

Abstract
Nitric oxide production by the endothelium is required for normal vascular homeostasis; however, in conditions of oxidative stress, interactions of nitric oxide with reactive oxygen species (ROS) are thought to underlie endothelial dysfunction. Beyond canonical nitric oxide signaling pathways, nitric oxide production results in the post-translational modification of protein thiols, termed S-nitrosation. The potential interplay between S-nitrosation and ROS remains poorly understood and is the focus of the current study. The effects of the S-nitrosating agent S-nitrosocysteine (CysNO) in combination with redox-cycling agents was examined in bovine aortic endothelial cells (BAEC). CysNO significantly impairs mitochondrial function and depletes the NADH/NAD+ pool; however, these changes do not result in cell death. When faced with the additional stressor of a redox-cycling agent used to generate ROS, further loss of NAD+ occurs, and cellular ATP pools are depleted. Cellular S-nitrosothiols also accumulate, and cell death is triggered. These data demonstrate that CysNO sensitizes endothelial cells to redox-cycling agent-dependent mitochondrial dysfunction and cell death and identify attenuated degradation of S-nitrosothiols as one potential mechanism for the enhanced cytotoxicity.
Abbreviations: BAEC, Bovine aortic endothelial cells; BSO, Buthioninesulphoximine; cGMP, Cyclic guanosine monophosphate; CysNO, S-nitrosocysteine; DMNQ, 2,3-dimethoxy-1,4-naphthoquinone; DMSO, Dimethyl sulfoxide; DPBS, Dulbecco’s phosphate buffered saline; DTPA, Diethylenetriaminepentaacetic acid; DTT, Dithiothreitol; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; GSHee, Glutathione Ethyl Ester; LDH, Lactate Dehydrogenase; NAC, N-acetyl cysteine; N.D., Not detectable; NOS, Nitric oxide synthase; OCR, Oxygen consumption rate; ROS, Reactive oxygen species; SEM, Standard error of the mean.
Keywords: S-nitrosation, S-nitrosylation, Thiol, Reactive oxygen species, Nitric oxide, Mitochondria
Graphical abstract
Highlights
► S-Nitrosothiols sensitize to ROS-dependent bioenergetic dysfunction and death. ► This results from changes in adenine nucleotide pools and ATP homeostasis. ► Metabolic changes may indirectly enhance S-nitrosothiol-dependent cellular effects. ► These findings aid in defining beneficial and deleterious effects of S-nitrosothiols.
Introduction
Conserved cysteine residues occur in almost all classes of proteins and are often critical for protein function [1–3]. Additionally, modification of protein cysteine residues, the primary mechanism for redox signaling events, occurs in a specific manner. While cysteine is present in most proteins, it is the second least abundant amino acid in proteins (approximately 1.9% of total amino acid composition), and only a small percentage of cysteine is susceptible to modification [4]; thus, thiols are poised to mediate diverse and specific redox signaling responses and control important aspects of cell growth, differentiation, stress responses, and cell death (reviewed in [3,5]). For these reasons, there is significant interest in developing therapeutic agents which target redox-sensitive pathways (termed ‘redox therapeutics’) for multiple diseases including cardiovascular disease, diabetes, neurodegeneration, and cancer [6].
In the vasculature, significant emphasis has been put on understanding the specific role for S-nitrosation, a nitric oxide-dependent cysteine modification that results in the addition of a nitroso moiety to a protein thiol [7]. The endothelium represents an ideal site for S-nitrosation-dependent signals, as it constitutively expresses the nitric oxide producing enzyme endothelial nitric oxide synthase (NOS), and in inflammatory conditions, is also exposed to nitric oxide-derived from the inducible NOS isoform [8]. Although canonical nitric oxide signaling occurs through its interaction with soluble guanylyl cyclase and control of cyclic guanosine monophosphate (cGMP)-dependent signaling [9,10], some have suggested that S-nitrosation is an essential component of vascular (dys)function [11].
Vasculature pathologies such as atherosclerosis are characterized by endothelial dysfunction. In this case, the endothelium is exposed to both nitric oxide and reactive oxygen species (ROS), such as hydrogen peroxide and superoxide, as the result of infiltrating inflammatory cells [12]. Environmental exposure to redox-cycling agents also represents an important source of ROS in vivo. Environmental pollutants such as polycyclic aromatic hydrocarbons [13] and the herbicide paraquat redox-cycle [14,15] with aldo-ketoreductases and components of the mitochondrial electron transport chain (e.g., NADH dehydrogenase and ubiquinol:cytochrome c oxidoreductase), respectively. Dietary polyphenols [16] and some anti-cancer agents (e.g., doxorubicin) [17] also possess this activity. The endothelium is acutely sensitive to nitric oxide, its downstream mediators, and ROS. It is well-established that nitric oxide reacts readily with superoxide to produce the highly oxidizing species peroxynitrite [18], and many of the deleterious effects of combined exposure to nitric oxide/superoxide have been attributed to peroxynitrite formation. However, far less is understood regarding the potential interplay between S-nitrosation-dependent thiol modification and ROS.
A key action of S-nitrosation in the vasculature is the control of metabolic pathways. This is highlighted in work examining the effects of S-nitrosothiols in preclinical models of ischemia-reperfusion injury. Here, administration of S-nitrosothiols such S-nitrosocysteine (CysNO) [19] and mitochondrial-targeted S-nitroso-N-acetylpenicillamine [20,21] significantly improves functional recovery of the heart upon reperfusion. Protection from ischemia-reperfusion injury is thought to occur through the S-nitrosation of several metabolic enzymes including glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Complex I of the mitochondrial electron transport chain which inhibits their activity and mitigates oxidant production during reperfusion [22]. We have also shown that exposure of endothelial cells to S-nitrosothiols significantly down-regulates mitochondrial function [23], and increasing evidence implicates mitochondria as both sources and targets of ROS [24,25]. Thus, coupled with the growing appreciation for exogenous redox-cycling agents as sources of ROS in (patho)physiology, a further understanding of the effects of S-nitrosothiols on ROS-dependent alterations in cellular bioenergetics is needed.
Here, we examined the effects of the S-nitrosothiol CysNO on cellular bioenergetics, nucleotide pools, and cell death in bovine aortic endothelial cells (BAEC). CysNO significantly impairs mitochondrial function and depletes the NADH/NAD+ pool; however, these changes do not result in cell death. In contrast, when faced with the additional stressor of a redox-cycling agent used to generate ROS, further loss of NAD+ occurs, and cellular ATP pools are depleted. Concomitantly, cellular S-nitrosothiols accumulate, and cell death is triggered. These data demonstrate that CysNO sensitizes endothelial cells to redox cycling agent-dependent mitochondrial dysfunction and cell death and identify attenuated degradation of S-nitrosothiols as a potential mechanism for the enhanced cytotoxicity.
Materials and methods
Materials
All materials were of analytical grade and obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. 2,3-Dimethoxy-1,4-naphthoquinone (DMNQ) was purchased from Enzo Life Sciences (Farmingdale, NY). CysNO was synthesized by incubating cysteine with acidified nitrite as described previously [26].
Cell culture
Bovine aortic endothelial cells (BAEC) were purchased from Lonza (Walkersville, MD) and cultured in high glucose (25 mM) DMEM supplemented with 10% fetal bovine serum (FBS), 200 units/mL penicillin, and 200 μg/mL streptomycin (Invitrogen; Carlsbad, CA). Cells were treated with CysNO and DMNQ in Dulbecco's phosphate buffered saline (DPBS) supplemented with 5.5 mM glucose and 1 mM sodium pyruvate.
Lactate dehydrogenase (LDH) release
After treatment, aliquots of media were taken, and then cells were harvested by scraping and lysed in PBS containing 0.1% Triton-X 100. Samples were centrifuged at 10,000×g for 10 min, and supernatants were used for LDH analysis. LDH activity in media and cell lysates was monitored as the oxidation of NADH (0.3 mM) at 334 nm as described previously [27].
S-nitrosothiol measurements
After treatment, cells were washed twice with DPBS, and then scraped into 250 μL of lysis buffer (50 mM phosphate, 1 mM diethylene triamine penta acetic acid (DTPA), and 50 mM N-ethyl maleimide, pH 7.4). Samples were sonicated for 15 s prior to centrifugation (10,000×g for 10 min). Supernatants were used for tri-iodide-based chemiluminescence detection of S-nitrosothiols as described previously [28–30]. Mercuric chloride (5 mM for 10 min) was used to verify the presence of S-nitrosothiols, and a standard curve was generated using S-nitrosoglutathione.
HPLC analysis of adenine and pyridine nucleotides
Adenine (ATP, ADP, and AMP) and oxidized pyridine (NAD+) nucleotides were extracted using perchloric acid precipitation. NADH was extracted using alkaline conditions (0.5 M KOH/Hank's balanced salt solution, 3:1), and the pH of lysates was adjusted to ∼8 using 6 M HCl and ammonium acetate (1 M, pH 4.7) [31,32]. All samples were filtered prior to HPLC analysis on a Kinetex C-18 column (2.6 μm, 100 mm×4.6 mm internal diameter) and separated using solvent A (0.1 M potassium phosphate, 4 mM tetrabutyl ammonium bisulfate, pH 6.0, diluted v/v in water 64:36) and solvent B (0.1 M potassium phosphate, 4 mM tetrabutyl ammonium bisulfate, pH 6.0, diluted v/v in methanol 64:36) with a flow rate of 1 mL/min. HPLC peaks were measured for each sample, compared with standards, and normalized to total protein. Importantly, this protocol specifically distinguishes NADH/NAD+ from NADPH/NADP+.
Mitochondrial function analysis
Activity of mitochondrial dehydrogenases was assessed using the MTT assay [33]. Briefly, after treatment, complete culture medium containing 0.4 mg/mL thiazolyl blue tetrazolium was added, and cells were incubated for 2 h at 37 °C. Formazan crystals were then solubilized in dimethyl sulfoxide (DMSO), and the absorbance was measured at 590 nm with background reference at 620 nm. A Seahorse Bioscience XF24 Analyzer (North Billerica, MA) [34] was used to measured basal oxygen consumption rates (OCRs) from BAEC. BAEC were seeded in specialized microplates 24 h prior to treatment. Cells were then treated as described prior to be being assayed in the XF24 Analyzer. DPBS supplemented with 5.5 mM glucose and 1 mM sodium pyruvate was used as assay media, and protein levels were assessed at the end of the assay using the Bradford method (Bio-Rad, Hercules, CA).
Statistical analysis
Results are reported as means±SEM for n≥3 as indicated in the figure legends. Statistical significance was evaluated by Student's t-test. The minimum level of significance was set at p<0.05.
Results
Morphological changes and LDH release after exposure to CysNO and DMNQ
We first defined the cytotoxic effects of treatment with the S-nitrosating agent CysNO alone and in combination with DMNQ in BAEC. Treatment with CysNO or DMNQ alone did not result in marked changes in cell morphology; however, pretreatment with CysNO for 1 h followed by DMNQ exposure for a further 4 h caused cell-rounding and loss of cell adhesion to the tissue culture plate (Fig. 1A), indicating acute effects of this treatment. Moreover, incubation in complete culture media for an additional 12 h resulted in a significant increase in LDH release from cells exposed to both CysNO and DMNQ (Fig. 1B).
Fig. 1.
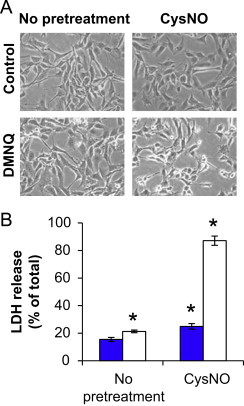
Morphological changes and LDH release after exposure to CysNO and DMNQ. BAEC were pretreated with CysNO (100 μM) for 1 h prior to treatment with (open bars) or without (blue bars) DMNQ (20 μM) for an additional 4 h. Representative light micrographs are shown in Panel A. Treatment media was removed, and cells were incubated for 12 h in complete culture media prior to harvesting cells and media for analysis of LDH release spectrophotometrically (B). Values represent mean ±SEM, n=3. ⁎p<0.05 compared to control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
S-nitrosothiol levels after exposure to CysNO and DMNQ
The primary mechanism by which CysNO is thought to control cellular responses is through transnitrosation to protein thiols, thereby initiating redox-dependent signals. We next examined the levels of S-nitrosothiols after treatment with CysNO and DMNQ. As expected, CysNO exposure increased S-nitrosothiol levels over that of control to approximately 670 pmol/mg protein (Fig. 2). The addition of DMNQ after CysNO treatment significantly enhanced S-nitrosothiol levels to more than 3 times that of CysNO alone.
Fig. 2.
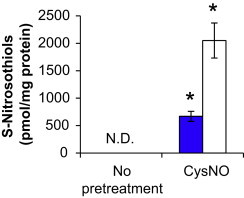
S-Nitrosothiol levels after exposure to CysNO and DMNQ. BAEC were pretreated with CysNO (100 μM) for 1 h prior to treatment with (open bars) or without (blue bars) DMNQ (20 μM) for an additional 4 h. Cells were harvested for tri-iodide-dependent chemiluminescent detection of S-nitrosothiols, and results were normalized to total protein per sample. Values represent mean ±SEM, n=3. The level of S-nitrosothiols in control samples was not detectable (N.D.). ⁎p<0.05 compared to control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Effects of CysNO and DMNQ on the NADH/NAD+levels
A key mechanism for degradation of S-nitrosothiols in cells is through the activity of the NADH-dependent enzyme alcohol dehydrogenase 3, also known as S-nitrosoglutathione (GSNO) reductase [35], and we have previously demonstrated that BAEC possess an NADH-dependent mechanism for the consumption of GSNO [36]. Because protein S-nitrosothiols are in equilibrium with low-molecular weight S-nitrosothiol such as GSNO through transnitrosation, changes in alcohol dehydrogenase 3 activity can affect the entire S-nitrosothiol pool [37]. Thus, the effect of combined treatment with CysNO and DMNQ on NADH and NAD+ was examined. Interestingly, DMNQ alone resulted in a major decrease in NADH levels (Fig. 3A), likely due to the requirement of NADH for redox cycling [38]. However, decrease of NADH was accompanied by a decrease rather than an increase in NAD+, suggesting the occurrence of oxidant-mediated DNA-damage and the initiation of poly (ADP-ribosy)lation as others have shown previously [39]. CysNO alone resulted in loss of NADH (Fig. 3A; consistent with a role for enzymatic GSNO reduction) which was enhanced in the presence of DMNQ. Disruptions in the NADH/NAD+ redox couple provide a possible explanation for the enhanced S-nitrosothiol levels observed above through attenuated alcohol dehydrogenase 3 activity and degradation of S-nitrosothiols. More surprising was the synergistic loss of NAD+ that occurred upon combined treatment of CysNO and DMNQ (Fig. 3B). This suggests that the combination of CysNO and DMNQ cause a severe metabolic dysfunction in the cells. For this reason, we examined the effect of these agents on mitochondrial function.
Fig. 3.
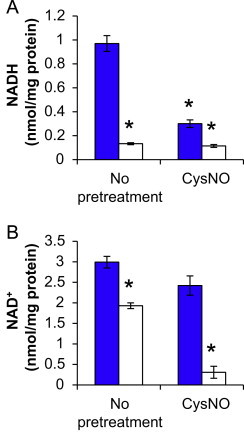
Effects of CysNO and DMNQ on the NADH/NAD+ ratio. BAEC were pretreated with CysNO (100 μM) for 1 h prior to treatment with (open bars) or without (blue bars) DMNQ (20 μM) for an additional 4 h. Cells were harvested using alkaline (A) or acidic (B) sample processing for HPLC analysis of NADH and NAD+ levels, respectively, and results were normalized to total protein per sample. Values represent mean ±SEM, n=3. ⁎p<0.05 compared to control.
Mitochondrial function in response to CysNO and DMNQ
Through the activity of Complex I, NADH dehydrogenase, mitochondria are a major NADH-consuming organelle [24]. Due to this and the critical role for ATP in de novo biosynthesis of NADH, the effects of CysNO and DMNQ on mitochondrial function in BAEC were determined. BAEC were pretreated with CysNO for 1 h, and then increasing concentrations of DMNQ (10–50 μM) were added for an additional 4 h. DMNQ caused a concentration-dependent decline in mitochondrial dehydrogenase activity as measured by the reduction of MTT to formazan. Treatment with CysNO alone resulted in significant impairment of mitochondrial dehydrogenase activity that was not further inhibited upon the addition of DMNQ (Fig. 4A).
Fig. 4.
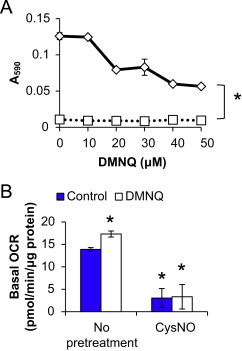
Mitochondrial function in response to CysNO and DMNQ. BAEC were either pretreated with CysNO (100 μM, 1 h; square symbols) or not (diamond symbols) prior to the addition of DMNQ (10–50 μM) for an additional 4 h. Activity of mitochondrial dehydrogenases was assessed by MTT assay (A). Basal oxygen consumption rate (OCR) was also measured using an XF24 Analyzer after exposure to CysNO (100 μM) with (open bars) or without (blue bars) DMNQ (20 μM) as described above (B). Values represent mean±SEM, n=8 for MTT assay and 3–4 for extracellular flux analysis. ⁎p<0.05 compared to control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Changes in mitochondrial function were confirmed using extracellular flux analysis to monitor basal oxygen consumption rate (OCR) after treatment. Again, CysNO alone or in combination with DMNQ inhibited basal OCR (Fig. 4B); however, here we observed a slight, but statistically significant increase in OCR with DMNQ alone. This likely represents oxygen consumption which occurs through the redox-cycling activity of DMNQ as has been reported previously [40] and not a stimulation of basal mitochondrial respiration.
Effects of CysNO and DMNQ on adenine nucleotide pools
To extend the mitochondrial function findings, the effects of CysNO and DMNQ on adenine nucleotide pools were next examined. CysNO or DMNQ alone caused a modest decrease in ATP levels (Fig. 5A), but this did not translate into increases in ADP or AMP (Fig. 5B,C). In contrast, the combined treatment of CysNO and DMNQ resulted in a nearly complete loss of ATP and significant increases in ADP and AMP. Interestingly, there is an approximately 39 nmol/mg protein decrease in ATP, but the sum of ADP and AMP pools only increase by approximately 6 nmol/mg protein. This suggests that further catabolism of adenine nucleotides may occur in endothelial cells under conditions of nitrosative and oxidative stress.
Fig. 5.
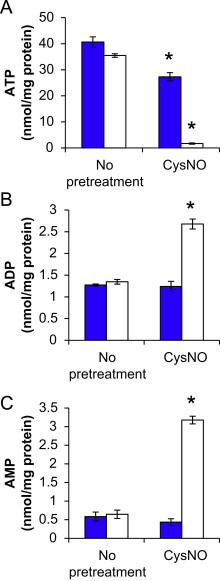
Effects of CysNO and DMNQ on adenine nucleotide pools. BAEC were pretreated with CysNO (100 μM) for 1 h prior to treatment with (open bars) or without (blue bars) DMNQ (20 μM) for an additional 4 h. Cells were harvested for HPLC analysis of ATP (A), ADP (B), and AMP (C), and results were normalized to total protein per sample. Values represent mean±SEM, n=3. ⁎p<0.05 compared to control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Effect of thiol supplementation on nucleotide pools and mitochondrial function following CysNO and DMNQ treatment
Because of the primary mechanism of action for CysNO and other S-nitrosothiols is through transnitrosation to protein thiols, we investigated whether bolstering the cellular thiol pool in BAEC could restore mitochondrial function and nucleotide pools. When BAEC were pretreated with glutathione ethyl ester (GSHee) or N-acetyl cysteine (NAC) for 1 h prior to the addition of DMNQ, the DMNQ-dependent depletion of ATP and NAD+ as well as impairment of mitochondrial dehydrogenase activity was restored to levels similar to control (Fig. 6). ADP and AMP levels did not change in these conditions. However, in cells exposed to both CysNO and DMNQ, supplementation with GSHee or NAC only partially mitigated the loss of ATP and NAD+, and these nucleotides were not restored to control levels (Fig. 6A,B). This also resulted in no change in the CysNO and DMNQ-dependent decreases in mitochondrial dehydrogenase activity (Fig. 6C). Further studies to address specific role of GSH in the maintenance of nucleotide pools demonstrated that, unlike CysNO, depletion of glutathione using buthioninesulphoximine (BSO) did not exacerbate DMNQ-dependent changes in nucleotide pools (Supplementary Fig. 1).
Fig. 6.
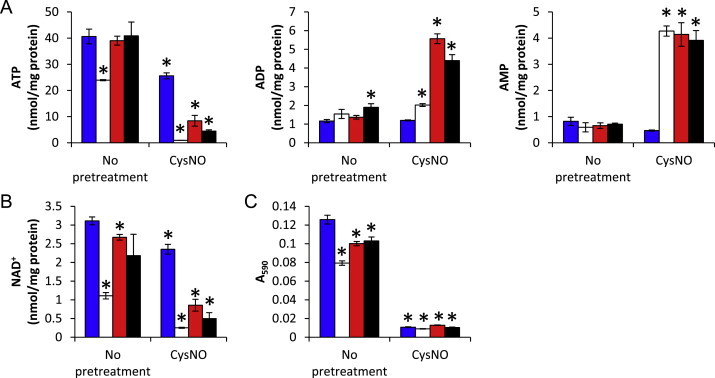
Effect of NAC and GSHee supplementation on nucleotide pools and mitochondrial function following CysNO and DMNQ treatment. BAEC were pretreated with CysNO (100 μM) for 1 h prior to treatment with (open bars) or without (blue bars) DMNQ (20 μM) for an additional 4 h in the presence of NAC (200 μM; red bars) or GSHee (200 μM; black bars). Cells were harvested for HPLC analysis of ATP (A), ADP (B), and AMP (C), and results were normalized to total protein per sample. Identical samples were also analyzed for mitochondrial dehydrogenase activity using the MTT assay (D). Values represent mean±SEM, n=3 for adenine nucleotides and 8 for MTT assay. ⁎p<0.05 compared to control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Discussion
Understanding the interplay between S-nitrosation-dependent thiol modification and ROS is critical to define biological action of nitric oxide in vascular pathologies. Here, we have used the S-nitrosothiol CysNO as a cell-transportable S-nitrosating agent and examined its effects in the presence of the redox-cycling agent DMNQ. As expected, treatment with CysNO causes an increase in cellular S-nitrosothiols (Fig. 2) which is accompanied by significant decreases in NADH levels and a more modest depletion of NAD+ (Fig. 3). Loss of NADH is likely due to the activity of S-nitrosothiol degradation pathways through alcohol dehydrogenase 3, an NADH-dependent enzyme [35]. Our data suggest that although S-nitrosothiol degradation is occurring, it is not sufficient to reduce all cellular S-nitrosothiols since total S-nitrosothiols are still elevated over control levels during the course of our experiments. CysNO also readily impairs mitochondrial function resulting in loss in activity of mitochondrial dehydrogenases and basal oxygen consumption (Fig. 4). This is consistent with the modification of several metabolic enzymes known to be targets of S-nitrosation, including GADPH and Complex I of the mitochondrial electron transport chain [41,42]. In fact, S-nitrosation of these enzymes is thought to play an important role in the therapeutic effects of S-nitrosothiols in ischemia-reperfusion injury [19–21]. Despite these marked changes in mitochondrial function, only modest changes in adenine nucleotide pools occur (Fig. 5), and no cytotoxicity is observed (Fig. 1), suggesting that compensatory mechanisms are sufficient to maintain cellular energy balance.
The effects of the redox-cycling agent DMNQ alone are somewhat similar to those of CysNO. DMNQ also affects the NADH/NAD+ redox couple by decreasing total levels of both the reduced and oxidized nucleotides (Fig. 3). It is well-established that DMNQ requires NADH or another electron source to redox-cycle [43,44]; thus, a constant ‘pull’ on this nucleotide pool provides an explanation for the observed effects. DMNQ also impairs the activity of mitochondrial dehydrogenases, but to a lesser extent than CysNO and without diminishing basal oxygen consumption (Fig. 4). Interestingly, these DMNQ-dependent effects are completely reversible upon the addition of the thiol-containing agents NAC and GSHee (Fig. 6), indicating the mechanism of action for DMNQ in this context may also be through redox processes. Wright et al. reported that administration of DMNQ to intact skeletal muscles significantly inhibited protein phosphatases through dithiothreitol (DTT)-reversible thiol modification [45]. This is just one of many instances which demonstrate effects of redox-cycling agents on redox-sensitive cellular processes. Nonetheless, DMNQ itself does not result in cell death in BAEC under these experimental conditions (Fig. 1).
CysNO significantly sensitized BAEC to the effects of DMNQ. Combined treatment with CysNO and DMNQ caused metabolic stress characterized by mitochondrial dysfunction (Fig. 4), nearly complete depletion of ATP and NAD+ pools (Figs. 3 and 5), and cell death (Fig. 1). Surprisingly, the synergistic effects of CysNO and DMNQ were only partially reversed by supplementation of thiol-containing agents (Fig. 6), indicating that mechanisms beyond reversible thiol modification may also be important.
There are several possible explanations for the synergistic activity of CysNO and DMNQ. The first is that CysNO exposure, and subsequent S-nitrosation of protein thiols, limits the cell's ability to respond to oxidative stress imposed by DMNQ. Oxidative stress can significantly impact the glutathione pool, the major redox buffer in cells, and affect redox processes. For example, Circu et al. demonstrated that the redox-cycling agents menadione and DMNQ oxidize GSH. Reduction of glutathione disulfide back to GSH requires the activity of glutathione reductase, an NADPH-dependent enzyme [38]. In this study, these authors demonstrated that as the NADPH/NADP+ pool is tapped for detoxification pathways. NADH and NAD+are then used as substrates to produce additional NADP+ through the activity NAD+ kinase; thus, also linking nucleotide pools to detoxification processes. However, this is an unlikely explanation for our studies, since attempts to replete the GSH pool using NAC and GSHee had minimal effects on the synergistic loss of ATP, NAD+, and activity of mitochondrial dehydrogenases. Moreover, depletion of glutathione using BSO followed by a DMNQ challenge did not alter nucleotide levels (Supplementary Fig. 1).
A second explanation for the synergy between CysNO and DMNQ may be through reactions between the free radicals generated by these agents, nitric oxide and superoxide, respectively. Nitric oxide and superoxide react at diffusion-limited rates to form peroxynitrite, a highly oxidizing species [18]. Thus, in this case, it is the production of a new species that accounts for synergistic effects on cellular bioenergetics and cell death. Peroxynitrite is known to oxidatively inactivate several mitochondrial proteins including citric acid cycle dehydrogenases and components of the mitochondrial electron transport chain [18,46], and we have shown that nitric oxide can be released from CysNO in the intracellular environment [47]. Our recent study examining the combine exposure to nitric oxide and redox cycling agents in BAEC demonstrates the synergistic loss of ATP and NAD+ (manuscript in preparation) further supporting a role for peroxynitrite in this context.
A final explanation for how CysNO sensitizes to DMNQ-dependent cellular dysfunction focuses on the fact that both compounds target the NADH/NAD+ redox couple through S-nitrosothiol degradation pathways and redox-cycling, respectively. The inability to degrade S-nitrosothiols through NADH-dependent alcohol dehydrogenase 3 is consistent with the accumulation of S-nitrosothiols observed in the combined treatment of CysNO and DMNQ (Fig. 2). Alternatively, changes in the redox state of the electron transport chain are known to impact endogenous cytochrome c-dependent S-nitrosothiol production [48]. In either case, increased S-nitrosation of protein thiols, particularly metabolic enzymes, is likely to result in enhanced metabolic dysfunction, depletion of adenine and pyridine nucleotide pools, and ultimately cell death (Fig. 7). We have previously shown that S-nitrosation of GAPDH results in subsequent covalent inactivation of the enzyme [42]. This irreversible, S-nitrosation-dependent modification requires the synthesis of new protein to restore GAPDH activity in cells. Irreversible protein modification may be an important mechanism for enhanced cytotoxicity in cells treated with both CysNO and DMNQ in this study and would also explain the inability of thiol supplementation to restore cellular function.
Fig. 7.
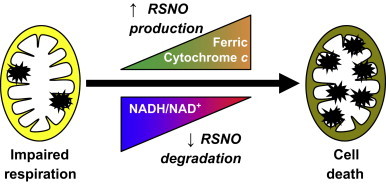
Working model for enhanced S-nitrosothiols (RSNO) levels after exposure to CysNO and redox cycling agents. Modest impairment of respiration by CysNO does not elicit cell death. However, in the presence of redox cycling agents, changes in electron transport chain activity may cause increased ferric cytochrome c-dependent RSNO production. Concomitantly, depletion of the NADH/NAD+ pool may limit RSNO degradation through GSNO reductase. This culminates in the accumulation of RSNO (denoted with the starburst symbol) and ultimately results in cell death.
Conclusions
Taken together, these results demonstrate that S-nitrosothiols sensitize endothelial cells to redox-cycling agent-dependent bioenergetic dysfunction and death with a concomitant elevation of cellular S-nitrosothiol levels. The mechanism for this sensitization is uncertain, but likely involves effects on adenine nucleotide pools and ATP homeostasis. We have also defined an important interaction between adenine and pyridine nucleotide pools and S-nitrosothiol clearance mechanisms, highlighting the fact that changes in metabolism may indirectly enhance S-nitrosothiol-dependent cellular effects. These findings may be particularly relevant in defining potential beneficial and deleterious effects of S-nitrosothiols in cardiovascular (patho)physiology.
Sources of funding
This research was supported by the Redox Biology Program at the Medical College of Wisconsin (N.H.), an Interdisciplinary Cancer Research Post-Doctoral Fellowship from the Cancer Center of the Medical College of Wisconsin (A.R.D.), and National Institutes of Health grant R01-GM-55792 (N.H.).
Acknowledgments
The authors would like to acknowledge Drs. B. Kalyanaraman and Brian P. Dranka for important support for these studies.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2012.11.003.
Appendix A. Supplementary materials
Supplementary data
References
- 1.Claiborne A., Yeh J.I., Mallett T.C. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38(47):15407–15416. doi: 10.1021/bi992025k. 23. [DOI] [PubMed] [Google Scholar]
- 2.Satoh T., Lipton S.A. Redox regulation of neuronal survival mediated by electrophilic compounds. Trends in Neurosciences. 2007;30(1):37–45. doi: 10.1016/j.tins.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Trachootham D., Lu W., Ogasawara M.A., Nilsa R.D., Huang P. Redox regulation of cell survival. Antioxidants and Redox Signaling. 2008;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landar A., Oh J.Y., Giles N.M. A sensitive method for the quantitative measurement of protein thiol modification in response to oxidative stress. Free Radical Biology and Medicine. 2006;40(3):459–468. doi: 10.1016/j.freeradbiomed.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 5.Wall S.B., Oh J.Y., Diers A.R., Landar A. Oxidative modification of proteins: an emerging mechanism of cell signaling. Frontiers in Physiology. 2012;3:369. doi: 10.3389/fphys.2012.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schopfer F.J., Cipollina C., Freeman B.A. Formation and signaling actions of electrophilic lipids. Chemical Reviews. 2011;111(10):5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broniowska K.A., Hogg N. The chemical biology of S-nitrosothiols. Antioxidants and Redox Signaling. 2012;17(7):969–980. doi: 10.1089/ars.2012.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffith O.W., Stuehr D.J. Nitric oxide synthases: properties and catalytic mechanism. Annual Review of Physiology. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 9.Denninger J.W., Marletta M.A. Guanylate cyclase and the NO/cGMP signaling pathway. Biochimica Biophysica Acta. 1999;1411(2–3):334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 10.Waldman S.A., Murad F. Biochemical mechanisms underlying vascular smooth muscle relaxation: the guanylate cyclase–cyclic GMP system. Journal of Cardiovascular Pharmacology. 1988;12(Suppl 5):S115–S118. [PubMed] [Google Scholar]
- 11.Hess D.T., Matsumoto A., Kim S.O., Marshall H.E., Stamler J.S. Protein S-nitrosylation: purview and parameters. Nature Reviews Molecular Cell Biology. 2005;6(2):150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 12.Lee M.Y., Griendling K.K. Redox signaling, vascular function, and hypertension. Antioxidants and Redox Signaling. 2008;10(6):1045–1059. doi: 10.1089/ars.2007.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L., Jin Y., Huang M., Penning T.M. The role of human aldo-keto reductases in the metabolic activation and detoxication of polycyclic aromatic hydrocarbons: interconversion of PAH catechols and PAH o-quinones. Frontiers in Pharmacology. 2012;3:193. doi: 10.3389/fphar.2012.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank D.M., Arora P.K., Blumer J.L., Sayre L.M. Model study on the bioreduction of paraquat, MPP+, and analogs. Evidence against a “redox cycling” mechanism in MPTP neurotoxicity. Biochemical and Biophysical Research Communications. 1987;147(3):1095–1104. doi: 10.1016/s0006-291x(87)80183-3. [DOI] [PubMed] [Google Scholar]
- 15.Castello P.R., Drechsel D.A., Patel M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. Journal of Biological Chemistry. 2007;282(19):14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perron N.R., Garcia C.R., Pinzon J.R., Chaur M.N., Brumaghim J.L. Antioxidant and prooxidant effects of polyphenol compounds on copper-mediated DNA damage. Journal of Inorganic Biochemistry. 2011;105(5):745–753. doi: 10.1016/j.jinorgbio.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Kalyanaraman B., Perez-Reyes E., Mason R.P. Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs. Biochimica Biophysica Acta. 1980;630(1):119–130. doi: 10.1016/0304-4165(80)90142-7. [DOI] [PubMed] [Google Scholar]
- 18.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proceedings of the National Academy of Sciences of USA. 2004;101(12):4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogg N., Broniowska K.A., Novalija J., Kettenhofen N.J., Novalija E. Role of S-nitrosothiol transport in the cardioprotective effects of S-nitrosocysteine in rat hearts. Free Radical Biology and Medicine. 2007;43(7):1086–1094. doi: 10.1016/j.freeradbiomed.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Nadtochiy S.M., Burwell L.S., Brookes P.S. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. Journal of Molecular and Cellular Cardiology. 2007;42(4):812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prime T.A., Blaikie F.H., Evans C. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proceedings of the National Academy of Sciences of USA. 2009;106(26):10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burwell L.S., Nadtochiy S.M., Brookes P.S. Cardioprotection by metabolic shut-down and gradual wake-up. Journal of Molecular and Cellular Cardiology. 2009;46(6):804–810. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diers A.R., Broniowska K.A., Darley-Usmar V.M., Hogg N. Differential regulation of metabolism by nitric oxide and S-nitrosothiols in endothelial cells. American Journal of Physiology—Heart and Circular Physiology. 2011;301(3):H803–H812. doi: 10.1152/ajpheart.00210.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramachandran A., Levonen A.L., Brookes P.S. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radical Biology and Medicine. 2002;33(11):1465–1474. doi: 10.1016/s0891-5849(02)01142-5. [DOI] [PubMed] [Google Scholar]
- 25.Ballinger S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radical Biology and Medicine. 2005;38(10):1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y., Hogg N. The mechanism of transmembrane S-nitrosothiol transport. Proceedings of the National Academy of Sciences of USA. 2004;101(21):7891–7896. doi: 10.1073/pnas.0401167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diers A.R., Higdon A.N., Ricart K.C. Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signalling mechanisms. Biochemical Journal. 2010;426(1):31–41. doi: 10.1042/BJ20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rassaf T., Bryan N.S., Kelm M., Feelisch M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. Free Radical Biology and Medicine. 2002;33(11):1590–1596. doi: 10.1016/s0891-5849(02)01183-8. [DOI] [PubMed] [Google Scholar]
- 29.Samouilov A., Zweier J.L. Development of chemiluminescence-based methods for specific quantitation of nitrosylated thiols. Analytical Biochemistry. 1998;258(2):322–330. doi: 10.1006/abio.1998.2609. [DOI] [PubMed] [Google Scholar]
- 30.Yang B.K., Vivas E.X., Reiter C.D., Gladwin M.T. Methodologies for the sensitive and specific measurement of S-nitrosothiols, iron-nitrosyls, and nitrite in biological samples. Free Radical Research. 2003;37(1):1–10. doi: 10.1080/1071576021000033112. [DOI] [PubMed] [Google Scholar]
- 31.Perez J., Hill B.G., Benavides G.A., Dranka B.P., Darley-Usmar V.M. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochemical Journal. 2010;428(2):255–267. doi: 10.1042/BJ20100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diers A.R., Broniowska K.A., Chang C.F., Hogg N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. Biochemical Journal. 2012;444(3):561–571. doi: 10.1042/BJ20120294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 34.Ferrick D.A., Neilson A., Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discovery Today. 2008;13(5–6):268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Staab C.A., Hellgren M., Hoog J.O. Medium- and short-chain dehydrogenase/reductase gene and protein families: dual functions of alcohol dehydrogenase 3: implications with focus on formaldehyde dehydrogenase and S-nitrosoglutathione reductase activities. Cellular and Molecular Life Sciences. 2008;65(24):3950–3960. doi: 10.1007/s00018-008-8592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Broniowska K.A., Zhang Y., Hogg N. Requirement of transmembrane transport for S-nitrosocysteine-dependent modification of intracellular thiols. Journal of Biological Chemistry. 2006;281(45):33835–33841. doi: 10.1074/jbc.M603248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Hogg N. S-Nitrosothiols: cellular formation and transport. Free Radical Biology and Medicine. 2005;38(7):831–838. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Circu M.L., Maloney R.E., Aw T.Y. Disruption of pyridine nucleotide redox status during oxidative challenge at normal and low-glucose states: implications for cellular adenosine triphosphate, mitochondrial respiratory activity, and reducing capacity in colon epithelial cells. Antioxidants and Redox Signaling. 2011;14(11):2151–2162. doi: 10.1089/ars.2010.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loor G., Kondapalli J., Schriewer J.M., Chandel N.S., Vanden Hoek T.L., Schumacker P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radical Biology and Medicine. 2010;49(12):1925–1936. doi: 10.1016/j.freeradbiomed.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dranka B.P., Hill B.G., Darley-Usmar V.M. Mitochondrial reserve capacity in endothelial cells: the impact of nitric oxide and reactive oxygen species. Free Radical Biology and Medicine. 2010;48(7):905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burwell L.S., Nadtochiy S.M., Tompkins A.J., Young S., Brookes P.S. Direct evidence for S-nitrosation of mitochondrial complex I. Biochemical Journal. 2006;394(Pt 3):627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Broniowska K.A., Hogg N. Differential mechanisms of inhibition of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosothiols and NO in cellular and cell-free conditions. American Journal of Physiology—Heart and Circular Physiology. 2010;299(4):H1212–H1219. doi: 10.1152/ajpheart.00472.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe N., Forman H.J. Autoxidation of extracellular hydroquinones is a causative event for the cytotoxicity of menadione and DMNQ in A549-S cells. Archives of Biochemistry and Biophysics. 2003;411(1):145–157. doi: 10.1016/s0003-9861(02)00716-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadenas E. Antioxidant and prooxidant functions of DT-diaphorase in quinone metabolism. Biochemical Pharmacology. 1995;49(2):127–140. doi: 10.1016/s0006-2952(94)00333-5. [DOI] [PubMed] [Google Scholar]
- 45.Wright V.P., Reiser P.J., Clanton T.L. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. Journal of Physiology. 2009;587(Pt 23):5767–5781. doi: 10.1113/jphysiol.2009.178285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J.H., Yang E.S., Park J.W. Inactivation of NADP+-dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. Journal of Biological Chemistry. 2003;278(51):51360–51371. doi: 10.1074/jbc.M302332200. [DOI] [PubMed] [Google Scholar]
- 47.Riego J.A., Broniowska K.A., Kettenhofen N.J., Hogg N. Activation and inhibition of soluble guanylyl cyclase by S-nitrosocysteine: involvement of amino acid transport system L. Free Radical Biology and Medicine. 2009;47(3):269–274. doi: 10.1016/j.freeradbiomed.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broniowska K.A., Keszler A., Basu S., Kim-Shapiro D.B., Hogg N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochemical Journal. 2012;442(1):191–197. doi: 10.1042/BJ20111294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data