

Abstract
Manganese superoxide dismutase, encoded by the Sod2 gene, is a ubiquitously expressed mitochondrial antioxidant enzyme that is essential for mammalian life. Mice born with constitutive genetic knockout of Sod2 do not survive the neonatal stage, which renders the longitudinal study of the biochemical and metabolic effects of Sod2 loss difficult. However, multiple studies have demonstrated that tissue-specific knockout of Sod2 in murine liver yields no observable gross pathology or injury to the mouse. We hypothesized that Sod2 loss may have sub-pathologic effects on liver biology, including the acquisition of reactive oxygen species-mediated mitochondrial DNA mutations. To evaluate this, we established and verified a hepatocyte-specific knockout of Sod2 in C57/B6 mice using Cre-LoxP recombination technology. We utilized deep sequencing to identify possible mutations in Sod2−/− mitochondrial DNA as compared to wt, and both RT-PCR and traditional biochemical assays to evaluate baseline differences in redox-sensitive pathways in Sod2−/− hepatocytes. Surprisingly, no mutations in Sod2−/− mitochondrial DNA were detected despite measurable increases in dihydroethidium staining in situ and concomitant decreases in complex II activity indicative of elevated superoxide in the Sod2−/− hepatocytes. In contrast, numerous compensatory alterations in gene expression were identified that suggest hepatocytes have a remarkable capacity to adapt and overcome the loss of Sod2 through transcriptional means. Taken together, these results suggest that murine hepatocytes have a large reserve capacity to cope with the presence of additional mitochondrial reactive oxygen species.
Keywords: SOD, Superoxide, Liver, Mitochondrial DNA, Redox compensation
Graphical abstract
Genetic elimination of SOD2 expression in murine hepatocytes resulted in increased pro-oxidant production and a concomitant increase in antioxidant defenses, resulting in no observed mutational damage to mitochondrial DNA.
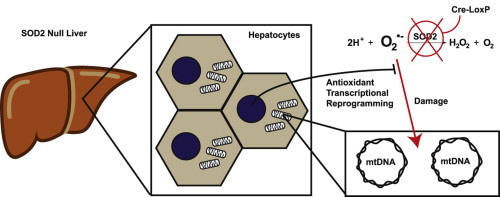
Highlights
► Sod2 was deleted in murine hepatocytes, increasing prooxidant production. ► No overt pathologic changes were seen, confirming previous results. ► There were no observed mutational events in mitochondrial DNA. ► Multiple compensatory alterations in redox and iron biology were identified.
Introduction
The liver is a highly metabolically active organ, and is heavily dependent on mitochondrial energy production for appropriate functionality. Hepatocytes thus require a high intrinsic capacity for superoxide removal in the mitochondrial compartment to mitigate free radical production from the electron transport chain. Moreover, as part of the blood detoxification and filtration system, hepatocytes encounter an enormous number of extrinsic pro-oxidant species. Not surprisingly, reactive oxygen species (ROS) have a demonstrable role in pathogenesis for a number of significant liver diseases, including those associated with alcohol intoxication, acetaminophen overdose, and hepatitis viral infection [1–4]. It has additionally been demonstrated that both the iron response pathway and the hypoxia-inducible factor pathway are perturbed in the presence of pro-oxidant stress [5,6]. These findings collectively underscore the importance of maintaining an appropriate redox balance in hepatocytes, and illustrate the potential health benefits associated with further investigation in this area.
Manganese superoxide dismutase (SOD2) is the solitary mitochondrial superoxide dismutase, and is highly abundant in mitochondrion-rich hepatocytes. Previously, multiple groups have studied the effects of SOD2 loss in murine models. Nonspecific genetic deletion of the Sod2 locus was not an effective method for study of liver biology, as it proved to be lethal in the neonatal period. Pups born without functional SOD2 protein died within 3 weeks of birth from massive multisystem failure [7,8]. Subsequent use of the Cre-LoxP recombination system enabled the tissue-specific deletion of Sod2 in murine hepatocytes, circumventing the neonatal lethality of the total-animal knockout. To date, two studies have demonstrated that albumin-Cre-mediated excision of Sod2 produces no overt liver pathology by traditional histology or any measurement in markers indicative of oxidative stress [9,10]. A third study, published by Lenart et al, excised Sod2 at an earlier developmental stage with an α-fetoprotein-Cre, and reported distinct changes in hepatocyte physiology and disrupted zonal patterns of gene expression in the murine liver [11]. From these prior results, it appears clear that SOD2 plays an important role in liver development, because albumin-Cre mediates post-natal excision of the Sod2 locus. However, we remained intrigued by the absence of overt liver pathology in the absence of SOD2 protein in murine hepatocytes, given both the intrinsic and extrinsic pro-oxidant stresses that the liver is subjected to on a regular basis. We hypothesized that sub-pathologic effects, such as oxidative damage to mitochondrial DNA, may be accumulating in these hepatocytes, without leading to overt organ system damage.
To address this, we utilized Cre-LoxP technology with an albumin-Cre-mediated knockout to generate Sod2−/− hepatocytes in C57/B6 mice. Confirming previous studies with this model, these mice did not suffer any adverse pathologic consequences from the loss of SOD2 mRNA transcript, protein, or activity in hepatocytes. To evaluate mitochondrial DNA integrity, we deep sequenced mitochondrial DNA from both Sod2−/− animals and wt controls. We additionally investigated a representative series of both antioxidant and iron response pathways via RT-PCR, western blotting, and biochemical assays to identify potential compensatory mechanisms to account for the lack of gross pathology. Our results demonstrate that the loss of mitochondrial superoxide dismutase alone is an insufficient stimulus to promote an accumulation of mitochondrial DNA mutations in murine hepatocytes, secondary to a remarkable reserve capacity of hepatocytes to accommodate enhanced pro-oxidant production.
Materials and methods
Animal colony maintenance and use
Mice homozygous for the loxP-flanked Sod2 allele (B6.Cg-Sod2tm1, henceforth Sod2L/L) were obtained from Dr. Takuji Shirasawa of the Tokyo Metropolitan Institute of Gerontology and maintained as previously described [12]. B6.Cg-Tg-AlbCre21Mgn/J (henceforth Alb-Cre) animals were donated by Dr. Curt Sigmund (The University of Iowa) and were previously described [13,14]. Experimental animals were generated using an F3 breeding scheme and littermate controls were utilized for all studies. Genotyping was accomplished with DNA isolated from tail snips taken at the time of weaning, with the following primer sequences: Sod2-P1:5′-CGAGGGGCATCTAGTGGAGAAG-3′, Sod2-P2: 5′-TTAGGGCTCAGGTTTGTCCAGAA-3′, Cre-F: 5′-CGATGCAACGAGTGATGAGGT-3′, Cre-R: 5′-GTGAAACAGCATTGCTGTCACTT-3′. Animals were cared for and handled in accordance with the appropriate Institutional Animal Care and Use Committee guidelines at the University of Iowa. Experimental animals were sacrificed using CO2 asphyxiation followed by cervical dislocation. All animals were 10 weeks of age unless otherwise noted. Livers were dissected out, washed in ice-cold PBS, and weighed before individual portions were cut and processed for different experimental analysis. Portions that were unused at the time of harvest were snap frozen in liquid nitrogen and stored at −80 °C.
Histological analysis and immunohistochemistry
Samples used for histological analysis of liver pathology were placed in 10% neutral buffered formalin for fixation and preservation. Samples were embedded in paraffin, sectioned, and stained at the Comparative Histopathology Core Facility at the University of Iowa, under the direction of Dr. David Meyerholz, DVM, PhD, and Dr. Alicia Olivier, DVM, PhD. Immunohistochemical analysis for the presence of SOD2 in liver sections was accomplished using SOD2 antibody (AB10346, Millipore, Billerica, MA, USA).
Quantitative RT-PCR analysis of gene expression
Snap-frozen liver pieces were ground in liquid nitrogen using a mortar and pestle, and a portion of the pulverized liver material was used for RNA isolation by the TRIZOL method, followed by quantitation on a Nanodrop ND-1000. One microgram of total RNA was converted to cDNA using the cDNA archive kit (Applied Biosystems). Quantitative real-time PCR was then performed using 10 ng total of cDNA with an ABI Fast 7500 system thermal cycler running in standard mode. Both SYBR green and Taqman primer probe technologies were utilized for gene analysis as specified in the text. Fold changes were calculated using the ΔΔCt method, using the 18S ribosomal RNA as a control and Sod2L/L animals as references.
Protein immunoblotting
Snap-frozen liver pieces were prepared as above with a mortar and pestle under liquid nitrogen and suspended in a minimal volume of standard RIPA buffer supplemented with protease inhibitors (Product P8340, Sigma Aldrich), 100 μM PMSF, 1 mM sodium vanadate, 1 mM sodium fluoride, 1 mM DTT. Samples were snap frozen in liquid nitrogen and thawed 3 times, and cellular debris was pelleted by centrifugation at 20,000g for 10 min at 4 °C. Protein concentration was determined by the Bradford method, and equivalent protein amounts were run on 10% SDS-PAGE gels before transfer to nitrocellulose. Blots were probed using primary antibodies followed by HRP-conjugated secondary antibodies with appropriate species specificity. Antibodies used for these analyses include SOD2 (Millipore), SDHB (Abcam), HMOX1 (Abcam), ALAS2 (Abcam), TFR1 (Abcam), FTH (Abcam), and β-actin (Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City). Quantitation of blots was done utilizing ImageJ software.
Antioxidant enzyme activity assays
SOD activity was measured in liver homogenates using the assay developed by Spitz and Oberley [15]. SOD2 activity was measured in the presence of 5 mM sodium cyanide to inhibit SOD1 activity. Total GSH levels were determined using a recycling assay that spectrophotometrically measures dTNB reduction to TNB in the presence of glutathione reductase according to protocols described by Griffith [16] and Anderson [17]. Glutathione peroxidase activity was measured by monitoring NADPH oxidation in the presence of reduced glutathione, glutathione reductase, and either H2O2 or cumene hydroperoxide [18]. All sample measurements were normalized to protein content as evaluated by the Bradford method (enzyme activities) or the BCA method (total glutathione). All statistics were calculated using Microsoft Excel.
Dihydroethidium staining of frozen sections
Samples were analyzed in a method adapted from use in cardiovascular studies [19–21]. Briefly, freshly dissected livers were frozen in OCT medium, and 10 μm sections were cut and placed on slides. In a dark room, several drops of 5 μM dihydroethidium (DHE) and 10 nM Mitotracker Green (Life Technologies) were placed on tissue sections and slides were incubated in a humidified chamber for 30 min at 37 °C. Slides were carefully rinsed with PBS, coverslipped, and examined with a Zeiss 710 confocal microscope with an excitation wavelength of 544 nm and an emission wavelength of 612 nm. Images were converted into TIFF format using ImageJ.
Mitochondrial complex II activity assay
Freshly isolated livers were dissociated in ice-cold mannitol homogenization buffer (0.3 M mannitol, 0.006 M KH2PO4, 0.014 M K2HPO4) using a Dounce homogenizer with 10 strokes of the loose and then 10 strokes of the tight pestle. Homogenates were spun at 600 g for 10 min at 4 °C to pellet nuclear and cellular debris. The resultant supernatants were spun at 5000 g for 10 min at 4 °C to crudely pellet mitochondria. Mitochondria were washed once and resuspended in 1 ml of ice-cold assay buffer (0.3 M mannitol, 0.006 M KH2PO4, 0.014 M K2HPO4, 0.01 M KCL, 0.005 M MgCl2, pH 6.2). Protein was quantified using the Bradford method. A total of 50 μg of protein were used per assay, and antimycin A (1 μg/μl), sodium azide (10 mM), and rotenone (1 μg/μl) were utilized to block other complex respiration. Complex II activity was calculated by observing the reduction of dichloroindophenol (DCIP) at 600 nm following the addition of 5 mM succinate to the reaction volume. The reaction was followed on a Tecan Infinite 200 PRO microplate spectrophotometer and final complex II activity was calculated using a molar extinction coefficient for DCIP of 19100. All statistical analysis was accomplished using Microsoft Excel.
Mitochondrial complex I respiration assay
Complex I activity was assessed using a Seahorse XF96 Extracellular Flux Analyzer, following the protocol established by Rogers et al. [22]. Mitochondrial fractions were isolated as described and 4 ug of total mitochondrial isolate was loaded per well in the XF96 plate format. Plates were spun at 4 °C at 2000 g for 20 min to affix mitochondria to the wells. The assay was run in the presence of 5 mM pyruvate and 5 mM malate, with sequential injections of FCCP and Antimycin A to final concentrations of 4 μM apiece. A minimum of 5 technical replicates was used for each mitochondrial isolation. Statistical analysis was accomplished using Microsoft Excel.
Mitochondrial genomic sequencing
Mitochondrial DNA was prepared for sequencing according to the Ion Torrent Ion Fragment Library Kit protocol, with minor modifications. DNA was sonicated to fragments between 150 and 200 bp and confirmed on a 1% agarose gel. Fragmented DNA was then subjected to the end-repair reaction, followed by ligation of sequencing adapters. The adapter-ligated library was size-separated on a 2% agarose gel and library products between the sizes of 170 bp and 225 bp were isolated using a modified siliconized glass wool extraction method and purified using the Agencourt AMPure XP PCR purification kit (Beckman Coulter, Beverly, MA). The purified library was subjected to a nick-translation and amplification for 5 cycles, quantitated on an Agilent High Sensitivity DNA chip, diluted to 8.4×107 molecules/μl, and 5 μl was added to the OneTouch emulsion PCR reaction. Beads with template were isolated using the Ion Torrent OneTouch ES and loaded on to a 316 sequencing chip. Sequencing was performed according to manufacturer's protocols and the resulting data downloaded as a FASTQ file. Sequences were loaded into SoftGenetic's NextGene software version 2.0.0 and filtered for length (minimum 25 called bases) and quality (median Q12 or higher, trim after 1 base less than Q12). Filtered reads were then aligned to mouse mitochondrial sequence from assembly NCBI37/mm9 and examined for variants.
Results and discussion
Establishment of hepatocyte-specific loss of Sod2 in conditional knockout mice
LoxP-flanked mice harboring albumin-Cre by PCR analysis were identified as potential hepatocyte-specific Sod2−/− candidates. This was verified by assaying liver homogenates in both Sod2L/L and Sod2−/− animals for evidence of Sod2 expression (Fig. 1A–C). Sod2−/− animals had a significant reduction in SOD2 protein, Sod2 mRNA, and SOD2 activity when compared with Sod2L/L littermate controls. In situ immunohistochemical analysis for SOD2 demonstrated near total pan-hepatocellular reduction in SOD2 protein in Sod2−/− livers compared to Sod2L/L livers, with residual signal remaining in both endothelial cells and occasional Kupffer cells (Fig. 1D). In situ staining with DHE was utilized to evaluate the production of pro-oxidant species in frozen sections. Sod2−/− sections demonstrated enhanced DHE positivity with a high degree of nuclear localization when compared with Sod2L/L control mice, suggesting increased production of pro-oxidant species (Fig. 1E). Notably, no overt structural or other phenotypic differences were seen when comparing Sod2−/− livers to Sod2L/L livers using a variety of other histochemical techniques, including Masson's Trichrome for aberrant collagen deposition, hematoxylin and eosin staining for basic structural abnormalities, or Prussian blue for iron accumulation (data not shown). Additionally, no differences in overall mitochondrial number were observed following quantitative morphometric analysis using transmission electron microscopy (data not shown). Collectively, these results confirm both a successful knockout of Sod2 in murine hepatocytes and affirm the results from previous studies using albumin-Cre as a driver for Sod2 excision.
Fig. 1.
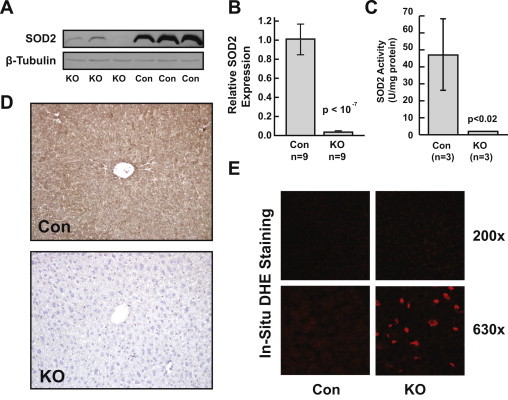
Establishment ofSod2hepatocyte-specific knockout. (A) Western blot for SOD2 demonstrating loss of SOD2 protein in Sod2−/− (KO) livers compared to Sod2L/L (Con) livers. (B and C) Quantification of significant reduction in Sod2 message (B) and SOD2 activity (C) in KO livers compared to control. (D) Immunohistochemical staining for SOD2 demonstrates pan-hepatocellular knockout in Sod2−/− livers compared to control. (E) Representative in situ DHE staining demonstrating enhanced nuclear positivity in Sod2−/− livers compared to control.
Evaluation of complex I and complex II in Sod2−/− hepatocytes
To address the hypothesis that SOD2 loss in murine hepatocytes might be promoting sub-pathological damage accumulation, several aspects of mitochondrial biology were evaluated. Complex I activity was evaluated using isolated mitochondria with a Seahorse XF96 extracellular flux analyzer, and demonstrated no differences between Sod2L/L and Sod2−/− mitochondria (Fig. 2A). In contrast, complex II activity was markedly different between Sod2L/L and Sod2−/− mitochondria. Sod2−/− samples had an average activity of 0.76 nm DCIP reduced mg protein−1 s−1 compared to 2.31 nm DCIP reduced mg protein−1 s−1 in Sod2L/L samples, demonstrating a significant reduction (p=0.004) in overall complex II activity. This was accompanied by a concomitant loss of immunoreactive succinate dehydrogenase subunit B (SDHB), which harbors the critical iron–sulfur core of complex II and is known to be sensitive to perturbation by superoxide (Fig. 2B). This observation, coupled with the enhanced DHE positivity seen in Sod2−/− frozen sections (Fig. 1E), strongly suggest that the loss of Sod2 is promoting an increase in superoxide in Sod2−/− mitochondria.
Fig. 2.
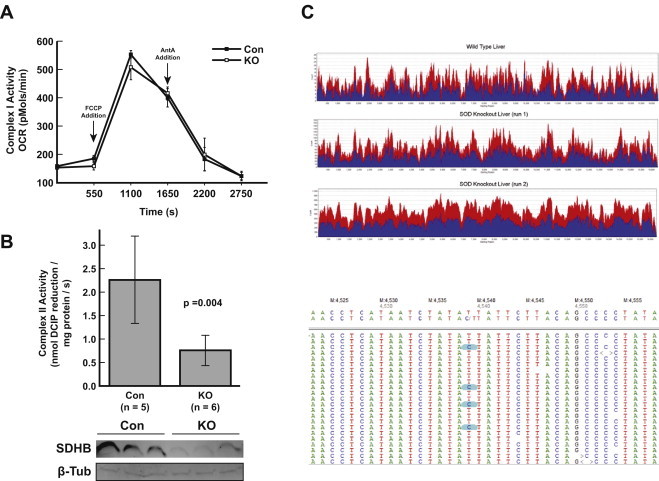
Measurement of mitochondrial complex activity and mtDNA integrity. (A) Complex I activity assay as run on Seahorse extracellular flux analyzer, with no significant differences noted between KO (empty squares) and control (filled squares) samples (n=3 mice per group). (B) Complex II activity assay demonstrating significant reduction in complex II in Sod2−/− samples compared to control, with an accompanying reduction in SDHB protein on western blotting. (C) Graph of sequencing coverage for Sod2+/+ and Sod2−/− mitochondrial genomes. The number of reads at each position of the mitochondrial genome is shown for forward (red) and reverse (blue) reads. Below: sequence variant at position 4539 in the mitochondrial genome. Screen capture showing representative reads from the second sequencing run of the Sod2−/− mitochondrial genome aligned to reference (top sequence), the variant is shown as a C/T transition just below the reference sequence. Reads containing the variant are highlighted in blue. Mitochondrial genome position is indicated above the reference sequence. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Mitochondrial genomic sequencing
It is commonly accepted that mitochondrial DNA (mtDNA) is more susceptible to oxidative modification than its nuclear counterpart, due to both its environment in the redox active mitochondrial matrix and the lack of involvement with histones [23]. We thus hypothesized that the enhanced pro-oxidant production in the mitochondrial compartment would lead to an accumulation of oxidative damage to mtDNA. Mitochondrial DNA from the livers of 10-week Sod2L/L and Sod2−/− mice was subjected to deep sequencing to determine if any mutations were accumulating in Sod2−/− mice (Fig. 2C). MtDNA sequence was compared to reference sequence (NCBI37/mm9 assembly) for all sequencing runs. In both the Sod2+/+ and Sod2−/− samples a variant (T>C) was found at position 4539 within the NADH dehydrogenase subunit 2 sequence that changes an isoleucine to threonine (Fig. 2C). Based on its presence in both Sod2−/− and SodL/L sequences, this variant appears unrelated to SOD status. In both Sod2−/− sequencing runs a variant (C>T) was found at position 11828 within the NADH dehydrogenase subunit 5 that is synonymous. There were insufficient reads in the wild type sequence to confirm presence of the position 11828 variant in the wild type mouse. Otherwise, no mutations were found in the mitochondrial DNA of the SOD KO−/− mouse's mitochondrial DNA relevant to the wild type sample. This was a remarkable result—despite being essentially devoid of SOD activity and thus exposed to elevated levels of pro-oxidant production sufficient to promote biochemical defects, mtDNA integrity was preserved in Sod2−/− hepatocytes and indistinguishable from normal hepatocytes. We thus hypothesized that a compensatory mechanism was being implemented by Sod2−/− hepatocytes to enable them to escape damage both to liver parenchyma and mtDNA.
Gene expression and pathway analysis
To initially address possible compensatory mechanisms at play in Sod2−/− hepatocytes, we designed a custom quantitative RT-PCR array to examine a variety of distinct genes chosen for their importance in several critical redox systems. To provide robust results for these experiments, template cDNA was pooled from 9 Sod2−/− and 9 Sod2L/L livers for group comparison. These animals ranged from 65 to 87 weeks and had genotypes confirmed based on Sod2 mRNA expression. Analysis demonstrated that 13 of the 44 genes on the array had at least a twofold difference in expression based on Sod2 status (Fig. 3A). Critically, these genes fell into several pathways, including those involved in iron homeostasis such as heme oxygenase (Hmox1), ferritin light chain 1 (Ftl1), and ferrocheletase (Fech); several involved in antioxidant response pathways such as Cu/Zn superoxide dismutase (Sod1), peroxiredoxin 1 (Prdx1), thioredoxin 2 (Txn2), and glutathione synthase (Gss); and others involved in other aspects of liver biology such as deleted in malignant brain tumors 1 (Dmbt1). Notably, Sdhb transcript was elevated in Sod2−/− hepatocytes, suggesting a compensatory transcriptional upregulation secondary to the loss of SDHB protein established in Fig. 2B.
Fig. 3.
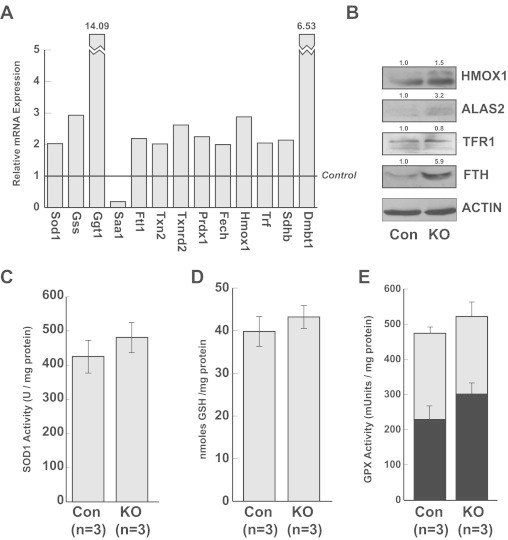
Compensatory mechanisms inSod2−/−hepatocytes. (A) Results from gene array detailing 13 genes with greater than twofold expression difference in Sod2−/− compared with control. Differences are derived from pooled populations of 9 mice per group. (B) Representative iron homeostasis genes are altered in Sod2−/− hepatocytes. HMOX1—heme oxygenase 1, ALAS2—aminolevulinic acid synthase 2, TFR1—transferrin receptor 1, FTH—ferritin heavy chain. Numbers above blots indicate quantitation using ImageJ software. (C) SOD1 activity assay demonstrating elevated SOD1 activity in Sod2−/− liver homogenates compared to control. (D) Total GSH is slightly elevated in Sod2−/− liver homogenates compared to control. (E) Sod2−/− liver homogenates have increased selenium-dependent (black bar), selenium independent (gray partition), and total GPx (overall height) compared to control. Results in (C), (D), and (E) are not statistically significant.
To evaluate the downstream effects of these transcriptionally up-regulated genes several biochemical assays were utilized. Using the results of the expression array as a guide, several iron homeostasis elements were probed using western blotting. These genes all are regulated at the post-transcriptional level through the iron response protein system, which is sensitive to both free iron levels and oxidative modification. Tellingly, HMOX1, aminolevulinic acid synthase 2 (ALAS2), and ferritin heavy chain (FTH) were all upregulated at the protein level to at least some degree in response to Sod2 loss, suggesting that Sod2−/− hepatocytes are actively engaging in a redistribution of iron (Fig. 3B).
To test the contribution of representative antioxidant response pathways, several elements were examined. SOD1, though predominantly localized to the cytoplasm, does reside in the intermembrane space of mitochondria [24] and could contribute to mitochondrial protection. Accordingly, a small increase of 56 U/mg was seen in SOD1 activity in Sod2−/− hepatocytes, though this was not statistically significant (Fig. 3C). As multiple pathways in the array analysis utilized glutathione, total GSH was measured in both Sod2−/− and Sod2L/L samples. Sod2−/− samples contained an average of 43.2 nmoles GSH/mg protein, whereas Sod2L/L samples contained an average of 39.8 nmoles GSH/mg protein (Fig. 3D). These same samples were additionally assayed for glutathione peroxidase (GPx) activity. Again, small but statistically not significant increases were seen in Sod2−/− hepatocytes in both selenium-dependent (p=0.07) and selenium-independent GPx activities as well as total GPx activity (Fig. 3E). While none of these results reached statistical significance, obvious trends in multiple different assays spanning multiple redox pathways were seen. The likelihood of these subtle results being randomly aligned in this way is slim, especially given the diverse approaches taken to measure them.
Conclusions
Previous studies have demonstrated that Sod2−/− murine livers suffer no observable pathologic damage at baseline. We hypothesized that sub-pathologic damage, primarily in the form oxidative damage to mtDNA, may be accumulating in these animals without reaching a threshold for overt pathology. Surprisingly, this was not the case; Sod2−/− liver mtDNA was qualitatively indistinguishable from Sod2L/L liver mtDNA at 10 weeks of age, suggesting that Sod2−/− hepatocytes are engaging in a significant compensatory effort to overcome measurable increases in pro-oxidant production. Here, we demonstrated that a constellation of subtle changes in redox biology pathways is indeed taking place in Sod2−/− hepatocytes, with the end result being a functional murine liver with no evidence of microscopic damage. The Sod2−/− murine liver thus represents a tremendous model for evaluation of the reserve capacity of hepatocytes to deal with exogenous pro-oxidant stress. Continued evaluation of this model may yield important insights into further hepatoprotective mechanisms that could be extended to clinical situations in humans.
Acknowledgements
The authors thank Dr. Curt Sigmund for the gift of Alb-Cre mice, Drs. David Meyerholz and Alicia Olivier for outstanding pathology support, and the outstanding team at the Arizona Cancer Center Shared Genomic Service. This work was supported by NIH grants R01 CA115438 (FED), P30 CA086862, R01 CA133114 (DRS), and P42 ES004940 (BWF).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Anthony R. Cyr, Email: anthony-cyr@uiowa.edu.
Kyle E. Brown, Email: kyle-brown@uiowa.edu.
Michael L. McCormick, Email: michael-mccormick@uiowa.edu.
Mitchell C. Coleman, Email: mitchell-coleman@uiowa.edu.
George S. Watts, Email: gwatts@azcc.arizona.edu.
Bernard W. Futscher, Email: bfutscher@azcc.arizona.edu.
Douglas R. Spitz, Email: douglas-spitz@uiowa.edu.
Frederick E. Domann, Email: frederick-domann@uiowa.edu.
References
- 1.Das S.K., Vasudevan D.M. Alcohol-induced oxidative stress. Life Sciences. 2007;81(3):177–187. doi: 10.1016/j.lfs.2007.05.005. [Epub 2007/06/16] [DOI] [PubMed] [Google Scholar]
- 2.Albano E. Alcohol, oxidative stress and free radical damage. Proceedings of the Nutrition Society. 2006;65(3):278–290. doi: 10.1079/pns2006496. [Epub 2006/08/23] [DOI] [PubMed] [Google Scholar]
- 3.Koike K. Hepatitis B virus X gene is implicated in liver carcinogenesis. Cancer Letters. 2009;286(1):60–68. doi: 10.1016/j.canlet.2009.04.010. [Epub 2009/05/26] [DOI] [PubMed] [Google Scholar]
- 4.Chun L.J., Tong M.J., Busuttil R.W., Hiatt J.R. Acetaminophen hepatotoxicity and acute liver failure. Journal of Clinical Gastroenterology. 2009;43(4):342–349. doi: 10.1097/MCG.0b013e31818a3854. [Epub 2009/01/27] [DOI] [PubMed] [Google Scholar]
- 5.Peyssonnaux C., Zinkernagel A.S., Schuepbach R.A., Rankin E., Vaulont S., Haase V.H. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) Journal of Clinical Investigation. 2007;117(7):1926–1932. doi: 10.1172/JCI31370. [Epub 2007/06/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muckenthaler M.U., Galy B., Hentze M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annual Review of Nutrition. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [Epub 2008/05/21] [DOI] [PubMed] [Google Scholar]
- 7.Li Y., Huang T.T., Carlson E.J., Melov S., Ursell P.C., Olson J.L. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [Epub 1995/12/01] [DOI] [PubMed] [Google Scholar]
- 8.Lebovitz R.M., Zhang H., Vogel H., Cartwright J., Jr., Dionne L., Lu N. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9782–9787. doi: 10.1073/pnas.93.18.9782. [Epub 1996/09/03] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikegami T., Suzuki Y., Shimizu T., Isono K., Koseki H., Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochemical and Biophysical Research Communications. 2002;296(3):729–736. doi: 10.1016/s0006-291x(02)00933-6. [Epub 2002/08/15] [DOI] [PubMed] [Google Scholar]
- 10.Shimizu T., Nojiri H., Kawakami S., Uchiyama S., Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase gene. Geriatrics and Gerontology International. 2010;10(Suppl. 1):S70–S79. doi: 10.1111/j.1447-0594.2010.00604.x. [Epub 2010/07/16] [DOI] [PubMed] [Google Scholar]
- 11.Lenart J., Dombrowski F., Gorlach A., Kietzmann T. Deficiency of manganese superoxide dismutase in hepatocytes disrupts zonated gene expression in mouse liver. Archives of Biochemistry and Biophysics. 2007;462(2):238–244. doi: 10.1016/j.abb.2007.02.007. [Epub 2007/03/21] [DOI] [PubMed] [Google Scholar]
- 12.Case A.J., McGill J.L., Tygrett L.T., Shirasawa T., Spitz D.R., Waldschmidt T.J. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free radical Biology and Medicine. 2011;50(3):448–458. doi: 10.1016/j.freeradbiomed.2010.11.025. [Epub 2010/12/07] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Postic C., Magnuson M.A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26(2):149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [Epub 2000/03/21] [DOI] [PubMed] [Google Scholar]
- 14.Postic C., Shiota M., Niswender K.D., Jetton T.L., Chen Y., Moates J.M. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. Journal of Biological Chemistry. 1999;274(1):305–315. doi: 10.1074/jbc.274.1.305. [Epub 1998/12/29] [DOI] [PubMed] [Google Scholar]
- 15.Spitz D.R., Oberley L.W. An assay for superoxide dismutase activity in mammalian tissue homogenates. Analytical Biochemistry. 1989;179(1):8–18. doi: 10.1016/0003-2697(89)90192-9. (Epub 1989/05/15) [DOI] [PubMed] [Google Scholar]
- 16.Griffith O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Analytical Biochemistry. 1980;106(1):207–212. doi: 10.1016/0003-2697(80)90139-6. [Epub 1980/07/15] [DOI] [PubMed] [Google Scholar]
- 17.Anderson M.E. Determination of glutathione and glutathione disulfide in biological samples. Methods in Enzymology. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [Epub 1985/01/01] [DOI] [PubMed] [Google Scholar]
- 18.Lawrence R.A., Burk R.F. Glutathione peroxidase activity in selenium-deficient rat liver. Biochemical and Biophysical Research Communications. 1976;71(4):952–958. doi: 10.1016/0006-291x(76)90747-6. [Epub 1976/08/23] [DOI] [PubMed] [Google Scholar]
- 19.Robinson K.M., Janes M.S., Beckman J.S. The selective detection of mitochondrial superoxide by live cell imaging. Nature Protocols. 2008;3(6):941–947. doi: 10.1038/nprot.2008.56. [Epub 2008/06/10] [DOI] [PubMed] [Google Scholar]
- 20.Dikalova A.E., Bikineyeva A.T., Budzyn K., Nazarewicz R.R., McCann L., Lewis W. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation Research. 2010;107(1):106–116. doi: 10.1161/CIRCRESAHA.109.214601. [Epub 2010/05/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikalov S., Griendling K.K., Harrison D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49(4):717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [Epub 2007/02/14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rogers G.W., Brand M.D., Petrosyan S., Ashok D., Elorza A.A., Ferrick D.A. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PloS One. 2011;6(7):e21746. doi: 10.1371/journal.pone.0021746. [Epub 2011/07/30] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee H.C., Wei Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. International Journal of Biochemistry and Cell Biology. 2005;37(4):822–834. doi: 10.1016/j.biocel.2004.09.010. [Epub 2005/02/08] [DOI] [PubMed] [Google Scholar]
- 24.Kawamata H., Manfredi G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxidants and Redox Signaling. 2010;13(9):1375–1384. doi: 10.1089/ars.2010.3212. [Epub 2010/04/07] [DOI] [PMC free article] [PubMed] [Google Scholar]