

Abstract
Alzheimer disease (AD) is an age-related neurodegenerative disease characterized by the presence of three pathological hallmarks: synapse loss, extracellular senile plaques (SP) and intracellular neurofibrillary tangles (NFTs). The major component of SP is amyloid β-peptide (Aβ), which has been shown to induce oxidative stress. The AD brain shows increased levels of lipid peroxidation products, including 4-hydroxy-2-nonenal (HNE). HNE can react covalently with Cys, His, or Lys residues on proteins, altering structure and function of the latter. In the present study we measured the levels of the HNE-modified lipoic acid in brain of subjects with AD and age-matched controls. Lipoic acid is a key co-factor for a number of proteins including pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, key complexes for cellular energetics. We observed a significant decrease in the levels of HNE-lipoic acid in the AD brain compared to that of age-matched controls. To investigate this phenomenon further, the levels and activity of lipoamide dehydrogenase (LADH) were measured in AD and control brains. Additionally, LADH activities were measured after in-vitro HNE-treatment to mice brains. Both LADH levels and activities were found to be significantly reduced in AD brain compared to age-matched control. HNE-treatment also reduced the LADH activity in mice brain. These data are consistent with a two-hit hypothesis of AD: oxidative stress leads to lipid peroxidation that, in turn, causes oxidative dysfunction of key energy-related complexes in mitochondria, triggering neurodegeneration. This study is consonant with the notion that lipoic acid supplementation could be a potential treatment for the observed loss of cellular energetics in AD and potentiate the antioxidant defense system to prevent or delay the oxidative stress in and progression of this devastating dementing disorder.
Abbreviations: AD, Alzheimer disease; HNE, 4-hydroxy-2-trans nonenal; HNE-LA, HNE-bound lipoic acid; IPL, inferior parietal lobule; LA, lipoic acid; LADH, lipoamide dehydrogenase/dihydrolipoamide dehydrogenase
Keywords: Alzheimer disease, Lipid peroxidation, 4-hydroxy-2-trans nonenal (HNE), Lipoic acid, Lipoamide dehydrogenase
Graphical abstract
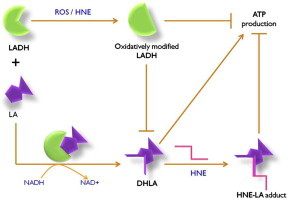
The NADH-dependent oxido-reductase enzyme lipoamide dehydrogenase (LADH) is an important member of the mitochondrial energy generation complex. Alteration of the structure and activity of LADH by elevated reactive oxygen species (ROS) may hamper energy metabolism and ATP production. Lipoic acid (LA) must be in the reduced form as part of its co-factor function for mitochondrial TCA complexes such as α-ketoglutarate dehydrogenase. However, oxidized LADH is unable to reduce LA to DHLA, and therefore HNE is unable to bind to DHLA efficiently. Consequently, in this study, decreased LA-HNE binding was observed in Alzheimer disease brain. Severe effects on learning, memory, and higher executive functioning, all significantly lost in AD patients, would be expected. Supplementation of LA conceivably may protect LADH from ROS or end products of ROS (e.g., HNE) by self-sacrifice mechanism, potentially providing protection against dementia or slowing the rate of progression of AD.
Highlights
► HNE-bound lipoic acid (HNE-LA) levels were decreased in the IPL region of AD brain. ► The TCA enzyme LADH converts inactive lipoic acid to its active-antioxidant form. ► LADH levels and activity were deceased in AD in IPL brain region. ► In-vitro HNE-treatment to mouse brain reduced LADH activity. ► The results fit the two-hit hypothesis of AD.
Introduction
Oxidative stress occurs due to an imbalance in the levels of antioxidant defense systems and production of reactive oxygen/reactive nitrogen species. Oxidative stress has reported to be important in the pathophysiology of a number of age-related diseases, including Alzheimer disease (AD). AD is characterized by the presence of three principal pathological hallmarks: synapse loss, extracellular senile plaques (SP), and intracellular neurofibrillary tangles (NFTs). The major component of SP is amyloid β-peptide (Aβ), a 40–42 amino acid peptide that is derived from proteolytic cleavage of an integral membrane protein, i.e., amyloid precursor protein (APP), by the action of beta- and gamma-secretases [1,2].
Shown to induce oxidative stress, Aβ1–42 can insert as oligomers into the lipid bilayer and initiate lipid peroxidation [3–8], resulting in the formation of lipid peroxidation products including 4-hydroxy-2-nonenal (HNE), malondialdehyde, F2-isoprostanes, and 2-propen-1-al (acrolein), among others. Protein-bound HNE is one of the important markers used for studying the lipid peroxidation process, and has been known to be involved in depleting cellular nucleophilic compounds such as thioredoxin, glutathione, lipoic acid, etc.[5,9–13].
Protein-bound HNE as well as free HNE, TBARS, MDA, and isoprostanes (F2isoP) levels are increased in plasma, urine, and CSF in AD and amnestic mild cognitive impairment (aMCI-arguably the earliest form of AD), compared to healthy controls [5,8,14–17]. The increased HNE formation of covalent Michael adducts account for one means of increased formation of protein carbonyls [18]. Brains from both AD and aMCI subjects show increased levels of protein carbonyls in AD in affected brain regions, while the cerebellum, largely devoid of Aβ pathology, remained relatively untouched [19]. The anti-oxidant system, both enzymatic and non-enzymatic, shows an inverse correlation with increased oxidative stress markers suggesting that free radicals are important in the progression and pathogenesis of AD [20–22].
Lipoic acid is an important co-factor for multi-enzyme complexes such as α-ketoglutarate dehydrogenase (KGDH), pyruvate dehydrogenase (PDH), branched oxo-acid or α-ketoacid dehydrogenase complex, and glycine decarboxylase complex or glycine cleavage system [23]. Lipoic acid also is unique among endogenous antioxidants, in that it can scavenge free radicals in aqueous as well as in lipid or membrane phase [23]. Lipoic acid in its endogenous form contains a disulfide bond and it remains inactive unless reduced to dihydrolipoic acid (DHLA) in a reaction catalyzed by lipoamide dehydrogenase (LADH), using NADH as the source of reducing equivalents [24]. The antioxidant and functional activity of lipoic acid as a co-factor is attributed to its reduced DHLA form [23]. The sulfhydryl groups on DHLA act similar to –SH group from glutathione or cysteine and help in reduction of free radicals by providing –H or an electron. Hence, covalent modification of reduced lipoic acid by HNE could have detrimental effects on cellular energetics and the antioxidant defense system [9].
In the present study, we determined the levels of HNE-bound lipoic acid and also measured the levels and activity of the enzyme LADH in AD and age-matched control brain. In addition, we investigated the alterations in LADH enzyme activity in presence of the lipid peroxidation end product-HNE.
Materials and methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. Nitrocellulose membranes and the electrophoresis transfer system employing a Trans-Blot semidry transfer cell were obtained from Bio-Rad (Hercules, CA, USA).
Control and AD brains
Frozen inferior parietal lobule (IPL) specimens from ten control and twelve AD subjects were obtained from the University of Kentucky Rapid Autopsy Program of the Alzheimer's Disease Clinical Center (UK ADC). All the subjects were longitudinally followed and underwent neurological and physical examinations and annual neuropsychological testing. The control subjects used in the current study were without history of dementia or other neurological disorders and with intact activities of daily living (ADLs).
Sample preparation
Briefly, the frozen brain tissue samples were weighed, and homogenized via polytron at 200 mg/mL in ice-cold PBS with a complete cocktail of protease inhibitors (from Amresco, in Solon, OH). An aliquot of the raw homogenate was centrifuged at 20,800×g for 30 mins at 4 °C. The supernatants were collected and stored at −80 °C until the time of assay, which was then used for slot blot analysis. For Western blot and enzyme assays raw homogenates were used.
HNE-bound lipoic acid (HNE-LA) levels
Levels of HNE-LA were measured by the slot blot technique as previously described with some modification in the immunochemical detection part of the technique [25,26]. Protein (1 μg) from AD-IPL and age matched control samples were loaded onto nitrocellulose membranes using the slot blot technique. After the transfer of proteins on the nitrocellulose membrane, the membrane was incubated in a solution of 250 mM sodium borohydride in 100 mM MOPS buffer at pH 8.0 for 15 min. Sodium borohydride chemically reduced the aldehyde moiety on HNE to an alcohol, since the primary antibody would recognize only 4-hydroxy-2-trans-nonanol bound lipoic acid. The reducing solution was prepared fresh and the pH of the MOPS was adjusted to 8.0 prior to addition of sodium borohydride. After 15 min, the membrane was washed three times with ultra pure H2O followed by washing three times with PBS for 5 min each and blocked for 30 min in 5% milk in 1x PBS. Then the membrane was subsequently incubated with primary antibody (1:2000) followed by HRP-conjugated secondary antibody as described previously [25,26] for the detection of the HNE-LA levels.
Lipoamide dehydrogenase (LADH) levels
LADH levels were estimated by the Western blot technique as described previously [20]. Protein (75 μg) from controls and AD samples were loaded side by side on SDS-PAGE gels. Separated proteins were then transferred to the nitrocellulose membrane and detected immunochemically using the specific primary and secondary antibodies.
In-vitro HNE treatment to mice brain
Whole brains from male CD-1/129 background mice (n=4) were homogenized separately in isolation buffer (0.32 M sucrose, 2 mM EDTA, 2 mM EGTA, 20 mM HEPES, 0.2 mM PMSF, 4 μg/ml leupeptin, 4 μg/ml pepstatin, 5 μg/ml aprotinin, pH 7.4). The raw homogenates were centrifuged at 2500xg, for 10 min at 4 °C and supernatants were collected. The supernatant solutions were treated with HNE as described previously [8]. 1 mg of the supernatant from a sample was incubated with 10 μM of HNE (prepared in 10 mM, pH 8.0 PBS) and for control, another 1 mg was incubated with the same volume of PBS alone. The HNE concentration was chosen based on previous studies [8,27–31]. The incubation was carried out for 3 h at 37 °C on an orbital shaker. After the incubation, control and HNE-treated samples were centrifuged at 10, 000×g for 10 min at 4 °C. The resultant pellets were separately washed with sucrose buffer three times and finally suspended in ∼150 μL of the same sucrose buffer. Protein estimation was performed prior to measuring LADH enzyme activity and HNE-LA levels (as described above) in these samples.
Lipoamide dehydrogenase activity assay
The enzymatic activity of LADH (EC 1.8.1.4) was measured by decreased absorbance caused by oxidation of NADH. The assay protocol was provided by Sigma-Aldrich. In brief, a mixture of 2% bovine serum albumin with 30 mM EDTA, 2 mM NAD+, 1.5 mM NADH disodium salt, along with 20 mM lipoic acid as a substrate was added to 20 μg of sample. Sodium phosphate buffer (50 mM, pH 6.5) was used as an assay buffer. The rate of the reaction was monitored spectrophotometrically at 340 nm wavelength for 10 min.
Statistical and data analysis
All the data obtained from different biochemical assays were subjected to statistical analysis. Significant difference in AD-IPL and age-matched control samples as well as HNE-treated and age-matched controls was determined by employing the Student's unpaired t-test with significance at p<0.05.
Results
The levels of HNE-bound lipoic acid (HNE-LA) were detected in the IPL region of AD and age-matched control brain samples using a specialized antibody against HNE-LA modification (produced in Dr. Luke Szweda's laboratory). Significantly decreased HNE-LA levels in AD samples were observed (Fig. 1-left). Previous studies from our laboratory and other showed that HNE levels are elevated in AD, and the oxidative modification of the proteins affects the structure and function of proteins [3,8,17,32,33]. HNE can react only with the reduced form of lipoic acid-DHLA [34], and the enzyme responsible for reduction of LA to DHLA is lipoamide dehydrogenase. Therefore, to test the hypothesis that reduced levels of HNE bound to lipoic acid could be due to alterations in lipoamide dehydrogenase, we measured activities and levels of LADH in AD and control IPL brain region. Both the levels and activities of LADH were significantly reduced in AD-IPL compared to age-matched controls (Fig. 1-center, Fig. 2-left), consistent with the notion that LADH has decreased ability to reduce LA in AD brain. To investigate further if HNE itself can inhibit enzyme activity of LADH, an in-vitro HNE treatment was performed using mice brain homogenates. LADH enzyme activity in HNE-treated mice brain samples was significantly decreased compared to their age-matched controls (Fig. 2-right), consistent with the notion that HNE can covalently modify LADH. However, HNE-LA levels were increased in mice brain after HNE-treatment (Fig. 1-right).
Fig. 1.
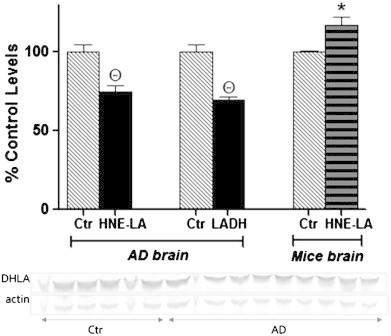
Left: percent control HNE-LA levels estimated by the slot blot technique in IPL brain region of control (n=10) and AD (n=12) samples. Center: percent control lipoamide dehydrogenase (LADH) levels measured by Western blot analysis in IPL brain region of control (n=10) and AD (n=12) samples. Right: percent control HNE-LA levels measured by the slot blot technique in mice brain samples (n=3) after in-vitro HNE-treatment. The bar graphs are accompanied by Western blot bands of LADH and actin, the later used as loading control to which the intensity of each LADH band was normalized. All the values are expressed as mean±SEM, *p<0.05, Θp<0.001 compared to mean of respective control samples.
Fig. 2.
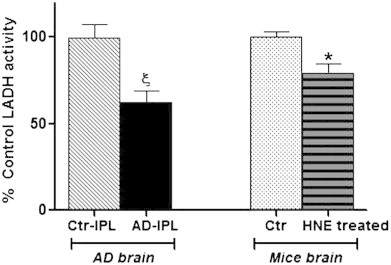
Left: lipoamide dehydrogenase (LADH) activity in control and AD-IPL-human brain region; control n=10, AD n=12. Right: LADH activity measured in control and HNE-treated mice brain; n=4. All the values are expressed as percent control mean±SEM; *p<0.05, ξp<0.01 compared to mean of control samples.
Discussion
Glucose metabolism is essential for proper brain function; a minimum interruption of glucose metabolism causes brain dysfunction and memory loss [35]. Positron emission tomography [36] scanning shows a consistent pattern of reduced cerebral glucose utilization in AD brain [37]. Mitochondria are a main source for free radical production and are also critically important for production of the cellular currency, ATP. AD brain shows abnormal mitochondrial morphology and impaired mitochondrial energy metabolism [38–43]. A number of studies suggest that the rate of cerebral metabolism is reduced in AD, MCI and early AD [44–47], and redox proteomics studies from our laboratory identified a large number of glycolytic, TCA, and mitochondrial enzymes as oxidatively dysfunctional [48]. A recent study employing a triple transgenic mouse model of AD showed that mitochondrial bioenergetics deficits precede AD pathology [49].
Decreased cerebral energy metabolism correlates with the altered expression and decreased activity of mitochondrial energy-related proteins such as the pyruvate dehydrogenase complex, and alpha-ketoglutarate dehydrogenase complex (KGDHC). As mentioned earlier, both these proteins have lipoic acid as a co-factor that is critical for proper function of the protein. The modification of lipoic acid by HNE would likely lead to altered energy metabolism, consequently leading to decreased levels of cellular ATP at nerve terminals promoting loss of synapses and synaptic function. Detrimental downstream consequences of ATP loss include decreased propagation of action potentials, decreased long-term potentiation, and loss of memory and learning, likely contributing to these clinical observations exhibited by AD and MCI patients. As noted above, synapse loss is considered an early feature of AD progression.
The treatment of rat mitochondria with HNE leads to selective inhibition of KGDH and PDH leading to the loss of activity. These results were echoed when purified forms of KGDH and PDH were treated with HNE suggesting that HNE modification of lipoic acid might contribute to their reduced activity [34]. Further, Korotchkina et al. [50] showed that HNE inactivates KGDH and PDH in human HepG2 cells. However, successive treatment with lipoic acid and other thiol supplements protected these complexes from HNE modification. Further, the same study reported that inhibition of PDH was observed at its subunit E2E3-BP. More extensive damage was in the subunit E3-dihydrolipoamide dehydrogenase (reduced form of LADH) when NADH was present and sulfhydryl groups on lipoic acid were in the reduced form [50].
Similar to lipoic acid, LADH is also shared as a subunit E3 by four multi-enzyme complexes: KGDH, PDH, the branched oxo acid or α-ketoacid dehydrogenase complex, and the glycine decarboxylase complex or glycine cleavage system [51–53]. Inhibition of the levels and activity of LADH may reflect the loss of enzymatic activity of these multi-enzyme complexes in AD. Our results are consonant with previous findings of decreased LADH levels and activity in AD brain [53]. Furthermore, decreased LADH activity after in-vitro HNE-treatment suggests oxidative inhibition of LADH can be induced by HNE as observed previously in case of other TCA enzymes [34]. There is substantial evidence that alterations in the enzymes related to mitochondrial energetics play a role in pathogenesis of neurodegenerative diseases [54]. Studies with caucasians [55] and Ashkenazi Jewish populations [56] have associated the gene for LADH with reduced risk of AD. In other research, mice deficient in LADH had altered vulnerability to the dopaminergic neurotoxin MPTP used in Parkinson disease models and to malonate and 3-nitropropionic acid, which have been used in models of Huntington disease [36]. However, post HNE-treatment increased HNE-LA levels are contrary to our observation in AD brain. While we do not have a definitive explanation for this observation, several differences in mouse vs. human brain may be involved, three of which may include: (a) involvement of multiple signaling pathways that could affect HNE-LA levels; (b) involvement of some but not all lipoic acid dependent enzyme complexes in mouse brain following HNE addition, which would lead to elevated HNE-LA; and/or (c) because of the extensive oxidative stress present in AD brain a less reductive environment in AD brain exists than in mouse brain, the latter perhaps permitting more disulfide bond breakage of LA via means other than LADH with consequent elevated HNE-LA. Such possibilities are the subject of future studies in our laboratory.
In AD, oxidative stress can alter the functions of TCA enzymes as well as induce intracellular accumulation of calcium, which may lead to cellular death [33,57,58]. Dysfunction of TCA enzymes may compromise cellular energetics and elevate oxidative stress further [55,59]. If not intervened, this feed-forward loop may accelerate the progression of the disease. Various studies have proposed using antioxidant therapeutics to prolong the time of onset of AD or retard the rate of its progression [60–65]. Treatment with lipoic acid reduced the lipid peroxidation marker-HNE and apoptotic markers in AD fibroblasts cultures [66]. In the same study, co-treatment of cells with N-acetyl cysteine showed much better protection from oxidative stress than lipoic acid or N-acetyl cysteine alone [66]. Apart from acting as an antioxidant itself, lipoic acid in the reduced form can also reinforce other water-or lipid-soluble antioxidants such as glutathione, ascorbate, and vitamin E by scavenging their radicals [23]. A previous study from our laboratory showed that pretreatment of cortical neurons with acetyl l-carnitine and lipoic acid protected cells against HNE-mediated oxidative stress and neurotoxicity by inducing GSH levels and reducing protein and lipid oxidation and inducing protective pathways involving ERK1/2 [67]. The current studies have provided a rationale (Fig. 3) for studies underway in our laboratory to better understand the mechanism(s) and develop therapeutic(s) to prevent Aβ-induced oxidative stress and neurotoxicity, relevant to slowing or inhibition of the progression and pathogenesis of AD.
Fig. 3.
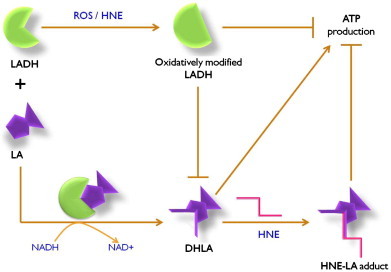
The NADH-dependent oxido-reductase enzyme lipoamide dehydrogenase (LADH) is also an important member of the mitochondrial energy generation complex. Alteration of the structure and activity of LADH by elevated reactive oxygen species (ROS) may hamper energy metabolism and ATP production. Lipoic acid (LA) must be in the reduced form as part of its co-factor function for mitochondrial TCA complexes such as α-ketoglutarate dehydrogenase. However, oxidized LADH is unable to reduce LA to DHLA, and therefore HNE is unable to bind to DHLA efficiently. Consequently, in AD brain decreased LA-HNE binding was observed. Severe effects on learning, memory, and higher executive functioning, all significantly lost in AD patients would be expected. Supplementation of LA conceivably may protect LADH from ROS or end products of ROS (e.g., HNE) by self-sacrifice mechanism, potentially providing protection against dementia or slowing the rate of progression of AD.
Finally, the results of this paper fit the two-hit hypothesis of AD [68–72], namely that oxidative stress may combine with other events to push the patient to AD. In this case, the well-known oxidative stress in AD, which occurs early in the disease, i.e., at the aMCI stage if not earlier [3,4], could lead to loss of ATP production by oxidatively modifying numerous enzymes of the glycolytic and TCA cycles and the ETC, among which is LADH. The consequent decreased levels of DHLA would decrease ATP production, while simultaneously having a less reduced environment with consequent more free radical oxidative stress. The resulting loss of neurons could then contribute to the clinical presentation, progression, and pathology of AD. Studies to test this idea are underway in our laboratory.
Contribution of authors
Dr. Hardas carried out the experiment. Drs. Butterfield, Hardas and Sultana contributed to the experimental design, interpretation of the data and manuscript preparation. Ms. Clark contributed in the experimental work. Dr. Sweda generated antibody against lipoic acid, and helped in experimental design. Drs. Murphy and Beckett provided the tissue resources and helped in the experimental design. Dr. Butterfield additionally supervised the entire study.
Conflict of interest statement
No conflict of interest exists for any of the authors.
Acknowledgments
This work was supported, in part, by a NIH grant to D. A. Butterfield (AG-05119). We are grateful to the Neuropathology Core of the University of Kentucky Alzheimer Disease Clinical Center for providing us well-characterized specimens to carry out this research.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Duyckaerts C., Delatour B., Potier M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathology. 2009;118(1):5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 2.Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y.C., Zaidi M.S., Wisniewski H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. Journal of Biological Chemistry. 1986;261(13):6084–6089. [PubMed] [Google Scholar]
- 3.Markesbery W.R. Oxidative stress hypothesis in Alzheimer's disease. Free Radical Biology and Medicine. 1997;23(1):134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield D.A., Drake J., Pocernich C., Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends in Molecular Medicine. 2001;7(12):548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 5.Butterfield D.A., Castegna A., Lauderback C.M., Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiology of Aging. 2002;23(5):655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 6.Mark R.J., Lovell M.A., Markesbery W.R., Uchida K., Mattson M.P. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. Journal of Neurochemistry. 1997;68(1):255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 7.Mattson M.P. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiological Reviews. 1997;77(4):1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 8.Lauderback C.M., Hackett J.M., Huang F.F., Keller J.N., Szweda L.I., Markesbery W.R. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of a beta 1-42. Journal of Neurochemistry. 2001;78(2):413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 9.Higdon A.N., Landar A., Barnes S., Darley-Usmar V.M. The electrophile responsive proteome: integrating proteomics and lipidomics with cellular function. Antioxidants and Redox Signaling. 2012;17(11):1580–1589. doi: 10.1089/ars.2012.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mello C.F., Sultana R., Piroddi M., Cai J., Pierce W.M., Klein J.B. Acrolein induces selective protein carbonylation in synaptosomes. Neuroscience. 2007;147(3):674–679. doi: 10.1016/j.neuroscience.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norton N.D., Mamiya B.M., Kehrer J.P. Relationships between cell density, glutathione and proliferation of A549 human lung adenocarcinoma cells treated with acrolein. Toxicology. 1997;122(1-2):111–122. doi: 10.1016/s0300-483x(97)00086-3. [DOI] [PubMed] [Google Scholar]
- 12.Pocernich C.B., Butterfield D.A. Acrolein inhibits NADH-linked mitochondrial enzyme activity: implications for Alzheimer's disease. Neurotoxicity Research. 2003;5(7):515–519. doi: 10.1007/BF03033161. [DOI] [PubMed] [Google Scholar]
- 13.Yang X., Wu X., Choi Y.E., Kern J.C., Kehrer J.P. Effect of acrolein and glutathione depleting agents on thioredoxin. Toxicology. 2004;204(2-3):209–218. doi: 10.1016/j.tox.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 14.Keller J.N., Schmitt F.A., Scheff S.W., Ding Q., Chen Q., Butterfield D.A. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64(7):1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 15.Yao Y., Clark C.M., Trojanowski J.Q., Lee V.M., Pratico D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Annals of Neurology. 2005;58(4):623–626. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- 16.Lovell M.A., Ehmann W.D., Mattson M.P., Markesbery W.R. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiology of Aging. 1997;18(5):457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 17.Markesbery W.R., Lovell M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiology of Aging. 1998;19(1):33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield D.A., Stadtman E.R. Protein Oxidation Processes in Aging Brain. In: Paula S.T., Bittar E.E., editors. Advances in Cell Aging and Gerontology. Elsevier; 1997. pp. 161–191. [Google Scholar]
- 19.Hensley K., Hall N., Subramaniam R., Cole P., Harris M., Aksenov M. Brain regional correspondence between Alzheimers-disease histopathology and biomarkers of protein oxidation. Journal of Neurochemistry. 1995;65(5):2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 20.Sultana R., Piroddi M., Galli F., Butterfield D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochemical Research. 2008;33(12):2540–2546. doi: 10.1007/s11064-008-9593-0. [DOI] [PubMed] [Google Scholar]
- 21.Guidi I., Galimberti D., Lonati S., Novembrino C., Bamonti F., Tiriticco M. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer's disease. Neurobiology of Aging. 2006;27(2):262–269. doi: 10.1016/j.neurobiolaging.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Butterfield D.A., Poon H.F., St., Clair D., Keller J.N., Pierce W.M., Klein J.B. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiology of Disease. 2006;22(2):223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Kagan V.E., Shvedova A., Serbinova E., Khan S., Swanson C., Powell R. Dihydrolipoic acid—a universal antioxidant both in the membrane and in the aqueous phase: reduction of peroxyl, ascorbyl and chromanoxyl radicals. Biochemical Pharmacology. 1992;44(8):1637–1649. doi: 10.1016/0006-2952(92)90482-x. [DOI] [PubMed] [Google Scholar]
- 24.Biewenga G.P., Dorstijn M.A., Verhagen J.V., GRMM Haenen, Bast A. Reduction of lipoic acid by lipoamide dehydrogenase. Biochemical Pharmacology. 1996;51(3):233–238. doi: 10.1016/0006-2952(95)02124-8. [DOI] [PubMed] [Google Scholar]
- 25.Butterfield D.A. Beta-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chemical Research in Toxicology. 1997;10(5):495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 26.Sultana R., Mecocci P., Mangialasche F., Cecchetti R., Baglioni M., Butterfield D.A. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer's disease: insights into the role of oxidative stress in Alzheimer's disease and initial investigations into a potential biomarker for this dementing disorder. Journal of Alzheimer's Disease. 2011;24(1):77–84. doi: 10.3233/JAD-2011-101425. [DOI] [PubMed] [Google Scholar]
- 27.Carbone D.L., Doorn J.A., Kiebler Z., Petersen D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chemical Research in Toxicology. 2005;18(8):1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 28.Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radical Biology and Medicine. 2009;47(5):469–484. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 29.Carini M., Aldini G., Facino R.M. Mass spectrometry for detection of 4-hydroxy-trans-2-nonenal (HNE) adducts with peptides and proteins. Mass Spectrometry Reviews. 2004;23(4):281–305. doi: 10.1002/mas.10076. [DOI] [PubMed] [Google Scholar]
- 30.Poli G., Dianzani M.U., Cheeseman K.H., Slater T.F., Lang J., Esterbauer H. Separation and characterization of the aldehydic products of lipid peroxidation stimulated by carbon tetrachloride or ADP-iron in isolated rat hepatocytes and rat liver microsomal suspensions. Biochemical Journal. 1985;227(2):629–638. doi: 10.1042/bj2270629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poli G., Schaur J.R. 4-hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life. 2000;50(4/5):315–321. doi: 10.1080/713803726. [DOI] [PubMed] [Google Scholar]
- 32.Subramaniam R., Roediger F., Jordan B., Mattson M.P., Keller J.N., Waeg G. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. Journal of Neurochemistry. 1997;69(3):1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- 33.Beal M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Annals of Neurology. 1995;38(3):357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 34.Humphries K.M., Szweda L.I. Selective inactivation of α-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37(45):15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 35.Meier-Ruge W., Bertoni-Freddari C., Iwangoff P. Changes in brain glucose metabolism as a key to the pathogenesis of Alzheimer's disease. Gerontology. 1994;40(5):246–252. doi: 10.1159/000213592. [DOI] [PubMed] [Google Scholar]
- 36.Klivenyi P., Starkov A.A., Calingasan N.Y., Gardian G., Browne S.E., Yang L. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. Journal of Neurochemistry. 2004;88(6):1352–1360. doi: 10.1046/j.1471-4159.2003.02263.x. [DOI] [PubMed] [Google Scholar]
- 37.Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. European Journal of Pharmacology. 2004;490(1-3):115–125. doi: 10.1016/j.ejphar.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 38.Manczak M., Park B.S., Jung Y., Reddy P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. NeuroMolecular Medicine. 2004;5(2):147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 39.Wang X., Su B., Siedlak S.L., Moreira P.I., Fujioka H., Wang Y. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences USA. 2008;105(49):19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aleardi A.M., Benard G., Augereau O., Malgat M., Talbot J.C., Mazat J.P. Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. Journal of Bioenergetics and Biomembranes. 2005;37(4):207–225. doi: 10.1007/s10863-005-6631-3. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Gracia E., Torrejon-Escribano B., Ferrer I. Dystrophic neurites of senile plaques in Alzheimer's disease are deficient in cytochrome c oxidase. Acta Neuropathology. 2008;116(3):261–268. doi: 10.1007/s00401-008-0370-6. [DOI] [PubMed] [Google Scholar]
- 42.Kim S.H., Vlkolinsky R., Cairns N., Fountoulakis M., Lubec G. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Science. 2001;68(24):2741–2750. doi: 10.1016/s0024-3205(01)01074-8. [DOI] [PubMed] [Google Scholar]
- 43.Sultana R., Butterfield D.A. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. Journal of Bioenergetics and Biomembranes. 2009;41(5):441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease. FDG-PET studies in MCI and AD. European Journal of Nuclear Medicine and Molecular Imaging. 2005;32(4):486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- 45.Rapoport S.I., Hatanpaa K., Brady D.R., Chandrasekaran K. Brain energy metabolism, cognitive function and down-regulated oxidative phosphorylation in Alzheimer disease. Neurodegeneration. 1996;5(4):473–476. doi: 10.1006/neur.1996.0065. [DOI] [PubMed] [Google Scholar]
- 46.Atamna H., Frey W.H., 2nd Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer's disease. Mitochondrion. 2007;7(5):297–310. doi: 10.1016/j.mito.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Mosconi L., Pupi A., De Leon M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Annals of the New York Academy of Sciences. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butterfield D.A., Perluigi M., Reed T., Muharib T., Hughes C.P., Robinson R.A. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxidants and Redox Signaling. 2012;17(11):1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao J., Irwin R.W., Zhao L., Nilsen J., Hamilton R.T., Brinton R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences USA. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korotchkina L.G., Yang H.-S., Tirosh O., Packer L., Patel M.S. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radical Biology and Medicine. 2001;30(9):992–999. doi: 10.1016/s0891-5849(01)00491-9. [DOI] [PubMed] [Google Scholar]
- 51.Lindsay J.G. Targeting of 2-oxo acid dehydrogenase complexes to the mitochondriona. Annals of the New York Academy of Sciences. 1989;573(1):254–266. doi: 10.1111/j.1749-6632.1989.tb15002.x. [DOI] [PubMed] [Google Scholar]
- 52.Hunter A., Lindsay J.G. Immunological and biosynthetic studies on the mammalian 2-oxoglutarate dehydrogenase multienzyme complex. European Journal of Biochemistry. 1986;155(1):103–109. doi: 10.1111/j.1432-1033.1986.tb09464.x. [DOI] [PubMed] [Google Scholar]
- 53.Mastrogiacomo F., Lindsay J.G., Bettendorff L., Rice J., Kish S.J. Brain protein and α-ketoglutarate dehydrogenase complex activity in Alzheimer's disease. Annals of Neurology. 1996;39(5):592–598. doi: 10.1002/ana.410390508. [DOI] [PubMed] [Google Scholar]
- 54.Gibson G.E., Park L.C.H., Sheu K.-F.R., Blass J.P., Calingasan N.Y. The α-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochemistry International. 2000;36(2):97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- 55.Sheu K.-F.R., Cooper A.J.L., Koike K., Koike M., Lindsay J.G., Blass J.P. Abnormality of the α-ketoglutarate dehydrogenase complex in fibroblasts from familial Alzheimer's disease. Annals of Neurology. 1994;35(3):312–318. doi: 10.1002/ana.410350311. [DOI] [PubMed] [Google Scholar]
- 56.Brown A.M., Gordon D., Lee H., Caudy M., Hardy J., Haroutunian V. Association of the dihydrolipoamide dehydrogenase gene with Alzheimer's disease in an Ashkenazi Jewish population. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2004;131B(1):60–66. doi: 10.1002/ajmg.b.30008. [DOI] [PubMed] [Google Scholar]
- 57.Fu W., Luo H., Parthasarathy S., Mattson M.P. Catecholamines potentiate amyloid beta-peptide neurotoxicity: involvement of oxidative stress, mitochondrial dysfunction, and perturbed calcium homeostasis. Neurobiology of Disease. 1998;5(4):229–243. doi: 10.1006/nbdi.1998.0192. [DOI] [PubMed] [Google Scholar]
- 58.Gibson G.E., Zhang H., Xu H., Park L.C.H., Jeitner T.M. Oxidative stress increases internal calcium stores and reduces a key mitochondrial enzyme. Biochimica Biophysica Acta. 2002;1586(2):177–189. doi: 10.1016/s0925-4439(01)00091-6. [DOI] [PubMed] [Google Scholar]
- 59.Smith M.A., Rottkamp C.A., Nunomura A., Raina A.K., Perry G. Oxidative stress in Alzheimer's disease. Biochimica Biophysica Acta. 2000;1502(1):139–144. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 60.Opii W.O., Joshi G., Head E., Milgram N.W., Muggenburg B.A., Klein J.B. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiology of Aging. 2008;29(1):51–70. doi: 10.1016/j.neurobiolaging.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mancuso C., Bates T.E., Butterfield D.A., Calafato S., Cornelius C., Lorenzo A.D. Natural antioxidants in Alzheimer's disease. Expert Opinion on Orphan Drugs. 2007;16(12):1921–1931. doi: 10.1517/13543784.16.12.1921. [DOI] [PubMed] [Google Scholar]
- 62.Yatin S.M., Varadarajan S., Butterfield D.A. Vitamin E prevents Alzheimer's amyloid β-peptide (1–42)-induced neuronal protein oxidation and reactive oxygen species production. Journal of Alzheimer's Disease. 2000;2(2):123–131. doi: 10.3233/jad-2000-2212. [DOI] [PubMed] [Google Scholar]
- 63.Behl C., Moosmann B. Antioxidant neuroprotection in Alzheimer's disease as preventive and therapeutic approach. Free Radical Biology and Medicine. 2002;33(2):182–191. doi: 10.1016/s0891-5849(02)00883-3. [DOI] [PubMed] [Google Scholar]
- 64.Praticò D. Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy. Annals of the New York Academy of Sciences. 2008;1147(1):70–78. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- 65.Pitchumoni S.S., Doraiswamy P.M. Current status of antioxidant therapy for Alzheimer's Disease. Journal of the American Geriatrics Society. 1998;46(12):1566–1572. doi: 10.1111/j.1532-5415.1998.tb01544.x. [DOI] [PubMed] [Google Scholar]
- 66.Moreira P.I., Harris P.L.R., Zhu X., Santos M.S., Oliveira C.R., Smith M.A. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. Journal of Alzheimer Disease. 2007;12(2):195–206. doi: 10.3233/jad-2007-12210. [DOI] [PubMed] [Google Scholar]
- 67.Hafiz M.A., Butterfield D.A. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by cotreatment of acetyl-l-carnitine and α-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Free Radical Biology and Medicine. 2007;42(3):371–384. doi: 10.1016/j.freeradbiomed.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu X., Lee H-g, Perry G., Smith M.A. Alzheimer disease, the two-hit hypothesis: an update. Biochimica Biophysica Acta. 2007;1772(4):494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 69.Reddy V.P., Zhu X., Perry G., Smith M.A. Oxidative stress in diabetes and Alzheimer's disease. Journal of Alzheimer Disease. 2009;16(4):763–774. doi: 10.3233/JAD-2009-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagy Z. The last neuronal division: a unifying hypothesis for the pathogenesis of Alzheimer's disease. Journal of Cellular and Molecular Medicine. 2005;9(3):531–541. doi: 10.1111/j.1582-4934.2005.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu X., Castellani R.J., Takeda A., Nunomura A., Atwood C.S., Perry G. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mechanisms of Ageing and Development. 2001;123(1):39–46. doi: 10.1016/s0047-6374(01)00342-6. [DOI] [PubMed] [Google Scholar]
- 72.Zhu X., Raina A.K., Perry G., Smith M.A. Alzheimer's disease: the two-hit hypothesis. Lancet Neurology. 2004;3(4):219–226. doi: 10.1016/S1474-4422(04)00707-0. [DOI] [PubMed] [Google Scholar]