

Abstract
Mechanistic investigations coupled with epidemiology, case-control, cohort and observational studies have increasingly linked isocyanate exposure (both chronic and acute) with pulmonary morbidity and mortality. Though ascribed for impairment in endothelial cell function, molecular mechanisms of these significant adverse pulmonary outcomes remains poorly understood. As preliminary studies conducted in past have failed to demonstrate a cause-effect relationship between isocyanate toxicity and compromised pulmonary endothelial cell function, we hypothesized that direct exposure to isocyanate may disrupt endothelial structural lining, resulting in cellular damage. Based on this premise, we comprehensively evaluated the molecular repercussions of methyl isocyanate (MIC) exposure on human pulmonary arterial endothelial cells (HPAE-26). We examined MIC-induced mitochondrial oxidative stress, pro-inflammatory cytokine response, oxidative DNA damage response and apoptotic index. Our results demonstrate that exposure to MIC, augment mitochondrial reactive oxygen species production, depletion in antioxidant defense enzymes, elevated pro-inflammatory cytokine response and induced endothelial cell apoptosis via affecting the balance of mitochondrial-nuclear cross talk. We herein delineate the first and direct molecular cascade of isocyanate-induced pulmonary endothelial cell dysfunction. The results of our study might portray a connective link between associated respiratory morbidities with isocyanate exposure, and indeed facilitate to discern the exposure-phenotype relationship in observed deficits of pulmonary endothelial cell function. Further, understanding of inter- and intra-cellular signaling pathways involved in isocyanate-induced endothelial damage would not only aid in biomarker identification but also provide potential new avenues to target specific therapeutic interventions.
Keywords: Mitochondrial oxidative stress, Endothelial cell apoptosis, Pulmonary toxicity, Occupational health, Environmental medicine
Graphical abstract

Highlights
► Pulmonary endothelial cells accomplish a vast variety of specialized functions. ► A balance in mitochondrial-nuclear cross talk is essential for endothelial integrity. ► Methyl isocyanate impairs redox balance in endothelial milieu. ► We profess a corollary between respiratory morbidities and methyl isocyanate exposure.
Introduction
Lungs are invariably a major target of environmental toxin exposure. Although pulmonary endothelium is believed to be a quiescent tissue with the potential to exhibit rapid and effective damage repair but a wide variety of genotoxins act differently to trigger a common downstream event insinuating tissue injury [1]. These can cause irreversible structural and functional changes manifested as obstructive pulmonary disease, chronic bronchitis, emphysema and other detectable forms. When the induced-exposure is substantive, a large amount of endothelial lining is destroyed which subsequently affects the elasticity of the lung and pose additional health ailments. About 1–6×1013 endothelial monolayer cells play a vital role in maintaining vascular tone, permeability, blood flow, thrombolysis, inflammation, tissue repair, and growth during normal homeostasis [2]. Both chronic (long-term; occupational) or acute (short-term; accidental) environmental exposure have the potential to perturb vasculo-endothelial environment and assert toxico-genomic implications at a molecular level [3,4].
There has been a resurgence of interest in isocyanates as an important industrial entity with diverse applications [5]. The largest volume of isocyanate is consumed in the production of polyurethane foams. Besides, isocyanate is increasingly used as a raw material in pesticide, insecticide, paints and varnishes and elastomer industries [6]. Isocyanates are a group of low molecular weight aromatic and aliphatic compounds containing one or more 3NåCåO groups that can combine with other compounds containing alcohol groups. Preliminary reports have linked isocyanate exposure to pulmonary fibrosis, bronchial asthma, chronic obstructive pulmonary disease (COPD), emphysema, pneumonitis, recurrent chest infections, severe dry or wet cough, pulmonary edema and distress [7–10]. Some of these associations were attributed to a compromised endothelial function; however, the defined mechanisms by which isocyanates mediate injury to pulmonary endothelial cells are not clear [11]. Earlier we speculated that isocyanates due to their biotransformation ability can activate the immune system which in turn induces a pro-inflammatory response that might possibly impair pulmonary endothelial function [12–15]. On this premise, the present study aimed at evaluating a direct cause-effect relationship of isocyanate exposure on endothelial cell damage using human pulmonary arterial endothelial cells (HPAE-26) as a model. Apparently, a balance in the interface between leukocytes and endothelial cells is crucial for lung function [16,17], therefore, understanding the effect of an important class of chemicals such as isocyanates might help to identify biomarkers that can predict toxin induced pulmonary endothelial injury, in general.
Materials and methods
Cell lines and culture conditions
HPAE-26 (ATCC No, CRL-2598) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Further, they were seeded at the density of 1×105 cells in Ham's F12K complete growth medium. Supplemented with 2 mM l-glutamine adjusted to contain 1.5 g/L sodium bicarbonate, 0.1 mg/ml heparin, 0.03 mg/ml endothelial cell growth supplement (Millipore, Billerica, MA, USA), 10% fetal bovine serum as per ATCC catalog instructions. Cells after attaining optimum confluence were treated with N-succinimidyl N-methylcarbamate, a surrogate chemical equivalent to methyl isocyanate (MIC) [12,14]. At the onset of the experiments, the cells were at an exponential and asynchronous phase of growth.
Reagents
N-succinimidyl N-methylcarbamate [CAS No. 18342-66-0] (Sigma Aldrich Laboratories, St. Louis, USA) stock dissolved in 2 mM dimethylsulfoxide (DMSO) with final concentration of 0.005 μM was used for investigations. The culture petri-dishes were procured from BD Falcon Discovery Labware (Franklin Lakes, NJ, USA). Bradford reagent for protein estimation was procured from Amresco Inc. (Solon, OH, USA). Evaluation of reactive oxygen species (ROS) was performed by CM-H2DCFDA from Molecular Probes, Life Technologies Co. (Carlsbad, CA, USA). Formation of 8-hydroxy-2′deoxyguanosine (8-oxo-dG) and depletion of superoxide dismutase (SOD) and glutathione reductase (GR), markers of oxidative stress, were evaluated using ELISA kits procured from Trevigen Inc. (Gaithersburg, MD, USA). Inflammatory cytokine levels were assessed using a multiplex cytometric bead array (CBA) assay kit custom synthesized from BD Biosciences (San Diego, CA, USA). Evaluation of qualitative DNA damage response was assessed by immune-labeling using polyclonal antibodies to p-ATM and γH2AX, p-p53 and p-p21 from Abcam (Cambridge, UK) and Calbiochem (Nottingham, UK). Secondary antibodies from Santacruz Biotechnology Inc. (Santa Cruz, CA, USA) were used with appropriate dilution in 1x PBS. Mitochondrial depolarization was studied by mitochondrial membrane potential detection kit from BD Biosciences. Caspase-3 activation was determined using PE active caspase-3 apoptosis kit from BD Biosciences.
Study design
Studies were conducted at different sampling intervals (n=3) for time course studies ranging from 0 to 72 h. The final concentration of the chemical used for the treatment was 0.005 μM (1 μg/1 μL). The selection of the dose was performed on the basis of authors' previous observations in which this concentration was found optimum to induce DNA damage, apoptosis, oxidative stress and inflammation in cultured mammalian cells [12,18,19]. Controls were untreated normal HPAE-26 cells.
Assessment of ROS generation
A fresh stock solution of CM-H2DCFDA (5 mM) was prepared in DMSO and diluted to a final concentration of 1 mM in PBS. The cells were washed with PBS followed by incubation with 50 μL of working solution of flurochrome marker CM-H2DCFDA (final working concentration adjusted to 2.5 μg/50 μL) for 2 h. The cells were harvested, washed in PBS and cell-associated fluorescence was qualitatively assessed by applied spectral bio-imaging system with nucleus counterstained by Hoechst and images were acquired immediately through the spectral bio-imaging system and analyzed with version 4.0 software (Applied Spectral Imaging, Edingen Neckarhausen, Germany). Quantitative measurement of DCF fluorescence in cells was performed by flow cytometry in FL-1channel [20].
Evaluation of oxidative damage by 8-oxo-dG
8-oxo-dG is a modified nucleoside base, which is the most commonly studied and detected byproduct of DNA damage that is excreted upon DNA repair. ELISA for quantification of 8-oxo-dG in culture supernatant was performed according to the manufacturer instructions and optical density was measured at 450 nm on an ELISA reader (Tecan Sunrise, Männedorf, Austria) [21,22].
Estimation of antioxidant enzymes
SODs are a group of isozymes functioning as superoxide radical scavenger in the living organisms. While, SOD catalyze the dismutation of the superoxide radical into H2O2 and elemental oxygen which diffuses into the inter-membrane space or mitochondrial matrix, GR, on other hand, plays an essential role in maintaining the appropriate levels of intracellular reduced GSH [23]. SOD and GR assays were performed as per manufacturer's instructions and absorbance kinetics was measured at 450 nm and 340 nm respectively through an ELISA reader [24,25].
Examination of inflammatory cytokine response
Supernatants collected from cultures were subjected for measuring inflammatory response by determining levels of cytokines, interleukin (IL)-2, IL-6, tumor necrosis factor (TNF) and NF-kβ. The assay was performed as per the manufacturer's instructions. Data acquisition and analysis were carried out on the flow cytometric platform using BD™ CBA software [15,26].
Qualitative analysis of DNA damage response
DNA damage response was analyzed qualitatively for ATM and γH2AX phosphorylation through indirect immunofluorescence method. Cells were grown overnight in Dulbecco's modified eagles medium (DMEM) and exposed to N-succinimidyl N-methylcarbamate treatment. The cells were fixed in 4% para-formaldehyde for 1 h and permeabilized with 0.1% Triton-X-100 solution for 30 min. Blocking was done using 3% BSA for 3 h, and then incubated with anti-pATM/anti-H2AX antibodies (dilution 1:1000) for 3 h followed by FITC/Texas Red conjugated secondary antibodies labeling (dilution 1:200) for 1 h. For qualitative analysis, nuclei were counterstained with Hoechst (Sigma Aldrich Laboratories) and the cytoskeleton with phalloidin red (Molecular Probes, Life Technologies Co.). Preparations were immediately mounted with Vecta Shield (Vector Laboratories, Burlingame, CA, USA) and stored in the dark at 4 °C. Images were acquired through the spectral bio-imaging system and analyzed with version 4.0 software (Applied Spectral Imaging, Edingen Neckarhausen, Germany) [27].
Quantitation of phosphorylation kinetics
For quantitation of phosphorylated states of ATM, H2AX, p53 and p21 proteins, indirect immunofluorescence method was followed. Briefly, cells were washed with 1x PBS following treatment and fixed in BD Cytofix solution (BD Biosciences) for 1 h, permeabilized in 500 μL of 1x BD Cytoperm buffer (BD Biosciences) for 30 min and blocked in 3% BSA for 2 h. Immuno-labeling was performed using primary polyclonal rabbit antibody with dilution 1:1000 and was incubated for 1 h, followed by staining with secondary polyclonal goat anti rabbit antibody in 1:200 dilution conjugated to FITC and incubated in dark for half an hour. The immuno-stained cells were then acquired through flow cytometer (FACS Calibur, BD IS, San Jose, CA, USA). Cell-associated fluorescence in FL1 channel was analyzed for 10,000 total events [28].
Measurement of mitochondrial trans-membrane potential
Mitochondrial membrane potential of the cells was detected by staining cells with JC-1 (5, 5′, 6, 6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazol carbocyanine iodide). The cells were washed with PBS followed by incubation with 200 μL of working solution of flurochrome marker JC-1 for 20 min. The cells were harvested, washed in PBS and cell-associated fluorescence was qualitatively assessed by applied spectral bio-imaging system with nucleus counterstained by Hoechst and images were acquired immediately through the spectral bio-imaging system and analyzed as mentioned above. While, the quantitative flow cytometric JC-1 assay was carried out as per supplier's instructions. The gate was applied in the FSC/SSC dot plot to restrict the analysis to HPAE-26 cells only. For the gated cells, the ratio of FL1/FL2 was evaluated. In each case, a total of 10,000 events were recorded in HI mode through flow cytometry [29,30].
Activation of caspase-3
The expression of caspase-3, a marker of mitochondrial mediated apoptosis, was measured qualitatively through immunocytochemistry using monoclonal anti-caspase-3 antibody. Nuclei were counterstained using Hoechst and images were acquired through the spectral bio-imaging system as mentioned above. The activity of active caspase-3 was measured flow cytometrically by washing the cells with cold 1x PBS, re-suspended in BD cytofix/cytoperm solution at a concentration of 1×106 cells/mL, followed by incubation of 20 min on ice. The cells were then harvested and washed, followed by incubation with antibody for 30 min at RT. The cells were washed and analyzed by flow cytometry in FL2 channel [29,30].
Statistical analysis
Student's t-test was employed for statistical analysis using SPSS software (SPSS Inc. Chicago, IL, USA) package and p≤0.05 was considered to be statistically significant.
Results
Increased ROS generation
Generation of intracellular ROS in terms of DCFH oxidation (marked by DCF fluorescence) was measured as an initiator for MIC-induced oxidative stress. An early and time dependent enhancement in the ROS generation in the form of H2O2, was observed in the treated cells as against the controls until 72 h of exposure (Fig. 1A). A highest value for DCF fluorescence 52.89±4.12 was observed at 24 h after treatment as against in untreated control cells 16.1±5.13 (Fig. 1B).
Fig. 1.

Increased ROS generation. (A) Representative fluorescent microphotographs (original magnification 200x) showing production of ROS as depicted by increased DCF fluorescence (green) in control and MIC treated HPDE-6 cells. Nuclei counterstained with DAPI (blue). Scale bar, 5 μm. (B). Flow cytometric analysis (n=3) showing formation of reactive oxygen species in control and treated HPAE-26. Percent DCF fluorescence recorded a consistent increase at 6 h, 12 h, 24 h, 48 h and 72 h following treatment with 0.005 μM MIC. n=3 and values expressed as mean±SE (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Augmentation in oxidative damage
In nuclear DNA, 8-oxo-dG is one of the predominant forms of free radical-induced oxidative lesions, and has been widely used as a biomarker for oxidative stress and to assess genotoxic implication upon exposure to various environmental agents. Notably, a significant augmentation in formation of 8-oxo-dG was observed along time-course experiments with utmost 104.2±16.29 ng/mL of accumulated 8-oxo-dG after 24 h in comparison to control cells 9.93±2.58 (Fig. 2).
Fig. 2.
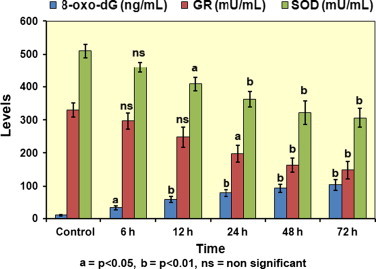
Augmentation in oxidative damage and depletion of antioxidant enzyme levels. Bar diagram depicting levels of accumulated 8-oxo-dG (ng/mL) and status of levels of antioxidant defense enzymes GR and SOD (mU/mL) in HPAE-26 cells prior and along the time course following treatment with 0.005 μM concentration of MIC. n=3 and values expressed as mean±SE.
Depletion of antioxidant enzyme levels
GR and SOD are key components of the cellular antioxidant defense mechanisms. Diminution in these antioxidant enzyme levels results in cell injury. In the present study, a consistent depletion of both SOD and GR enzyme activities was noted from 6 to 72 h with the maximum inhibition of SOD and GR activity being 306.3±29.19 mU/mL and 148.2±26.18 mU/mL, respectively, at 72 h in cells incubated with MIC (Fig. 2).
Elevated levels of pro-inflammatory cytokines
The inflammation process is characterized by the activation of pro-inflammatory cytokines in response to tissue injury. Multiplex CBA assay for human inflammatory cytokines in culture supernatant displayed time-dependent increase in levels of IL-2, TNF, IL-6 and NF-кβ cytokines in treated cells as compared with control cells. Mean levels of secreted cytokines IL-2, TNF, IL-6 and NF-кβ at 72 h were 380.1±30.7, 296.4±31.5, 136.2±26.5 and 160.2±23.8 pg/mL, respectively (Fig. 3).
Fig. 3.

Elevated levels of pro-inflammatory cytokines. Bar diagram showing secreted levels of pro-inflammatory cytokines measured through multiplex CBA flow cytometric assay. Concentration (pg/mL) of IL-1, IL-6, TNF and NF-кβ cytokines displayed a time dependent increase following treatment with 0.005 μM concentration of MIC. n=3 and values expressed as mean±SE.
Induction of DNA damage response
Double strand breaks (DSBs) are efficient indicators of DNA damage. Immunofluorescence analysis illustrated the DNA damage response in control and treated HPAE-26 cells qualitatively through hyper-phosphorylation profiles of signaling factors of DSBs, pATM at Ser 1981, γH2AX (phospho-H2AX) at Ser139, p-p53(phospho-p53) at Ser 15 and p-p21 at Thr145, suggestive of increased nuclear retention of these factors in terms of foci formation (Fig. 4A). Quantitative analysis performed by flow cytometry measured the time kinetics of percent p-ATM, γ-H2AX, p-p53 and p-p21 proteins in cultured HPAE-26 cells. In contrast to controls, a kinetics measurement through flow cytometry showed a steady increase in p-ATM, γ-H2AX, p-p53 and p-p21 positive cells, reaching a maximum of 30.1±2.9, 36.3±3.1, 71.9±5.8 and 64.8±4.4%, respectively, up to 72 h of the study period (Fig. 4B).
Fig. 4.

Induction of DNA damage response. (A). Representative microphotographs (original magnification 400x) showing immunofluorescence analysis of ATM and H2AX phosphorylation in nuclei of control and MIC treated HPAE-26 cells. A distinct interspersed pattern of foci was observed in the nuclei of treated cells in comparison with respective controls. Nucleus counterstained with Hoechst (blue), ATM and γH2AX foci with FITC (green), and cytoskeleton with phalloidin (red). Scale bar, 2 μm. (B). Time-kinetics showing percent phosphorylation states of ATM, H2AX, p53 and p21 proteins evaluated through flow cytometry in HPAE-26 cells following treatment with 0.005 μM concentration of MIC at 0, 6, 12, 24, 48 and 72 h. n=3 and values expressed as mean±SE (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Perturbation in mitochondrial trans-membrane potential
Membrane-permeable lipophilic cationic fluorochrome JC-1 (5, 5, 6, 6-tetrachloro-1, 1, 3, 3- tetraethyl-benzimidazolcarbocyanine iodide) is used as a probe of trans-membrane potential (ψ). JC-1 penetrates into cells and its fluorescence is a reflection of ψ. Evaluation of mitochondrial depolarization in treated cells and controls was performed through epifluorescence microscopy and flow cytometry. In our study, we visualized an increased shift from JC-1 aggregate positive (intact ψ) cells towards JC-1 monomers (depolarized ψ ) positive cells after MIC exposure until 48 h. Quantitative flow cytometric measurement of JC-1 fluorescence presented a time kinetics of JC-1 monomerisation suggestive of consistent depolarization of mitochondria (loss of ψ). The percentage of cells with dissipated mitochondrial membrane potential levels at 72 h were (78.4±9.5) in contrast to the levels of the untreated control cells (10.9±3.2) (Fig. 5A and B).
Fig. 5.

Perturbation in mitochondrial trans-membrane potential. (A). Fluorescent microphotographs (original magnification 200x) showing qualitative analysis of mitochondrial trans-membrane potential (ψ) in control and MIC treated HPAE-26 cells. In treated cells, an increase in formation of JC-1 monomers (depolarized ψ; green) was observed with respect to JC-1 aggregates (intact ψ; red) in control cells. Nucleus counterstained with Hoechst (blue). Scale bar, 5 μm. (B). Quantitative flow cytometric measurement of JC-1 fluorescence depicted a time dependent increase of JC-1 monomerisation (% depolarized cells), suggestive of loss of mitochondria membrane potential in treated cells with respect to controls. n=3 and values expressed as mean±SE (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Activation of caspase-3
Active caspase-3, a indicator for cells undergoing apoptosis, comprises a hetero-dimer of 17- and 112 kDa subunits, which in turn are derived from a 32 kDa pro-enzyme. Active caspase-3 proteolytically cleaves and activates other caspases and relevant targets in the cytoplasm. The apoptotic activity was measured in MIC treated cells through qualitative immunocytochemical and quantitative flow cytometric analysis of caspase-3. Qualitative immunocytochemical observations using anti-active caspase-3 antibody show a higher nuclear accumulation of active caspase-3 in MIC treated cells (Fig. 6A). Quantitative measurement of the caspase-3 activity in MIC-treated cells through flow cytometry, showed an incremental trend along the time course post-treatment. After 72 h, HPAE-26 cells treated with MIC exhibited maximum activation of caspase-3 (80.6±10.8%) as compared to controls (9.2±3.3%) (Fig. 6B). These results possibly indicate that in treated cells, caspase-3 is translocated from cytosol into nucleus through active nuclear transport mechanism during the induction of mitochondrial mediated apoptosis.
Fig. 6.

Activation of caspase-3. (A). Control and MIC treated HPAE-26 cells illustrating qualitative analysis of active caspase-3 expression (original magnification 200x). Higher nuclear accumulation of active caspase-3, a marker of mitochondrial-mediated apoptosis, in MIC treated cells was observed. Nucleus counterstained with Hoechst (blue). Scale bar, 2 μm. (B). Expression of active caspase-3 measured through flow cytometry demonstrated an upsurge of apoptotic index (% positive cells) following treatment with 0.005 μM concentration of MIC at 0, 6, 12, 24, 48 and 72 h. n=3 and values expressed as mean±SE (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Discussion
There has been recent surge in interest towards ascertaining the molecular mechanisms involved in environmental chemicals mediated pulmonary damage. Epidemiologic studies have firmly linked isocyanate exposure to increased pulmonary morbidity and mortality [31–34]. Despite strong epidemiologic evidence, our understanding of the biological mechanisms for these adverse outcomes remains incomplete. As preliminary studies conducted in past have failed to demonstrate a cause-effect relationship between isocyanate toxicity and compromised pulmonary functions, we hypothesized that direct exposure to this chemical moiety may disrupt lung endothelial structural integrity, resulting in cellular dysfunction. As human pulmonary endothelial cells are considered to be an unique tissue that reflect the molecular repercussions on host following toxin exposure [35–37], we examined MIC-induced mitochondrial oxidative stress, pro-inflammatory cytokine response, oxidative DNA damage response and apoptotic index using HPAE-26 cells for our study.
Mitochondria are often susceptible to damage due to various exogenous ROS-producing stresses/agents that compromise cells ability to produce energy owing to impaired electron transport chain [38]. Due to anatomical position, lung is a susceptible target organ of free radical injury and is particularly sensitive to environmental toxicants [39]. Predominantly, the pulmonary endothelial cell lining is continuously exposed to large amounts of exogenous agents, thereby making it prone to injuries [40]. Previous studies have highlighted many aspects of endothelial function that can be disrupted by oxidative stress [41]. An imbalance between increased oxidative stress and impaired antioxidant defense may not only affect endothelial function, but may also impact pulmonary functions [42]. In the present study, oxidative injury was assessed by direct measurement of intracellular ROS production and status of anti-oxidant defense system enzymes. A time-dependent increase in DCF fluorescence, a marker for ROS generation, was observed in the MIC treated HPAE-26 cells indicative of oxidative stress condition through excessive H2O2 production (Fig. 1). The enzymes SOD and GR are the most important intracellular anti-oxidants in the metabolism of ROS. While SOD is among the first line of defense in the detoxification of products resulting from oxidative damage, GR plays a vital role in defense against the toxicity of superoxide radicals. Depletion in both the activities of SOD and GR can result in the breakdown of antioxidant defense system and indirectly increase isocyanate-mediated oxygen radical injury [20]. Correspondingly, inhibition of SOD and GR activities in MIC exposed HPAE-26 cells; suggest mitochondrial mediated oxidative stress as a contributory factor for endothelial damage (Fig. 2).
Cytosolic and nuclear events initiated by toxic oxidants might also contribute to inflammatory injury and be amenable to increased cellular demise in the pulmonary milieu [43,44]. Elevation of cytokines and chemokines such as tumor necrosis factor-α, interleukin (IL)-1β, IL-2, IL-6, and IL-8 is a feature common to lung injury of diverse etiologies [45,46]. NF-κB, a DNA-binding factor stimulates transcription of many different cytokines involved in acute lung inflammation [47]. The most important finding of this study is that up-regulation of inflammatory cytokines IL-2, IL-6 and TNF occurred in concert with NF-κB, reiterating an integral role of NF-κB in the feed-forward pro-inflammatory loop in MIC treated HPAE-26 cells (Fig. 3).
DNA damage response initiates with myriad signaling network that include cell cycle arrest and altered regulation of cell cycle checkpoints [48]. Following induction of double strand DNA breaks, ATM that belongs to the phosphoinositide 3-kinase (PI3-kinase)-related protein kinase (PIKK) family is auto-phosphorylated [49]. ATM has been invariably implicated in playing a central role in mediating the isocyanate-induced DNA damage response in human peripheral blood lymphocytes and neutrophils [12,14]. Therefore, it is not beyond reason to postulate that ATM and its subsequent down-stream signaling are involved in the pulmonary endothelial damage as a response to mitochondrial oxidative stress and inflammation albeit unexpected and unappreciated. Results of our study show up-regulation of ATM in HPAE-26 cells treated with 0.005 μM MIC (Fig. 4A and B). We further showed that γH2AX that lies downstream is phosphorylated along with key cell cycle regulatory proteins p53 and p21 [50,51]. The findings presented here clearly indicate that ATM plays an instructive role in activation of isocyanate-induced pulmonary endothelial cell dysfunction orchestrated by mitochondrial oxidative stress.
Pulmonary endothelial injury caused by oxidative damage and inflammation have been increasingly linked with cellular apoptosis. The tumor-suppressor protein, p53, is a key regulator of multiple critical cellular processes, including cell–cycle regulation, DNA repair, and apoptosis [52]. Previous reports have documented the increase in p53 and other cell cycle regulatory protein expression, professing a corollary between pulmonary disease and protein expression upon exposure to exogenous chemicals [53]. In this perspective, our in vitro study demonstrates the contribution of p53 and p21 to human endothelial apoptosis directly in response to isocyanate exposure (Fig. 4B). In addition, we also observed abrupt caspase-3 activation and mitochondrial depolarization (Figs. 5 and 6), the hallmark biochemical events, as an ancillary pathway to p53 dependent apoptosis.
In summary, our results demonstrate that exposure to MIC, augment mitochondrial reactive oxygen species production, depletion in antioxidant defense enzymes, elevated pro-inflammatory cytokine response and induced endothelial cell apoptosis via affecting the balance of mitochondrial-nuclear cross talk. We herein delineate the first and direct molecular cascade of isocyanate-induced pulmonary endothelial cell dysfunction. The results of our study might portray a connective link between associated respiratory morbidities with isocyanate exposure, and indeed facilitate to discern the exposure-phenotype relationship in observed deficits of pulmonary endothelial cell function. Further, understanding of inter- and intra-cellular signaling pathways involved in isocyanate-induced endothelial damage would not only aid in biomarker identification but also provide potential new avenues to target specific therapeutic interventions.
Acknowledgments
The investigations were partly supported by extra-mural grants received from Department of Science & Technology (DST) and Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India, New Delhi.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Hogg J.C., van Eeden S. Pulmonary and systemic response to atmospheric pollution. Respirology. 2009;14:336–346. doi: 10.1111/j.1440-1843.2009.01497.x. [DOI] [PubMed] [Google Scholar]
- 2.Emmerechts J., Hoylaerts M.F. The effect of air pollution on homeostasis. Hamostaseologie. 2012;32:5–13. doi: 10.5482/ha-1179. [DOI] [PubMed] [Google Scholar]
- 3.Samal A., Honovar J., White C.R., Patel R.P. Potential for chlorine gas-induced injury in the extrapulmonary vasculature. Proceedings of the American Thoracic Society. 2010;7:290–293. doi: 10.1513/pats.201001-006SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aung H.H., Lame M.W., Gohil K., He G., Denison M.S., Rutledge J.C., Wilson D.W. Comparative gene responses to collected ambient particles in vitro: endothelial responses. Physiological Genomics. 2011;43:917–929. doi: 10.1152/physiolgenomics.00051.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabin B.R., Grammer L.C. Occupational immunologic lung disease. Allergy and Asthma Proceedings. 2012;33:58–60. doi: 10.2500/aap.2012.33.3550. [DOI] [PubMed] [Google Scholar]
- 6.Fisseler-Eckhoff A., Bartsch H., Zinsky R., Schirren J. Environmental isocyanate-induced asthma: morphologic and pathogenetic aspects of an increasing occupational disease. International Journal of Environmental Research and Public Health. 2011;8:3672–3687. doi: 10.3390/ijerph8093672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crabb C. Revisiting the Bhopal tragedy. Science. 2004;306:1670–1671. doi: 10.1126/science.306.5702.1670. [DOI] [PubMed] [Google Scholar]
- 8.Report on health effects of the toxic gas leak from the methyl isocyanate plant in Bhopal. Indian Council of Medical Research, 2004.
- 9.Sriramachari S. The Bhopal gas tragedy: an environmental disaster. Current Science. 2004;86:905–920. [Google Scholar]
- 10.Mishra P.K. A pragmatic & translational approach of human biomonitoring to methyl isocyanate exposure in Bhopal. Indian Journal of Medical Research. 2012;135:479–484. [PMC free article] [PubMed] [Google Scholar]
- 11.Mishra P.K., Samarth R.M., Pathak N., Jain S.K., Banerjee S., Maudar K.K. Bhopal gas tragedy: review of clinical and experimental findings after 25 years. International Journal of Occupational and Environmental Health. 2009;22:193–202. doi: 10.2478/v10001-009-0028-1. [DOI] [PubMed] [Google Scholar]
- 12.Mishra P.K., Panwar H., Bhargava A., Gorantla V.R., Jain S.K., Banerjee S., Maudar K.K. Isocyanates induces DNA damage, apoptosis, oxidative stress, and inflammation in cultured human lymphocytes. Journal of Biochemical and Molecular Toxicology. 2008;22:429–440. doi: 10.1002/jbt.20260. [DOI] [PubMed] [Google Scholar]
- 13.Mishra P.K., Dabadghao S., Modi G.K., Desikan P., Jain A., Mittra I., Gupta D., Chauhan C., Jain S.K., Maudar K.K. In utero exposure to methyl isocyanate in the Bhopal gas disaster: evidence of persisting hyperactivation of immune system two decades later. Occupational and Environmental Medicine. 2009;66:279. doi: 10.1136/oem.2008.041517. [DOI] [PubMed] [Google Scholar]
- 14.Mishra P.K., Khan S., Bhargava A., Panwar H., Banerjee S., Jain S.K., Maudar K.K. Regulation of isocyanate-induced apoptosis, oxidative stress, and inflammation in cultured human neutrophils: isocyanate-induced neutrophils apoptosis. Cell Biology and Toxicology. 2010;26:279–291. doi: 10.1007/s10565-009-9127-9. [DOI] [PubMed] [Google Scholar]
- 15.Bhargava A., Punde R.P., Pathak N., Dabadghao S., Desikan P., Jain A., Maudar K.K., Mishra P.K. Status of inflammatory biomarkers in the population that survived the Bhopal gas tragedy: a study after two decades. Industrial Health. 2010;48:204–208. doi: 10.2486/indhealth.48.204. [DOI] [PubMed] [Google Scholar]
- 16.Mehta D., Malik A.B. Signaling mechanisms regulating endothelial permeability. Physiological Reviews. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 17.S.Y. Yuan, R.R. Rigor, Regulation of Endothelial Barrier Function. Source San Rafael (CA): Morgan & Claypool Life Sciences. Integrated systems physiology: from molecule to function to disease, 2010. [PubMed]
- 18.Mishra P.K., Bhargava A., Raghuram G.V., Jatawa S.K., Akhtar N., Khan S., Tiwari A., Maudar K.K. Induction of genomic instability in cultured human colon epithelial cells following exposure to isocyanates. Cell Biology International. 2009;33:675–683. doi: 10.1016/j.cellbi.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Mishra P.K., Gorantla V.R., Akhtar N., Tamrakar P., Jain S.K., Maudar K.K. Analysis of cellular response to isocyanates using N-succinimidyl N-methylcarbamate exposure in cultured mammalian cells. Environmental and Molecular Mutagenesis. 2009;50:328–336. doi: 10.1002/em.20469. [DOI] [PubMed] [Google Scholar]
- 20.Mishra P.K., Raghuram G.V., Panwar H., Jain D., Pandey H., Maudar K.K. Mitochondrial oxidative stress elicits chromosomal instability after exposure to isocyanates in human kidney epithelial cells. Free Radical Research. 2009;43:718–728. doi: 10.1080/10715760903037699. [DOI] [PubMed] [Google Scholar]
- 21.Raghuram G.V., Pathak N., Jain D., Panwar H., Pandey H., Jain S.K., Mishra P.K. Molecular mechanisms of isocyanate induced oncogenic transformation in ovarian epithelial cells. Reproductive Toxicology. 2010;30:377–386. doi: 10.1016/j.reprotox.2010.05.087. [DOI] [PubMed] [Google Scholar]
- 22.Jain D., Pathak N., Khan S., Raghuram G.V., Bhargava A., Samarth R., Mishra P.K. Evaluation of cytotoxicity and anticarcinogenic potential of mentha leaf extracts. International Journal of Toxicology. 2010;30:225–236. doi: 10.1177/1091581810390527. [DOI] [PubMed] [Google Scholar]
- 23.Mishra P.K., Raghuram G.V., Bhargava A., Ahirwar A., Samarth R., Upadhyaya R., Jain S.K., Pathak N. In vitro and in vivo evaluation of the anticarcinogenic and cancer chemopreventive potential of a flavonoid-rich fraction from a traditional Indian herb Selaginella bryopteris. British Journal of Nutrition. 2011;116:1154–1168. doi: 10.1017/S0007114511001498. [DOI] [PubMed] [Google Scholar]
- 24.Bhargava A., Pathak N., Panwar H., Khan S., Punde R.P., Varshney S., Mishra P.K. Occult hepatitis B virus infection with low viremia induces DNA damage, apoptosis, oxidative stress and inflammation in peripheral blood lymphocytes. Virus Research. 2010;153:143–150. doi: 10.1016/j.virusres.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 25.Khan S., Bhargava A., Pathak N., Maudar K.K., Varshney S., Mishra P.K. Circulating biomarkers and their possible role in pathogenesis 3 of chronic hepatitis B and C viral infections. Indian Journal of Clinical Biochemistry. 2010;26:161–168. doi: 10.1007/s12291-010-0098-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra P.K., Bhargava A., Raghuram G.V., Gupta S., Tiwari S., Upadhyaya R., Jain S.K., Maudar K.K. Inflammatory response to isocyanates and onset of genomic instability in cultured human lung fibroblasts. Genetics and Molecular Research. 2009;8:129–143. doi: 10.4238/vol8-1gmr544. [DOI] [PubMed] [Google Scholar]
- 27.Raghuram G.V., Pathak N., Jain D., Pandey H., Panwar H., Jain S.K., Banerjee S., Mishra P.K. Molecular characterization of isocyanate-induced male germ-line genomic instability. Journal of Environmental Pathology, Toxicology and Oncology. 2010;29:213–234. doi: 10.1615/jenvironpatholtoxicoloncol.v29.i3.50. [DOI] [PubMed] [Google Scholar]
- 28.H. Panwar, G.V. Raghuram, D. Jain, A.K. Ahirwar, S. Khan, S. Banerjee, S.K. Jain, P.K. Mishra, Cell cycle deregulation by methyl isocyanate: implications in liver carcinogenesis, Environmental Toxicology, 10.1002/tox.21757, in press. [DOI] [PubMed]
- 29.Khan S., Raghuram G.V., Bhargava A., Pathak N., Chandra D.H., Jain S.K., Mishra P.K. Role and clinical significance of lymphocyte mitochondrial dysfunction in type 2 diabetes mellitus. Translational Research. 2011;158:344–359. doi: 10.1016/j.trsl.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Bhargava A., Raghuram G.V., Pathak N., Varshney S., Jatawa S.K., Jain D., Mishra P.K. Occult hepatitis C virus elicits mitochondrial oxidative stress in lymphocytes and triggers PI3-kinase-mediated DNA damage response. Free Radical Biology and Medicine. 2011;51:1806–1814. doi: 10.1016/j.freeradbiomed.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Cullinan P., Acquilla S., Dhara V.R. Respiratory morbidity 10 years after the Union Carbide gas leak at Bhopal: a cross sectional survey. The International Medical Commission on Bhopal. British Medical Journal. 1997;314:338–342. doi: 10.1136/bmj.314.7077.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckett W.S. Persistent respiratory effects in survivors of the Bhopal disaster. Thorax. 1998;2:S43–S46. doi: 10.1136/thx.53.2008.s43. 53 Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vijayan V.K. Methyl isocyanate (MIC) exposure and its consequences on human health at Bhopal. International Journal of Environmental Studies. 2010;67:637–653. [Google Scholar]
- 34.De S. Retrospective analysis of lung function abnormalities of Bhopal gas tragedy affected population. Indian Journal of Medical Research. 2012;135:193–200. [PMC free article] [PubMed] [Google Scholar]
- 35.Castell J.V., Donato M.T., Gómez-Lechón M.J. Metabolism and bioactivation of toxicants in the lung. The in vitro cellular approach. Environmental Toxicology and Pharmacology. 2005;57(Suppl 1):189–204. doi: 10.1016/j.etp.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Fadini G.P., Avogaro A. Cell-based methods for ex vivo evaluation of human endothelial biology. Cardiovascular Research. 2010;87:12–21. doi: 10.1093/cvr/cvq119. [DOI] [PubMed] [Google Scholar]
- 37.Jugg B.J., Smith A.J., Rudall S.J., Rice P. The injured lung: clinical issues and experimental models. Philosophical Transactions of the Royal Society London B Biological Science. 2011;366:306–309. doi: 10.1098/rstb.2010.0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray P.D., Huang B.W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallyathan V., Shi X. The role of oxygen free radicals in occupational and environmental lung diseases. Environmental Health Perspectives. 1997;105:165–177. doi: 10.1289/ehp.97105s1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghio A.J., Carraway M.S., Madden M.C. Composition of air pollution particles and oxidative stress in cells, tissues, and living systems. Journal of Toxicology and Environmental Health Part B: Critical Reviews. 2012;15:1–21. doi: 10.1080/10937404.2012.632359. [DOI] [PubMed] [Google Scholar]
- 41.Widlansky M.E., Gutterman D.D. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxidants and Redox Signalling. 2011;15:1517–1530. doi: 10.1089/ars.2010.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas S.R., Witting P.K., Drummond G.R. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxidants and Redox Signalling. 2008;10:1713–1765. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 43.Frampton M.W. Inflammation and airborne particles. Clinics in Occupational and Environmental Medicine. 2006;5:797–815. doi: 10.1016/j.coem.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Bhalla D.K., Hirata F., Rishi A.K., Gairola C.G. Cigarette smoke, inflammation, and lung injury: a mechanistic perspective. Journal of Toxicology and Environmental Health, Part B. Critical Reviews. 2009;12:45–64. doi: 10.1080/10937400802545094. [DOI] [PubMed] [Google Scholar]
- 45.Tamagawa E., Bai N., Morimoto K., Gray C., Mui T., Yatera K., Zhang X., Xing L., Li Y., Laher I., Sin D.D., Man S.F., van Eeden S.F. Particulate matter exposure induces persistent lung inflammation and endothelial dysfunction. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2008;295:L79–L85. doi: 10.1152/ajplung.00048.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davel A.P., Lemos M., Pastro L.M., Pedro S.C., de André P.A., Hebeda C., Farsky S.H., Saldiva P.H., Rossoni L.V. Endothelial dysfunction in the pulmonary artery induced by concentrated fine particulate matter exposure is associated with local but not systemic inflammation. Toxicology. 2012;295:39–46. doi: 10.1016/j.tox.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Chow C., Abreu M.T.H., Suzuki T., Downey G.P. Oxidative stress and acute lung injury. American Journal of Respiratory Cell and Molecular Biology. 2003;29:427–431. doi: 10.1165/rcmb.F278. [DOI] [PubMed] [Google Scholar]
- 48.Jones R.M., Petermann E. Replication fork dynamics and the DNA damage response. Biochemical Journal. 2012;443:13–26. doi: 10.1042/BJ20112100. [DOI] [PubMed] [Google Scholar]
- 49.Ditch S., Paull T.T. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends in Biochemical Sciences. 2012;37:15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mishra P.K., Jatawa S.K., Raghuram G.V., Pathak N., Jain A., Tiwari A., Varshney S., Maudar K.K. Correlation of aberrant expression of p53, Rad50, and cyclin-E proteins with microsatellite instability in gallbladder adenocarcinomas. Genetics and Molecular Research. 2009;8:1202–1210. doi: 10.4238/vol8-4gmr653. [DOI] [PubMed] [Google Scholar]
- 51.Mishra P.K., Raghuram G.V., Jatawa S.K., Bhargava A., Varshney S. Frequency of genetic alterations observed in cell cycle regulatory proteins and microsatellite instability in gallbladder adenocarcinoma: a translational perspective. Asian Pacific Journal of Cancer Prevention. 2011;12:573–574. [PubMed] [Google Scholar]
- 52.Nadadur S.S., Haykal-Coates N., Mudipalli A., Costa D.L. Endothelial effects of emission source particles: acute toxic response gene expression profiles. Toxicology In Vitro. 2009;23:67–77. doi: 10.1016/j.tiv.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Damico R., Simms T., Kim B.S., Tekeste Z., Amankwan H., Damarla M., Hassoun P.M. p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: inhibitory effects of macrophage migration inhibitor factor. American Journal of Respiratory Cell and Molecular Biology. 2011;44:323–332. doi: 10.1165/rcmb.2009-0379OC. [DOI] [PMC free article] [PubMed] [Google Scholar]