

Abstract
There is increasing evidence that non-enzymatic post-translational protein modifications might play key roles in various diseases. These protein modifications can be caused by free radicals generated during oxidative stress or by their products generated during lipid peroxidation. 4-Hydroxynonenal (HNE), a major biomarker of oxidative stress and lipid peroxidation, has been recognized as important molecule in pathology as well as in physiology of living organisms. Therefore, its detection and quantification can be considered as valuable tool for evaluating various pathophysiological conditions.
The HNE-protein adduct ELISA is a method to detect HNE bound to proteins, which is considered as the most likely form of HNE occurrence in living systems. Since the earlier described ELISA has been validated for cell lysates and the antibody used for detection of HNE-protein adducts is non-commercial, the aim of this work was to adapt the ELISA to a commercial antibody and to apply it in the analysis of human plasma samples.
After modification and validation of the protocol for both antibodies, samples of two groups were analyzed: apparently healthy obese (n=62) and non-obese controls (n=15). Although the detected absolute values of HNE–protein adducts were different, depending on the antibody used, both ELISA methods showed significantly higher values of HNE–protein adducts in the obese group.
Abbreviations: ACR, Acrolein; BSA, Bovine serum albumin; c-Ab, Commercial antibody; Cys, Cysteine; DEPC, Diethyl pyrocarbonate; ELISA, Enzyme-linked immunosorbent assay; HCl, Hydrochloric acid; His, Histidine; HNE, 4-Hydroxy-trans-2-nonenal; HPLC, High performance liquid chromatography; HRP, Horseradish peroxidase; KLH, Keyhole limpet hemocyanin; LOD, limit of detection; LOQ, Limit of quantification; Lys, Lysine; MDA, Malondialdehyde; nc-Ab, Non-commercial antibody; PQL, Practical quantitation limit; PUFA, Polyunsaturated fatty acid; ROS, Reactive oxygen species
Keywords: HNE, MDA, Oxidative stress, Lipid peroxidation, ELISA, Antibodies, Human plasma, Obesity
Graphical abstract
Highlights
► ELISA allows determination of HNE–protein adducts in plasma and serum. ► Comparison: non-commercial (nc-Ab) vs. commercial antibody (c-Ab). ► nc-Ab shows higher specificity for HNE–Histidine adducts. ► c-Ab is not specific for HNE–Histidine adducts. ► HNE–protein adducts are elevated in obese patients in comparison to normal weight controls.
Introduction
In recent decades evidence has arisen that not only genetic but also epigenetic, mostly stress-related, mechanisms are involved in pathophysiology of aging and age-associated disorders. Among these mechanisms are non-enzymatic post-translational protein modifications which may alter structural and biological properties of proteins in living organisms. These alterations might be recognized as key events in certain diseases like degenerative diseases associated with protein storage. Such modifications of proteins are often influenced by their environment, their structural features, by protein folding states, as well as by free radicals generated in their vicinity. Abundant generation of reactive oxygen species (ROS) causes oxidative stress, which represents an imbalance between production of ROS and their elimination through antioxidative defense mechanisms [1]. ROS can attack all cellular macromolecules of which lipids are particularly susceptible. Since cholesterol esters, phospholipids, and triglycerides all contain polyunsaturated fatty acids (PUFAs) they are subject to free radical attack. Lipid peroxidation is marked by the breakdown of PUFAs yielding oxidation products such as reactive aldehydes of 3–9 carbons length of which 4-hydroxynonenal (HNE), malondialdehyde (MDA) and acrolein (ACR) are the most studied ones. These bioactive compounds can react with all major biomolecules of the cell, thus changing their structure and function and consequently influencing cellular physiology and pathophysiology.
Compared to free radicals, aldehydic products such as MDA and HNE are relatively stable and are able to roam freely and attack molecules, e.g. DNA, proteins, lipids far from the site of their origin. Amongst others, these aldehydic fragments may modify proteins and alter protein function, but are also considered as cytotoxic second messengers of oxidative stress which makes them highly utilized biomarkers in biological research [2,3]. Therefore, developing new methods for detection and quantification of these compounds in all kinds of biological samples, as well as improving the current ones, is of the utmost importance in the field of research related to oxidative stress.
4-Hydroxynonenal (HNE), a well known biomarker of oxidative stress and lipid peroxidation, is derived from ω-6 PUFAs such as arachidonic acid and linoleic acid and has been recognized as important molecule in pathology, as well as in physiology of living organisms [3–5]. Intracellular HNE reacts rapidly with thiol groups of glutathione and cysteine, with ε-amino groups of lysine, and with histidine residues of proteins [6,7]. Increased concentrations of HNE–modified proteins have been detected in various diseases, among them cancer, atherosclerosis, neurodegenerative disorders, ischemia, inflammation, diabetes, autoimmune diseases, bone diseases and aging [8–13].
As already mentioned, HNE can bind to proteins, forming relatively stable adducts, which are considered as the most likely form of its occurrence in living systems. We have previously reported that these HNE–protein adducts can be measured and quantified by an ELISA method using a mouse monoclonal antibody that specifically recognizes the HNE-histidine epitope [14]. Taking into consideration that ELISA methods require only micrograms of protein, they are suitable for clinical trials where only small amounts of material is available. Because it is becoming of increasing interest to evaluate the level of HNE-protein adducts in different human pathologies as well as in healthy human subjects, our aim was to establish a method that can be widely applied. Up to now, the only reliable and convenient method was the ELISA established for cell lysates by Borovic et al. [14]. To allow other researchers that are not able to use the non-commercial antibody to measure protein adducts of HNE by the ELISA used there, we selected one of the few available monoclonal antibodies against adducts of HNE. By comparing these two antibodies, it allows to compare results obtained with the commercial antibody in the future with those obtained with the non-commercial antibody.
On the other hand, the existing HNE–protein adduct ELISA was originally developed and validated for cell lysates and since the primary antibody used is non-commercial, we wanted to accomplish two goals: (a) to validate and if necessary modify the current protocol to be applicable for testing plasma/serum samples and (b) to compare the original non-commercial antibody (nc-Ab) with a commercial antibody (c-Ab), thus developing an ELISA method that differs only in the primary antibody used and that could be applied in all research laboratories related to lipid peroxidation and oxidative stress research. The here presented method is applicable to serum and plasma samples, when using a non-commercial as well as a commercially available antibody.
Materials and methods
Chemicals of best analytical grade available were purchased from Sigma-Aldrich (Taufkirchen, Germany) or Carl Roth (Karlsruhe, Germany), unless otherwise stated. Distilled water was used throughout for preparation of all solutions.
Primary antibodies used throughout were: commercial Ab (c-Ab): mouse monoclonal anti-4-Hydroxynonenal antibody (clone HNEJ-2; Abcam, Cambridge, UK); and non-commercial Ab (nc-Ab): genuine monoclonal mouse anti-HNE-His antibody in cell culture supernatant (clone HNE 1g4; produced and provided by Dr. Georg Waeg, Karl-Franzen's University in Graz, Institute of Molecular Biosciences, Austria). Secondary antibody was EnVision+ System-HRP Labeled Polymer anti-mouse (Dako, Denmark).
HNE–BSA standard preparation
HNE-dimethylacetal (Enzo Life Sciences) solution was dried under nitrogen flow and dissolved in 1 mM HCl at room temperature (RT) overnight. The concentration of this solution was determined by measuring the absorbance at 223 nm (ε=13,750 M−1 cm−1 [15]) with water as blank (Shimadzu, UV160-A). Afterwards, the solution was diluted to 1 mM with water.
For preparation of HNE–BSA standards, fatty acid free BSA was diluted in PBS to a protein concentration of 10 mg/ml. The zero standards consisted of fatty acid free BSA (no HNE). The highest standard concentration was prepared by adding different amounts of 1 mM HNE to fatty acid free BSA giving concentrations of 250 pmol/mg (ELISA with nc-Ab) and 5000 pmol/mg (ELISA with c-Ab). Prepared stock solutions were incubated for 3 h at 37 °C or overnight at 4 °C, to achieve complete binding of HNE to BSA, and stored at −20 °C until analysis.
ELISA procedure
This ELISA protocol was adapted from Borovic et al. [14]. Standards were prepared from stock solutions (stored at −20 °C) in concentrations ranging from 0 to 250 pmol/mg (ELISA with nc-Ab) or from 0 to 5000 pmol/mg (ELISA with c-Ab), respectively, by mixing varying proportions of HNE-BSA and fatty acid free BSA while the total protein concentration remained 10 mg/ml. Standards and diluted plasma/serum samples (10 mg/ml), respectively, were diluted 10-fold in 0.05 M carbonate binding buffer (pH 9.6; 0.015 M sodium carbonate, 0.035 M sodium bicarbonate). 100 μl of this mixture were transferred into wells of an ELISA plate (Nunc Immuno Maxisorp, Thermo Scientific) for triplicate analysis. Proteins were adsorbed to wells of the plate overnight at 4 °C. The following day, the plate was washed once with PBS (300 μl). Freshly prepared blocking solution (5% fat free dry milk in carbonate binding buffer) was transferred into wells and the plate was blocked for 2.5 h at RT followed by one washing step (0.1% Tween 20 in PBS). Primary antibody solution in 1% BSA in PBS (nc-Ab 1:100; c-Ab 1:500) was incubated for 2 h at 37 °C. To eliminate sample background values one well of each sample was incubated with 1% BSA in PBS (without primary antibody). After washing the plate seven times, it was incubated for 30 min with peroxidase blocking solution (3% H2O2 in PBS) at RT. Then it was again washed seven times. 100 μl of the goat anti-mouse secondary antibody solution in 1% BSA in PBS (1:100; Dako) were transferred into the wells and incubated for 1 h at RT, followed by a washing step (seven times). Freshly prepared TMB substrate solution (0.05 mg/ml) was transferred into the wells and after 30 min at 37 °C the reaction was stopped by adding 50 μl of stopping solution (2 M sulfuric acid). Absorption was read at 450 nm with the reference filter set to 620 nm. All analyses were performed in triplicate or quadruplicate and the amounts of HNE–protein adducts measured by the ELISA are expressed as pmol HNE/mg of proteins.
Analysis of samples
Sixty-two serum samples of morbidly obese candidates for surgical intervention were obtained from the Department of Surgery, University Hospital Mannheim (Germany) on the day before laparoscopic Roux-en-Y gastric bypass surgery. Oxidative stress markers HNE and MDA were compared with those in Na-heparin plasma obtained from 15 healthy controls which were kind gifts from Dr. Ina Bergheim from the University of Hohenheim (Germany). The study was approved by the Ethics Committees of the Institutions and informed consent was obtained from all participants. Blood samples were collected into appropriate tubes and serum or plasma, respectively, was separated from cells. Samples were stored at −80 °C until shipment and analysis, respectively.
Sample preparation
Total protein concentration of standards and plasma/serum, respectively was determined by the BioRad assay according to Bradford [16].
HNE-ELISA
Plasma and serum samples where diluted to a protein concentration of 10 mg/ml with PBS for ELISA analysis. HNE-protein adducts were analyzed by the ELISA as described above.
HPLC-analysis of malondialdehyde (MDA)
Malondialdehyde (MDA) in serum of obese patients and plasma of normal weight controls was determined by HPLC-analysis after derivatization with thiobarbituric acid (TBA) as describe by Wong et al. [17]. Quantification is done based on the area of the peaks compared to standards and expressed in μM.
Evaluation of differences observed in ELISA sensitivity and specificity
Capping experiment
BSA was treated with diethyl pyrocarbonate (DEPC) to block the amino acid Histidine. Histidine derivatization: 0.1% DEPC, a histidine-specific alkylating reagent was added to BSA (fatty acid free; 10 mg/ml) resuspended in 0.1 M sodium phosphate (pH 6.5) for 10 min at RT. The efficiency of histidine modification following DEPC treatment was proved by a shift in absorbance at 242 nm [18].
Dot Blots of capping experiments
Previously prepared samples (10 mg/ml) were diluted 20-fold and 100 μl were spotted onto nitrocellulose membrane (Amersham). The membrane was incubated in blocking solution (2% nonfat milk powder in PBS) for 1 h at RT and subsequently incubated overnight with primary antibodies (nc-Ab and c-Ab) diluted 1:100. After washing, the blots were incubated with 3% H2O2 to block endogenous peroxidases. Following another washing step, they were incubated with the secondary antibody for 1 h. Immune complexes were visualized using 3, 3′-diaminobenzidine tetrahydrochloride (Dako) staining and scanned for quantification of signals. Blanks were included in all experiments in which the antibody was omitted and replaced by control diluent (1% BSA in PBS).
Immunoblot analysis (western blot)
Prior to detection of nc-Ab and c-Ab binding, respectively, to HNE-protein adducts in HNE-treated BSA standards (120, 1250 pmol/mg) and human plasma, the protein concentration was measured with the Bradford assay. Samples were prepared with Laemmli buffer (v/v) and heated at 95 °C for 10 min. Twenty (for nc-Ab) or 30 μg (for c-Ab) of protein per lane in Laemmli buffer were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE, 10%). After electrophoresis the separated proteins were transferred to PVDF membrane. Membranes were blocked in 2% nonfat milk powder/TBS-Tween (50 mM Tris-base, pH 7.5, 200 mM sodium chloride, 0.05% Tween 20) overnight at 4 °C under constant agitation, followed by incubation with primary antibodies (nc-Ab: 1:100; c-Ab: 1:250) for 2 h at RT. After being washed three times, the blots were incubated with secondary antibody (1:100) for 2 h at RT. Blots were washed and exposed to the substrate ECL Reagent (Calbiochem). Visualization of bound secondary antibody was detected by enhanced chemiluminescence.
Statistical analysis
Basic descriptive statistics means, standard deviation (S.D.) were used. Means were compared using Student's t-test.
Results
Comparison of ELISA methods
Both methods showed linear standard curves in the range in which serum and plasma samples were expected (Fig.1).
Fig. 1.
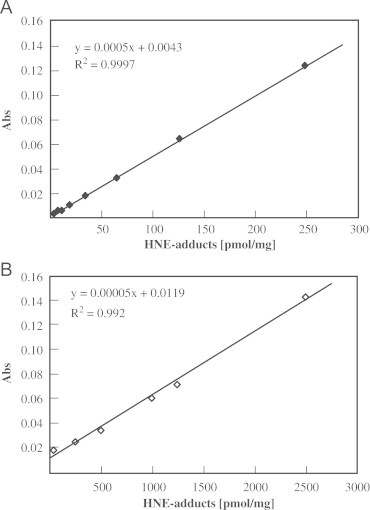
ELISA standard curves for both ELISA methods. (A) Typical ELISA standard curve using non-commercial antibody (nc-Ab). Standards were measured in quadruplicate in concentrations ranging from 0 to 250 pmol/mg. (B) Typical ELISA standard curve using commercial antibody (c-Ab). Standards were measured in quadruplicate in concentrations ranging from 0 to 2500 pmol/mg. Both curves were repeated multiple times.
Recovery
For determining the recovery of HNE-modified BSA with the nc-Ab, plasma and serum samples were spiked with standards by adding one volume of standard to one volume of sample, resulting in additional concentrations of 250, 62.5, and 7.8 pmol/mg, respectively. The mean recovery for the nc-Ab was measured to be between 100.0±3.3% (250 pmol/mg), 96.5±14.4% (62.5 pmol/mg) and 99.2±4.5% (7.8 pmol/mg). Mean recovery was 98.6±6.1% (Fig. 2). The recovery of the ELISA applying the c-Ab was determined by spiking samples with standard by adding one volume of standard to nine volumes of sample, resulting in additional concentrations of 250, 100, and 0 pmol/mg, respectively. The mean recovery was calculated to be between 108.6±18.5% (250 pmol/mg) and 98.1±17.4% (100 pmol/mg). Mean recovery was 103.3±18.4% (Fig. 2).
Fig. 2.
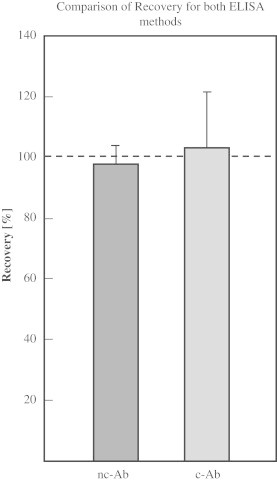
Comparison of recovery for both ELISA methods. Recovery of HNE–BSA standards in plasma samples was determined for both ELISA methods (c-Ab and nc-Ab). Plasma and serum samples were spiked with different concentrations of HNE–BSA and analyzed in triplicate. Recovery is expressed in percent. Columns are mean±S.D, n=3.
Limit of detection (LOD), practical quantitation limit (PQL), and limit of quantification (LOQ)
Limit of detection (LOD), practical quantitation limit (PQL), and limit of quantification (LOQ) were calculated as described previously [19,20]. The LOD for the nc-Ab is 9.7±2.5 pmol/mg, the PQL is 17.2±5.0 pmol/mg, and the LOQ is 24.6±7.4 pmol/mg. For the c-Ab, the limit of detection (LOD) is 405.4±48.0 pmol/mg, the practical quantitation limit (PQL) is 657.2±86.0 pmol/mg, and the limit of quantification (LOQ) is 909.0±157.6 pmol/mg (Fig.3).
Fig. 3.
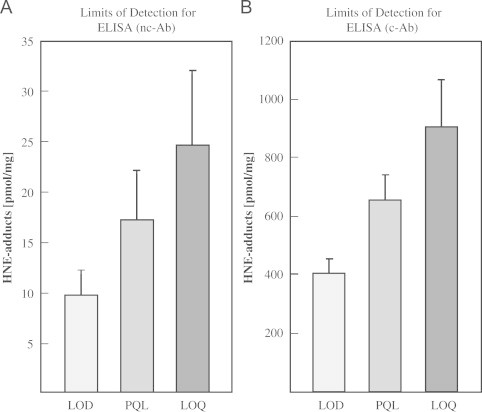
Limits of detection for both ELISA methods. Concentration limits for HNE-ELISA. Shown are the limits of detection (LOD), the practical quantitation limit (PQL), and the limit of quantification (LOQ) for the nc-AB (A) and c-Ab (B). Data represent mean±S.D, n=3.
Inter-day variance and inter-assay variance
As shown in Table 1, lower inter-day and inter-assay variations were calculated for the ELISA with nc-Ab compared to the method with the c-Ab, respectively. Regardless of the antibody used for either variance the variation was in the range between 13% and 22%.
Table 1.
Inter-day and inter-assay variation.
| Inter-day variation (%) | Inter-assay variation (%) | |
|---|---|---|
| nc-Ab | 13.6 | 13.8 |
| c-Ab | 22.6 | 16.6 |
HNE-protein adducts in samples of obese patients and normal weight controls
Since both ELISA tests worked satisfactorily using HNE-modified BSA standards, human blood was employed to perform an analysis of HNE-modified proteins. HNE-protein adduct concentrations in obese patient (serum, n=62) and normal weight control (plasma, n=15) samples were measured by both ELISA methods. Fig. 4 shows that obese patients have significantly elevated levels of HNE–protein adducts (nc-Ab: p<0.001; c-Ab: p<0.05).
Fig. 4.
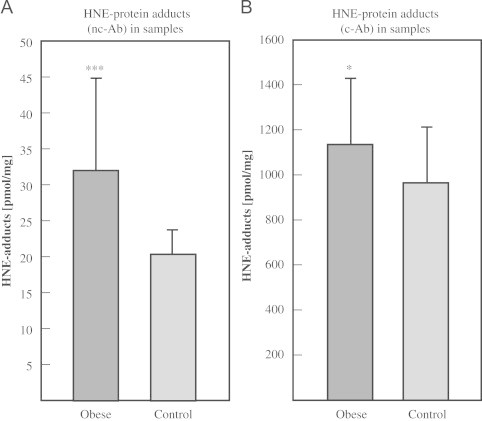
HNE–protein adducts in samples. Concentration of HNE–protein adducts in samples of obese patients (n=62) and normal weight controls (n=15). (A) ELISA using nc-Ab was applied to 62 serum samples of obese persons and 15 plasma samples of non-obese subjects. Obese subjects show significantly higher levels of HNE–protein adducts (p<0.001). (B) ELISA using c-Ab was applied to 62 serum samples of obese and 15 plasma samples of non-obese subjects. Obese subjects show significantly higher levels of HNE–protein adducts (p<0.05). Columns represent means±S.D. of triplicate measurement.
Correlation of both ELISA methods
Fig. 5 demonstrates the correlation between the measured HNE-protein adduct concentrations detected by both ELISA methods (r=0.305, p<0.01) using the 77 samples of obese (n=62) and normal weight persons (n=15) (Fig. 5).
Fig. 5.
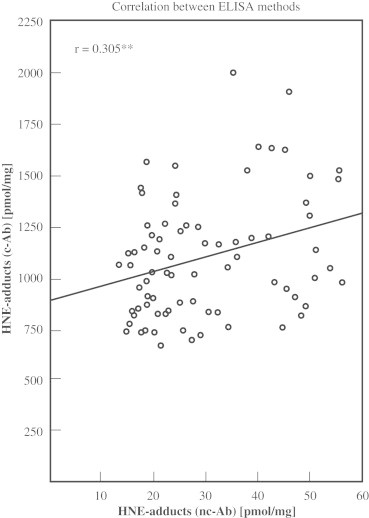
Correlation between ELISA methods. Concentrations measured in 77 samples with the ELISA applying c-Ab or nc-Ab, respectively, correlated significantly (r=0.305, p<0.01).
Correlation with MDA
HNE–protein adduct concentrations of obese and control samples determined by each of the ELISA methods were plotted against MDA concentrations of the same samples. The HNE-protein adducts in plasma/serum determined using the nc-Ab correlate highly significantly with MDA (r=0.779, p<0.001), HNE–protein adducts in plasma/serum determined using the c-Ab also correlate highly significantly with MDA (r=0.456, p<0.001, see Fig. 6).
Fig. 6.
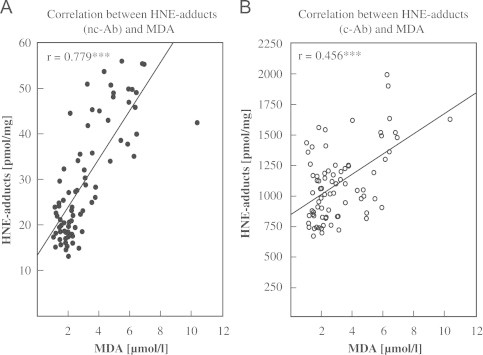
Correlation between HNE–protein adducts and MDA. HNE–protein adducts and malondialdehyde (MDA) was measured in 77 samples of obese patients (n=62, serum) and normal weight controls (n=15, plasma). (A) HNE–protein adducts measured with the nc-Ab were significantly correlated with MDA (r=0.779, p<0.001). (B) HNE–protein adducts measured with the c-Ab were significantly correlated with MDA (r=0.456, p<0.001).
Dot blot
Interestingly, both ELISAs show essentially the same results in correlation with the independent parameter MDA. It is remarkable that the absolute detected levels differ approximately some 40-fold. This is most likely due to the specificity of the antibody. Therefore, the specificity of antibody binding to HNE–His epitopes was tested. Since it is known that HNE binds to Cys, His, and Lys, and as both antibodies are declared to be specific for HNE-His epitope, we chemically blocked His and tested – after HNE-treatment – whether the antibodies recognize the modified protein to the same extent.
Dot blot analysis using the nc-Ab confirmed that His capping reduced the signal to 11.7%, whereas the binding of the c-Ab was reduced only to 90.9% compared to HNE-BSA without capping (Fig. 7). The negative control, BSA without HNE added, was negative for both antibodies, indicating no or very low signal could be detected when HNE-modified BSA is not present.
Fig. 7.
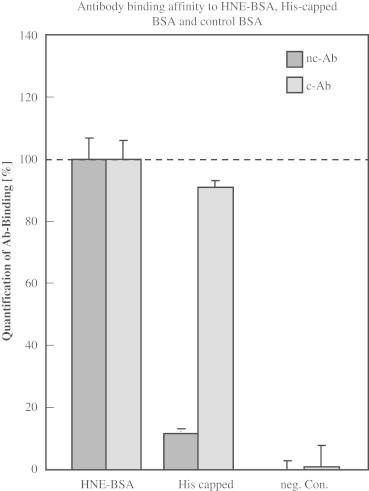
Dot Blot to determine binding affinity of antibodies to capped HNE–BSA. Dot Blot was carried out as described in the Materials and Methods section. Antibody binding to HNE–BSA (5000 pmol/mg) was set to 100%. Histidine side chains were capped on BSA using DEPC which was then treated with 5000 pmol/mg HNE (His capped). 50 μg of samples were loaded onto membranes. BSA without HNE-treatment was loaded as negative control (neg. Con.).
Western blot analysis of HNE–BSA standards and plasma sample
We decided to further test the sensitivity of both antibodies in immunoblots. While both primary antibodies detect the same band in human plasma samples (molecular weight of approximately 50 kDa; Fig. 8B), differences in their sensitivity towards HNE-BSA standards were observed (Fig. 8). Band signal intensity of standards obtained with the nc-Ab, was many times higher than for the c-Ab (Fig. 8A).
Fig. 8.
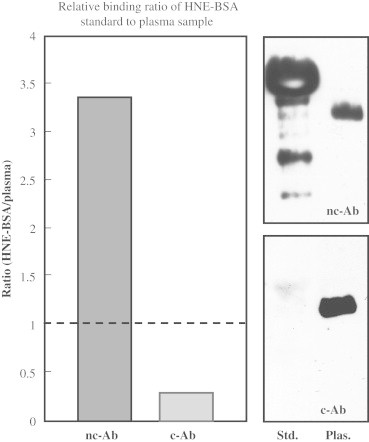
Western Blot of HNE–BSA standards and plasma. Quantitative analysis of Western Blots of HNE-protein adducts in HNE–BSA standards and plasma (120 pmol/mg with nc-Ab; 1250 pmol/mg with c-Ab) and a representative Western Blot analysis of HNE–protein adducts. Plasma sample was diluted to 10 mg/ml and 20 μg of protein (standards and plasma) were loaded onto gels.
Discussion
The present work compares two antibodies against HNE–protein adducts in an ELISA method to determine protein-bound HNE in human plasma and serum. The performance of both methods was evaluated for linearity, recovery, inter-day and inter-assay precision, and limit of detection. Regardless which antibody was used (nc-Ab and/or c-Ab), this method has shown great linearity (R²>0.99), in each and repeated measurements. Recovery for both antibodies, even on its limit of detection was 99.2±4.5% for nc-Ab and 98.1±17.4% for c-Ab, giving the overall recovery of 98.6±6.1% for nc-Ab and 103.3±18.4% for c-Ab, respectively. Inter-day and especially inter-assay precision, expressed as coefficient of variance (CV) does not exceed 22%, for both tested antibodies, which are acceptable criteria for ELISA with biological samples. Therefore, the main conclusion of this validation is that the method is working for both antibodies used and it allows the measurement of HNE-protein adducts in human plasma/serum.
Furthermore, the results obtained with each antibody in 77 samples (62 obese serum samples and plasma samples of 15 controls) show significant positive correlation with MDA, another lipid peroxidation marker. Correlation between both ELISAs (nc-Ab vs. c-Ab) was also positive and significant. The results from the different methods demonstrate the same tendency, which is a significant increase of HNE–protein adducts in the obese group. The correlation between obesity, oxidative stress and lipid peroxidation has already been well established. One characteristic feature of obesity is low-grade chronic inflammation of the adipose tissue with its concomitant oxidative stress [21]. Authors have reported increased oxidative stress and lipid peroxidation (MDA levels) are related to fat accumulation and BMI [22–24]. Many studies have demonstrated some role of HNE in the pathophysiology of obesity [25–28]. Since our results are in accordance with previous findings, we conclude that both methods are working but surprisingly, differences in quantification range of HNE–protein adducts in plasma/serum between both ELISAs were observed (nc-Ab:low; c-Ab:high).
When analyzing human blood samples it is important to take into account the specimen, the kind of sample, e.g. serum, EDTA-plasma, heparin or citrate plasma. The main difference between serum and plasma lies in clotting factors which are still present in plasma due to the addition of anticoagulants like EDTA, heparin or citrate. In serum, these clotting factors are consumed and therefore missing. For clinical trials, making sure that the specimen within the cohort is of the same kind is essential.
However, in a recent review, Giustarini et al. have demonstrated the effect of blood anticoagulation on MDA levels analyzed by GC–MS/MS in a group of 18 healthy young female volunteers [29]. Blood was taken using EDTA, lithium heparin, or serum monovettes. Concentrations of MDA in serum and heparin plasma were in the same range. Furthermore, a positive significant correlation (correlation coefficient 0.96) for MDA between serum and heparin plasma samples was reported (n=14) [30]. For reasons of sample availability, we compared serum samples with heparinized plasma samples of controls. Samples were diluted to the same protein concentration (10 mg/ml) to generally compare the amount of HNE bound to total proteins and to overcome the effect of clotting factors present/missing.
Both used antibodies are stated to be specific for the HNE-His epitope, however this seems to be unlikely given the different absolute levels. Therefore, experiments where BSA was treated in order to cap His prior to addition of HNE were undertaken. Thus prepared samples were further analyzed by dot blot. The dot blots confirm that the nc-Ab preferentially, and the c-Ab only partly recognizes the HNE-His epitope.
However, differences were observed when His was capped. While nc-Ab exhibits only slight binding (11.7±1.6%), results obtained with c-Ab display rather high binding (90.9±2.2%) compared to binding towards non-capped BSA, giving evidence that this latter antibody is not specific to the HNE-His epitope but may also recognize HNE bound to Cys and Lys, and possibly even other amino acid residues like tyrosine, serine, arginine, proline, proposing one of possible explanations [14,31,32].
The antigenic sites recognized by the two antibodies on modified albumin are apparently markedly different. However, both antibodies were produced with the use of keyhole limpet hemocyanin (KLH) modified by HNE and there were small differences in the ratio of HNE:immunogen used (2000 nmol/mg HNE-KLH for nc-Ab [33] and 1740 nmol/mg HNE-KLH for c-Ab [34]), therefore neither the degree of modification of the immunogen itself nor the antigene used can be the reason for the difference seen in affinity and sensitivity of antibodies.
Furthermore, we tested whether there were any qualitative differences between the two antibodies in the nature of plasma or serum proteins detected. Both antibodies specifically recognize preferentially one band in HNE-BSA standards and one in plasma and serum, as seen in Western Blots. This supports our intention to use BSA as a model protein, because albumin is one of the most abundant plasma proteins and is modified by HNE.
Because of the different results obtained, we suggest using the terms HNE-His-ELISA for the nc-Ab, and HNE-protein adduct ELISA for the c-Ab, respectively.
In the present work we show elevated levels of HNE–protein adducts and MDA in morbidly obese subjects for each of the applied primary antibodies, showing excellent correlation between the MDA levels and the HNE-His-ELISA, thus confirming the data obtained on human UV irradiated plasma and complementarities of the two methods (the MDA HPLC and the HNE-His-ELISA) [35].
As we were able to show, the non-commercial anti-HNE antibody appears to recognize only histidine epitopes, while the c-Ab recognized various amino acid residues. This fact likely explains why the detection of HNE adducts with the commercial Ab leads to quantitative values of about 40-fold higher than those achieved when using the non-commercial Ab and showed less statistical power (results reported in Fig. 4). Furthermore, besides the net difference in specificity, the two antibodies showed a quite different sensitivity as well (data reported in Fig. 8). As evidently shown, the nc-Ab is more specific and also more sensitive than the c-Ab. Both antibodies definitely do not measure the same epitopes, clearly indicating the requirement of testing the epitopes before using the antibody. We postulate that the c-Ab also measures other HNE adducts than those to histidine. This accounts for the 40-fold difference in absolute concentrations.
In conclusion, oxidative stress or more precisely the level of oxidative damage to lipids and consequently proteins, as measured by free MDA and by HNE–protein adducts, was significantly increased in obese humans if compared to healthy controls. We are certain that the presented method is suitable for clinical application to evaluate lipid peroxidation in human serum or plasma. Since this ELISA is not time-consuming, it has a high sample throughput and only essential routine equipment is needed. Therefore, it is an appropriate alternative as well as complementary method to the HNE-HPLC, which is not only time-consuming, but also requires costly equipment.
Acknowledgments
We thank Dr. Till Hasenberg, Department of Surgery, University Medical Centre Mannheim, Medical Faculty Mannheim of the University of Heidelberg, Mannheim, Germany for providing serum samples of obese patients and Dr. Ina Bergheim, Department of Nutritional Medicine (180a), University of Hohenheim, Stuttgart, Germany for providing plasma samples of normal weight controls.
We thank Dr. Georg Waeg, Institute of Molecular Biosciences, Karl-Franzen's University Graz, Austria for providing the non-commercial antibody.
This work was supported by the European Cooperation in the field of Scientific and Technical Research (COST) Action CM1001 (to D. Weber and L. Milkovic).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Halliwell B., Gutteridge J.M.C. Claredon Press; Oxford: 1989. The Chemistry of Oxygen Radicals and Other Oxygen-Derived Species. In: Free Radicals in Biology and Medicine. pp. 29–32. [Google Scholar]
- 2.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Progress in Lipid Research. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 3.Zarkovic N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Molecular Aspects of Medicine. 2003;24:281–291. doi: 10.1016/s0098-2997(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 4.Dianzani M.U. 4-hydroxynonenal from pathology to physiology. Molecular Aspects of Medicine. 2003;24:263–272. doi: 10.1016/s0098-2997(03)00021-9. [DOI] [PubMed] [Google Scholar]
- 5.Mrakovcic L., Wildburger R., Jaganjac M., Cindric M., Cipak A., Borovic-Sunjic S., Waeg G., Milankovic A.M., Zarkovic N. Lipid peroxidation product 4-hydroxynonenal as factor of oxidative homeostasis supporting bone regeneration with bioactive glasses. Acta Biochimica Polonica. 2010;57:173–178. [PubMed] [Google Scholar]
- 6.Uchida K., Szweda L.I., Chae H.Z., Stadtman E.R. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proceedings of the National Academy of Science USA. 1993;90:8742–8746. doi: 10.1073/pnas.90.18.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poli G., Schaur R.J. 4-Hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life. 2000;50:315–321. doi: 10.1080/713803726. [DOI] [PubMed] [Google Scholar]
- 8.Skrzydlewska E., Sulkowski S., Koda M., Zalewski B., Kanczuga-Koda L., Sulkowska M. Lipid peroxidation and antioxidant status in colorectal cancer. World Journal of Gastroenterology. 2005;11:403–406. doi: 10.3748/wjg.v11.i3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonarduzzi G., Chiarpotto E., Biasi F., Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Molecular Nutrition & Food Research. 2005;49:1044–1049. doi: 10.1002/mnfr.200500090. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt H., Grune T., Muller R., Siems W.G., Wauer R.R. Increased levels of lipid peroxidation products malondialdehyde and 4-hydroxynonenal after perinatal hypoxia. Pediatric Research. 1996;40:15–20. doi: 10.1203/00006450-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Podborska M., Sevcikova A., Trna J., Dite P., Lojek A., Kubala L. Increased markers of oxidative stress in plasma of patients with chronic pancreatitis. Neuroendocrinology Letters. 2009;30(Suppl 1):116–120. [PubMed] [Google Scholar]
- 12.Gil L., Siems W., Mazurek B., Gross J., Schroeder P., Voss P., Grune T. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radical Research. 2006;40:495–505. doi: 10.1080/10715760600592962. [DOI] [PubMed] [Google Scholar]
- 13.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Molecular Aspects of Medicine. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 14.Borovic S., Rabuzin F., Waeg G., Zarkovic N. Enzyme-linked immunosorbent assay for 4-hydroxynonenal-histidine conjugates. Free Radical Research. 2006;40:809–820. doi: 10.1080/10715760600693422. [DOI] [PubMed] [Google Scholar]
- 15.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology & Medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 16.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 17.Wong S.H., Knight J.A., Hopfer S.M., Zaharia O., Leach C.N., Jr., Sunderman F.W., Jr. Lipoperoxides in plasma as measured by liquid-chromatographic separation of malondialdehyde-thiobarbituric acid adduct. Clinical Chemistry. 1987;33:214–220. [PubMed] [Google Scholar]
- 18.Muhlrad A., Hegyi G., Horanyi M. Studies on the properties of chemically modified actin. 3. Carbethoxylation. Biochimica Biophysica Acta. 1969;181:184–190. doi: 10.1016/0005-2795(69)90240-2. [DOI] [PubMed] [Google Scholar]
- 19.Weber D., Kneschke N., Grimm S., Bergheim I., Breusing N., Grune T. Rapid and sensitive determination of protein-nitrotyrosine by ELISA: Application to human plasma. Free Radical Research. 2012;46:276–285. doi: 10.3109/10715762.2011.652627. [DOI] [PubMed] [Google Scholar]
- 20.L.R. Ellison, M. Rousslein, A. Williams, EURACHEM/CITAC Guide. Quantifying Uncertanity in Analytical Measurement, 2000.
- 21.Wellen K.E., Hotamisligil G.S. Obesity-induced inflammatory changes in adipose tissue. The Journal of Clinical Investigation. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furukawa S., Fujita T., Shimabukuro M., Iwaki M., Yamada Y., Nakajima Y., Nakayama O., Makishima M., Matsuda M., Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. The Journal of Clinical Investigation. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olusi S.O. Obesity is an independent risk factor for plasma lipid peroxidation and depletion of erythrocyte cytoprotectic enzymes in humans. International Journal of Obesity and Related Metabolic Disorders. 2002;26:1159–1164. doi: 10.1038/sj.ijo.0802066. [DOI] [PubMed] [Google Scholar]
- 24.Prazny M., Skrha J., Hilgertova J. Plasma malondialdehyde and obesity: is there a relationship? Clinical Chemistry and Laboratory Medicine. 1999;37:1129–1130. doi: 10.1515/CCLM.1999.164. [DOI] [PubMed] [Google Scholar]
- 25.Devries M.C., Hamadeh M.J., Glover A.W., Raha S., Samjoo I.A., Tarnopolsky M.A. Endurance training without weight loss lowers systemic, but not muscle, oxidative stress with no effect on inflammation in lean and obese women. Free Radical Biology and Medicine. 2008;45:503–511. doi: 10.1016/j.freeradbiomed.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 26.Singh S.P., Niemczyk M., Saini D., Awasthi Y.C., Zimniak L., Zimniak P. Role of the electrophilic lipid peroxidation product 4-hydroxynonenal in the development and maintenance of obesity in mice. Biochemistry. 2008;47:3900–3911. doi: 10.1021/bi702124u. [DOI] [PubMed] [Google Scholar]
- 27.Grimsrud P.A., Picklo M.J., Sr., Griffin T.J., Bernlohr D.A. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Molecular and Cellular Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Singh S.P., Niemczyk M., Zimniak L., Zimniak P. Fat accumulation in Caenorhabditis elegans triggered by the electrophilic lipid peroxidation product 4-hydroxynonenal (4-HNE) Aging (Albany NY) 2009;1:68–80. doi: 10.18632/aging.100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giustarini D., Dalle-Donne I., Tsikas D., Rossi R. Oxidative stress and human diseases: origin, link, measurement, mechanisms, and biomarkers. Critical Reviews in Clinical Laboratory Science. 2009;46:241–281. doi: 10.3109/10408360903142326. [DOI] [PubMed] [Google Scholar]
- 30.Steghens J.P., van Kappel A.L., Denis I.C. Diaminonaphtalene a new highly specific reagent for HPLC-UV measurement of total and free malondialdehyde in human plasma or serum. Free Radical Biology & Medicine. 2001;31:242–249. doi: 10.1016/s0891-5849(01)00578-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhao J., Chen J., Zhu H., Xiong Y.L. Mass spectrometric evidence of malonaldehyde and 4-hydroxynonenal adductions to radical-scavenging soy peptides. Journal of Agriculture and Food Chemistry. 2012;60:9727–9736. doi: 10.1021/jf3026277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurgens G., Lang J., Esterbauer H. Modification of human low-density lipoprotein by the lipid peroxidation product 4-hydroxynonenal. Biochimica Biophysica Acta. 1986;875:103–114. doi: 10.1016/0005-2760(86)90016-0. [DOI] [PubMed] [Google Scholar]
- 33.Waeg G., Dimsity G., Esterbauer H. Monoclonal antibodies for detection of 4-hydroxynonenal modified proteins. Free Radical Research. 1996;25:149–159. doi: 10.3109/10715769609149920. [DOI] [PubMed] [Google Scholar]
- 34.Toyokuni S., Miyake N., Hiai H., Hagiwara M., Kawakishi S., Osawa T., Uchida K. The monoclonal antibody specific for the 4-hydroxy-2-nonenal histidine adduct. FEBS Letters. 1995;359:189–191. doi: 10.1016/0014-5793(95)00033-6. [DOI] [PubMed] [Google Scholar]
- 35.Breusing N., Grune T., Andrisic L., Atalay M., Bartosz G., Biasi F., Borovic S., Bravo L., Casals I., Casillas R., Dinischiotu A., Drzewinska J., Faber H., Fauzi N.M., Gajewska A., Gambini J., Gradinaru D., Kokkola T., Lojek A., Luczaj W., Margina D., Mascia C., Mateos R., Meinitzer A., Mitjavila M.T., Mrakovcic L., Munteanu M.C., Podborska M., Poli G., Sicinska P., Skrzydlewska E., Vina J., Wiswedel I., Zarkovic N., Zelzer S., Spickett C.M. An inter-laboratory validation of methods of lipid peroxidation measurement in UVA-treated human plasma samples. Free Radical Research. 2010;44:1203–1215. doi: 10.3109/10715762.2010.499907. [DOI] [PubMed] [Google Scholar]