR agonists. We report herein that PPAR agonists increased catalase gene expression and enzymatic activity in primary cultures of normal rat astrocytes. In contrast, PPAR agonists failed to increase catalase expression in glioma cells. To gain insight into a potential mechanism, Cos-1 cells were transfected with a PPARγ-dominant negative (PPARγ-d.n.) plasmid, revealing that PPARγ plays a significant role in the regulation of catalase expression. The ability of PPAR agonists to selectively upregulate catalase expression in normal astrocytes but not glioma cells suggests that these compounds might represent novel adjuvant therapeutic agents for the treatment of gliomas.
Methods
Materials
Tissue culture materials were obtained from the following manufacturers: Dulbecco's Modified Eagle's Medium (DMEM/F12), Minimum Essential Medium (MEM), trypsin/EDTA penicillin and streptomycin (Gibco, Grand Island, NY); fetal bovine serum (Hyclone; Logan, Utah); L-glutamine and gentamycin (Amersham; Arlington Heights, IL). Pioglitazone, Troglitazone, WY-14,643, and 9 cis Retinoic Acid (9cRA) were purchased from BioMol (Plymouth Meeting, PA); Rosiglitazone was purchased from Cayman Chemical (Ann Arbor, MI). The PPARγ agonists CP-086,325-02 (CP086; Darglitazone) and CP-096,125 (CP096) were kindly donated by Michael Gibbs at Pfizer (Groton, CT). The PPARα specific agonist GW3276 and GR-259662 were kind gifts from Tim Willson at Glaxo Wellcome (Research Triangle Park, NC).
Cell culture
Primary rat astrocytes were isolated from 1 to 2 day old Sprague-Dawley rat pups [23]. The cells were cultured in MEM containing 10% FBS, 2 mM L-glutamine, 6 g/L glucose (33 mM) and 50 μg/mL gentamycin. The cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2. Astrocytes were purified from other glial cells by shaking the flasks before each media change. The media was changed every fourth day and the cells were used within a week of reaching confluence. Immunofluorescence staining with GFAP was carried out to characterize the cell purity (>98% astrocytes).
The rat malignant glioma cell line C6 was grown in DMEM/F12 containing 10% FBS, 50 IU/mL penicillin, and 50 IU/mL streptomycin. Cells were maintained at 37 °C as a monolayer in 75 cm2 tissue culture flasks in a humidified atmosphere containing 5% CO2. Stock cultures were passaged twice weekly by trypsinization. For experiments, the cultures were between passage 12 and 20, and the incubations started when the cells were roughly 40% confluent.
Cloning of promoter deletion constructs
Total genomic DNA from primary rat astrocytes or rat brain microvessel endothelial cells (RBMECs) was isolated as previously reported [24]. Rat catalase promoter deletion constructs were generated by PCR using the following primers: -1046, 5′-ACAGCCCACAGCCCATAATC-3′ (3641–3660); -938, 5′-ATTGATTAAAATGAAAAATAAGCGAC-3′ (3751–3776); and -207, 5′-CTCCTTCCAATCCTGTCCC-3′ (4481–4499). The number represents the location of the rat catalase promoter construct with respect to the translational start site. The common downstream primer, 5′-CAGATGAAGCAGTGGAAGGA-3′ (4719–4738), was used with all the primers listed above. The numbers inside the parentheses indicate the location of primers with reference to the published sequence beginning with the first nucleotide of exon 1 [GenBank accession # M25669].
PCR was performed using Taq DNA polymerase (Perkin-Elmer; Emeryville CA). The PCR conditions consisted of 94 °C for 4 min, followed by 35 cycles of 94 °C for 1 min, 58 °C for 50 s, and 72 °C for 1.5 min, followed by a final extension at 72 °C for 5 min. The expected sizes of the PCR products were verified by 1 kb and 100 bp DNA markers (Promega; Madison, WI) on a 1% TAE gel. The PCR products were ligated into the pCR2.1 TA-TOPO cloning vector and transformed into competent cells as described by the manufacturer (Invitrogen; Carlsbad, CA). DNA was isolated using the gel extraction kit (Qiagen), digested with EcoRI (Holden Cancer Center; University of Iowa) and the identity was verified by sequencing (DNA Core Facility; University of Iowa). The constructs were engineered by subcloning the promoter deletion fragments from pCR2.1 (Invitrogen) into a luciferase-based plasmid pGL3-basic (Promega). The constructs were verified by sequencing (DNA Core Facility; The University of Iowa).
The tandem tripeat of the catalase PPRE (DR1x3) was synthesized using an oligonucleotide consisting of TAATCAAGGTGAAAGTTGAGAAG with KpnI and XhoI restriction sites on its 5′- and 3′-ends, respectively. The oligonucleotide was restricted with KpnI and XhoI, gel isolated, and cloned into pGL3-basic. The DR1x3 was verified by sequencing.
Expression vectors
The full length RXRα was synthesized as previously reported [24]. PPARα expression vector was synthesized from mouse liver RNA by PCR using the following primers: 5′-GTGGCTGGTCAAGTTCGG-3′ (upstream) and 5′-CTCGGAGGTCCCTGAACAG-3′ (downstream) for PPARα, [GenBank Acc# X57638]. Mouse liver was dounce homogenized and total RNA was isolated. Total RNA was reversed transcribed using the Superscript II Reverse Transcription kit (Roche) according to the manufacturer's specifications. The cDNA generated was PCR amplified under the conditions of 94 °C for 4 min, followed by 35 cycles of 94 °C for 1 min, 62.5 °C for 1 min, and 72 °C for 1.5 min, followed by a final extension at 72 °C for 5 min. The expected size of the PCR product was verified on a 1% low melting point TAE gel. A single band of the expected size of the PPARα PCR product was isolated using the gel extraction kit (Qiagen) and cloned into the pTargeT mammalian expression vector (Promega) according to the manufacturer's directions. The PPARα expression vector was verified by sequencing.
Transfection and reporter luciferase assay
C6 rat glioma and Cos-1 cells were transiently transfected using SuperFect Reagent (Qiagen; Valencia, CA) according to the manufacturer's directions. Primary rat astrocytes were transiently transfected with Effectene (Qiagen). Cells were transfected with 0.25–1 μg of rat catalase reporter deletion constructs or the tandem tripeat of the catalase PPRE (DR1x3) and co-transfected with a plasmid containing 0.5–3 μg CMV SPORT-β-galactosidase (Invitrogen) for 18–24 h depending on cell type. In a subset of experiments, cells were additionally transiently transfected with 0.1–1 μg of pTargeT, mRXRα, mPPARγ2, and mPPARα expression vectors. Following transfection, cells were rinsed and treated with PPAR and/or RXR agonists for 24 h. The cells were then rinsed with phosphate buffered saline (PBS), lysed with Passive Lysis Buffer (Promega), and luciferase activity was measured using Luciferase Assay System (Promega) according to manufacturer's instructions. To control for transfection efficiency, β-galactosidase activity was measured at 420 nm using o-nitrophenyl β-d-galactopyranoside (Sigma; St. Louis, MO) as substrate.
Protein sample preparation
Normal rat astrocyte and C6 glioma cell cultures were incubated in the presence of the various PPAR and RXR agonists for 48 h and were confluent at the time of harvest. Cells were rinsed in PBS twice and harvested by scraping and centrifugation. The cell pellets were then stored at −20 °C. Upon use, the cell pellets were resuspended in 50 mM potassium phosphate buffer (pH 7.8) and then sonicated on ice with four bursts of 20 seconds each using a Vibra Cell sonicator (Sonics and Materials, Inc.) with a cup horn at full power. Total protein concentrations were determined by the Bradford method [25] using BSA as a standard. For nuclear protein extraction, cells were rinsed in PBS and nuclear protein was then isolated as described previously [26].
Catalase activity assay
Catalase enzymatic activity was determined by a modified version of the method described by Beers and Sizer [27]. Briefly, 300 μg of primary rat astrocyte and C6 glioma extract were added to 30 mM H2O2 in 50 mM of potassium phosphate buffer (pH 7.8), and the consumption of H2O2 was measured at 240 nm for 120 s at 15 s intervals. Catalase activity was expressed in K units per g protein per second (K/g/s).
Western blot analysis
Western blot analysis was performed using anti-IgG catalase antibody (Athens BioTech.; Athens, GA), anti-PPAR α antibodies (Santa Cruz, Santa Cruz, CA), anti-PPAR γ antibodies (Wak-Chemie; Sulzbacherstasse, Germany and Santa Cruz), anti-RXRα antibody and anti-Actin antibody (Santa Cruz). Briefly, 10–20 μg of whole cell lysate or nuclear protein were separated by polyacrylamide gel electrophoresis (PAGE) on a 10–12% gel by a modified version described previously [23]. Proteins were transferred to a nitrocellulose membrane (Schleicher and Schuell; Keene, NH) for 1 h at 100 V and then blocked with 5% dry milk in TBST (0.02 M Tris/0.15 M NaCl buffer, pH 7.45 and 0.05% Tween 20). The gel was stained by Gel Code (Pierce Chem.; Rockford, IL) as a control for loading variation. Membranes were incubated with catalase antibody (1:1000 dilution), PPARα antibodies (1:500 dilution), PPARγ antibodies (1:500 dilution) or Actin antibody (1:400) for 2–3 h, rinsed with TBST and then incubated with a horseradish peroxidase-conjugated secondary antibody (Sigma; St. Louis, MO) for 1 h. Bands were visualized by ECL chemiluminescence (Amersham) and exposed to film.
Generation of a rat catalase partial cDNA probe
Total mRNA was extracted according to the directions provided by the manufacturer (Tel-Test B Inc.; Friendswood, TX). Briefly, the cells were homogenized into RNA-STAT-60 and then the total RNA was extracted into chloroform. The RNA was removed, placed into a fresh tube, and allowed to precipitate with isopropanol. Following the precipitation, the total RNA was washed in 75% ethanol and quantified using UV spectroscopy. Total RNA from primary rat astrocytes was used to synthesize the catalase cDNA by RT-PCR using the following primers: 5′-AAACCCGATGTCCTGACCAG-3′ and 5′-CCTTTGCCTTGGAGTA-TCTGG-3′. These primers were located in exons 2 and 3 [GenBank Accession #M25670 and M25671, respectively]. The predicted product was 228 bp in length. The RT-PCR reactions were carried out using one-step RT-PCR kit (Qiagen; Santa Clarita, CA) in 50 μL reaction solution containing 1 μg total RNA, 10 μL of 10x PCR buffer, 2 μL of 10 mM dNTP, 1 μM of sense and antisense primers, 8 units of RNase inhibitor (Promega; Madison, WI), and 2 μL enzyme mixture. The following PCR conditions were used: 94 °C for 5 min, then 35 cycles of 94 °C for 1 min, 64 °C for 30 s, and 72 °C for 1 min, followed by a final extension step at 72 °C for 5 min. The 228 bp PCR products of catalase were cloned into the pCR2.1 TA-TOPO cloning vector and transformed into competent cells as described by the manufacturer (Invitrogen; Carlsbad, CA, USA). DNA was isolated (Promega) and sequenced by the DNA Core at the University of Iowa.
RNA isolation and Northern blot analysis
Total mRNA was extracted as described above. Five to 10 μg of RNA were resolved by electrophoresis in a 1% agarose gel containing 2.2% formaldehyde using a running buffer of 20 mM MOPS, 5 mM sodium acetate, and 1 mM EDTA at pH 7. RNA was transferred to a Nytran membrane using a TurboBlotter transfer system (Schleicher and Schuell) for 2 h and then UV cross-linked (Stratagene; LaJolla, CA). The catalase partial cDNA probe described above was labeled with [α32P]dCTP (NEN Life Science Products; Boston, MA) using a random primed labeling kit according to the manufacturer's instructions (Roche Diagnostics; Mannheim, Germany). Membranes were prehybridized with PerfectHyb Plus Hybridization Buffer (Sigma; St. Louis, MO) for 1 h and then hybridized with the same buffer for 6–8 h at 68 °C. Membranes were washed and scanned using a Typhoon 8600 PhosphoImager (Molecular Dynamics; Piscataway, NJ) and densitometry was performed using ImageQuant 5.1. Membranes were stripped and reprobed with a radiolabeled cyclophilin cDNA probe (Ambion; Austin, TX).
Electrophoretic mobility shift assay (EMSA)
The following top and bottom strand complementary oligonucleotides were hybridized to generate a double stranded DNA probe of the same sequence as the PPRE found in the rat catalase promoter: 5′-AGCTTAATCAAGGTGAAAGTTGAGAAG-3′ and 5′-GATCCTTCTCAACTTTCACCT TGATTA-3′. The oligonucleotide was fill-in end-labeled with Klenow DNA polymerase, α32P-dCTP, and dNTPs for 5 min at 37 °C. One to 10 μg of nuclear protein, gel shift buffer [10 mM Tris (pH 7.5), 4% glycerol, 50 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol (DTT)], poly dIdC (Pharmacia, Piscataway, NJ), and the 32P labeled probe described above were incubated at room temperature for 15–30 minutes. Following the incubation, antibodies were added for 1.5 h. Antibodies were purchased from Santa Cruz Biotechnology, Inc.: anti-PPARα (H-98X), anti-PPARγ (H-100X), anti-RXRα (D-20X) and normal rabbit IgG (sc2027). The bound DNA complexes with antibody were separated from free probe by PAGE on a 5% native gel between 15–20 mA in 1x TBE. Gels were exposed to X-ray film at −80 °C.
Statistics
Data were expressed as the mean±SEM. Both Student's t-test and ANOVA were used in the comparison of different groups. Within ANOVA, the weighted least squares method was used to adjust for unequal variance among the groupings [28]. Dunnit's multiple comparison procedure was performed to determine significance.
Results
PPARα, PPARγ and RXR agonists upregulate catalase activity in normal rat astrocytes but not in rat malignant glioma cells. Primary rat astrocytes treated with PPARα or PPARγ agonists (10 μM) for 48 h increased catalase activity compared to the vehicle control (Fig. 1A). The maximal increase in catalase activity was observed in primary cultures of rat astrocytes treated with the PPARγ agonist CP096 (5.82±0.77 k/g/s), a value significantly greater than that observed in primary cultures of rat astrocytes treated with DMSO vehicle alone (2.78±0.23 k/g/s, p>0.0001). There was no difference between vehicle control (0.02% vol/vol of DMSO) and untreated confluent primary cultures of rat astrocytes. Significant increases in catalase activity were also observed in astrocytes incubated with PPARγ agonists rosiglitazone (Rosi), troglitazone or RXR agonist (9cRA). Less pronounced increases were observed following treatment of astrocytes with PPARα agonists GW3276 or WY14643. In contrast, treating C6 glioma cells with PPARα, PPARγ or RXR agonists (10 μM) for 48 h failed to enhance catalase activity in C6 glioma cells (Fig. 1B). Moreover, C6 glioma cells (4.54±0.09 k/g/s) expressed higher constitutive levels of catalase activity as compared to the primary rat astrocytes (2.78±0.23 k/g/s, p=0.002, t-test).
Fig. 1.
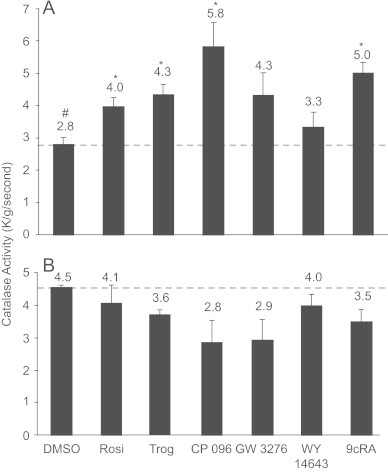
PPARα and PPARγ agonists increased catalase enzymatic activity in astrocytes but not in glioma cells. Primary rat astrocytes (A) and C6 glioma cells (B) were supplemented for 48 h with Rosi, Troglitazone, and CP096 (PPARγ), GW3276 and WY-14,643 (PPARα), 9cRA (RXR) and vehicle (DMSO) at concentration of 10 μM. Protein was then harvested and catalase activity was determined spectrophotometrically at 240 nm. For astrocytes, one-way analysis of variance (ANOVA) was used to compare the groups. Within ANOVA, the weighted least squares method was used to adjust for the heteroskedasticity (that is, the unequal variance among the groupings) [28]. The overall test of differences among the groups was statistically significant (p<0.0001 by ANOVA). Rosi, troglitazone, CP096 and 9cRA were significantly different from the DMSO group by Dunnett's multiple comparison procedure as indicated by ⁎. For C6 glioma cells, the overall test of differences among the groups was not statistically significant (p<0.30 by ANOVA). No groups were significantly different from the DMSO group at the 0.05 level. There is a significant difference in the mean values for the outcome variable between the astrocyte control (DMSO) vs C6 control (DMSO) (p=0.002) as indicated by #. Results are derived from at least six independent experiments and data are expressed as mean±SEM.
PPARα, PPARγ and RXR agonists upregulate catalase mRNA and protein expression in primary rat astrocytes but not in C6 glioma cells
Agonist treatment resulted in 1.5- to 2-fold increase in steady-state catalase mRNA levels compared to the vehicle control (Fig. 2A). Conversely, C6 glioma cells incubated with the same set of PPAR and RXR agonists failed to alter catalase mRNA expression (Fig. 2B) over the same time. There was no difference between untreated cells compared to DMSO-treated cells for both primary rat astrocytes and C6 rat glioma cells. PPARα agonist (GW3276) and RXR agonist (9cRA) increased catalase protein in primary rat astrocytes 1.9- and 1.6-fold, respectively (Fig. 2C). Additionally, catalase immunoreactive protein levels were increased 1.4- and 1.5-fold following Rosi and troglitazone treatment, respectively (Fig. 2C). In contrast, malignant glioma cells treated with the same agonists failed to increase catalase protein levels (Fig. 2D).
Fig. 2.
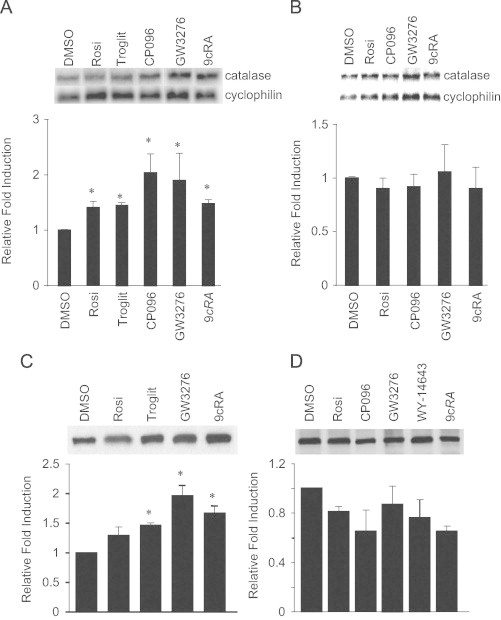
PPAR and RXR agonists increase catalase expression in astrocytes but not in glioma cells. Primary rat astrocytes and C6 rat glioma cells were supplemented for 48 h with the following agonists (10 μM). Representative Northern blots for primary rat astrocytes (A) and C6 rat glioma cells (B) probed for catalase and cyclophillin. Densitometric analysis was performed using Typhoon 8600 Phosphoimager and software package ImageQuant 5.1 (below blots). Representative Western blot for primary rat astrocytes (C) and C6 glioma cells (D) probed for catalase. Quantification of catalase protein levels were determined (below). Northern and Western blots are representatives of at least four independent experiments and data are expressed as mean±SEM. Treatments were significantly different from the DMSO group by Dunnett's multiple comparison procedure as indicated by ⁎, p<0.01.
PPAR agonist-mediated increase in catalase mRNA is not caused increased mRNA stability
Changes in steady-state mRNA levels may be attributable to alterations in the degradation rate of a transcript and/or changes in the rate of transcription. The relative contribution of a post-transcriptional mechanism in the PPAR agonist-mediated modulation of catalase mRNA levels was determined using the transcription inhibitor actinomycin D (ActD). Primary rat astrocytes were supplemented with 10 μM CP096 (as it showed a>2-fold increase in catalase mRNA), Rosi, or DMSO for 24 h and then treated with 5 μg/mL ActD. Cells were harvested at 0, 4, 8, 12 and 24 h time-intervals after addition of ActD. The amount of catalase mRNA was normalized using the expression of cyclophilin (Fig. 3A). The mRNA decay rates were comparable for both the PPARγ agonists and DMSO-treated cells, suggesting that the PPAR-mediated increase in catalase was independent of transcript stability (Fig. 3B).
Fig. 3.
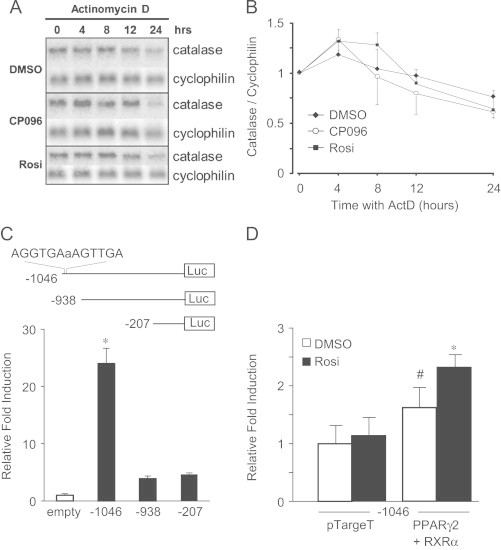
PPAR-induced catalase mRNA in astrocytes is not due to increased message stability. Primary rat astrocytes were supplemented for 24 h with 10 M CP096, Rosi, or DMSO (vehicle control) and then treated up to an additional 24 h with 5 μg/mL ActD (transcription inhibitor). Total RNA was harvested at 0, 4, 8, 12, and 24 h post ActD (A). Densitometric analysis was performed using Typhoon 8600 Phosphoimager and software package ImageQuant 5.1. Catalase mRNA was normalized to the housekeeping gene, cyclophilin, within each time point. This Northern blot is a representative of at least three independent experiments and data are expressed as mean±SEM (B). Primary rat astrocytes were transiently transfected with the indicated rat catalase promoter deletion constructs (-1046, -938, -207). The β-gal expression vector was used to control for transfection efficiency. Luciferase activity was normalized to β-gal and then all the catalase promoter deletion constructs were compared to the activity of the empty vector control (pGL3-basic) (C). Primary rat astrocytes were transiently transfected with the rat catalase promoter deletion construct containing the PPRE (-1046), PPARγ2, RXRα, or the empty vector (pTargeT) control and then treated with 5 μM Rosi for 24 h. Luciferase activity was normalized to β-gal and then all the catalase promoter deletion constructs were normalized to the activity of primary rat astrocytes treated transfected with -1046 and pTargeT treated with vehicle (DMSO) (D). Results of the luciferase promoter reporter assays shown represent mean±SEM of at least five independent experiments performed in triplicate. ⁎, p<0.01 compared to all groups; #, p<0.01 compared to pTargeT treated with DMSO.
PPAR agonists induce de novo transcription of catalase mRNA
We previously identified a PPRE in the rat catalase promoter located at nucleotide (nt) −1027 to −1015 with respect to the translation start site [24]. To examine whether PPAR agonists led to transcriptional transactivation of catalase gene expression, promoter deletion constructs of the rat catalase promoter were transiently transfected in to primary rat astrocytes. The promoter deletion construct containing the PPRE (-1046) increased promoter activity >20-fold compared to empty vector control (Fig. 3C). The deletion of the PPRE significantly diminished reporter activity following the transfection of rat catalase deletion constructs (-938 and -207). The luciferase activity was normalized to β-gal and then all the catalase promoter deletion constructs were normalized to the activity of the empty vector control (pGL3-basic). Additionally, transiently transfected primary rat astrocytes had significantly increased promoter activity with the rat catalase promoter deletion construct containing the PPRE (-1046) with expression plasmids PPARγ2 and RXRα compared to empty vector (pTargeT) control (Fig. 3D, open bars). Moreover, the treatment of 5 μM Rosi for 24 h further increased promoter activity compared to DMSO-treated primary rat astrocytes transfected with PPARγ2 and RXRα (Fig. 3D, dark bars). Taken together, our results support the hypothesis that the PPAR-mediated increase in steady-state catalase mRNA observed in primary rat astrocytes is due to de novo catalase transcript rather than increased catalase mRNA stability.
PPARγ regulates catalase expression. To gain better insight into mechanism, Cos-1 cells were used. Cos-1 cells exhibited low endogenous levels of RXRα protein determined by Western blot analysis. In contrast, PPARα and PPARγ protein were not detectable. Transient transfection of mPPARγ2 and RXRα expression vectors into Cos-1 cells resulted in a robust expression of PPARγ and RXRα protein compared to the empty vector pTargeT control (Supplemental data S1). To demonstrate binding and formation of the PPAR and RXR heterodimer to the PPRE, Cos-1 cells were transiently transfected with PPARγ2 and RXRα and nuclear extracts were isolated for EMSAs. The rat catalase PPRE (AGGTGA-a-AGTTGA) was end-labeled with 32P and incubated with nuclear extracts. The nuclear extracts isolated from Cos-1 cells transfected with PPARγ2 and RXRα exhibited pronounced binding to the catalase PPRE compared to cells transfected with the pTargeT vector alone (Fig. 4A). Supershift experiments were performed to confirm specific PPAR binding to the catalase PPRE. The incubation of anti-PPARγ antibody resulted in a dose-dependent supershift (Fig. 4A, lanes 6 and 9) in the binding of nuclear extract isolated from Cos-1 cells transfected with PPARγ2 and RXRα.
Fig. 4.
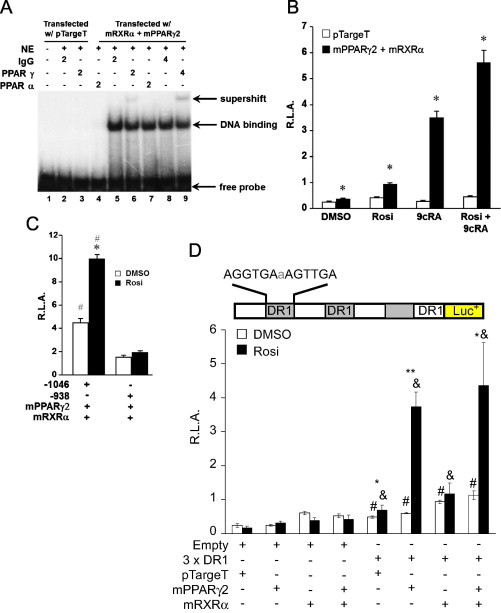
PPARγ plays a significant role in the regulation of catalase. Transfected PPARγ and RXRα bind to the PPRE in the rat catalase promoter determined by EMSA. Cos-1 cells were transfected (supplemental S1), nuclear protein was isolated, incubated with 32P-labeled catalase PPRE, and electrophoresed on a 5% nondenaturing polyacylamide gel. Supershift assay were performed by incubating 2 or 4 μL anti-PPARγ and -PPARα antibodies with reaction mixture (Santa Cruz) (A). Cos-1 cells were transiently transfected with the catalase promoter deletion construct containing the PPRE (-1046), mPPARγ2, mRXRα or pTargeT (empty vector control for the NHR expression plasmids) and then incubated with Rosi and 9cRA for 24 h. Transfected cells were lysed, luciferase activity was measured, and then normalized to β-gal activity (B). Results of the promoter reporter assays shown represent mean±SEM of at least five independent experiments performed in triplicate (⁎, p<0.006 for comparison between open bars vs shaded bars). Cos-1 cells were transiently transfected with β-gal, mPPARγ2, mRXRα, and either the catalase promoter deletion construct containing the PPRE (-1046) or lacking the PPRE (-938). Transfected cells were treated with Rosi for 24 h and promoter activity was determined. Results of the promoter reporter assays shown represent mean±SEM of at least seven independent experiments performed in triplicate (⁎, p<0.002 compared to DMSO; #, p<0.001 compared to catalase promoter construct lacking PPRE, -938) (C). Cos-1 cells were transiently transfected with the DR1x3 (inset) or empty vector (pGL3-promoter) in the presence of mPPARγ2+mRXRα or pTargeT. Transfected cells were treated with Rosi for 24 h and promoter activity was determined. Results of the promoter reporter assays shown represent mean±SEM of at least four independent experiments performed in triplicate (#, p<0.002 for comparison between DR1x3 vs empty for DMSO treatment; &, p<0.006 compared to respective empty vector treated with Rosi; ⁎⁎, p<0.0001 compared between Rosi vs DMSO; ⁎, p<0.05 compared between Rosi vs DMSO) (D).
Although, the findings indicate that PPAR/RXRα heterodimers present in the nuclear extract of transfected Cos-1 cells can bind to the catalase PPRE, this does not demonstrate that the catalase PPRE is functional. To confirm the functionality of the PPRE located in the rat catalase promoter, Cos-1 cells were transfected with the catalase promoter deletion construct containing the PPRE (-1046), PPARγ2, RXRα or pTargeT (empty vector control for the NHR expression plasmids) and then incubated with Rosi and 9cRA. PPAR-RXR heterodimers have been shown to synergistically activate reporter genes when both receptors are activated by their respective agonists [29,30]. A similar synergistic increase in reporter activity was observed in Cos-1 cells transfected with the -1046 rat catalase promoter deletion construct, PPARγ2, RXRα and incubated with Rosi and 9cRA, or both agonists together showed a 2.3-, 9.5- and 15-fold increase, respectively (Fig. 4B). Moreover, Cos-1 cells transfected with PPARγ2, RXRα and a catalase promoter deletion construct (-938) lacking the PPRE failed to increase promoter activity in response to Rosi treatment (Fig. 4C). To determine whether the catalase PPRE alone was sufficient to enhance PPAR-mediated promoter activity, we constructed a promoter containing a tandem tripeat of the catalase PPRE (DR1x3). Cos-1 cells were transfected with the DR1x3 or empty vector (pGL3-promoter) in the presence of PPARγ2/RXRα or pTargeT. Cells transfected with the DR1x3 exhibited a 2-fold (#, p=0.002) induction in reporter activity compared with empty vector (Fig. 4D). Moreover, Cos-1 cells transfected with DR1×3 containing PPARγ2, RXRα or both expression plasmids significantly increased promoter activity 2.5-, 3.8- and 4.6-fold, respectively (&, p<0.001). Rosi treatment resulted in a significant increase in promoter activity only in cells transfected with exogenous PPARγ alone (6.2-fold, ⁎⁎, p<0.0001) or in combination with RXRα (3.9-fold, ⁎, p=0.021) compared to their respective DMSO treated controls (Fig. 4D). A similar response was observed in Cos-1 cells transfected with the ACOX-PPRE, which was used as a positive control (not shown).
Pioglitazone (Pio)-mediated induction of catalase activity is dependent on PPARγ
Primary rat astrocytes were transfected with PPARγ-d.n. and then treated with a similar PPARγ agonist Pio (10 μM) for 48 h. Primary rat astrocytes treated with Pio for 48 h resulted in a 1.8-fold increase in catalase immunoreactive protein levels. However, primary rat astrocytes transfected with the PPARγ-d.n. construct failed to exhibit a Pio-mediated increase in catalase protein (Supplemental data S2). More importantly, Pio (10 μM) significantly increased catalase activity in primary rat astrocytes compared to vehicle alone (⁎, p<0.001). The Pio-mediated increase in catalase activity was completely abolished in cells transfected with the PPARγ-d.n. construct (Fig. 5A).
Fig. 5.
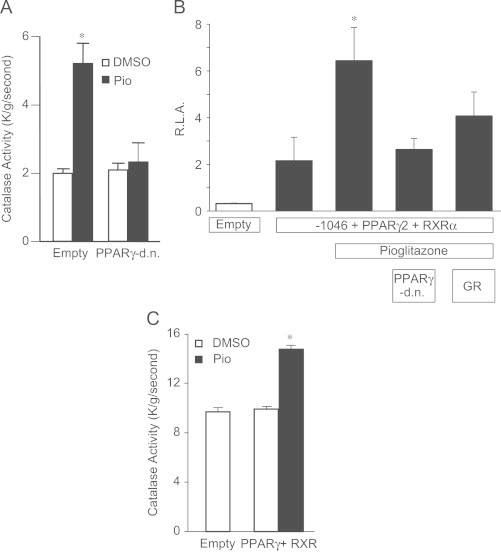
Pioglitazone-mediated induction of catalase activity is dependent on PPARγ. Primary rat astrocytes were transiently transfected with PPARγ-d.n. or empty vector and then treated with Pio (10 μM) for 48 h. Transfected cells were scrape harvested and catalase activity was determined spectrophotometrically at 240 nm. Results are derived from at least three independent experiments and data are expressed as mean±SEM (⁎, p<0.001 compared to all groups) (A). Cos-1 cells were transiently transfected with mPPARγ2, mRXRα, and either rat catalase promoter deletion construct containing the PPRE (-1046) or empty vector (pGL3-basic) and then treated with Pio for 24 h. Additionally, Cos-1 cells were also transiently co-transfected (as described above) with PPARγ-d.n. or co-treated with the PPARγ specific antagonist GR-259662 plus Pio for 24 h. Cells were lysed and promoter activity was determined. Results of the promoter reporter assays are mean±SEM of at least three independent experiments performed in triplicate (⁎, p<0.01 compared to all groups) (B). Catalase activity was measured using protein samples from the Cos-1 cells transfected with mPPARγ2 and mRXRα treated with 10 μM Pio for 24 h. Results are derived from at least three independent experiments and data are expressed as mean±SEM (⁎, p<0.01 compared to all groups) (C).
Pioglitazone treatment increased promoter activity 3-fold (⁎, p<0.01) in Cos-1 cells transfected with the rat catalase promoter deletion construct containing the PPRE (-1046), PPARγ2, and RXRα. The Pio-mediated increase in promoter activity was ablated by the co-transfection of PPARγ-d.n. or with the incubation of the PPARγ specific inhibitor GR-259662 (Fig. 5B). Lastly, catalase enzymatic activity was significantly increased in Cos-1 cells transfected with PPARγ2 and RXRα treated with Pio (10 μM) for 24 h (Fig. 5C).
Discussion
Treatment of primary cultures of normal rat astrocytes with PPARα, PPARγ and RXR agonists increased steady-state levels of catalase mRNA, immunoreactive protein, and enzymatic activity. In contrast, treatment of the C6 rat malignant glioma cell line with a similar panel of PPAR and RXR agonists failed to increase catalase mRNA, protein levels, and catalase enzymatic activity. Promoter analysis and cell treatment with ActD indicated that the increase in astrocyte catalase mRNA was a result of new mRNA synthesis. In support of this, EMSA analysis revealed that Cos-1 cells transfected with PPARγ and RXRα showed these transcriptional regulatory proteins bound to the PPRE of the rat catalase promoter and increased catalase promoter activity, as compared to empty (pTargeT) control vector. Finally, the transfection of PPARγ-d.n. completely blocked Pio-induced catalase activity. In aggregate, these studies reveal that PPARγ is responsible for the upregulation of catalase gene expression and enzymatic activity in normal rat astrocytes treated with PPARγ agonists.
Previous studies have demonstrated the presence of PPAR and RXR isotypes in the rat CNS. The degree of expression and tissue localization varies between these different lipid receptors. In the adult rat brain, PPARα, PPARγ and RXRα mRNA have all been identified in cortical astrocytes and to a lesser extent in cerebellar granule neurons [33]. In the rat spinal cord, PPARα mRNA and protein are expressed homogeneously in the gray matter, but in the white matter are exclusively localized to the astrocytes; PPARγ was not detected in cervical, thoracic or lumbar segments of the spinal cord [34]. Herein, Western blot analysis revealed the presence of PPARα and PPARγ but not RXRα protein in normal rat astrocytes, with the constitutive levels of expression for PPARα and PPARγ being markedly less than in C6 glioma cells. These results are consistent with previous observations showing a similar increase in constitutive expression of PPARγ in human glioma cells as compared to normal human astrocytes [31].
PPARα protein was expressed in Lipari human glioblastoma cell line, but no comparison was made with levels in normal astrocytes at that particular time [32]. In a separate study, all PPAR isotypes and RXRβ were detected in rat astrocytes by immunoblotting [33]. The present inability to identify RXRα protein in primary cultures of normal rat astrocytes confirms previous studies in which RXRα expression could only be observed using RT-PCR [34]. RXRα has been detected in human and rat glioma cells and 20 primary cultures established from biopsies from patients with glioblastomas multiforme [35]. Thus, the constitutive expression of not only PPARγ, but also of PPARα and RXRα is greater in glioma cells than in normal cell counterparts. This increased expression of PPARs and RXRs in glioma cells compared to the astrocytes may account for the increased catalase gene expression and enzymatic activity observed in glioma cells (Figs. 1 and 2).
Despite this increased expression of PPAR and RXR isotypes, C6 glioma cells incubated with PPAR and RXR agonists failed to exhibit an increase in catalase expression and/or activity. This failure to modulate catalase levels might reflect glioma cell cytotoxicity. PPARγ agonists are selectively cytotoxic to human U87 and A172 glioma cells and to C6 glioma cells by not only inhibiting proliferation but also by inducing apoptosis. In contrast, primary murine astrocytes were unaffected by PPARγ agonist treatment [36]. However, this cytotoxic effect was only observed in C6 cells incubated with concentrations ranging from 30 to 100 μM of the PPARγ agonist ciglitazone. A similar study was performed treating C6 glioma cells with ciglitazone and Rosi for 48 h at a concentration of 10 μM and there was roughly 60% and 35% reduction in cell viability determined by MTT assay, respectively [20]. We failed to detect significant levels of PPAR and/or RXR agonist-mediated cytotoxicity at 48 h, assessed in terms of esterase activity using the fluorescent dye Calcein AM in primary rat astrocytes and C6 glioma cells (not shown). One potential reason for the discrepancy at 48 h was that we treated the cells with media containing 10% serum whereas the former study was conducted in the absence of serum-containing medium. Thus, the selective ability of PPAR and RXR agonists to increase catalase expression in normal astrocytes but not in glioma cells does not appear to reflect glioma cell cytotoxicity after 48 h of treatment.
Most of the antineoplastic effects of PPARγ agonists occur with concentrations that typically are much higher than what is required to bind to and activate PPARγ suggesting PPARγ-dependent effects may be in part a consequence of off-target drug effects. Moreover, the concentrations of PPARγ agonists required to induce cytotoxicity and antineoplastic effects is at least a thousand times greater than drug concentrations used to treat type 2 diabetes mellitus [37]. PPARγ agonist-dependent and -independent effects inhibit cell proliferation, suppress inflammation, and block angiogenesis, thereby altering tumor microenvironments [38]. These actions can ultimately alter tumor cell viability and metastatic potential. As noted previously, the present experiments treated C6 glioma cells and astrocytes with PPAR agonists in serum-containing culture conditions, a scenario that best models in vivo conditions.
Besides the differences in constitutive levels of catalase enzymatic activity between primary cultures of normal rat astrocytes and rat malignant glioma cell line C6 (Fig. 1), there was also a greater expression of catalase mRNA and protein in the glioma cells compared to the astrocytes. These results are consistent with previous observations made in the 36B10 rat glioma cell line [23]. The relationship between this apparent increase in catalase activity in these two rat glioma cell lines and the resistance of glioma cells to redox-related anticancer therapies such as ionizing radiation remains unclear. However, it is notable that a comparison of antioxidant enzyme activities in 10 human and rat glioma cell lines with sensitivity to 1,3-bis (2-chloroethyl)-1-nitrosourea (BCNU, a chemotherapeutic and glutathione modulating drug used in the treatment of brain cancer), revealed a significant positive correlation with catalase activity alone [39]. Additionally, catalase inhibition sensitizes 36B10 glioma cells to oxidative stress. Catalase was inhibited in 36B10 glioma cells by two methods: (1) biochemically, using 3-amino-1,2,4-triazole as a non-competitive inhibitor and (2) using a catalase shRNA knockdown approach. Both methods reduced catalase enzymatic activity by ∼75% thereby increasing extracellular H2O2 and oxidative stress in the glioma cells. These catalase deficient glioma cells had increased sensitivity to ionizing radiation, supporting a regulatory role for catalase in the resistance of glioma cells to oxidative stress [40].
The current data support the concept that the selective manipulation of catalase gene expression and/or activity may improve normal tissue survival during radiation therapy. Catalase expression is regulated at the level of tissue-specific transcription, post-transcription, and post-translation [41,42]. The identification of functional PPRE in the 5′-flanking region of the rat catalase promoter (located between nt-1027 and -1015 with respect to the translation start site) supports that there is a transcriptional mechanism for regulation of catalase gene expression that could be mediated by TZDs, a drug class presently used to treat diabetes. This is reinforced by ActD (Fig. 3A), promoter-based studies (Figs. 3–5) and EMSA (Fig. 4A) revealing that PPAR agonist-induced upregulation of catalase expression in primary rat astrocytes is due, at least in part, to agonist binding and activation of PPAR/RXR heterodimers to the PPRE in the rat catalase promoter. Additionally, this observed differential increase in catalase expression and activity in astrocytes by PPAR agonists would suggest that astrocytes have increased protection from H2O2-induced damage. Experiments to test this would be of significant value. Moreover, an in vivo experiment testing whether this observed increase in catalase expression and activity in astrocytes resulted in a potential adjuvant therapy in treating malignant gliomas would be of great interest as most patients with these tumors have a poor prognosis.
Conclusions
In summary, PPARα and PPARγ agonists selectively increase catalase expression in normal astrocytes but not in glioma cells. The ability of PPARα and PPARγ agonists to selectively upregulate catalase expression in microvascular endothelial cells and astrocytes, which are both critical modulators of the response of the brain to radiation-induced injury, is promising. Since PPAR agonists did not increase catalase enzymatic activity or expression in glioma cells, a potential therapeutic strategy is revealed for reducing the severity of normal brain injury in glioma patients, without compromising tumor cell killing. These findings offer the promise of reducing the severity of normal brain injury in glioma patients without compromising tumor cell kill. This concept is reinforced by the growing evidence that PPARγ agonists inhibit proliferation of human and rat glioma cells. Thus, cancer therapeutic strategies utilizing PPAR agonists could protect normal tissue through the upregulation of catalase activity and increase the susceptibility of glioma cells. These agonists might have significant therapeutic impact as novel agents for improving the outcome for what remains an aggressive and all too often fatal neoplasm.
Acknowledgments
The authors would like to dedicate this work in memory of Mike E. Robbins and Larry W. Oberley. This work was supported by National Institutes of Health grants CA82722 (to M.E. Robbins), CA73612 and CA66081 (to F.E. Domann) and NS24621 (to S.A. Moore).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Nicholas K.H. Khoo, Email: nkhoo@pitt.edu.
Sachin Hebbar, Email: hebbar.sachin@gmail.com.
Weiling Zhao, Email: wzhao@wfubmc.edu.
Steven A. Moore, Email: steven-moore@uiowa.edu.
Frederick E. Domann, Email: frederick-domann@uiowa.edu.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2012.12.006.
Appendix A. Supporting information
Fig. S1.
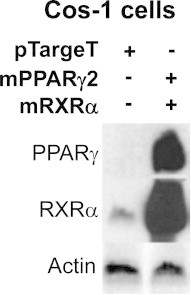
Cos-1 cells were transiently transfected with mPPARγ2, mRXRα, or empty control (pTargeT) expression vectors. Nuclear protein extracts were isolated 48 h after transfection and expression levels of PPARγ and RXRα were determined by Western blot analysis. The blot was then stripped and reprobed for Actin. This Western blot is a representative of at least four separate experiments
Fig. S2.
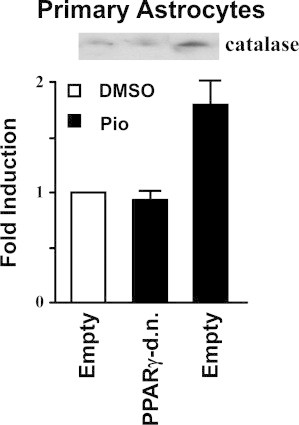
Primary rat astrocytes were transiently transfected with PPARγ-d.n. or empty vector and then treated with Pio (10 μM) for 48 h. Transfected cells were scrape harvested and Western blot analysis was performed. This Western blot is a representative of two independent experiments
References
- 1.National Cancer Institute and Central Brain Tumor Registry, [〈http://www.cbtrus.org/factsheet/factsheet.html〉].
- 2.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Davis F.G., Freels S., Grutsch J., Barlas S., Brem S. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: an analysis based on surveillance, epidemiology, and end results (SEER) data, 1973–1991. Journal of Neurosurgery. 1998;88:1–10. doi: 10.3171/jns.1998.88.1.0001. [DOI] [PubMed] [Google Scholar]
- 4.Schultheiss T.E., Stephens L.C. Invited review: permanent radiation myelopathy. British Journal of Radiology. 1992;65:737–753. doi: 10.1259/0007-1285-65-777-737. [DOI] [PubMed] [Google Scholar]
- 5.Imperato J.P., Paleologos N.A., Vick N.A. Effects of treatment on long-term survivors with malignant astrocytomas. Annals of Neurology. 1990;28:818–822. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 6.Crossen J.R., Garwood D., Glatstein E., Neuwelt E.A. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. Journal of Clinical Oncology. 1994;12:627–642. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 7.Clarke D.D., Sokoloff L. Lippincott-Raven; Philadelphia: 1999. Circulation and Energy Metabolism of the Brain. [Google Scholar]
- 8.Dringen R., Gutterer J.M., Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. European Journal of Biochemistry. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 9.Peuchen S., Bolanos J.P., Heales S.J., Almeida A., Duchen M.R., Clark J.B. Interrelationships between astrocyte function, oxidative stress and antioxidant status within the central nervous system. Progress in Neurobiology. 1997;52:261–281. doi: 10.1016/s0301-0082(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 10.Smith K.J., Kapoor R., Felts P.A. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathology. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spector A. Essentiality of fatty acids. Lipids. 1999;34:S1–S3. doi: 10.1007/BF02562220. [DOI] [PubMed] [Google Scholar]
- 12.Gottlicher M., Widmark E., Li Q., Gustafsson J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proceedings of the National Academy of Sciences USA. 1992;89:4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kliewer S.A., Sundseth S.S., Jones S.A., Brown P.F., Wisely G.B., Koble C.S., Devchand P., Wahli W., Willson T.M., Lenhard J.M., Lehmann J.M. Fatty Acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and g. Proceedings of the National Academy of Sciences USA. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kliewer S.A., Umesono K., Noonan D.J., Heyman R.A., Evans R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.A unified nomenclature system for the nuclear receptor superfamily. Cell 97 (1999) 161–163 [DOI] [PubMed]
- 16.Yoon M. The role of PPAR[alpha] in lipid metabolism and obesity: Focusing on the effects of estrogen on PPAR[alpha] actions. Pharmacological Research. 2009;60:151–159. doi: 10.1016/j.phrs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Tontonoz P., Spiegelman B.M. Fat and beyond: the diverse biology of PPARγ. Annual Review of Biochemistry. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 18.Reilly S.M., Lee C.-H. PPAR[delta] as a therapeutic target in metabolic disease. FEBS Letters. 2008;582:26–31. doi: 10.1016/j.febslet.2007.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grommes C., Landreth G.E., Sastre M., Beck M., Feinstein D.L., Jacobs A.H., Schlegel U., Heneka M.T. Inhibition of in vivo glioma growth and invasion by peroxisome proliferator-activated receptor γ agonist treatment. Molecular Pharmacology. 2006;70:1524–1533. doi: 10.1124/mol.106.022194. [DOI] [PubMed] [Google Scholar]
- 20.Perez-Ortiz J.M., Tranque P., Vaquero C.F., Domingo B., Molina F., Calvo S., Jordan J., Cena V., Llopis J. Glitazones differentially regulate primary astrocyte and glioma cell survival. Journal of Biological Chemistry. 2004;279:8976–8985. doi: 10.1074/jbc.M308518200. [DOI] [PubMed] [Google Scholar]
- 21.Montgomery D.L. Astrocytes: form, functions, and roles in disease. Veterinary Pathology. 1994;31:145–167. doi: 10.1177/030098589403100201. [DOI] [PubMed] [Google Scholar]
- 22.Vartak S., McCaw R., Davis C.S., Robbins M.E.C., Spector A.A. g-linolenic acid (GLA) is cytotoxic to 36B10 malignant rat astrocytoma cells but not to ‘normal’ rat astroxytes. British Journal of Cancer. 1998;77:1612–1620. doi: 10.1038/bjc.1998.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Preuss M., Girnun G.D., Darby C.J., Khoo N., Spector A.A., Robbins M.E. Role of antioxidant enzyme expression in the selective cytotoxic response of glioma cells to gamma-linolenic acid supplementation. Free Radical Biology and Medicine. 2000;28:1143–1156. doi: 10.1016/s0891-5849(00)00210-0. [DOI] [PubMed] [Google Scholar]
- 24.Girnun G.D., Domann F.E., Moore S.A., Robbins M.E. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Molecular Endocrinology. 2002;16:2793–2801. doi: 10.1210/me.2002-0020. [DOI] [PubMed] [Google Scholar]
- 25.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Marx N., Schonbeck U., Lazar M.A., Libby P., Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circulation Research. 1998;83:1097–1103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beers R.F., Sizer I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. Journal of Biological Chemistry. 1952;195:133–140. [PubMed] [Google Scholar]
- 28.Draper N.R., Smith H. John Wiley & Sons, Inc; New York: 1998. Applied Regression Analysis. ,pp. 221–229. [Google Scholar]
- 29.DiRenzo J., Soderstrom M., Kurokawa R., Ogliastro M.H., Ricote M., Ingrey S., Horlein A., Rosenfeld M.G., Glass C.K. Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Molecular and Cellular Biology. 1997;17:2166–2176. doi: 10.1128/mcb.17.4.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulman I., Shao G., Heyman R. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor γ (PPARγ) heterodimers: intermolecular synergy requires only the PPARγ hormone-dependent activation function. Molecular Cell Biology. 1998;18:3483–3494. doi: 10.1128/mcb.18.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nwankwo J.O., Robbins M.E. Peroxisome proliferator-activated receptor-gamma expression in human malignant and normal brain, breast and prostate-derived cells. Prostaglandins, Leukotrienes and Essential Fatty Acids. 2001;64:241–245. doi: 10.1054/plef.2001.0266. [DOI] [PubMed] [Google Scholar]
- 32.Cimini A., Cristiano L., Bernardo A., Farioli-Vecchioli S., Stefanini S., Ceru M.P. Presence and inducibility of peroxisomes in a human glioblastoma cell line. Biochimica Biophysica Acta. 2000;1474:397–409. doi: 10.1016/s0304-4165(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 33.Cristiano L., Bernardo A., Ceru M.P. Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. Journal of Neurocytology. 2001;30:671–683. doi: 10.1023/a:1016525716209. [DOI] [PubMed] [Google Scholar]
- 34.Cullingford T.E., Bhakoo K., Peuchen S., Dolphin C.T., Patel R., Clark J.B. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. Journal of Neurochemistry. 1998;70:1366–1375. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- 35.Bouterfa H., Picht T., Ke D., Herbold C., Noll E., Black P.M., Roosen K., Tonn C. Retinoids inhibit human glioma cell proliferation and migration in primary cell cultures but not in established cell lines. Neurosurgery. 2000;46:419. doi: 10.1097/00006123-200002000-00029. [DOI] [PubMed] [Google Scholar]
- 36.Zander T., Kraus J.A., Grommes C., Schlegel U., Feinstein D., Klockgether T., Landreth G., Koenigsknecht J., Heneka M.T. Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARgamma. Journal of Neurochemistry. 2002;81:1052–1060. doi: 10.1046/j.1471-4159.2002.00899.x. [DOI] [PubMed] [Google Scholar]
- 37.Panigrahy D., Singer S., Shen L.Q., Butterfield C.E., Freedman D.A., Chen E.J., Moses M.A., Kilroy S., Duensing S., Fletcher C. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. Journal of Clinical Investigation. 2002;110:923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panigrahy D., Huang S., Kieran M.W., Kaipainen A. PPARgamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biology and Therapy. 2005;4:687–693. doi: 10.4161/cbt.4.7.2014. [DOI] [PubMed] [Google Scholar]
- 39.Zhong W., Yan T., Lim R., Oberley L.W. Expression of superoxide dismutases, catalase, and glutathione peroxidase in glioma cells. Free Radical Biology and Medicine. 1999;27:1334–1345. doi: 10.1016/s0891-5849(99)00181-1. [DOI] [PubMed] [Google Scholar]
- 40.Smith P.S., Zhao W., Spitz D.R., Robbins M.E. Inhibiting catalase activity sensitizes 36B10 rat glioma cells to oxidative stress. Free Radical Biology and Medicine. 2007;42:787–797. doi: 10.1016/j.freeradbiomed.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 41.Van Remmen H., Williams M.D., Yang H., Walter C.A., Richardson A. Analysis of the transcriptional activity of the 5′-flanking region of the rat catalase gene in transiently transfected cells and in transgenic mice. Journal of Cell Physiology. 1998;174:18–26. doi: 10.1002/(SICI)1097-4652(199801)174:1<18::AID-JCP3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 42.Cao C., Leng Y., Liu X., Yi Y., Li P., Kufe D. Catalase is regulated by ubiquitination and proteosomal degradation. Role of the c-Abl and Arg tyrosine kinases. Biochemistry. 2003;42:10348–10353. doi: 10.1021/bi035023f. [DOI] [PubMed] [Google Scholar]