

Abstract
Cigarette smoking remains a major health concern worldwide, and many of the adverse effects of cigarette smoke (CS) can be attributed to its abundant electrophilic aldehydes, such as acrolein (2-propenal). Previous studies indicate that acrolein readily reacts with thioredoxin reductase 1 (TrxR1), a critical enzyme involved in regulation of thioredoxin (Trx)-mediated redox signaling, by alkylation at its selenocysteine (Sec) residue. Because alkylation of Sec within TrxR1 has significant implications for its enzymatic function, we explored the potential importance of TrxR1 alkylation in acrolein-induced activation or injury of bronchial epithelial cells. Exposure of human bronchial epithelial HBE1 cells to acrolein (1–30 μM) resulted in dose-dependent loss of TrxR thioredoxin reductase activity, which coincided with its alkylation, as determined by biotin hydrazide labeling, and was independent of initial GSH status. To test the involvement of TrxR1 in acrolein responses in HBE1 cells, we suppressed TrxR1 using siRNA silencing or augmented TrxR1 by cell supplementation with sodium selenite. Acrolein exposure of HBE1 cells induced dose-dependent activation of the MAP kinases, extracellular regulated1 kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, and activation of JNK was markedly enhanced after selenite-mediated induction of TrxR1, and was associated with increased alkylation of TrxR1. Conversely, siRNA silencing of TrxR1 significantly suppressed the ability of acrolein to activate JNK, and also appeared to attenuate acrolein-dependent activation of ERK and p38. Alteration of initial TrxR1 levels by siRNA or selenite supplementation also affected initial Trx1 redox status and acrolein-mediated alkylation of Trx1, but did not significantly affect acrolein-mediated activation of HO-1 or cytotoxicity. Collectively, our findings indicate that alkylation of TrxR1 and/or Trx1 may contribute directly to acrolein-mediated activation of MAP kinases such as JNK, and may therefore be important in acrolein-induced alterations in airway epithelial function, as a contributing mechanism in tobacco-related respiratory disease.
Keywords: Cigarette smoke, Epithelium, Electrophile, Michael addition, c-Jun N-terminal kinase, GSH
Graphical abstract
Highlights
► Acrolein is an important thiol-reactive electrophile in cigarette smoke. ► Acrolein-induced alterations in redox systems in airway epithelial cells include inactivation of thioredoxin reductase (TrxR). ► Acrolein-induced alkylation of TrxR results in both loss and gain-of-function. ► Acrolein-dependent alkylation of TrxR and thioredoxin depends on TrxR expression. ► TrxR expression dictates the ability of acrolein to activate MAPK signaling pathways.
Introduction
Cigarette smoking is recognized to have profound human health implications, and is strongly associated with the development of chronic respiratory diseases such as chronic obstructive pulmonary disease (COPD), lung cancer, and asthma [1–3]. The main proximal target of inhaled CS is the respiratory epithelium, and CS-mediated effects on the respiratory epithelium include alterations in epithelial integrity [4,5], production of various inflammatory cytokines [6,7], and induction of squamous or mucus metaplasia [8,9], as major events that culminate in chronic lung diseases associated with smoking. Although the mechanisms involved in CS-related disease are highly complex, various lines of evidence indicate the importance of electrophilic aldehydes such as the α,β-unsaturated aldehyde, acrolein. Indeed, CS-related epithelial injury or activation is often associated with alterations in cellular glutathione (GSH) status, which are primarily due to its conjugation to acrolein or related electrophilic aldehydes in CS [10,11]. Moreover, CS-mediated effects on epithelial integrity, cytokine production, or mucus metaplasia can be replicated by comparable exposure to acrolein [12]. Finally, genetic determinants of chronic lung diseases such as COPD or lung cancer include polymorphisms in one of the main enzymes involved in acrolein metabolism, GSH S-transferase P1 [13,14], which is prominently expressed within the respiratory epithelium [15].
The mechanisms by which acrolein affects biological systems are still incompletely understood, but many of its biological effects have been attributed to activation of mitogen-activated protein kinases (MAPKs), such as c-Jun N-terminal kinase (JNK) or p38 [16,17], which mediate acrolein-induced epithelial chemokine production [18], alterations in epithelial/endothelial integrity [19] or induction of apoptosis/oncosis [16,20]. However, the precise mechanisms by which acrolein activates these MAPK pathways still remain to be identified. Acrolein is the simplest and most reactive α,β-unsaturated aldehyde and primarily reacts with biological targets by Michael addition to nucleophilic amino acid residues (Cys, Lys, His) [21–24] or DNA bases [22,25]. Proteomic studies indicate that acrolein preferably reacts with protein cysteine residues, and thereby affects enzymes involved in cellular redox signaling [26,27]. For example, acrolein can directly alkylate selected cysteines within Kelch-like ECH-associated protein 1(KEAP1), which promotes the activation of nuclear erythroid-2 related factor 2 (Nrf2) and induces adaptive responses to acrolein or other electrophiles by induction of antioxidant (electrophile) response element (ARE) genes [28–30].
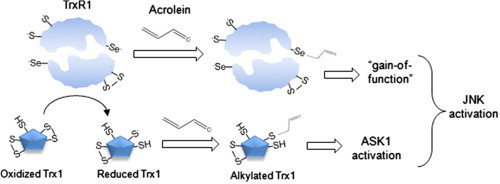
Recent studies indicate that acrolein can interact with various redox signaling proteins, including peroxiredoxins, thioredoxin (Trx) and thioredoxin reductase (TrxR) [27,31–33]. TrxR is a homodimeric selenoprotein that exists as two isoforms (cytosolic TrxR1 and mitochondrial TrxR2) and utilizes reducing equivalents from NADPH to reduce thioredoxin (Trx) 1 or Trx2 (and possibly other substrates) using a mechanism that is dependent on the selenocysteine (Sec)-containing C-terminal redox center, and thereby plays a central role in controlling redox processes involved in mitochondrial function or in regulating cell cycle and survival pathways [34,35]. The biological significance of the unusual Sec residue within TrxR is debated, but its unique electrochemical properties and strong nucleophilic character make it a highly susceptible target for alkylation by soft electrophiles such as acrolein [21,34,36,37]. While such alkylation of Sec in TrxR inhibits its Trx reductase activity [32,38], a number of studies have indicated that adduction of electrophiles to Sec results in a functionally altered protein with enhanced NADPH oxidase activity and increased pro-apoptotic properties, and has been referred to as “selenium compromised thioredoxin reductase-derived apoptotic protein” (SecTRAP) [39,40]. Our recent studies established selective alkylation of Sec within TrxR by acrolein, resulting in suppressed Trx reductase activity [27], although it is still unclear whether acrolein induces a similar gain-of-function of TrxR as was reported for other electrophiles. Therefore, the present studies were designed to address the importance of TrxR1 and its alkylation in cellular responses to acrolein in bronchial epithelial cells, and our findings indicate that TrxR1 controls the susceptibility of Trx1 to acrolein-induced alkylation, and that alkylation of TrxR1/Trx1 actively contributes to MAPK activation by acrolein and potentially to functional alterations related to these signaling pathways.
Methods
Cell culture and treatments
Human bronchial epithelial (HBE1) cells (generously provided by Dr. Reen Wu at the University of California, Davis [41]) were cultured at 37 °C in 95% humidified air containing 5% CO2 using Dulbecco's Modified Eagle's Medium (DMEM/F-12) supplemented with 50 U/mL penicillin, 50 μg/mL streptomycin, 10 ng/mL choleratoxin (List Biological Laboratories, Inc.), 10 ng/mL epidermal growth factor (Calbiochem), 15 μg/mL bovine pituitary extract, 0.5 mg/mL bovine serum albumin (Invitrogen), 5 μg/mL insulin, 5 μg/mL transferrin, and 0.1 μM dexamethasone (Sigma).
For experimentation, cells were plated at near-confluence in 12-well plates and placed in Hank's Balanced Salt Solution (HBSS) for treatments with acrolein (to avoid unwanted reactions of acrolein with other constituents present with the culture media), and collected after 30 min for various biochemical analyses, or cells were placed in full culture media for continued incubation. At indicated time points, cells were collected in appropriate lysis buffer, and total protein was quantified using the BCA protein assay kit (Pierce). Conditioned media was collected after 24 h for analysis of cytokine secretion or cell viability.
Alteration and analysis of GSH
GSH was analyzed in cell lysates by derivatization with 2 mM monobromobimane (mBrB), and analyzed by HPLC with fluorescence detection, as previously described [42]. Where indicated, GSH levels were repressed by 24-h preincubation with 100 μM buthionine sulfoximine (BSO), or augmented by 4-h pretreatment with 5 mM glutathione ethyl ester (GEE) (Sigma), prior to acrolein treatment.
Manipulation of TrxR1 status
To increase TrxR protein levels prior to acrolein treatment, cells were cultured in the presence of 50 nM sodium selenite (Na2SeO3; Sigma) for 5 days. Conversely, to suppress endogenous TrxR1 levels, cells were seeded at 70% confluence in 24-well plates and transfected with 50 nM TXNRD1 Smartpool siRNA (Dharmacon) and DharmaFECT transfection reagent according to manufacturer's instructions. Media was replaced after 24 h and cells were used for experimentation 60 h after transfection.
Measurement of TrxR activity by insulin assay
Cells were lysed in 50 mM Tris/Cl (pH 7.4) containing 1 mM EDTA, and the reductase activity of TrxR was measured using a previously described end-point insulin assay [43,44]. Protein lysates (20 μg) were incubated with 2 mM NADPH, 20 μM E. coli thioredoxin (Trx), and 1.5 mg/ml insulin for 60 min at 37 °C after which the reaction was stopped with 8 M guanidine HCL containing 1 mM 5,5’-dithiobis(2-nitrobenzoic) acid (DTNB). Formation of 2-nitro-5-thiobenzoic acid (TNB) was measured at 412 nm using a BioMate 5 spectrophotometer (Thermo Spectronic), and Trx-dependent reductase activity was determined by calculating the difference in activity with and without Trx.
Assessment of Trx 1 redox status by redox western blot
Analysis of Trx redox status was performed as previously described [45]. Briefly, cell proteins were S-carboxymethylated in lysis buffer (50 mM Tris/Cl, pH 8.3; 3 mM EDTA; 6 M Guanidine HCL; 0.5% Triton X-100; 2% protease inhibitor) containing 5 mM iodoacetic acid (IAA) at 37 °C for 30 min in the dark, cell lysates were collected and filtered using centrifugal filter devices (3000 MWCO; Millipore) to remove excess IAA. Samples were mixed with 2× non-reducing sample buffer, containing 0.5 M Tris/Cl (pH 6.8), 20% v/v glycerol, and 0.02% bromophenol blue, and separated on 18% Native PAGE gels (Invitrogen, Carlsbad, CA) for analysis of various oxidation states of Trx 1, by Western blotting using a polyclonal rabbit antibody against Trx 1 (1:2000, Cell Signaling) and detection with SuperSignal West chemiluminescent substrate (Pierce). Band densities were quantified using ImageJ software (http://imagej.nih.gov/ij/), and expressed as a percentage of total Trx intensity for each treatment condition.
Studies with semi-synthetic mTrxR
Full length semisynthetic mitochondrial TrxR-GCUG and a truncated variant lacking the C-terminal CUG residues (mTrxRΔ3) were prepared as described [46], and incubated at a final concentration of 200 nM in 100 mM potassium phosphate buffer containing 1 mM EDTA with 30 μM acrolein for 15 min. After acrolein adduction, 20 nM mTrxR was incubated with 200 μM NADPH (Sigma) and 20 μM E. coli thioredoxin, and the Sec-dependent thioredoxin reductase activity was measured as a result of NADPH consumption by absorbance at 340 nm. In addition, NADPH oxidase activity, which may reflect activity at the N-terminal cysteines within mTrxR [37,47], was addressed by mixing 12 nM mTrxR with 200 μM NADPH and measurement of NADPH consumption at 340 nm as previously described [48], in the absence or presence of 1 mM α-lipoic acid (Sigma). The rate of reaction was evaluated from the linear portion of the curve, and expressed as ΔA340×60 s−1.
Detection of protein–acrolein adducts using biotin hydrazide labeling
Protein lysates (≥300 μg) were mixed with 5 mM biotin hydrazide (Thermo Scientific) solubilized in DMSO at room temperature for 2 h with constant rotation. This was followed by the addition of 30 mM sodium cyanoborohydride (NaCNBH4; Sigma) and incubation on ice for 60 min. Biotin-labeled proteins were purified by avidin chromatography as described previously [27,49,50]. Briefly, biotinylated lysates were washed three times with 300 μL of 20 mM Tris/Cl pH 7.4 using centrifugal filter devices (3000 MWCO; Millipore) at 10,000 rpm for 10 min, to remove excess biotin hydrazide and NaCNBH4. Labeled proteins were then isolated by affinity chromatography (batch method) by the addition of 50 μL of a 50% suspension of high capacity neutravidin agarose resin (Thermo Scientific). The beads were washed six times with 0.2 M glycine pH 2.8, centrifuged between each wash at≤2500 rpm, then washed once with 20 mM Tris/Cl pH 7.4. Proteins were eluted in 100 μL 2× reducing sample buffer and boiled at 95–100 °C for 5 min, and analyzed by 10% or 18% SDS-PAGE and Western blotting.
Western blotting
Cells were lysed in buffer containing 1% Triton X-100, 250 mM NaCl, 50 mM HEPES, 10% glycerol, 1.5 mM magnesium chloride (MgCl2), 1 mM polymethylsulfonyl chloride (PMSF), 1 mM ethylene glycol tetraacetic acid (EGTA), 2 mM sodium orthovanadate (Na3VO4), 10 μg/mL aprotinin, and 10 μg/mL leupeptin for protein analysis. Following 15 min incubation on ice, cells were sonicated for 20 pulses on ice using a sonic dismembrator (Fisher Scientific) and centrifuged at 14,000 rpm, 4 °C for 5 min to remove cell debris. Protein was quantified via the bicinchoninic acid assay (BCA protein assay kit; Pierce). Aliquots containing 15 μg protein were mixed with 2× reducing sample buffer (containing 4% SDS, 20% glycerol, and 10% β-mercaptoethanol), and separated by SDS-PAGE, transferred to nitrocellulose membranes, and blotted for specific proteins with the following primary antibodies: TrxR1: (1:1000, Abcam), Trx1 (1:2000, Cell Signaling), phospho SAPK/JNK1/2 (1:1000, Cell Signaling), SAPK/JNK1/2 (1:1000, Cell Signaling), phospho ERK1/2 (1:1000, Cell Signaling), ERK1/2 (1:1000, Cell Signaling), phospho P-38 (1:1000, Cell Signaling), P-38 (1:1000, Cell Signaling), HO-1 (1:250, Biovision), NQO1 (1:1000, Cell Signaling), β-actin (1:5000, Sigma). Primary antibodies were detected using HRP-conjugated anti-rabbit or anti-mouse IgG and visualized by SuperSignal West chemiluminescent substrate (Pierce). Band densities were evaluated using ImageJ software, and normalized to total levels of respective proteins or to β-actin.
Measurement of cell toxicity
Loss of cell viability was evaluated by analysis of lactate dehydrogenase (LDH) in conditioned media using LDH Cytotoxicity Detection Kit (Takara Bio Inc.), and expressed as a percentage of total cellular LDH which was estimated by cell lysis in 2% Triton X-100.
Statistical analyses
Data for each group were statistically analyzed via t-test or analysis of variance (ANOVA) depending on the amount of groups/experiment and significance was assigned at a cut off of p<0.05.
Results
Acrolein exposure inactivates TrxR independent of GSH depletion and in association with alkylation
To analyze the effects of acrolein exposure of HBE1 cells on various cellular redox systems, cells were treated with 3–30 μM acrolein for 30 min, and GSH levels and TrxR activity were determined. As shown in Fig. 1A, acrolein induces depletion of cellular GSH and inhibition of TrxR activity in a dose-dependent manner, but the effects on TrxR were more pronounced at lower concentrations. To determine whether cellular GSH status affects the sensitivity of TrxR to acrolein, GSH levels were augmented by pretreatment with GEE or depleted by BSO (Fig. 1B). However, neither treatment significantly affected acrolein-dependent inhibition of TrxR (Fig. 1C).
Fig. 1.
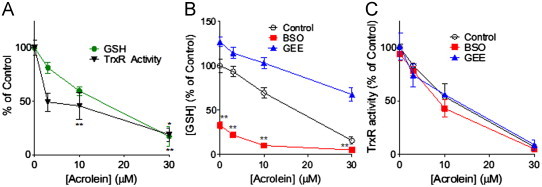
Acrolein-induced redox alterations in HBE1 cells. HBE1 cells were treated with acrolein at indicated concentrations for 30 min after which TrxR activity and GSH levels were measured as described in the Methods section (A). HBE1 cells were pretreated with BSO (24 h) or GEE (4 h) prior to acrolein treatment of indicated for 30 min, after which GSH levels (B) and TrxR1 activity (C) were measured. Results are expressed as mean±SEM, n=4. *: p<0.05, **: p<0.01 compared to control.
We also evaluated the effects of acrolein on Trx1 oxidation using redox western blots, which indicated increased formation of higher MW Trx1 bands, most likely reflecting partially and fully oxidized forms of Trx1, in response to acrolein (Fig. 2A). Alteration of cellular GSH levels using either BSO or GEE did not affect basal Trx1 oxidation status, but enhanced acrolein-dependent Trx1 oxidation was observed in BSO pretreated cells, whereas the extent of acrolein-induced Trx1 was reduced in GEE pretreated cells (Fig. 2B). We realize that these Trx1 redox blots do not distinguish between Trx1 oxidation or alkylation, but our findings indicate that acrolein-induced Trx1 oxidation/alkylation depends on initial GSH status, in contrast to TrxR1 inactivation which occurred regardless of initial GSH status. These findings are consistent with the notion that TrxR is highly susceptible to inhibition by acrolein, compared to other cellular redox systems.
Fig. 2.
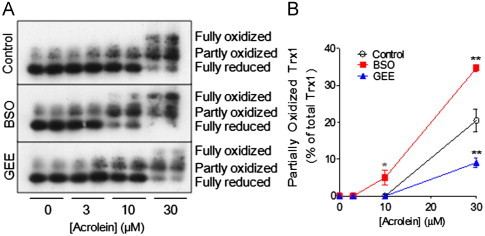
Acrolein-dependent Trx oxidation depends on GSH status. HBE1 cells were pretreated with BSO (24 h) or GEE (4 h) prior to acrolein treatment for 30 min, and Trx1 redox status was determined by S-carboxymethylation and non-reducing native PAGE. Representative redox Western blots are shown in (A) and relative band densities of partially oxidized Trx are summarized in (B). Results are expressed as mean±SEM, n=4. *: p<0.05, **: p<0.01 compared to control.
Acrolein inhibits thioredoxin reductase activity but enhances NADPH oxidase activity of mTrxR
Based on previous studies suggesting the ability of alkylating agents to induce a gain-of-function in TrxR [39], we investigated the effects of acrolein on semisynthetic full-length mitochondrial TrxR (mTrxR-GCUG) and a truncated version (mTrxRΔ3) that lacks the C-terminal CUG motif including Sec (U), and is relatively resistant to alkylation by acrolein [27]. As shown in Fig. 3A, acrolein treatment significantly inhibits thioredoxin reductase activity (measured using the insulin assay) in full-length mTrxR-GCUG, whereas truncated mTrxRΔ3 had little activity as it lacks the C-terminal Sec residue. In contrast, analysis of NADPH oxidase activity in the presence of alternative substrates such as α-lipoic acid, which do not require the C-terminal Sec residue [47], indicated that acrolein significantly enhanced NADPH oxidase activity of mTrxR-GCUG (Fig. 3B). The fact that the comparable activity of mTrxRΔ3 towards α-lipoic acid was not significantly affected by acrolein under similar conditions (Fig. 3B) is consistent with selective reaction of acrolein with the C-terminal Sec within the full-length enzyme which is absent in mTrxRΔ3 [27]. Presumably, Sec alkylation within the full-length enzyme may induce a conformational change that favors its ability to reduce alternative substrates such as α-lipoic acid (A. P. Lothrop and R. J. Hondal, unpublished). Overall, these data indicate that acrolein not only inhibits the reductase activity of TrxR but also induces a “gain-of-function” illustrated by enhanced NADPH oxidase activity in the presence of α-lipoic acid.
Fig. 3.
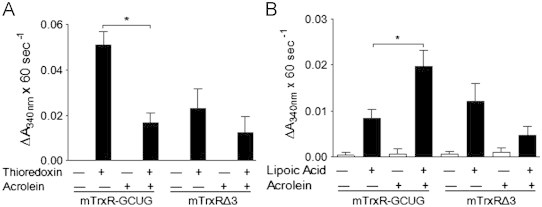
Effects of acrolein on enzymatic activity of mTrxR. Semisynthetic mTrxR-GCUG and mTrxRΔ3 (200 nM) were incubated with acrolein for 30 min, and thioredoxin reductase activity was assessed using the insulin assay (A), and NADPH oxidase activity was determined in the absence (white bars) or presence (black bars) of α-lipoic acid (B). Results expressed as the mean change in absorbance x 60 s−1±SEM, n=5. *: p<0.05 compared to control enzyme.
Involvement of TrxR1 in acrolein-mediated Trx1 oxidation and alkylation
Based on our observations that TrxR1 is highly sensitive to acrolein compared to e.g. GSH and that acrolein may induce a “gain-of-function” in TrxR by direct alkylation to its Sec residue, we wished to determine whether alkylation of TrxR may directly contribute to acrolein-induced cytotoxicity or cell activation. To address this, we used two different approaches to either augment or suppress TrxR levels in HBE1 cells, and evaluated its effects on acrolein-induced TrxR alkylation, Trx status and cytotoxicity. First, since TrxR expression is regulated by selenium availability [51–53], we cultured HBE1 cells in media supplemented with selenite (at non-toxic concentrations ranging from 20 to 100 nM), and determined that supplementation with 50 nM for 5 days resulted in optimal increased in TrxR1 protein and activity (Fig. S1). As illustrated in Fig. 4A and B, selenite-supplemented HBE1 cells contained about 2-fold more TrxR1 protein and also contained correspondingly increased levels of alkylated TrxR after acrolein treatment, compared with non-supplemented HBE1 cells (Fig. 4C). We realize that selenium supplementation may also affect expression of other selenoproteins such as glutathione peroxidases [54,55], and therefore cannot conclusively state that effects of selenium supplementation are exclusively due to increases in TrxR expression. As a converse approach, we suppressed TrxR1 using siRNA which resulted in approx. 80% knockdown of TrxR1 protein (Fig. 4D and E), with comparable increases in alkylated TrxR1 after acrolein treatment (Fig. 4F). Therefore, manipulation of TrxR1 levels resulted in corresponding changes in alkylated TrxR after acrolein exposure, allowing us to determine whether cellular changes induced by acrolein may be related to alkylation of TrxR.
Fig. 4.
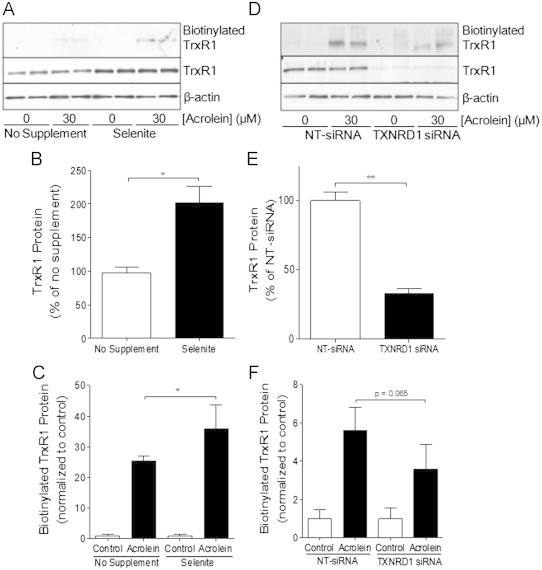
Alterations in TrxR by selenite supplementation or siRNA silencing. HBE1 cells were supplemented with 50 nM selenite (A, B and C) or transfected with TXNRD1 or NT siRNA (D, E and F), and treated with 30 μM acrolein for 30 min for Western blot analysis of total TrxR1 protein levels or β-actin in whole cell lysates (B and E), or analysis of acrolein-modified TrxR1 which was determined after labeling with biotin hydrazide and analysis of avidin-purified proteins by Western blot for TrxR1 (C and F). Densitometry of Western blots was analyzed and results are expressed as mean±SEM. *: p<0.05, **: p<0.01 compared to non-supplemented or NT-siRNA controls.
Although selenite supplementation of HBE1 cells did not affect overall GSH levels (not shown), it appeared to alter Trx1 redox status to a more reduced state (Fig. 5A and B). Conversely, siRNA silencing of TrxR1 appeared to result in slightly more oxidized Trx1 status, compared to NT-siRNA transfection (Fig. 5C and D). However, neither selenite supplementation nor TXNRD1 siRNA silencing appeared to significantly affect acrolein-induced oxidation/alkylation of Trx1, as the relative amounts of partially or fully oxidized Trx1 after treatment with 10–30 μM acrolein were highly similar (Fig. 5B and D). Because the electrophoretic mobility shift in Trx1 may also be due to alkylation rather than oxidation, we used biotin hydrazide labeling and neutravidin chromatography to more directly evaluate Trx1 alkylation in these cases. As illustrated in Fig. 6A and C, acrolein treatment resulted in significantly enhanced Trx1 alkylation in selenite-supplemented cells compared to non-supplemented cells, whereas markedly reduced Trx1 alkylation was observed in acrolein-treated TXNRD1 siRNA transfected cells compared to control transfected cells (Fig. 6B and D). Importantly, these changes in Trx1 alkylation were not related to altered overall Trx1 levels, and are most likely related to alterations in initial Trx1 redox status by either selenite supplementation or siRNA silencing (Fig. 5). Thus, the ability of acrolein to alkylate Trx1 appears to depend on the presence of TrxR1 and its impact on the reduction status of Trx1.
Fig. 5.
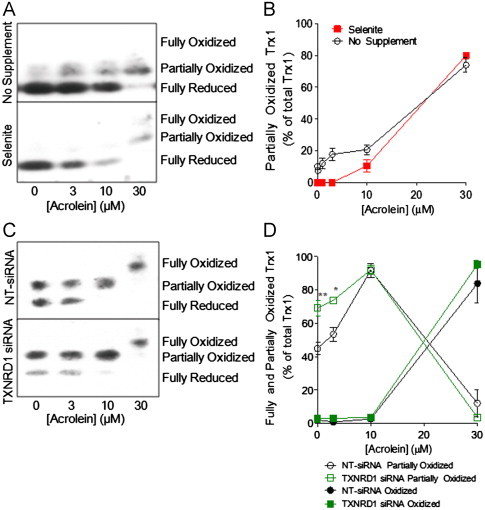
Effects of TrxR1 alterations on acrolein-induced Trx1 oxidation/alkylation. HBE1 cells were supplemented with selenite or transfected with TXNRD1 siRNA and treated with acrolein for 30 min, and the oxidation/alkylation status of Trx1 was determined by redox Western blot (A and C). Graphs show relative band densities of partially and/or fully oxidized Trx1 (B and D). Results are expressed as mean±SEM, n=4. *: p<0.05, **: p<0.01 non-supplemented or NT-siRNA controls.
Fig. 6.
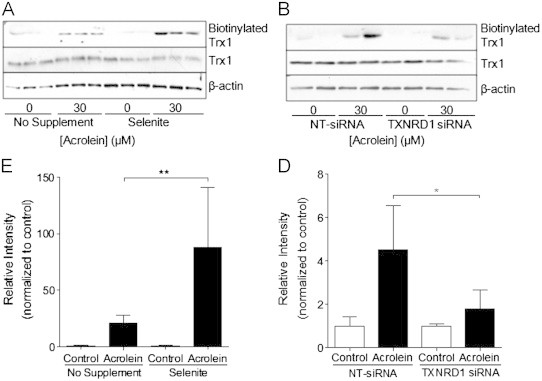
Alterations in Trx1 by selenite supplementation or siRNA silencing. HBE1 cells were supplemented with 50 nM selenite (A) or transfected with TXNRD1 or NT siRNA (B), and treated with 30 μM acrolein for 30 min for Western blot analysis of total Trx1 protein levels or β-actin in whole cell lysates, or analysis of acrolein-modified TrxR1 which was determined after labeling with biotin hydrazide and analysis of avidin-purified proteins by Western blot for Trx1. Graphs show relative band densities of Biotinylated Trx1 (C, D). Results are expressed as mean±SEM. *: p<0.05, **: p<0.01 compared to non-supplemented or NT-siRNA controls.
Effects of TrxR on acrolein-mediated cytotoxity
Since alkylation of TrxR by other electrophiles has been reported to enhance its pro-apoptotic properties [39], we determined whether TrxR and its alkylation may also be required for the pro-apoptotic properties of acrolein [20]. To test this, selenite-supplemented or TXNRD1 siRNA transfected HBE1 cells were treated with acrolein for 30 min and incubated for up to 24 h for analysis of cell viability using LDH release. No significant cytotoxicity was observed in non-supplemented or selenite-supplemented HBE1 cells by acrolein concentrations up to 30 min, and acrolein-induced loss of viability in siRNA transfected HBE1 cells was nearly identical between TXNRD1 siRNA and NT-siRNA transfected cells (Fig. S2). These data suggest that inhibition or alkylation of TrxR1 does not contribute significantly to the cytotoxic properties of acrolein under these conditions.
Involvement of TrxR1 in acrolein-induced MAPK signaling
Since acrolein is capable of activating various MAPKs [16,18], which may involve oxidation of Trx1 and dissociation from apoptosis signal-regulated kinase (ASK1) [26] or potentially the activation of oxidant mechanisms by alkylation of TrxR [39,40], we next explored the impact of selenite supplementation and TrxR1 siRNA silencing on the ability of acrolein to activate JNK1/2, ERK1/2, and p38. As expected, acrolein dose-dependently induces activation of JNK, ERK, and p38 in HBE1 cells, as indicated by phosphorylation of ERK at concentrations as low as 3 μM and phosphorylation of JNK and p38 at 10–30 μM (Figs. 7 and 8). Interestingly, the ability of acrolein to induce the phosphorylation of these MAPK appeared to be increased in selenite-supplemented cells (Fig. 7A), which was statistically significant in case of JNK (Fig. 7B and C). Conversely, acrolein-mediated phosphorylation of especially JNK1, but also ERK and p38, was suppressed in TXNRD1 siRNA transfected cells compared to NT-siRNA transfected cells (Fig. 8). Together, these data indicate that acrolein-induced activation of MAPK, primarily JNK1 and p38, is dependent on the presence of TrxR and is likely related to alkylation of TrxR1 and/or Trx1.
Fig. 7.
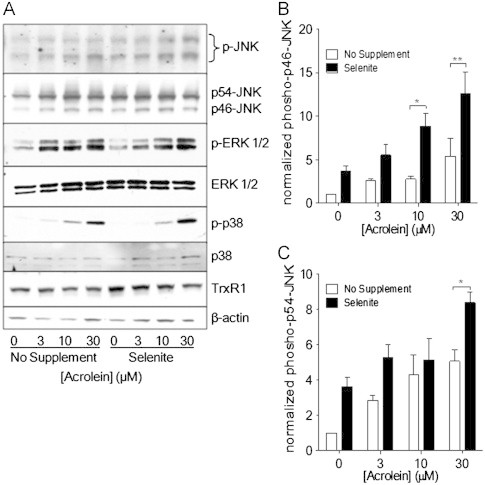
Effect of selenite supplementation on acrolein-induced MAPK activation. Non-supplemented or selenite-supplemented HBE1 cells were treated with indicated concentrations of acrolein for 30 min, and cell lysates were analyzed for p-JNK, p-ERK1/2, and p-p38 by Western blot (A). Band densities of phosphorylated p46-JNK (B) and p56-JNK (C) were quantified relative to total JNK protein. Results are expressed as mean±SEM, n=3. *: p<0.05, **: p<0.01 compared to non-supplemented controls.
Fig. 8.
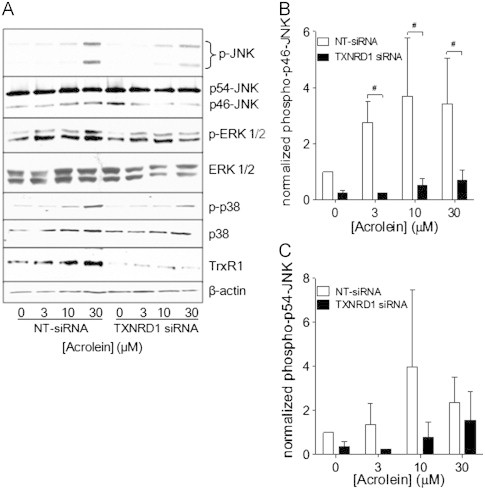
Effect of TrxR1 siRNA silencing on acrolein-induced MAPK activation. HBE1 cells were transfected with Non-targeting (NT-siRNA) or TXNRD1 siRNA and treated with 30 μM acrolein for 30 min, and cell lysates were analyzed for p-JNK, p-ERK, and p-p38 by Western Blot (A). Band densities of phosphorylated p46-JNK (B) and p54-JNK (C) were quantified relative to total JNK protein. Results expressed as mean±SEM, n=3. *: p<0.05 compared to NT-siRNA controls.
To address the possibility that TrxR1 alkylation might promote MAPK activation by increased NADPH oxidase activation and production of O2•− and H2O2, we performed some experiments in the presence of two scavengers of H2O2 and/or O2•−, ebselen (10 μM) or EUK134 (50 μM). However, both compounds themselves increased basal levels of JNK phosphorylation, and did not appear to significantly prevent JNK phosphorylation after subsequent exposure to acrolein (Fig. S3), although both compounds were shown to inhibit H2O2-dependent signaling under similar conditions [56,57]. Therefore, the ability of acrolein to activate JNK does not appear to involve H2O2 or O2•−.
TrxR1 does not mediate acrolein-dependent induction of phase II responses
Acrolein and related aldehydes can induce the expression of anti-inflammatory or phase II enzymes through activation of the antioxidant response element (ARE), and recent studies indicate that this may also involve MAPK pathways [29]. We therefore tested the effects of selenite supplemention or TrxR1 silencing on acrolein-mediated induction of two ARE genes, heme oxygenase 1 (HO-1) and NADPH:quinone oxidoreductase 1 (NQO1). However, although acrolein-induced dose-dependent increases in HO-1 expression in HBE1 cells, this was not affected by selenite supplementation or TrxR1 silencing (Fig. 9A and C). Somewhat surprisingly, acrolein did not appear to significantly affect NQO1 expression, irrespective of TrxR1 status (Fig. 9B and D). Therefore, acrolein-mediated adduction of TrxR1 and/or Trx1 does not significantly impact on induction of ARE-regulated genes.
Fig. 9.
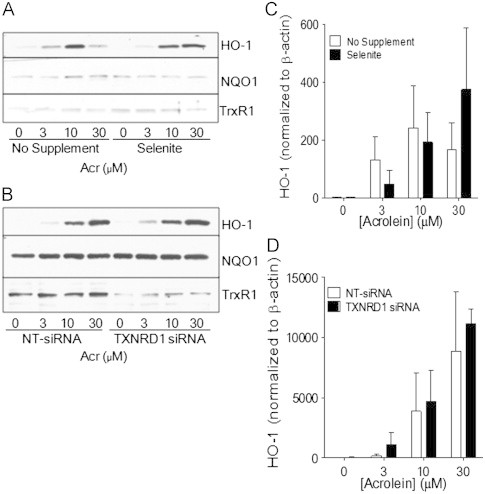
Effect of TrxR1 status on acrolein-induced phase II responses. Selenite-supplemented (A) or TXNRD1 siRNA transfected (B) HBE1 cells were treated with acrolein and expression of HO-1, NQO-1, and TrxR1 were analyzed after 8 h by Western blot. Quantified HO-1 band densities relative to β-actin are presented as mean±SEM (n=4) (C, D).
Discussion
Chronic lung diseases such as COPD and asthma have been strongly associated with cigarette smoking [2,3] and based on studies with thiol-based antioxidants, the adverse effects of smoking are largely mediated by thiol-reactive substances within tobacco smoke, of which acrolein is among the most abundant and reactive [10,58]. Our present studies were based on recent observations that TrxR is highly sensitive to direct modification by acrolein due to its highly nucleophilic Sec residue [26,27], and that functional alterations due to such alkylation by electrophiles can contribute to their cytotoxic properties or other cellular effects [34,39]. We confirmed that acrolein readily inactivates thioredoxin reductase activity in HBE1 cells, independent of depletion of GSH or other redox alterations, and corresponding with direct alkylation of TrxR1. Using purified semisynthetic mTrxR, we also demonstrated that acrolein-induced inhibition of thioredoxin reductase activity is associated with enhanced NADPH-dependent reductase activity towards other substrates, which does not require the C-terminal Sec residue [47]. Although such findings with mitochondrial mTrxR (TrxR2) may not be fully applicable to cytosolic TrxR1, because of potential structural differences and the presumption that the C-terminal tail in mTrxR exhibits greater flexibility and might thereby facilitate reduction of a larger variety of substrates at the N-terminal dithiol region if Sec is alkylated, they are consistent with other recent findings with TrxR1 indicating that electrophilic addition to Sec results in reduced thioredoxin reductase activity and increased NADPH oxidase-like activity [59,60].
We used two independent approaches to modulate TrxR1 expression levels in HBE1 cells to address the potential cellular consequences of TrxR1 alkylation. While these approaches did not significantly affect the overall cytotoxic effects of acrolein, they demonstrated that the ability of acrolein to activate various MAPK pathways, most notably JNK and p38, depend on the presence of TrxR1, suggesting that these pathways may be activated by a gain-of-function in TrxR1 due to its alkylation. One potential consequence of such a gain-of-function is increased production of O2•− and H2O2 due to enhanced NADPH oxidase activity [59,60], which may in turn be responsible for activation of JNK or p38 [61]. In an attempt address this possibility, we used two chemical catalysts of H2O2/O2•− decomposition, ebselen and EUK134, but neither compound clearly affected acrolein-induced JNK phosphorylation (Fig. S3). Thus, our findings suggest that activation of these signaling pathways may not involve H2O2/O2•− production as a result of TrxR1 alkylation, but rather depend on alternative functional consequences of such alkylation.
Alterations in TrxR1 activity might also affect acrolein-dependent signaling by more indirect effects on Trx redox status. While our studies did not indicate a significant impact of TrxR1 status on acrolein-dependent alterations in overall Trx1 redox status, direct alkylation of Trx by acrolein was proportional to initial TrxR1 expression levels, most likely related to corresponding changes in Trx redox status and relative availability of reduced Trx for alkylation. Indeed, various lines of evidence suggest a direct role for acrolein-induced Trx1 alkylation in downstream signaling events. For example, acrolein modification of Trx1 was recently linked to increased monocyte adhesion to endothelial cells, although the proximal signaling events in this adhesion were not examined [31]. More recently, Trx1 alkylation was suggested to promote dissociation of Trx1 from ASK1, which may lead to its activation and subsequent MAPK phosphorylation [26]. Our findings are consistent with such a notion, since alterations in acrolein-dependent MAPK signaling in relation to TrxR1 status were found to be proportional to the extent of alkylated Trx1, although they were also proportional to the amount of alkylated TrxR1, which may itself induce functional alterations. Future studies will be required to more definitively distinguish between these two scenarios.
We have attempted to address potential functional consequences of enhanced MAPK activation in relation to TrxR/Trx alkylation. While our studies did not indicate significant consequences of TrxR1 status for acrolein-mediated cytotoxicity, activation of MAPK pathways have also been implicated in acrolein-dependent induction of anti-inflammatory or phase II responses [29], epithelial production of cytokines such as interleukin-8 or thymic stromal lymphopoietin (TSLP) [7,18], or altered epithelial barrier integrity [62]. However, we did not observe significant effects of TrxR1 status on acrolein-dependent induction of anti-inflammatory responses such as HO-1, probably because such induction is initiated primarily by alkylation of Keap1 and activation of Nrf2, which is independent of TrxR1. Also, in apparent contrast with previous studies [7,18], we did not observe significant induction of either IL-8 or TSLP by acrolein under these conditions in HBE1 cells (results not shown). Unfortunately, we were not able to evaluate consequences for epithelial barrier function by e.g. transepithelial electrical resistance, because the HBE1 cell line is not suitable for this purpose. The potential involvement of TrxR1 in acrolein-mediated alterations in epithelial integrity will therefore have to be addressed in future studies in more appropriate airway epithelial systems.
In summary, our data suggest that the effects of acrolein on airway epithelial biology may be mediated in part by direct adduction to TrxR1 and/or Trx1, which appears to directly impact on acrolein-mediated activation of MAPKs, specifically JNK and p38. Although we did not definitively demonstrate a consequence for airway epithelial biology, our findings are likely to be relevant for epithelial alterations in smoking-related diseases such as COPD and asthma, and may implicate TrxR or Trx as potential therapeutic targets in treatment or management of these diseases.
Footnotes
This work was supported by research grants from NIH (R01 ES021476) and the Flight Attendant Medical Research Institute (FAMRI) to AvdV, and NIH grant R01 GM094172 to RJH. PCS was supported by a YCIA from FAMRI, and MJR received a Research Fellowship from the Department of Pathology.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
MAPK, mitogen-activated protein kinase; TrxR1, thioredoxin reductase 1; Trx1, thioredoxin; ASK1, apoptotic signaling kinase 1; JNK, c-jun N-terminal kinase; SecTRAP, selenium compromised thioredoxin reductase-derived apoptotic protein; Na2SeO3, sodium selenite; ERK, extracellular regulated signaling kinase; ARE, antioxidant response element; HO-1, heme oxygenase 1; NQO1, NADPH:quinone oxidoreductase 1; TSLP, thymic stromal lymphopoietin.
Appendix A. Supplementary Information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org.10.1016/j.ultramic.2011.01.020.
Appendix A. Supplementary materials
Fig. S1.
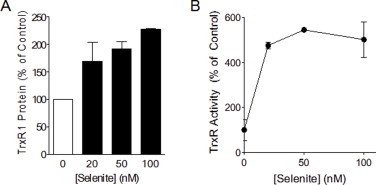
Selenite supplementation alters TrxR1 protein levels and enzyme activity. HBE1 cells were supplemented with selenite (20–100 nM) for a duration of 5 days after which cells were lysed for analysis of TrxR1 protein levels by Western blotting (A), and TrxR activity using the insulin assay as described in the Methods section (B). Results are expressed as mean±SEM.
Fig. S2.
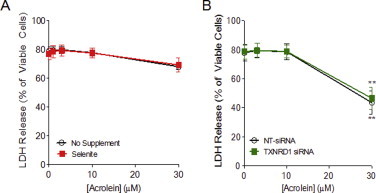
TrxR1 status does not affect acrolein-induced toxicity. Selenite-supplemented (A) or TXNRD1 siRNA transfected (B) HBE1 cells were treated with acrolein, and cell viability was assessed by analysis of LDH release in the culture media after 24 h. Results are expressed as mean±SEM, n=3. **: p<0.01 compared to control.
Fig. S3.
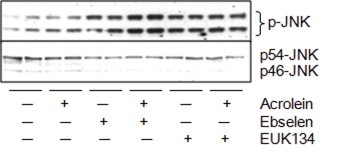
Effect of ROS scavengers on acrolein-mediated JNK phosphorylation. HBE1 cells were pretreated for 30 min with either ebselen (10 μM) or EUK134 (50 μM) followed by a 30 min exposure of acrolein (30 μM), after which cells were lysed and p-JNK and JNK were analyzed by Western blot. Representative blots of two experiments are shown.
References
- 1.Office of the Surgeon General. The Health Consequences of Smoking: A Report of the Surgeon General; 2004. [PubMed]
- 2.Saetta M. Airway inflammation in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 1999;160:S17–20. doi: 10.1164/ajrccm.160.supplement_1.6. [DOI] [PubMed] [Google Scholar]
- 3.Thomson N.C., Chaudhuri R. Asthma in smokers: challenges and opportunities. Current Opinion in Pulmonary Medicine. 2009;15:39–45. doi: 10.1097/MCP.0b013e32831da894. [DOI] [PubMed] [Google Scholar]
- 4.Heijink I.H., Brandenburg S.M., Postma D.S., van Oosterhout A.J. Cigarette smoke impairs airway epithelial barrier function and cell–cell contact recovery. European Respiratory Journal. 2012;39:419–428. doi: 10.1183/09031936.00193810. [DOI] [PubMed] [Google Scholar]
- 5.Xiao C., Puddicombe S.M., Field S., Haywood J., Broughton-Head V., Puxeddu I., Haitchi H.M., Vernon-Wilson E., Sammut D., Bedke N., Cremin C., Sones J., Djukanovic R., Howarth P.H., Collins J.E., Holgate S.T., Monk P., Davies D.E. Defective epithelial barrier function in asthma. Journal of Allergy and Clinical Immunology. 2011;128:549–556. doi: 10.1016/j.jaci.2011.05.038. e541-512. [DOI] [PubMed] [Google Scholar]
- 6.Mio T., Romberger D.J., Thompson A.B., Robbins R.A., Heires A., Rennard S.I. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. American Journal of Respiratory and Critical Care Medicine. 1997;155:1770–1776. doi: 10.1164/ajrccm.155.5.9154890. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura Y., Miyata M., Ohba T., Ando T., Hatsushika K., Suenaga F., Shimokawa N., Ohnuma Y., Katoh R., Ogawa H., Nakao A. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. Journal of Allergy and Clinical Immunology. 2008;122:1208–1214. doi: 10.1016/j.jaci.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 8.Shao M.X., Nakanaga T., Nadel J.A. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cells. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2004;287:L420–427. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 9.Araya J., Cambier S., Markovics J.A., Wolters P., Jablons D., Hill A., Finkbeiner W., Jones K., Broaddus V.C., Sheppard D., Barzcak A., Xiao Y., Erle D.J., Nishimura S.L. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. The Journal of Clinical Investigation. 2007;117:3551–3562. doi: 10.1172/JCI32526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy S., Finkelstein E.I., Wong P.S., Phung A., Cross C.E., van der Vliet A. Identification of glutathione modifications by cigarette smoke. Free Radical Biology and Medicine. 2002;33:1490–1498. doi: 10.1016/s0891-5849(02)01079-1. [DOI] [PubMed] [Google Scholar]
- 11.van der Toorn M., Smit-de Vries M.P., Slebos D.J., de Bruin H.G., Abello N., van Oosterhout A.J., Bischoff R., Kauffman H.F. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2007;293:L1156–1162. doi: 10.1152/ajplung.00081.2007. [DOI] [PubMed] [Google Scholar]
- 12.Bein K., Leikauf G.D. Acrolein—a pulmonary hazard. Molecular Nutrition & Food Research. 2011;55:1342–1360. doi: 10.1002/mnfr.201100279. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y.L., Lin Y.C., Lee Y.C., Wang J.Y., Hsiue T.R., Guo Y.L. Glutathione S-transferase P1 gene polymorphism and air pollution as interactive risk factors for childhood asthma. Clinical & Experimental Allergy. 2004;34:1707–1713. doi: 10.1111/j.1365-2222.2004.02099.x. [DOI] [PubMed] [Google Scholar]
- 14.Tamer L., Calikoglu M., Ates N.A., Yildirim H., Ercan B., Saritas E., Unlu A., Atik U. Glutathione-S-transferase gene polymorphisms (GSTT1, GSTM1, GSTP1) as increased risk factors for asthma. Respirology. 2004;9:493–498. doi: 10.1111/j.1440-1843.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 15.Reddy P.M., Tu C.P., Wu R. Glutathione S-transferases in tracheobronchial epithelium. American Journal of Physiology. 1995;269:L473–481. doi: 10.1152/ajplung.1995.269.4.L473. [DOI] [PubMed] [Google Scholar]
- 16.Tanel A., Averill-Bates D.A. P38 and ERK mitogen-activated protein kinases mediate acrolein-induced apoptosis in Chinese hamster ovary cells. Cell Signal. 2007;19:968–977. doi: 10.1016/j.cellsig.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 17.Ranganna K., Yousefipour Z., Nasif R., Yatsu F.M., Milton S.G., Hayes B.E. Acrolein activates mitogen-activated protein kinase signal transduction pathways in rat vascular smooth muscle cells. Molecular & Cellular Biochemistry. 2002;240:83–98. doi: 10.1023/a:1020659808981. [DOI] [PubMed] [Google Scholar]
- 18.Moretto N., Facchinetti F., Southworth T., Civelli M., Singh D., Patacchini R. alpha,beta-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2009;296:L839–848. doi: 10.1152/ajplung.90570.2008. [DOI] [PubMed] [Google Scholar]
- 19.Schweitzer K.S., Hatoum H., Brown M.B., Gupta M., Justice M.J., Beteck B., Van Demark M., Gu Y., Presson R.G., Jr, Hubbard W.C., Petrache I. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2011;301:L836–846. doi: 10.1152/ajplung.00385.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kehrer J.P., Biswal S.S. The molecular effects of acrolein. Toxicological Sciences: an Official Journal of the Society of Toxicology. 2000;57:6–15. doi: 10.1093/toxsci/57.1.6. [DOI] [PubMed] [Google Scholar]
- 21.Cai J., Bhatnagar A., Pierce W.M., Jr. Protein modification by acrolein: formation and stability of cysteine adducts. Chemical Research in Toxicology. 2009;22:708–716. doi: 10.1021/tx800465m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LoPachin R.M., Gavin T., Petersen D.R., Barber D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chemical Research in Toxicology. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens J.F., Maier C.S. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Molecular Nutrition & Food Research. 2008;52:7–25. doi: 10.1002/mnfr.200700412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aldini G., Orioli M., Carini M. Protein modification by acrolein: relevance to pathological conditions and inhibition by aldehyde sequestering agents. Molecular Nutrition & Food Research. 2011;55:1301–1319. doi: 10.1002/mnfr.201100182. [DOI] [PubMed] [Google Scholar]
- 25.Wang H.T., Zhang S., Hu Y., Tang M.S. Mutagenicity and sequence specificity of acrolein-DNA adducts. Chemical Research in Toxicology. 2009;22:511–517. doi: 10.1021/tx800369y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers C.R., Myers J.M., Kufahl T.D., Forbes R., Szadkowski A. The effects of acrolein on the thioredoxin system: implications for redox-sensitive signaling. Molecular Nutrition & Food Research. 2011;55:1361–1374. doi: 10.1002/mnfr.201100224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spiess P.C., Deng B., Hondal R.J., Matthews D.E., van der Vliet A. Proteomic profiling of acrolein adducts in human lung epithelial cells. Journal of Proteomics. 2011 doi: 10.1016/j.jprot.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tirumalai R., Rajesh Kumar T., Mai K.H., Biswal S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicology Letters. 2002;132:27–36. doi: 10.1016/s0378-4274(02)00055-3. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H., Forman H.J. Signaling pathways involved in phase II gene induction by alpha, beta-unsaturated aldehydes. Toxicology and Industrial Health. 2009;25:269–278. doi: 10.1177/0748233709102209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMahon M., Lamont D.J., Beattie K.A., Hayes J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proceedings of the National Academy of Sciences USA. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Go Y.M., Halvey P.J., Hansen J.M., Reed M., Pohl J., Jones D.P. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. American Journal of Pathology. 2007;171:1670–1681. doi: 10.2353/ajpath.2007.070218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myers C.R., Myers J.M. The effects of acrolein on peroxiredoxins, thioredoxins, and thioredoxin reductase in human bronchial epithelial cells. Toxicology. 2009;257:95–104. doi: 10.1016/j.tox.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X., Wu X., Choi Y.E., Kern J.C., Kehrer J.P. Effect of acrolein and glutathione depleting agents on thioredoxin. Toxicology. 2004;204:209–218. doi: 10.1016/j.tox.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 34.Cassidy P.B., Edes K., Nelson C.C., Parsawar K., Fitzpatrick F.A., Moos P.J. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27:2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- 35.Holmgren A., Lu J. Thioredoxin and thioredoxin reductase: current research with special reference to human disease. Biochemical and Biophysical Research Communications. 2010;396:120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 36.Arner E.S. Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine? Experimental Cell Research. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 37.Hondal R.J., Ruggles E.L. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2011 doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park Y.S., Misonou Y., Fujiwara N., Takahashi M., Miyamoto Y., Koh Y.H., Suzuki K., Taniguchi N. Induction of thioredoxin reductase as an adaptive response to acrolein in human umbilical vein endothelial cells. Biochemical and Biophysical Research Communications. 2005;327:1058–1065. doi: 10.1016/j.bbrc.2004.12.104. [DOI] [PubMed] [Google Scholar]
- 39.Anestal K., Prast-Nielsen S., Cenas N., Arner E.S. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS One. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Q., Antholine W.E., Myers J.M., Kalyanaraman B., Arner E.S., Myers C.R. The selenium-independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. The Journal of Biological Chemistry. 2010;285:21708–21723. doi: 10.1074/jbc.M110.117259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yankaskas J.R., Haizlip J.E., Conrad M., Koval D., Lazarowski E., Paradiso A.M., Rinehart C.A., Jr, Sarkadi B., Schlegel R., Boucher R.C. Papilloma virus immortalized tracheal epithelial cells retain a well-differentiated phenotype. American Journal of Physiology. 1993;264:C1219–1230. doi: 10.1152/ajpcell.1993.264.5.C1219. [DOI] [PubMed] [Google Scholar]
- 42.van der Vliet A., O’Neill C.A., Cross C.E., Koostra J.M., Volz W.G., Halliwell B., Louie S. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am J Physiol. 1999;276:L289–296. doi: 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- 43.Arner E.S., Holmgren A. Current Protocols in Toxicology. Wiley Online Library, Wiley and Sons; Hoboken, NJ: 2001. Measurement of thioredoxin and thioredoxin reductase. Chapter 7, Unit 7 4. [DOI] [PubMed] [Google Scholar]
- 44.Holmgren A., Bjornstedt M. Thioredoxin and thioredoxin reductase. Methods in Enzymology. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 45.Watson W.H., Heilman J.M., Hughes L.L., Spielberger J.C. Thioredoxin reductase-1 knock down does not result in thioredoxin-1 oxidation. Biochemical and Biophysical Research Communications. 2008;368:832–836. doi: 10.1016/j.bbrc.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eckenroth B., Harris K., Turanov A.A., Gladyshev V.N., Raines R.T., Hondal R.J. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry. 2006;45:5158–5170. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lothrop A.P., Ruggles E.L., Hondal R.J. No selenium required: reactions catalyzed by mammalian thioredoxin reductase that are independent of a selenocysteine residue. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang J., Lu J., Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. The Journal of Biological Chemistry. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 49.Oh J.Y., Giles N., Landar A., Darley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochemical Journal. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hristova M., Spiess P.C., Kasahara D.I., Randall M.J., Deng B., van der Vliet A. The tobacco smoke component, acrolein, suppresses innate macrophage responses by direct alkylation of c-Jun N-terminal kinase. American Journal of Respiratory Cell and Molecular Biology. 2012;46:23–33. doi: 10.1165/rcmb.2011-0134OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allan C.B., Lacourciere G.M., Stadtman T.C. Responsiveness of selenoproteins to dietary selenium. Annual Review of Nutrition. 1999;19:1–16. doi: 10.1146/annurev.nutr.19.1.1. [DOI] [PubMed] [Google Scholar]
- 52.Lu J., Holmgren A. Selenoproteins. The Journal of Biological Chemistry. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 53.Papp L.V., Lu J., Holmgren A., Khanna K.K. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxidants & Redox Signaling. 2007;9:775–806. doi: 10.1089/ars.2007.1528. [DOI] [PubMed] [Google Scholar]
- 54.Clarke C., Baghdadi H., Howie A.F., Mason J.I., Walker S.W., Beckett G.J. Selenium supplementation attenuates procollagen-1 and interleukin-8 production in fat-loaded human C3A hepatoblastoma cells treated with TGFbeta1. Biochimica et Biophysica Acta. 2010;1800:611–618. doi: 10.1016/j.bbagen.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Romanowska M., Kikawa K.D., Fields J.R., Maciag A., North S.L., Shiao Y.H., Kasprzak K.S., Anderson L.M. Effects of selenium supplementation on expression of glutathione peroxidase isoforms in cultured human lung adenocarcinoma cell lines. Lung Cancer. 2007;55:35–42. doi: 10.1016/j.lungcan.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Boots A.W., Hristova M., Kasahara D.I., Haenen G.R., Bast A., van der Vliet A. ATP-mediated activation of the NADPH oxidase DUOX1 mediates airway epithelial responses to bacterial stimuli. The Journal of Biological Chemistry. 2009;284:17858–17867. doi: 10.1074/jbc.M809761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorissen S.H., Hristova M., Habibovic A., Sipsey L.M., Spiess P.C., Janssen-Heininger Y.M., van der Vliet A. DUOX1 is required for airway epithelial cell migration and bronchiolar re-epithelialization following injury. American Journal of Respiratory Cellular and Molecular Biology. 2013;48:337–345. doi: 10.1165/rcmb.2012-0393OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology & Medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 59.Anestal K., Arner E.S. Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. The Journal of Biological Chemistry. 2003;278:15966–15972. doi: 10.1074/jbc.M210733200. [DOI] [PubMed] [Google Scholar]
- 60.Nordberg J., Zhong L., Holmgren A., Arner E.S. Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. The Journal of Biological Chemistry. 1998;273:10835–10842. doi: 10.1074/jbc.273.18.10835. [DOI] [PubMed] [Google Scholar]
- 61.Pantano C., Anathy V., Ranjan P., Heintz N.H., Janssen-Heininger Y.M. Nonphagocytic oxidase 1 causes death in lung epithelial cells via a TNF-RI-JNK signaling axis. American Journal of Respiratory Cellular and Molecular Biology. 2007;36:473–479. doi: 10.1165/rcmb.2006-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carrozzino F., Pugnale P., Feraille E., Montesano R. Inhibition of basal p38 or JNK activity enhances epithelial barrier function through differential modulation of claudin expression. American Journal of Physiology—Cell Physiology. 2009;297:C775–787. doi: 10.1152/ajpcell.00084.2009. [DOI] [PubMed] [Google Scholar]