

Abstract
Oxidative stress plays a crucial role in the development of the aging process and age dependent diseases. Both are closely connected to disturbances of proteostasis by protein oxidation and an impairment of the proteasomal system. The final consequence is the accumulation of highly cross-linked undegradable aggregates such as lipofuscin. These aggregates of damaged proteins are detrimental to normal cell functions. Here we provide an overview about effect of these aggregates on the proteasomal system, followed by transcription factor activation and loss of cell viability. Furthermore, findings on the mechanism of radical genesis, proteasomal inhibition and the required components of lipofuscin formation were resumed.
Keywords: Aging, Oxidative stress, Proteasome, Lipofuscin, Autophagy
Introduction
In unstressed situations protein homeostasis is balanced by folding and stabilization of proteins by chaperones of the Hsp family [1] and the controlled degradation of proteins by the proteasomal system. The proteasome exists in different forms, and its activity is modulated by multiple regulators. The 20S core proteasome contains the proteolytic activity and selectively degrades a multitude of oxidized proteins [2–5], as well as other substrates [6], in an ubiquitin- and ATP-independent manner. When the core 20S proteasome combines with two 19S regulators, the 26S proteasome is formed [7] which selectively removes polyubiquitinated proteins [8–10]. Under stress conditions and, therefore, most notably during aging the balance between protein damage and clearance of damaged proteins is disturbed leading to a malfunctioning of proteostasis and an accumulating mass of oxidized proteins, aggregate and aggresome formation and finally to the accumulation of highly cross-linked materials such as lipofuscin, compromising cell viability. Accumulation of aggregates in postmitotic cells seems to be especially dramatic, since they are not able to dilute this material by cell division.
Previous work has shown that especially the 26S proteasome is highly susceptible to inactivation during oxidative stress [11,12] and although inhibition of 26S proteasomes could be caused by oxidation products such as protein aggregates, it is most likely a stress-triggered disassembly [13,14] (Fig. 1A). It could be suspected, that this disassembly of 26S proteasomes serves to increase 20S proteasome abundance, allowing cells to clear irreparably damaged proteins more effectively [9,15,16]. But inhibition of 26S is accompanied with the accumulation of undegraded, polyubiquitinated proteins, which are sensed by the ubiquitinbinding domain of HDAC6 (Histone deacetylase 6) [17].
Fig. 1.
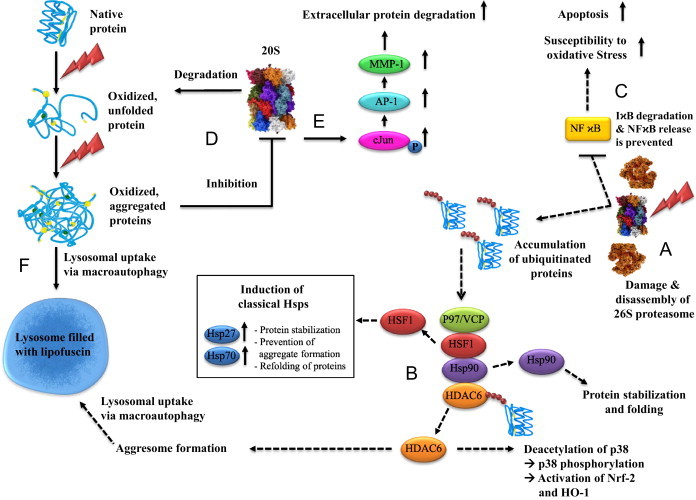
Consequences of oxidative stress on proteins, the proteasomal system and transcription factors. (A) The 26S proteasome is highly susceptible to inactivation during oxidative stress (figured as flash symbol) leading to an excessive accumulation of polyubiquitinated proteins. (B) Histone deacetylase 6 (HDAC6) has the ability to sense and bind ubiquitinated proteins via the BUZ (bound to ubiquitin zinc finger) domain which induces the dissociation of a complex formed out of Hsp90, HSF-1, p97/VCP and HDAC6 [35]. Following the liberation of HDAC6 and p97/VCP [17,36], the latter uses either its ATP dependent segregase activity to dissolve the bond between Hsp90 and HSF-1 directly or p97/VCP stimulates the Hsp90 ATPase activity, which results in the release of HSF-1 and the consequence up-regulation of several heat shock proteins, such as Hsp70 and Hsp 27 [17]. Beyond up-regulation of classical Hsps upon proteasome inhibition, HDAC6 is also involved in an induction of HO-1 after proteasome inhibition [18]. Initiation of this pathway is also the detection of ubiquitinated proteins and the release of HDAC6 and mediated by a p38/MAPK-dependent activation of Nrf-2, which is the most important transcriptional activator of HO-1 gene translation [171]. Further HDAC6 has the ability to favor the accumulation of polyubiquitinated proteins in cellular aggresomes by interaction with ubiquitin and dynein motors. Aggresomes are inclusion bodies next to the nucleus at the proximity end of the microtubule organizing center, finally eliminated by an autophagy-mediated mechanism. (C) Furthermore, a pivotal regulatory protein function is disrupted by 26S proteasomal inhibition, the transcription factor NFϰB. This factor binds to multiple DNA sequences, initiating the transcription of gene products including various cytokines, angiogenesis factors, cell adhesion molecules, enzymes and antiapoptotic factors [21]. NFϰB is located in the cytoplasm in an inactive form, bound to an inhibitor molecule IϰB. Stimulation of cells through a variety of mechanisms triggers a cascade of signaling events resulting in the degradation of IϰB by the proteasome. This degradation releases active NFϰB, which then translocates into the nucleus and binds to specific DNA sequences on its target genes [20]. Proteasomal inhibition blocks NFϰB activation and leads among others to increased susceptibility to oxidative stress and apoptosis. (D) The activity of the 20S proteasome, which is mostly responsible for the degradation of oxidized proteins and less susceptible to direct oxidative stress, can be nevertheless diminished by aggregated oxidized proteins. These protein aggregates are formed under stress conditions as complexes of unfolded proteins which do not normally interact with each other. It might require several steps depending on the nature of the initial conditions, leading to unfolding and aggregate formation. Due to the complex process of intermolecular interactions, such as during physiological aging, the process of aggregation is slow [30,37,38]. The aggregate is independent from the original structure of the protein and introduces a new toxic element into cellular metabolism, partly by inhibiting 20S proteasome. (E) Another side effect of proteasome inhibition is leading to a higher amount of phosphorylated c-Jun leading to an activation of AP-1, known to control the expression of MMP-1 [22] and numerous other genes. Subsequently increased extracellular protein degradation can be assumed, as observed in skin aging. (F) When proteolytic capacity declines below a critical threshold of activity required to cope with oxidative stress, the final consequence is the accumulation of aggregated proteins which may, instead, cross-link with one another or form extensive hydrophobic bonds. This material might undergo further reactions and finally form the age pigment lipofuscin which has toxic properties and accumulates in the lysosomal system..
Via HDAC6 this proteotoxic stress acts as a signal for a number of response mechanisms that cope with proteasomal inhibition, such as stabilization and prevention of aggregate formation by the induction of classical Hsps, elimination of polyubiquitinated proteins/aggregates by HDAC6 mediated aggresome formation and lysosomal uptake via autophagy, containment of inflammation by the induction of HO-1(heme oxygenase-1) and Nrf-2 (NF-E2-related factor 2) pathway and reduction of proteotoxic stress mediated apoptosis by the induction of classical Hsps and HO-1 [17–19] (Fig. 1B). Furthermore, nuclear translocation of NFϰB (Nuclear factor ϰB) requires degradation of ubiquitinated phospho-IϰB-a (inhibitor of ϰB) by the 26S proteasome [20]. NFϰB regulates critical survival pathways such as apoptosis in a variety of different cell types [21]. During proteasomal inhibition apoptosis is enhanced through inhibition of IϰB-a degradation (Fig. 1C). Although, the 20S proteasome is relatively resistant towards oxidants both in vitro and in vivo [11], it was shown that oxidized protein aggregates are able to inhibit the proteolytic activity of the 20S proteasome (Fig. 1D). So beside 26S disassembly as a consequence of oxidative stress the effectiveness of 20S is also diminished by accumulation of heavily damaged, oxidized, and aggregated proteins during postmitotic aging. It was further demonstrated that inhibition of 20S proteasome by UVA-irradiation followed aggregate formation subsequently leads to an accumulation of phosphorylated c-Jun and activation of activator protein-1 (AP-1), controlling MMP-1(matrix metalloproteinase-1) expression [22]. MMP-1 as a major protease of the extracellular matrix is thereby up-regulated causing increased extracellular protein degradation (Fig. 1E). Noteworthy, it can be expected that other AP-1 induced genes are also activated.
This accumulation of aggregates is the final fate of protein damage under stress conditions. Under such circumstances oxidized proteins may not undergo appropriate proteolytic digestion but instead, cross-link with one another or form extensive hydrophobic bonds. It is believed that the cross-linked proteins react further with other cellular components, forming a fluorescent material referred to as lipofuscin (Fig. 1F). Lipofuscin is accepted to consist of oxidized proteins (30–70%) as well as lipids (20–50%) [23] and from the fifth decade of life, bound sugar residues were also detected in human lipofuscin [24]. A hypothetical mechanism of lipofuscin formation was described in detail by Brunk and Terman in their widely accepted model known as “the mitochondrial–lysosomal axis theory of aging” [25]. According to this model, an intralysosomal accumulation of lipofuscin can be considered as the long-term result of a decreased degradation of oxidized proteins and an increase in intracellular free radical formation. Metals, including Fe, Cu, Zn, Al, Mn, and Ca, comprise up to 2% of lipofuscin [26] and especially catalytic iron seems to be an important factor in further oxidation reactions of the initial protein aggregate. In mammalian cells iron is the most abundant cellular transition metal and a fundamental player in the above mentioned mitochondrial–lysosomal axis theory of aging [25].
However, the detailed intracellular effects of lipofuscin in a cell are largely hypothetical and under discussion for a long time. By using an artificial lipofuscin it could be shown, that lipofuscin is a prominent source of oxidants and is able to incorporate iron in a redox-active manner (Fig. 2A). It could be shown that artificial lipofuscin and particularly iron-loaded artificial lipofuscin increase caspase-3 activity if taken up by cells and, therefore, apoptosis [27]. Furthermore, deferoxamine as chelating agent could reduce the effects of iron-loaded lipofuscin significantly. Thus, the amount of iron included in lipofuscin seems to play a crucial role in the intracellular effects of lipofuscin, particularly considering the ability to catalyze the formation of free radicals and the resulting cytotoxicity. These results match very well the hypothesis of Brunk and Terman [25], postulating that iron inclusions of lipofuscin result in a redox-active surface catalyzing the Fenton reaction.
Fig. 2.
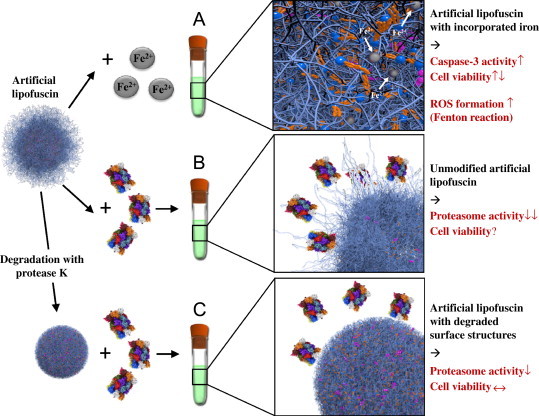
Properties of lipofuscin. Studies have suggested that lipofuscin is not an inert waste product, but rather an active component influencing the cellular metabolism, which is especially relevant in senescent cells. It was proposed that lipofuscin is cytotoxic because of its ability to incorporate redox-active transition metals, resulting in a redox-active surface, able to catalyze the Fenton reaction: in the presence of Fe2+, H2O2 is decomposed forming hydroxyl radicals (OH•) [39]. The resulting Fe3+ can be reduced by superoxide and the vicious cycle starts again. This ability to incorporate transition metals (iron) and form oxidants was tested by using artificial lipofuscin-like oxidized, cross-linked protein aggregates (“artificial lipofuscin”). Non-iron-loaded artificial lipofuscin was incubated with Fe2+ and was able to incorporate a maximum amount of 7 mass%, matching the characteristics of native lipofuscin very well [26]. In vitro this material is able to generate the formation of free radicals and initiates apoptotic cell death, resulting in a significant loss of cellular viability (A). Another major and already demonstrated characteristic of protein aggregates/lipofuscin is the ability to inhibit the degradation of oxidized proteins by competitively binding to the proteasome [40–42]. The proposed mechanism for proteasomal inhibition is binding to exposed hydrophobic amino acid structures on the surface of the highly oxidized and covalently cross-linked lipofuscin [5]. These structures seem to be main sites of proteasomal substrate recognition. After proteasomal binding to those structures the protease is unable to degrade the exposed structures completely because of sterical and/or mechanical inhibition by cross-links. So the proteasome is bound to the surface of lipofuscin in ineffective attempts of degradation detracting proteolytic capacity from other substrates and resulting in a measurable proteasomal inhibition. Furthermore, celluar viability is affected and significantly reduced (B). Degradation of these exposed structures by using the effective and nonspecific protease K demonstrated that such unfolded peptide binding sites are largely responsible for the inhibition of the proteasome. This degradation of these binding sites significantly reduces the proteasomal binding and the inhibition of the proteasome could be partially prevented. Consequently less proteasomal capacity is detracted in futile attempts of degradation and cellular viability was unaffected or increased referred to the undegraded material (C) [29].
Another major characteristic of lipofuscin is its ability to inhibit the degradation of oxidized proteins by competitively binding to proteolytic enzymes including the 20S proteasome as mentioned above as well as lysosomal proteases. How exactly the proteasomal substrate recognition functions is still under investigation, but one of the recognition motifs might be exposed hydrophobic patches from oxidatively damaged and partially unfolded proteins [5,28]. It seems likely that the proteasome also binds to such exposed hydrophobic oligopeptides on the lipofuscin surface but is then unable to completely degrade or to release these peptides, resulting in proteasomal inhibition (Fig. 2B). Evidence for this hypothetical mechanism could be recently shown by partial degradation of these hydrophobic surface patches in artificial lipofuscin with protease K, resulting in decreased proteasomal inhibition and reduced cytotoxicity (Fig. 2C) [29].
Furthermore there has been some speculation about dysfunction of the lysosomal proteases due to the accumulation of lipofuscin in lysosomes, but it is still unclear where the initial cross-linking reactions are taking place: in the cytosol and afterwards uptake by macroautophagy, or whether lysosomes are required components of lipofuscin formation [30,31]. Macroautophagy is a process, which is beside organelle degradation responsible for the uptake of larger aggregates. Normally aggregated proteins beyond proteasomal degradation are enclosed by a phagophore and mediated to the lysosomal system (Fig. 3). By application of oxidative stress to an ATG5 knockout model it was possible to manipulate the lipofuscin amount within lysosomes and the uptake of aggregates into lysosomes could be reduced. However this inhibition of macroautophagy actually did not prevent the formation of lipofuscin [32]. These findings indicate that in contrast to an earlier hypothesis [33,34] lipofuscin can also be formed in the cytosol (Fig. 4) and the uptake into an autophagosome seems to take place in a secondary step. Therefore, autophagosomes/lysosomes are not mandatory for the formation of lipofuscin but constitute a storage for aggregates reducing aggregate toxicity [32].
Fig. 3.
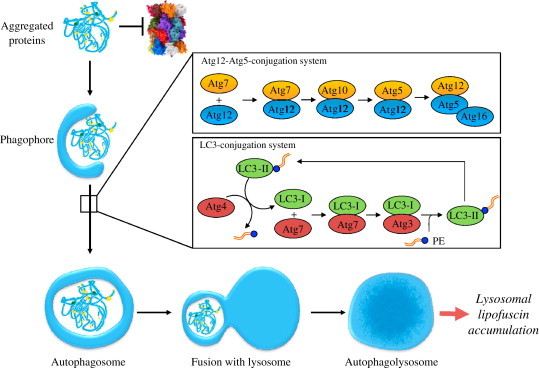
The fate of aggregated proteins during functional macroautophagy. Macroautophagy is a process, which is responsible for the uptake of larger aggregates. Aggregates which are no longer degradable by the proteasome, but rather inhibit its proteolytic function are enclosed by a phagophore and transported towards the lysosomal system. Macroautophagy comprise the formation of an autophagosome, a double-membrane vesicle that engulfs substrates trough the expansion of an isolation membrane, called phagophore. This isolation membrane is build, expanded and closed to an autophagosome and delivered to the endo/lysosomal system. Afterwards both structures fuse and the final structure is called autophagolysosome. The elongation of the phagophore requires two processes similar to ubiquitination: Atg12-Atg5-conjugation and LC3 (microtubule-associated-protein-light-chain-3)-modification (illustration of Atg12-Atg5-system and LC3 conjugation system according to Levine and Deretic [43]). In the first conjugation system (Atg12-Atg5-system) Atg12 is conjugated with Atg5, in a process assisted by an E1-ubiquitin-like activating enzyme: Atg7 and the E2-ubiquitin-like conjugation enzyme: Atg10. Afterwards the Atg12-Atg5 conjugate forms a protein complex with Atg16 on the outside of the nascent autophagosome [44,45]. This complex is functionally necessary for the end product of the secondary conjugation system, lipidated LC3. First the cysteine protease Atg4 eliminates the C-terminal tail of LC3I to expose a glycine residue. A G116 is activated by Atg7, and LC3I is conjugated via a thioester bond with Atg7. Afterwards LC3I is transferred on the E2-ubiquitin-like conjugation enzyme Atg3, and again connected via a thioester bond. Finally LC3I is bound to the amino group of phosphatidylethanolamine (PE). This complex of LC3 with PE is called LC3II and localized via its lipid part to the autophagosomal membrane enabling membrane-elongation. Finally LC3II is deconjugated from the phospholipid-anchor via Atg4 and the luminal associated LC3II is degraded. This cycle of conjugation and deconjugation is important for the normal progression of autophagy. Especially under conditions of oxidative stress a huge amount of aggregated proteins accumulate. These aggregates are taken up by macroautophagy and react further with other cellular components, forming a fluorescent material referred to as lipofuscin, which accumulates in the lysosomal system over time.
Fig. 4.
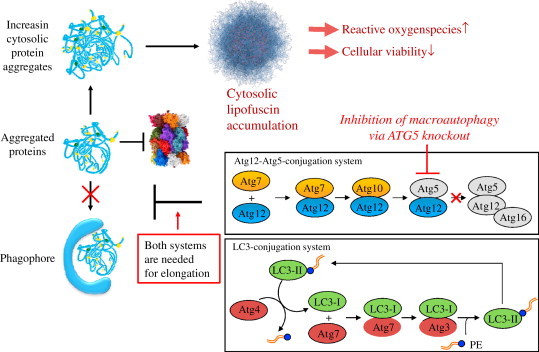
The fate of aggregated proteins during impaired or non-sufficient macroautophagy. Macroautophagy is maximally activated under stress conditions such as starvation, oxidative stress and conditions leading to enhanced protein misfolding [46–50]. This essential role of macroautophagy is underpinned by the fact that Atg5 or Atg7 null mice die a few hours after birth [51]. Nevertheless it is possible to investigate the role of macroautophagy in knockout models such as fibroblasts from mouse embryos deficient in Atg5 (Atg5−/− MEFs) under stress conditions. This ATG5−/− MEFs naturally show no Atg5 expression and thus no formation of the Atg12-Atg5-complex. As the Atg12-Atg5-conjugation system and LC3 modification (illustration according to Levine and Deretic [43]) are both mandatory for the formation of a phagophore, the uptake of aggregated proteins (and other substrates) is prevented in this knockdown system. The consequence is an increasing amount of cytosolic protein aggregates and finally a dramatically enhanced cytosolic lipofuscin accumulation. These elevated levels of extralysosomal lipofuscin are combined with a decline in cellular viability and an increased amount of ROS production.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Reference
- 1.Morimoto R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Development. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pickering A.M., Koop A.L., Teoh C.Y., Ermak G., Grune T., Davies K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochemical Journal. 2010;432:585–594. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ullrich O., Reinheckel T., Sitte N., Hass R., Grune T., Davies K.J. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proceedings of the National Academy of Sciences USA. 1999;96:6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grune T., Reinheckel T., Davies K.J. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. Journal of Biological Chemistry. 1996;271:15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- 5.Pacifici R.E., Kono Y., Davies K.J. Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. Journal of Biological Chemistry. 1993;268:15405–15411. [PubMed] [Google Scholar]
- 6.Reinheckel T., Grune T., Davies K.J. The measurement of protein degradation in response to oxidative stress. Methods in Molecular Biology. 2000;99:49–60. doi: 10.1385/1-59259-054-3:49. [DOI] [PubMed] [Google Scholar]
- 7.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 8.Ding Q., Dimayuga E., Keller J.N. Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxidants and Redox Signalling. 2006;8:163–172. doi: 10.1089/ars.2006.8.163. [DOI] [PubMed] [Google Scholar]
- 9.Davies K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 10.Hiller M.M., Finger A., Schweiger M., Wolf D.H. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- 11.Reinheckel T., Sitte N., Ullrich O., Kuckelkorn U., Davies K.J., Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochemical Journal. 1998;335(Pt 3):637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinheckel T., Ullrich O., Sitte N., Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Archives of Biochemistry and Biophysics. 2000;377:65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- 13.Seifert U., Bialy L.P., Ebstein F., Bech-Otschir D., Voigt A., Schroter F. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142:613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 14.Wang X., Yen J., Kaiser P., Huang L. Regulation of the 26S proteasome complex during oxidative stress. Science Signaling. 2010;3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grune T., Reinheckel T., Davies K.J. Degradation of oxidized proteins in mammalian cells. FASEB Journal. 1997;11:526–534. [PubMed] [Google Scholar]
- 16.Shringarpure R., Grune T., Mehlhase J., Davies K.J. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. Journal of Biological Chemistry. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 17.Boyault C., Zhang Y., Fritah S., Caron C., Gilquin B., Kwon S.H. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Development. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kästle M., Woschee E., Grune T. Histone deacetylase 6 (HDAC6) plays a crucial role in p38MAPK-dependent induction of heme oxygenase-1 (HO-1) in response to proteasome inhibition. Free Radical Biology and Medicine. 2012;53:2092–2101. doi: 10.1016/j.freeradbiomed.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Kawaguchi Y., Kovacs J.J., McLaurin A., Vance J.M., Ito A., Yao T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 20.Brown K., Gerstberger S., Carlson L., Franzoso G., Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 21.Baeuerle P.A., Henkel T. Function and activation of NF-kappa B in the immune system. Annual Review of Immunology. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 22.Catalgol B., Ziaja I., Breusing N., Jung T., Höhn A., Alpertunga B. The proteasome is an integral part of solar ultraviolet a radiation-induced gene expression. Journal of Biological Chemistry. 2009;284:30076–30086. doi: 10.1074/jbc.M109.044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Double K.L., Dedov V.N., Fedorow H., Kettle E., Halliday G.M., Garner B. The comparative biology of neuromelanin and lipofuscin in the human brain. Cellular and Molecular Life Sciences. 2008;65:1669–1682. doi: 10.1007/s00018-008-7581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benavides S.H., Monserrat A.J., Farina S., Porta E.A. Sequential histochemical studies of neuronal lipofuscin in human cerebral cortex from the first to the ninth decade of life. Archives of Gerontology and Geriatrics. 2002;34:219–231. doi: 10.1016/s0167-4943(01)00223-0. [DOI] [PubMed] [Google Scholar]
- 25.Brunk U.T., Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. European Journal of Biochemistry. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 26.Jolly R.D., Douglas B.V., Davey P.M., Roiri J.E. Lipofuscin in bovine muscle and brain: a model for studying age pigment. Gerontology. 1995;41(Suppl 2):283–295. doi: 10.1159/000213750. [DOI] [PubMed] [Google Scholar]
- 27.Höhn A., Jung T., Grimm S., Grune T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radical Biology and Medicine. 2010;48:1100–1108. doi: 10.1016/j.freeradbiomed.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 28.Levine R.L., Moskovitz J., Stadtman E.R. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50(4–5):301–307. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 29.Höhn A., Jung T., Grimm S., Catalgol B., Weber D., Grune T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radical Biology and Medicine. 2011;50:585–591. doi: 10.1016/j.freeradbiomed.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Grune T., Shringarpure R., Sitte N., Davies K. Age-related changes in protein oxidation and proteolysis in mammalian cells. Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2001;56:B459–B467. doi: 10.1093/gerona/56.11.b459. [DOI] [PubMed] [Google Scholar]
- 31.Grimm S., Ernst L., Grotzinger N., Höhn A., Breusing N., Reinheckel T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radical Biology and Medicine. 2010;44:1013–1026. doi: 10.3109/10715762.2010.495127. [DOI] [PubMed] [Google Scholar]
- 32.Höhn A., Sittig A., Jung T., Grimm S., Grune T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radical Biology and Medicine. 2012;53:1760–1769. doi: 10.1016/j.freeradbiomed.2012.08.591. [DOI] [PubMed] [Google Scholar]
- 33.Sohal R.S., Brunk U.T. Mitochondrial production of pro-oxidants and cellular senescence. Mutation Research. 1992;275:295–304. doi: 10.1016/0921-8734(92)90033-l. [DOI] [PubMed] [Google Scholar]
- 34.Brunk U.T., Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radical Biology and Medicine. 2002;33:611–619. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 35.Matthias P., Yoshida M., Khochbin S. HDAC6 a new cellular stress surveillance factor. Cell Cycle. 2008;7:7–10. doi: 10.4161/cc.7.1.5186. [DOI] [PubMed] [Google Scholar]
- 36.Boyault C., Gilquin B., Zhang Y., Rybin V., Garman E., Meyer-Klaucke W. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO Journal. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keller J.N., Gee J., Ding Q. The proteasome in brain aging. Ageing Research Reviews. 2002;1:279–293. doi: 10.1016/s1568-1637(01)00006-x. [DOI] [PubMed] [Google Scholar]
- 38.Merker K., Stolzing A., Grune T. Proteolysis, caloric restriction and aging. Mechanisms of Ageing and Development. 2001;122:595–615. doi: 10.1016/s0047-6374(01)00219-6. [DOI] [PubMed] [Google Scholar]
- 39.Kurz T., Terman A., Gustafsson B., Brunk U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochemistry and Cell Biology. 2008 doi: 10.1007/s00418-008-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friguet B., Stadtman E.R., Szweda L.I. Modification of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Formation of cross-linked protein that inhibits the multicatalytic protease. Journal of Biological Chemistry. 1994;269:21639–21643. [PubMed] [Google Scholar]
- 41.Sitte N., Merker K., Grune T., Von Z.T. Lipofuscin accumulation in proliferating fibroblasts in vitro: an indicator of oxidative stress. Experimental Gerontology. 2001;36:475–486. doi: 10.1016/s0531-5565(00)00253-9. [DOI] [PubMed] [Google Scholar]
- 42.Friguet B., Szweda L.I. Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Letters. 1997;405:21–25. doi: 10.1016/s0014-5793(97)00148-8. [DOI] [PubMed] [Google Scholar]
- 43.Levine B., Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nature Reviews Immunology. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizushima N. Autophagy: process and function. Genes Development. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 45.Codogno P., Meijer A.J. Atg5: more than an autophagy factor. Nature Cell Biology. 2006;8:1045–1047. doi: 10.1038/ncb1006-1045. [DOI] [PubMed] [Google Scholar]
- 46.Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular Biology of the Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiffin R., Christian C., Knecht E., Cuervo A.M. Activation of chaperone-mediated autophagy during oxidative stress. Molecular Biology of the Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cuervo A.M., Knecht E., Terlecky S.R., Dice J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. American Journal of Physiology. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 49.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO Journal. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwata A., Christianson J.C., Bucci M., Ellerby L.M., Nukina N., Forno L.S. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proceedings of the National Academy of Sciences USA. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]