

Abstract
In the newborn, alveolarization continues postnatally and can be disrupted by hyperoxia, leading to long-lasting consequences on lung function. We wanted to better understand the role of heme oxygenase (HO)-1, the inducible form of the rate-limiting enzyme in heme degradation, in neonatal hyperoxic lung injury and repair. Although it was not observed after 3 days of hyperoxia alone, when exposed to hyperoxia and allowed to recover in air (O2/air recovered), neonatal HO-1 knockout (KO) mice had enlarged alveolar spaces and increased lung apoptosis as well as decreased lung protein translation and dysregulated gene expression in the recovery phase of the injury. Associated with these changes, KO had sustained low levels of active β-catenin and lesser lung nuclear heterogeneous nuclear ribonucleoprotein K (hnRNPK) protein levels, whereas lung nuclear hnRNPK was increased in transgenic mice over-expressing nuclear HO-1. Disruption of HO-1 may enhance hnRNPK-mediated inhibition of protein translation and subsequently impair the β-catenin/hnRNPK regulated gene expression required for coordinated lung repair and regeneration.
Abbreviations: HO-1, heme oxygenase-1; KO, knockout; MEF, mouse embryonic fibroblasts; RAC, radial alveolar counts; O2/air, exposed to 95% oxygen for 3 days then recovered in air; hnRNPK, heterogeneous nuclear ribonucleoprotein protein K; SP-B, surfactant protein B; SP-C, surfactant protein C; OGG1, 8-oxoguanine DNA glycosylase
Keywords: Heme oxygenase-1, Neonatal hyperoxic lung injury and repair, β-catenin/hnRNPK, DNA damage and repair, Cell proliferation
Graphical abstract
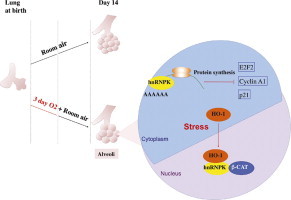
Highlights
► HO-1KO has worsened lung structure after hyperoxia and in room air recovery. ► Protein synthesis is inhibited and cell cycle gene expression is dysregulated in the KO. ► In the WT neonatal lung, HO-1 protein localizes to the nucleus in hyperoxia. ► HO-1 interacts with hnRNPK in vitro and in vivo. ► This modulates protein synthesis and gene expression explaining lung abnormalities.
Introduction
Due to limited oxygen-diffusing capacity, preterm infants may require supplemental oxygen during a period of continued postnatal lung development. Hyperoxia, along with mechanical ventilation, increases the risk for alveolar simplification and abnormal vascularization referred to as bronchopulmonary dysplasia (BPD) [2,15]. This disease poses a significant public health problem and is a leading cause of neonatal morbidity and mortality [30]. Well beyond the neonatal period, infants with BPD have impaired lung function [18]. In the neonatal rodent, hyperoxia results in lung injury resembling BPD [40] with long-term consequences on lung function [24,45] despite neonates being more resistant to hyperoxia than adults [12]. Because alveolarization continues postnatally, injury during this crucial period could lead to aberrant lung repair [3] and long-lasting consequences.
The neonatal lung is unique in that it is transitioning from a low oxygen environment in utero to a relatively oxygen rich environment at birth and there is a perinatal upregulation of many antioxidant genes [12] including heme oxygense-1 (HO-1), the rate limiting enzyme in bilirubin production [9]. Lung HO-1 mRNA levels subsequently decrease in the first weeks of life to reach adult values [9].
Many have implicated HO-1 in cytoprotection during oxidative stress [25,33] and have shown that HO-1 induction is a generalized response to oxidative stress [1]. Nevertheless, we have clearly shown that neonatal rodents exposed to hyperoxia do not upregulate HO-1 mRNA [10,16], nor do young adult HO-1 null mutants have increased susceptibility to a 3-day hyperoxic exposure compared to WT [10]. Moreover the HO-1 KO did not show loss of lung antioxidant capacity as compared to WT controls at baseline and did not have increased lung oxidative markers after hyperoxic exposure [9]. Interestingly, despite this relative tolerance to hyperoxia [10], HO-1 KO mice have alveolar simplification and reduced secondary crest formation as neonates [48]. Although some have reported that the HO-1 enzymatic by-products mediate its cytoprotection [17,34,47], even devoid of its enzymatic activity HO-1 has significant effects against oxidative stress in vitro [14,20]. HO-1 has also been shown to migrate to the nucleus during hyperoxia [19], and it also binds to other proteins [42]. Perhaps this could provide a signaling mechanism to modulate cytoprotective functions in the neonatal lung but it is not yet clear whether this occurs in vivo.
During development and with lung injury and repair, active Wnt signaling causes β-catenin to be hypo-phosphorylated, resulting in its stabilization with targeted downstream gene expression [26,11]. This modulates terminal differentiation of post-mitotic cells including alveolar Type II cells (ATII) [11], and therefore would significantly impact recovery from lung injury. It is not known whether HO-1 could modulate this pathway either directly or indirectly.
We have preliminarily shown that HO-1 can bind to hnRNPK [[19] and Table S1], an RNA binding protein with wide-ranging effects on cell cycle [5]. The pleiotropic hnRNPK protein is comprised of three conserved KH (K homology) nucleic acid binding domains and a kinase interacting region facilitating cross-talk between factors involved in regulating gene expression [5]. In addition, hnRNPK can interact with β-catenin to alter downstream gene expression [5]. It also can interact with the 3′UTR region of various mRNAs to regulate protein translation [13] as well as bind to other regions on mRNA [28]. Therefore, its modulation could have global effects on cellular function both at the gene and protein levels.
In this study, when neonatal HO-1 KO mice were exposed to hyperoxia and allowed to recover in room air (O2/air recovered), their lungs showed simplification, increased apoptosis and dysregulation of global protein translation and gene expression associated with changes in β-catenin and hnRNPK.
Methods
Animal model
HO-1 KO on a C57Bl/6 background (KO) and their WT littermates, as well as transgenic mice over-expressing HA-tagged nuclear HO-1 driven by the SP-C promoter (TR), were housed at Children's Hospital Research Institute Animal Laboratory Facility.
All protocols were reviewed and approved by the Children's Hospital Institutional Animal Care And Use Committee in accordance with the Animal Welfare Act of the NIH.
Hyperoxic exposure
Newborn mice (<12 h old) or adult mice (2 months old) were exposed to room air (RA) or hyperoxia for 72 h. In some experiments, neonatal mice were allowed to recover in RA for up to 14 days [44,48] after the hyperoxic exposure.
Isolation of nuclear proteins from lung tissue
Lung nuclear and cytosolic extracts from the neonatal and adult mice were obtained using previously described methods [19].
HO activity measurements
Total HO activity in the lung nuclear or cytosolic fractions was assessed by measuring CO levels using gas chromatograph as described previously [9]. Relative activity was calculated as the concentration of CO per HO-1 protein densitometry unit based on Western blots from the same lung.
Cell lines and infections
The KO and WT embryonic fibroblasts were generated from E13.5 embryos. Constructs of FLAG tagged full length (FL) and truncated HO-1 cDNA lacking the coding region for the 53 amino acids from the C-terminus (TR) were stably infected in the KO MEF cells using the p3xFLAG-CMV™-14 plasmid vector system (Sigma, Cat # E4901).
Hyperoxic exposure in vitro
MEF cells at 80% confluence were exposed to hyperoxia (95% O2 and 5% CO2) or air (5% CO2) for 18 h as previously described [19].
Lung tissue collection and evaluation of lung architecture and alveolarization
This was done as described in Refs. [8,44].
Immunohistochemical detection of protein signal
Immunoreactive protein signals were detected on 5 μm paraffin embedded tissue sections using the Tyramide Signal Amplification system (PerkinElmer, Waltham, MA) as described in [44]. Primary antibodies used were 1:100 β-catenin (#9582, Cell Signaling, Danvers, MA) and hnRNPk (#4675, Cell Signaling), 1:1000 HA (#MMS-101P, Covance, Princeton, NJ). Sections were visualized and images were captured as described [44].
Determination of PCNA positive cells on lung slides
These were performed as previously described [44].
Preparation of lung homogenate
The frozen tissue was homogenized in PBS with protease and phosphatase inhibitors as described [44].
Immunoprecipitation
Immunoprecipitated FLAG tagged HO-1 was washed and purified by an affinity competition with excess FLAG peptide (Sigma, Cat # F4799). The enriched supernatant was isolated and analyzed for detection of HO-1 and proteins co-immunoprecipitated by Western blot. Sample inputs (one tenth of the immunoprecipitation) were simultaneously loaded.
Western blot analysis
The cell lysates or lung homogenates were resolved by SDS-PAGE for the proteins as described [20] and probed using antibodies against active (#4270) and total β-catenin (#9582); Cyclin A1 (#4656); eIF2α total (#9722) and eIF2α (pSer-51; #9721) (all from Cell Signaling); E2F2 (sc-9967); p21 (sc-397 (both from Santa Cruz) and calnexin (Enzo Life Science SPA-860) as loading control. Image density of each band was measured using the Java-based Image J software program (rsbweb.nih.gov/ij).
RNA extraction, quantitative real-time PCR (qRT-PCR), and RT2-PCR arrays
Total RNA was extracted from lung tissue as described previously [44]. The effect of hyperoxia and recovery on 84 cell cycle regulated genes was evaluated using an RT2-PCR array (PAMM020, SABiosciences). Three lungs in each group were pooled and total RNA was extracted. PCR amplification was performed with ABI Prism SDS 7500 (Applied Biosystems). The Ct values on each of the 84 genes were obtained using SDS v1 software. The ΔCt and fold changes of each gene between groups were generated using the excel-based PCR array data analysis template provided. Because hyperoxia alters 4 of the manufacturer recommended housekeeping genes (Hprt1, Hsp90ab1, Gapdh and Actb), we chose to use Gusb for normalization since it was comparable in all 8 conditions. A 2-fold-change cut off was used to generate scatter plots for each comparison.
Statistical analysis
For comparison between treatment groups, the null hypothesis that there is no difference between treatment means was tested by a single factor analysis of variance (ANOVA) for multiple groups or unpaired t-test for two groups after testing for normal distribution (InStat 3, GraphPad Software). Statistical significance was accepted to be p<0.05. Comparison between and within groups was determined by Tukey's method of multiple comparisons. Each pup was considered as an n number throughout the experiments.
Results
Lung HO-1 distributes to the nucleus after hyperoxia in neonatal mice
In the neonates, in contrast to adults, lung nuclear fractions had enhanced immunoreactive HO-1 after hyperoxic exposure (Fig. 1). Total lung HO activity was then determined in the nuclear and cytoplasmic extracts. We verified that HO-2, the constitutive form of HO, did not change in any of the samples and therefore did not influence total HO activity (data not shown). Total HO activity was normalized to the densitometry values of HO-1 for each sample. HO activity per HO-1 protein was negligible in the neonatal lung nuclear extracts whereas neonatal lung cytoplasmic extracts had detectable HO activity per HO-1 protein (180 pmole CO/hr/HO-1 protein units) at 16 h of hyperoxic exposure. In contrast, the hyperoxia-exposed adults had a significantly higher lung cytoplasmic HO activity per HO-1 protein at this same time point (450 pmole CO/hr/HO-1 protein units). Overall these data illustrate that despite the lack of induction of lung HO-1 mRNA, lung HO-1 protein is redistributed after hyperoxic exposure in the newborns only and that the relative contribution of HO activity in the newborn is less than in adults.
Fig. 1.
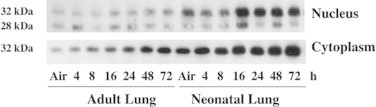
Distribution of lung HO-1 protein in adults and neonates after hyperoxia. Representative Western blot of immunoreactive HO-1 in cytoplasmic and nuclear fractions of lung homogenates exposed to >95% O2 for 0–72 h, controls were exposed to room air. Equal loading was verified with Ponceau Red.
HO-1 disruption alters lung developmental response to acute hyperoxic exposure in the neonates
As shown previously [48], neonatal HO-1 KO mice had mild alveolar disruption at birth. This was not visibly altered by 3 days of hyperoxia (results not shown). Radial alveolar counts (RAC), as indices of alveolarization, were significantly decreased in the HO-1 KO maintained in air as compared to WT (7.0±0.2 vs 8.2±0.3; p<0.05). However, there was no further decrease in RAC after 3 days of hyperoxia (7.0±0.2 vs 6.6±0.3; NS) in contrast to WT littermates (8.2±0.3 vs 6.8±0.2; p<0.05). This suggests that HO-1 participates in lung alveolar development and may mediate the lung's response to hyperoxic injury. Because hyperoxia targets alveolar Type II cell proliferation specific to the neonatal lung [46], we wanted to understand whether the differential response to hyperoxia could result from altered alveolar Type II cells. Therefore surfactant proteins B and C (SP-B and SP-C) were measured. There were no differences in these type cell markers in either WT or KO (data not shown). We next screened the group of genes related to cell proliferation and cell cycle in lung homogenate to understand whether HO-1 disruption had global changes in cell proliferation in the lung. Using an array assembly containing 86 genes, the expression levels in the WT and KO lungs were measured with and without hyperoxia, and the results compared (Fig. 2A and B). Few genes were upregulated, and these consisted mainly of DNA damage response genes. Likewise, few genes were downregulated, and these consisted mainly of genes involved in cell cycle regulation (Table S2). Of the genes affected p21, p53 and Ki-67 have been previously documented as modulated in the neonatal lung after hyperoxic exposure [23,31,37]. Although the KO response to hyperoxia was similar to the WT (Fig. 2C), five genes did not respond to hyperoxia in the KO, indicating that signal pathways regulating these genes may have been altered (Fig. 2C).
Fig. 2.
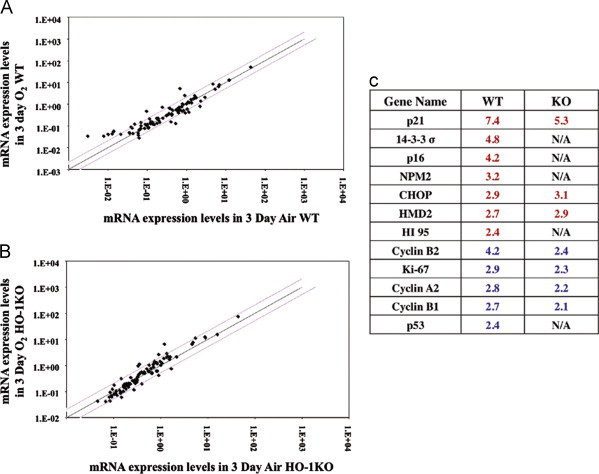
Steady state lung mRNA levels in the neonatal mice lung exposed to 3-day hyperoxia. (A) comparison of WT in 3 days hyperoxia (O2) exposure vs 3 days air (air) and, in (B) a similar comparison was made between O2 and air in the KO. Each dot represents one mRNA expression in cell cycle regulation. Red lines represent 2-fold over- or under-expression and black line represents equal expression. Three lung mRNAs were pooled for each gene expression profiling. In (C) comparison of fold changes in mRNA levels are shown between WT and KO in relative to their air controls. Red number indicates upregulation and blue number indicates downregulation (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
O2/air recovered KO mice have disrupted lung architecture
When the KO mice were allowed to recover in room air for 11 days (O2/air recovered), they had increased air spaces compared to similarly exposed WT controls (Fig. 3A). The RAC remained reduced in the 14 day air-exposed KO, but this was even more pronounced in the O2/air recovered KO vs similarly exposed WT (Fig. 3B).
Fig. 3.
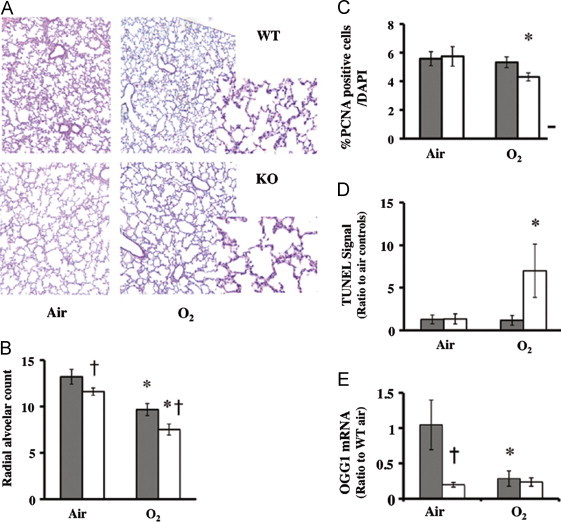
Disrupted lung architecture in O2/air recovered KO mice. (A) representative hematoxylin and eosin staining of lung slices from wild type (WT) or HO-1 knockout (KO) mice are shown at 20× magnification. Inset: higher magnification (40×). Shown in (B) radial alveolar counts, in (C) average of %PCNA positive cells over DAPI counted per 10 high powered fields, (D) number of TUNEL positive cells/hpf and in E, mRNA levels of OGG1 relative to the WT air. Grey bars: WT. Open bars: KO. Values are the mean±S.E. of five experiments in each group. *: p<0.05 vs air with the same strain. †: p<0.05 vs WT O2.
Disruption of HO-1 alters lung cell proliferation and DNA repair
Although there were no significant changes in SP-B and SP-C steady-state mRNA levels after 3 days of hyperoxic exposure, these were decreased in both WT and KO in the O2/air recovered animals, but there were no differences between the two groups (data not shown). This suggests that HO-1 does not specifically affect epithelial cell susceptibility to hyperoxic injury. When lung slides were screened for PCNA signal as an index of overall cell proliferation, the KO showed significant decrease in the number of PCNA positive cells after recovery (Fig. 3C). There was also an increase in TUNEL positive cells in the O2/air recovered KO compared to all other groups (Fig. 3D), indicating increased cell death and/or apoptosis. In addition, mRNA levels of 8-oxoguanine DNA glycosylase (OGG1), an important enzyme in oxidative DNA damage repair, was significantly reduced in the HO-1KO in air as compared to WT and remained low in the O2/air recovery (Fig. 3E). Furthermore, the O2/air recovered WT had downregulated lung cell cycle gene expression (Fig. 4A and Supplemental Table 3), whereas the KO had drastic up-regulation of 47 genes and down-regulation of 21 genes, suggesting global dysregulation of cell cycle and DNA repair gene expression (Fig. 4B and Table S3). Among the genes that differed in the WT compared to KO, six DNA damage-response genes were down regulated in the WT, whereas these were upregulated manyfold in the KO (Fig. 4C). This observation is consistent with increased cell death in the O2/air recovery KO (Fig. 3D). Overall these data demonstrate that HO-1 disruption exacerbates lung DNA damage and impairs the repair process during recovery from hyperoxia.
Fig. 4.
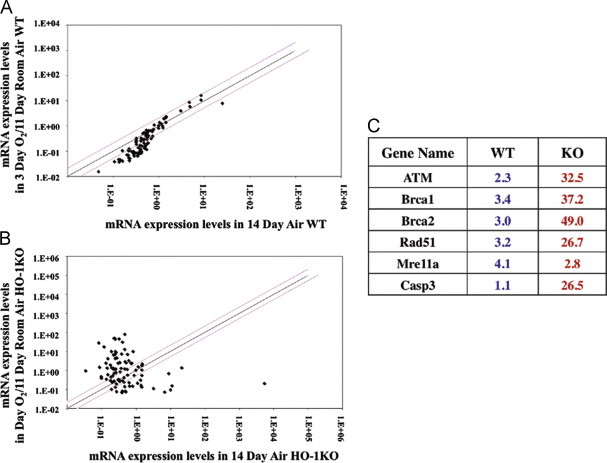
Steady state lung mRNA levels in O2/air recovered mice. In (A) comparison of WT in 3 days hyperoxia plus 11 days air recovery (O2) vs 14 days air (air) and in (B), comparison of KO in O2 vs air. Each dot represents one mRNA expression in cell cycle regulation. Red lines represent 2-fold over or under-expression and black line represents equal expression. Three lung mRNAs were pooled for each gene expression profiling. In (C) comparison of fold changes in mRNA levels are shown between WT and KO relative to their air controls. Red number indicates upregulation and blue number indicate downregulation (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Disruption of HO-1 modulates lung β-catenin signaling
To explain how HO-1 alters the response to hyperoxic injury and repair, we evaluated whether HO-1 could modulate lung β-catenin in our model since this protein is key to lung cell proliferation and differentiation during repair. In the KO lungs exposed to air, active β-catenin was significantly decreased vs WT, suggesting that disruption of HO-1 resulted in a reduced ability to proliferate. This reduced active β-catenin persisted after O2/air recovery but was also observed in the O2/air recovered WT, suggesting that hyperoxia itself also reduced the ability of the lung to repair (Fig. 5A).
Fig. 5.
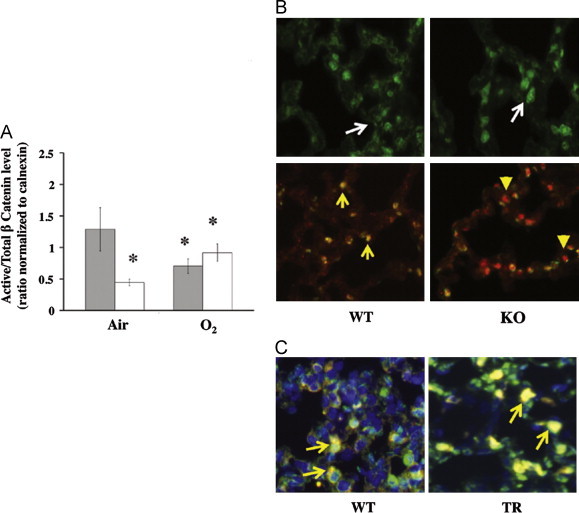
Effect of HO-1 on β-catenin cellular localization and activity. In (A) active β-catenin expressed as a ratio to the total in lung homogenates of O2/air recovered mice. Grey bars: WT, open bars: KO. Values are the mean±S.E. of 3 in each group. *: p<0.05 vs air; †: p<0.05 vs WT O2. (B) Representative immunohistochemical staining of β-catenin alone (Upper panel) or co-staining with hnRNPK (Lower panel) of WT and KO lungs is shown at 40x magnification. White arrows: cellular distributions of β-catenin; yellow arrows: co-localization of β-catenin (red) and hnRNPK (green) signals in the nucleus; yellow arrowheads: hnRNPK staining in the nucleus without β-catenin co-localization. In (C) representative lung slices from WT and transgenic mice over-expressing the nuclear form of HO-1 driven by an SP-C promoter were co-stained for HA for HO-1 transgene (red), hnRNPK (green) and DAPI (blue). Yellow arrows: co-localization of the two signals in the WT (overall distribution) and in the transgenic mice (predominantly in the nucleus) (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
To understand whether HO-1 might affect β-catenin localization and activities, we detected β-catenin immunoreactive signals in the WT and KO lung tissues. In corroborating with the Western analysis, lesser cells were stained positive for β-catenin in the KO lung as compared to the WT and these cells were clustered in only few selected areas of the lung (Fig. S1). However when these areas were viewed with higher magnification the β-catenin protein showed predominant localization in the nucleus (Fig. 5B upper panel).
Disruption of HO-1 dysregulates β-catenin/hnRNPK signaling
An association between β-catenin and heterogeneous nuclear ribonucleoprotein proteins (hnRNPs) has been described [6]. Having previously shown that hnRNPK protein was recovered (Table S1) when HO-1 was immunoprecipitated after hypoxia [19], and given that hnRNPK would interact with β-catenin [5,6], we hypothesized that HO-1 modulates β-catenin signaling through its interaction with hnRNPK. To this extent, we co-stained lung slices with hnRNPK and β-catenin. After O2/air recovery, the signals co-localized to the nucleus in the WT, but in the absence of HO-1 the hnRNPK signal was predominantly cytoplasmic (Fig. 5B lower panel), making it less likely that the two proteins would maintain their normal interaction in the absence of HO-1. To test whether HO-1 directly binds to β-catenin we immunoprecipitated HO-1 protein from HO-1KO MEF cells over expressing FLAG tagged-full length or truncated HO-1 (Fig. 6B). This verified that the full length HO-1 was exclusively found in the cytoplasm and the truncated HO-1 was exclusively found in the nucleus (Fig. S2A). When these constructs were immunoprecipitated with FLAG, β-catenin failed to co-immunoprecipate HO-1(Fig. S2B) obviating a direct HO-1/β-catenin–protein interaction.
Fig. 6.
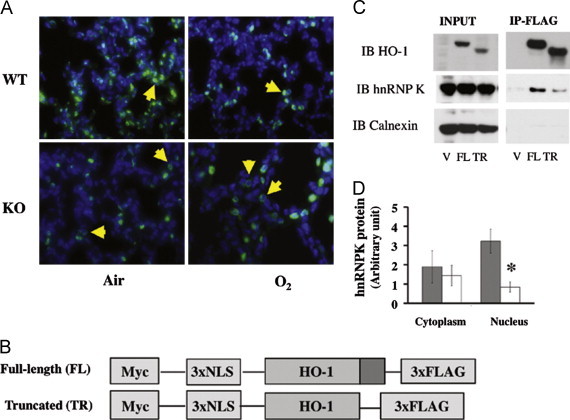
Effect of HO-1 on hnRNPK protein cellular distribution. (A) Representative hnRNPK immunohistochemical staining of WT and KO lungs exposed to 3 days of hyperoxia and recovered in air for 11 days is shown at 40x magnification. Yellow arrowheads indicate cellular distributions of hnRNPK. (B) Schematic representation of HO-1 constructs used in a tissue culture model. The full-length (FL) construct contains HO-1 cDNA whereas the truncated construct (TR) lacks the coding region for the terminal 53 amino acids, allowing the protein to migrate to the nucleus. There is a Myc tag at the 3′ end and 3 nuclear localization sequences (NLS) as well as a FLAG tag at the 5′ end. A construct with the empty vector (V) was used as control. (C) verification of HO-1/hnRNPK interaction in the cell lines over-expressing full length or truncated HO-1 by immunoprecipitation. (D) Comparison of cytoplasmic and nuclear hnRNPK protein levels in WT and HO-1KO MEF cells. Grey bars: WT; white bars: HO-1KO. N=3 experiments; *: p<0.05 vs WT nuclear extracts (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
We have previously documented that HO-1 can migrate to the nuclear compartment under oxidative stress [19]. To understand whether the nuclear portion of the HO-1 would retain the hnRNPK in the nucleus, we used a transgenic mouse model over-expressing nuclear HO-1 specifically in the lung. Indeed, lung nuclear hnRNPK was enhanced in these O2/air recovered transgenic mice (Fig. 5C) further suggesting that HO-1 is key to hnRNPK localization in vivo.
To further understand whether disruption of HO-1 would exclude hnRNPK from the nucleus, we stained lung slides from the O2/air recovered mice with anti-hnRNPK antibodies. Disruption of HO-1 significantly reduced the abundance of lung nuclear hnRNPK protein in the lung (Fig. 6A).
The interaction of HO-1 with hnRNPK in hyperoxia was further verified using immunoprecipitation in a cell model system consisting of KO MEF cells rescued with full length or nuclear HO-1 cDNA constructs (Fig. 6B). In fact, HO-1 was co-immunoprecipitated with hnRNPK (Fig. 6C) in both full length and truncated cDNA infected HO-1 KO MEF cells. When WT and KO MEF cells were fractioned into cytosolic and nuclear fractions, decreased nuclear hnRNPK signal was observed only in the KO MEFs, further supporting the hypothesis that HO-1 is key to retaining hnRNPK in the nucleus (Fig. 6D). Altogether, these data suggest that the nuclear form of HO-1 binds hnRNPK and retains this protein in the nuclear compartment.
Suppression of lung protein translation after hyperoxia is associated with altered β-catenin/hnRNP signaling
The β-catenin and hnRNP proteins work in concert to modify gene expression important to cellular proliferation and DNA repair [36]. Perhaps this could help explain the drastic alteration in cell proliferation and cell cycle genes observed in the O2/air recovered KO (Fig. 3B). We choose to evaluate several genes, in particular, E2F2 mRNA (upregulated 320-fold), Cyclin A1 mRNA (133-fold) and p21 (3 fold), as potential targets of β-catenin/hnRNPK signaling. Despite a 320-fold increase in E2F2 mRNA, protein levels were decreased in the O2/air recovered KO compared to air controls (Fig. 7A). Additionally, despite a 133-fold upregulation of cyclin A1 mRNA, cyclin A1 protein levels did not increase in O2/air recovered KO vs air controls (Fig. 7B). Furthermore, although p21 protein levels were higher in the KO than the WT at baseline, despite a 3-fold increase in p21 mRNA in the O2/air recovered KO, there was no further increase in p21 protein levels (Fig. 7C). Overall these observations support the idea that disruption of HO-1 in the lung led to an exaggerated increase in the mRNA of various genes involved in proliferation but this did not translate to changes in protein levels, suggesting a global suppression of protein expression in the HO-1 KO lung. Lastly, lung eIF2α phosphorylation was increased at baseline in the KO vs WT, and remained higher after O2/air recovery (Fig. 7D). This is a global index of suppressed protein translation [39,41]. As further evidence, eIF2α phosphorylation was enhanced in KO MEF cells vs WT both in air and hyperoxia (Fig. 7E).
Fig. 7.
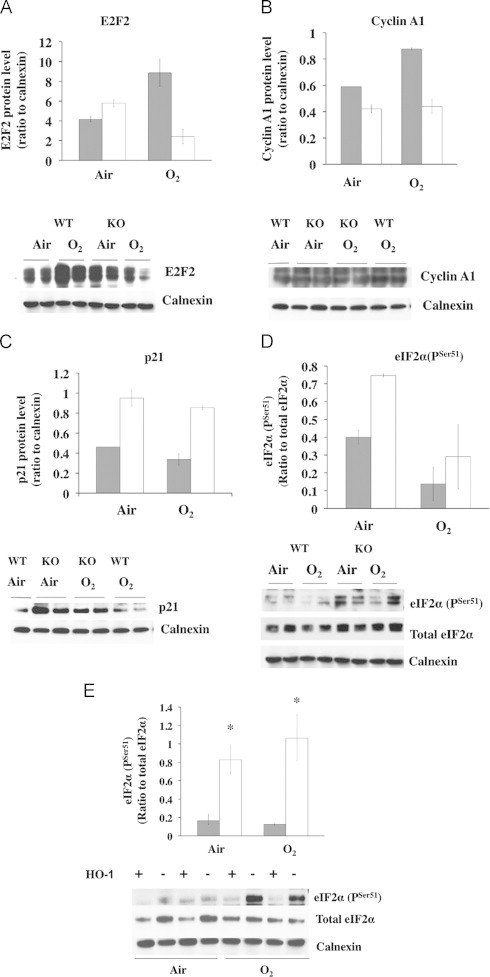
Lung protein content in O2/air recovered mice. In (A) lung E2F2, (B) cyclin A1, (C) p21, and in (D) phosphorylated eIF2α. Upper panels: densitometry values of WT (grey bars) and KO (open bars) normalized to Calnexin loading controls. Values are the mean±S.E. of 2. Lower panels: representative Western blots of each signal with calnexin as loading control. For eIF2α, the ratio of serine 51 phosphorylated signal over total protein content is shown. In E: level of serine 51 phosphorylated eIF2α over total in KO and WT MEF. Grey bars: WT, open bars: KO. Values are the mean±S.E. of 3 in each group. *: p<0.05 vs WT. Lower panel: representative Western blot of serine 51 positive signal over total. Calnexin is the loading control.
Discussion
It is well known that HO-1 has antioxidant properties ascribed to its enzymatic function [38]. More recently, non-enzymatic roles have been documented [20,21], and HO-1 nuclear translocation has been characterized in several models [21,32,35]. Whether nuclear HO-1 protects neonatal lung against hyperoxic injury and enhances repair is not known. Here we show that HO-1 is important to lung repair after hyperoxic injury and that nuclear HO-1 may be implicated in this process. Although the HO-1 KO model could not fully distinguish between the effects of absent nuclear (non-enzymatic) vs cytosolic (enzymatic) HO-1, several lines of evidence suggest a specific role of nuclear HO-1 during the neonatal period:
(1) In the neonates, there is redistribution of HO-1 protein to the nucleus after exposure to hyperoxia (Fig. 1); (2) lung HO activity is dramatically reduced in the lung nuclear fractions and the activity of cytosolic fractions is smaller than in adults as shown in this study; (3) the lungs of transgenic mice with HO-1cDNA specifically targeted to the nucleus show improved radial alveolar counts in hyperoxia after 4 days recovery (unpublished observations, F. Namba, 2012).
One of the proteins that associates with HO-1 in the nucleus during oxidative stress is hnRNPk (Supplemental material). Given its pleiotropic role in RNA and DNA binding, we focused on how hnRNPk distributes within cellular compartments in the presence or absence of HO-1 and when the nuclear form of HO-1 is highly expressed. Because hnRNPK recognizes specific RNA binding motifs on the 3′UTR of mRNAs in the cytoplasm [13] and serves as an RNA binding adaptor protein regulating gene transcription, we suspect that, when HO-1 is disrupted, preferential cytoplasmic distribution of hnRNPK results in global suppression of protein translation.
Since neonatal mice do not complete alveolarization until after the first two postnatal weeks [29], acute hyperoxia could lead to long-lasting alterations in lung structure and function. Interestingly, despite only subtle changes in KO after 3 days of hyperoxia, there were drastic differences after air recovery. Thus, susceptibility of the KO to hyperoxia was unmasked in the recovery period, suggesting that acute oxidative stress perturbs the ability of the developing lung to repair.
During lung injury and subsequent repair, re-establishment of a functional alveolar epithelium requires cellular proliferation, transdifferentiation, and migration [3]. Wnt/β-catenin signaling modulates cellular fate in various cell types [7,26]. Targeted loss of β-catenin in SP-C-expressing cells perturbs distal lung morphogenesis, demonstrating its importance in the formation of alveolar structure [27]. In the recovering KO lung, secondary septation was disrupted and β-catenin activity was decreased. This was not due to a direct interaction with HO-1 protein. Nevertheless, we verified that HO-1 binds to hnRNPK in the lung as in previous studies [6]. Others have also shown that hnRNPK interacts with β-catenin to both positively and negatively regulate gene transcription [5]. Therefore, HO-1 could influence β-catenin through hnRNPK. With loss of nuclear hnRNPK in the O2/air recovered KO, the normal hnRNPK/β-catenin interaction is disrupted, as shown here, and dysregulation of genes involved in DNA repair, cell proliferation and apoptosis would logically occur. The massively dysregulated lung gene expression observed in the O2/air recovered KO suggests that HO-1 participates in gene regulation during lung repair through these pathways. This remains to be fully evaluated.
In addition, with HO-1 disruption, redistribution of hnRNPK from the nucleus to the cytoplasm could result in inhibition of protective responses since accumulation of hnRNPK in the cytoplasm promotes inhibition of RNA translation [13]. In corroboration with this hypothesis, we observed decreased lung E2F2 and cyclin A1 proteins, despite increased steady state mRNA as well as increased eIF2α phosphorylation. The consequence of decreased E2F2 protein translation as well as decreased cyclin A1 translation would restrict cellular proliferation [22,43]. This could explain the abnormal lung repair seen in the O2/air recovered KO. In contrast, the suppressed p21 protein translation we observed would likely promote cell proliferation [4]. This paradoxical observation is not yet understood and we did not investigate whether the changes in p21 were cell type-specific.
In summary, HO-1 modulates cellular proliferation and differentiation in part through interaction with, and altered cellular distribution of hnRNPK. This results in altered β-catenin/hnRNPK signaling and significant gene dysregulation. In addition, the redistribution of hnRNPK was associated with a global decrease in protein synthesis. These pleiotropic effects of HO-1 disruption may lead to aberrant repair of the postnatal lung after hyperoxic injury.
Acknowledgments
We thank Patrick Amal Fernando for his expert technical assistance. This work was funded by the Lassin Funds of the Children's Hospital of Philadelphia (PAD), Grant HL-058752 (PAD) and a Fellowship Training Grant from the American Lung Association (FN).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2013.01.013.
Appendix A. Supporting information
Supplementary data
References
- 1.Applegate L.A., Frenk E. Oxidative defense in cultured human skin fibroblasts and keratinocytes from sun-exposed and non-exposed skin. Photodermatology, Photoimmunology and Photomedicine. 1995;11:95–101. doi: 10.1111/j.1600-0781.1995.tb00146.x. [DOI] [PubMed] [Google Scholar]
- 2.Bancalari E., Claure N., Sosenko I.R. Bronchopulmonary dysplasia: changes in pathogenesis, epidemiology and definition. Seminars in Neonatology. 2003;8:63–71. doi: 10.1016/s1084-2756(02)00192-6. [DOI] [PubMed] [Google Scholar]
- 3.Beers M.F., Morrisey E.E. The three R's of lung health and disease: repair, remodeling, and regeneration. Journal of Clinical Investigation. 2011;121:2065–2073. doi: 10.1172/JCI45961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blagosklonny M.V. A node between proliferation, apoptosis, and growth arrest. Bioessays. 1999;21:704–709. doi: 10.1002/(SICI)1521-1878(199908)21:8<704::AID-BIES10>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Bomsztyk K., Denisenko O., Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26:629–638. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- 6.M. Ciarlo, R. Benelli, O. Barbieri, S. Minghelli, P. Barboro, C. Balbi, N. Ferrari, Regulation of neuroendocrine differentiation by AKT/hnRNPK/AR/beta-catenin signaling in prostate cancer cells. The International Journal of Cancer, 2011. [DOI] [PubMed]
- 7.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Cooney T.P., Thurlbeck W.M. The radial alveolar count method of Emery and Mithal: a reappraisal 2-intrauterine and early postnatal lung growth. Thorax. 1982;37:580–583. doi: 10.1136/thx.37.8.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dennery P.A., Lee C.S., Ford B.S., Weng Y.H., Yang G., Rodgers P.A. Developmental expression of heme oxygenase in the rat lung. Pediatric Research. 2003;53:42–47. doi: 10.1203/00006450-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Dennery P.A., Visner G., Weng Y.H., Nguyen X., Lu F., Zander D., Yang G. Resistance to hyperoxia with heme oxygenase-1 disruption: role of iron. Free Radical Biology and Medicine. 2003;34:124–133. doi: 10.1016/s0891-5849(02)01295-9. [DOI] [PubMed] [Google Scholar]
- 11.Flozak A.S., Lam A.P., Russell S., Jain M., Peled O.N., Sheppard K.A., Beri R., Mutlu G.M., Budinger G.R., Gottardi C.J. Beta-catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. Journal of Biological Chemistry. 2010;285:3157–3167. doi: 10.1074/jbc.M109.070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frank L. Developmental aspects of experimental pulmonary oxygen toxicity. Free Radical Biology and Medicine. 1991;11:463–494. doi: 10.1016/0891-5849(91)90062-8. [DOI] [PubMed] [Google Scholar]
- 13.Habelhah H., Shah K., Huang L., Ostareck-Lederer A., Burlingame A.L., Shokat K.M., Hentze M.W., Ronai Z. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nature Cell Biology. 2001;3:325–330. doi: 10.1038/35060131. [DOI] [PubMed] [Google Scholar]
- 14.Hori R., Kashiba M., Toma T., Yachie A., Goda N., Makino N., Soejima A., Nagasawa T., Nakabayashi K., Suematsu M. Gene transfection of H25A mutant heme oxygenase-1 protects cells against hydroperoxide-induced cytotoxicity. Journal of Biological Chemistry. 2002;277:10712–10718. doi: 10.1074/jbc.M107749200. [DOI] [PubMed] [Google Scholar]
- 15.Jobe A.H., Ikegami M. Prevention of bronchopulmonary dysplasia. Current Opinion in Pediatrics. 2001;13:124–129. doi: 10.1097/00008480-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Kassovska-Bratinova S., Yang G., Igarashi K., Dennery P.A. Bach1 modulates heme oxygenase-1 expression in the neonatal mouse lung. Pediatric Research. 2009;65:145–149. doi: 10.1203/PDR.0b013e318191eedc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirkby K.A., Adin C.A. Products of heme oxygenase and their potential therapeutic applications. American Journal of Physiology—Renal Physiology. 2006;290:F563–571. doi: 10.1152/ajprenal.00220.2005. [DOI] [PubMed] [Google Scholar]
- 18.Landry J.S., Chan T., Lands L., Menzies D. Long-term impact of bronchopulmonary dysplasia on pulmonary function. Canadian Respiratory Journal. 2011;18:265–270. doi: 10.1155/2011/547948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Q., Weis S., Yang G., Weng Y.H., Helston R., Rish K., Smith A., Bordner J., Polte T., Gaunitz F., Dennery P.A. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. Journal of Biological Chemistry. 2007;282:20621–20633. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- 20.Lin Q.S., Weis S., Yang G., Zhuang T., Abate A., Dennery P.A. Catalytic inactive heme oxygenase-1 protein regulates its own expression in oxidative stress. Free Radical Biology and Medicine. 2008;44:847–855. doi: 10.1016/j.freeradbiomed.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin Y., Vreman H.J., Wong R.J., Tjoa T., Yamauchi T., Noble-Haeusslein L.J. Heme oxygenase-1 stabilizes the blood-spinal cord barrier and limits oxidative stress and white matter damage in the acutely injured murine spinal cord. Journal of Cerebral Blood Flow & Metabolism. 2007;27:1010–1021. doi: 10.1038/sj.jcbfm.9600412. [DOI] [PubMed] [Google Scholar]
- 22.McClellan K.A., Slack R.S. Specific in vivo roles for E2Fs in differentiation and development. Cell Cycle. 2007;6:2917–2927. doi: 10.4161/cc.6.23.4997. [DOI] [PubMed] [Google Scholar]
- 23.McGrath-Morrow S.A., Cho C., Soutiere S., Mitzner W., Tuder R. The effect of neonatal hyperoxia on the lung of p21Waf1/Cip1/Sdi1-deficient mice. American Journal of Respiratory Cell and Molecular Biology. 2004;30:635–640. doi: 10.1165/rcmb.2003-0049OC. [DOI] [PubMed] [Google Scholar]
- 24.McGrath-Morrow S.A., Ryan T., McGinley B.M., Okelo S.O., Sterni L.M., Collaco J.M. Polysomnography in preterm infants and children with chronic lung disease. Pediatric Pulmonology. 2012;47:172–179. doi: 10.1002/ppul.21522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morse D., Lin L., Choi A.M., Ryter S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radical Biology and Medicine. 2009;47:1–12. doi: 10.1016/j.freeradbiomed.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mucenski M.L., Nation J.M., Thitoff A.R., Besnard V., Xu Y., Wert S.E., Harada N., Taketo M.M., Stahlman M.T., Whitsett J.A. Beta-catenin regulates differentiation of respiratory epithelial cells in vivo. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2005;289:L971–979. doi: 10.1152/ajplung.00172.2005. [DOI] [PubMed] [Google Scholar]
- 27.Mucenski M.L., Wert S.E., Nation J.M., Loudy D.E., Huelsken J., Birchmeier W., Morrisey E.E., Whitsett J.A. beta-Catenin is required for specification of proximal/distal cell fate during lung morphogenesis. Journal of Biological Chemistry. 2003;278:40231–40238. doi: 10.1074/jbc.M305892200. [DOI] [PubMed] [Google Scholar]
- 28.Mukhopadhyay N.K., Kim J., Cinar B., Ramachandran A., Hager M.H., Di Vizio D., Adam R.M., Rubin M.A., Raychaudhuri P., De Benedetti A., Freeman M.R. Heterogeneous nuclear ribonucleoprotein K is a novel regulator of androgen receptor translation. Cancer Research. 2009;69:2210–2218. doi: 10.1158/0008-5472.CAN-08-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mund S.I., Stampanoni M., Schittny J.C. Developmental alveolarization of the mouse lung. Developmental Dynamics. 2008;237:2108–2116. doi: 10.1002/dvdy.21633. [DOI] [PubMed] [Google Scholar]
- 30.G. Natarajan, A. Pappas, S. Shankaran, D.E. Kendrick, A. Das, R.D. Higgins, A.R. Laptook, E.F. Bell, B.J. Stoll, N. Newman, E.C. Hale, R. Bara, M.C. Walsh, Outcomes of extremely low birth weight infants with bronchopulmonary dysplasia: impact of the physiologic definition. Early Human Development, 2012. [DOI] [PMC free article] [PubMed]
- 31.O'Reilly M.A., Staversky R.J., Stripp B.R., Finkelstein J.N. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. American Journal of Respiratory Cell and Molecular Biology. 1998;18:43–50. doi: 10.1165/ajrcmb.18.1.2950m. [DOI] [PubMed] [Google Scholar]
- 32.Pandya H.C., Snetkov V.A., Twort C.H., Ward J.P., Hirst S.J. Oxygen regulates mitogen-stimulated proliferation of fetal human airway smooth muscle cells. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2002;283:L1220–1230. doi: 10.1152/ajplung.00268.2001. [DOI] [PubMed] [Google Scholar]
- 33.Ryter S.W., Choi A.M. Heme oxygenase-1/carbon monoxide: from metabolism to molecular therapy. American Journal of Respiratory Cell and Molecular Biology. 2009;41:251–260. doi: 10.1165/rcmb.2009-0170TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryter S.W., Kim H.P., Nakahira K., Zuckerbraun B.S., Morse D., Choi A.M. Protective functions of heme oxygenase-1 and carbon monoxide in the respiratory system. Antioxidants and Redox Signaling. 2007;9:2157–2173. doi: 10.1089/ars.2007.1811. [DOI] [PubMed] [Google Scholar]
- 35.Sacca P., Meiss R., Casas G., Mazza O., Calvo J.C., Navone N., Vazquez E. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. The British Journal of Cancer. 2007;97:1683–1689. doi: 10.1038/sj.bjc.6604081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato S., Idogawa M., Honda K., Fujii G., Kawashima H., Takekuma K., Hoshika A., Hirohashi S., Yamada T. beta-catenin interacts with the FUS proto-oncogene product and regulates pre-mRNA splicing. Gastroenterology. 2005;129:1225–1236. doi: 10.1053/j.gastro.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 37.Silverman J.S., Hamilton J., Tamsen A. Benign recurring lipoblastoma in an adult vs well differentiated subcutaneous myxoid liposarcoma: clinicopathologic, immunohistochemical and molecular analysis of a unique case. Pathology—Research and Practice. 1999;195:787–792. doi: 10.1016/S0344-0338(99)80122-8. discussion 793. [DOI] [PubMed] [Google Scholar]
- 38.Suttner D.M., Sridhar K., Lee C.S., Tomura T., Hansen T.N., Dennery P.A. Protective effects of transient HO-1 overexpression on susceptibility to oxygen toxicity in lung cells. American Journal of Physiology. 1999;276:L443–451. doi: 10.1152/ajplung.1999.276.3.L443. [DOI] [PubMed] [Google Scholar]
- 39.van den Beucken T., Koritzinsky M., Wouters B.G. Translational control of gene expression during hypoxia. Cancer Biology and Therapy. 2006;5:749–755. doi: 10.4161/cbt.5.7.2972. [DOI] [PubMed] [Google Scholar]
- 40.Warner B.B., Stuart L.A., Papes R.A., Wispe J.R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. American Journal of Physiology. 1998;275:L110–117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 41.Wek R.C., Cavener D.R. Translational control and the unfolded protein response. Antioxidants and Redox Signaling. 2007;9:2357–2371. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 42.Weng Y.H., Yang G., Weiss S., Dennery P.A. Interaction between heme oxygenase-1 and -2 proteins. Journal of Biological Chemistry. 2003;278:50999–51005. doi: 10.1074/jbc.M307644200. [DOI] [PubMed] [Google Scholar]
- 43.Wu Z., Zheng S., Yu Q. The E2F family and the role of E2F1 in apoptosis. The International Journal of Biochemistry & Cell Biology. 2009;41:2389–2397. doi: 10.1016/j.biocel.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Yang G., Hinson M.D., Bordner J.E., Lin Q.S., Fernando A.P., La P., Wright C.J., Dennery P.A. Silencing hyperoxia-induced C/EBPalpha in neonatal mice improves lung architecture via enhanced proliferation of alveolar epithelial cells. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2011;301:L187–196. doi: 10.1152/ajplung.00082.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yee M., Chess P.R., McGrath-Morrow S.A., Wang Z., Gelein R., Zhou R., Dean D.A., Notter R.H., O'Reilly M.A. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2009;297:L641–649. doi: 10.1152/ajplung.00023.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yee M., Vitiello P.F., Roper J.M., Staversky R.J., Wright T.W., McGrath-Morrow S.A., Maniscalco W.M., Finkelstein J.N., O'Reilly M.A. Type II epithelial cells are critical target for hyperoxia-mediated impairment of postnatal lung development. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2006;291:L1101–1111. doi: 10.1152/ajplung.00126.2006. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X., Shan P., Otterbein L.E., Alam J., Flavell R.A., Davis R.J., Choi A.M., Lee P.J. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. Journal of Biological Chemistry. 2003;278:1248–1258. doi: 10.1074/jbc.M208419200. [DOI] [PubMed] [Google Scholar]
- 48.Zhuang T., Zhang M., Zhang H., Dennery P.A., Lin Q.S. Disrupted postnatal lung development in heme oxygenase-1 deficient mice. Respiratory Research. 2010;11:142. doi: 10.1186/1465-9921-11-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data