

Abstract
This article provides a succinct but limited overview of the protective and deleterious effects of reactive oxygen and nitrogen species in a clinical context. Reactive oxygen species include superoxide, hydrogen peroxide, single oxygen and lipid peroxides. Reactive nitrogen species include species derived from nitric oxide. This review gives a brief overview of the reaction chemistry of these species, the role of various enzymes involved in the generation and detoxification of these species in disease mechanisms and drug toxicity and the protective role of dietary antioxidants. I hope that the graphical review will be helpful for teaching both the first year medical and graduate students in the U.S. and abroad the fundamentals of reactive oxygen and nitrogen species in redox biology and clinical medicine.
Abbreviations: 4-HNE, hydroxynonenol; 8-OHdG, 8-hydroxy-2-deoxyguanosine; ATP, adenosine triphosphate; BH4, tetrahydrobiopterin; CAT, catalase; CGD, chronic granulomatous disease; cGMP, cyclic GMP; CKD, chronic kidney disease; CO2, carbon dioxide; CO3–, carbonate radical; Cu2+, cupric ion; DOX, doxorubicin; EDRF, endothelial-derived relaxing factor; eNOS, endothelial nitric oxide synthase or NOS-3; GPx, glutathione peroxidase; GSH, glutathione; GSSG, oxidized glutathione disulfide; GTP, guanosine triphosphate; H2O2, hydrogen peroxide; HOCl, hypochlorous acid; IC, intersystem crossing; iNOS, inducible nitric oxide synthase or NOS-2; Keap1, Kelch-like ECH-associated protein 1; LDL, low-density lipoprotein; LOO•, lipid peroxy radical; LOOH, lipid hydroperoxide; MC540, merocyanine 540; MnSOD, manganese superoxide dismutase; MPO, myeloperoxidase; nNOS, neuronal nitric oxide synthase or NOS-1; •NO, nitric oxide; NOS, •NO synthase; NOX, NADPH oxidase; O2•–, superoxide; •OH, hydroxyl radical; ONOO−, peroxynitrite; ONOOCO2−, nitrosoperoxycarbonate; ONOOH, peroxynitrous acid; OS, oxidative stress; PDT, photodynamic therapy; ROS, reactive oxygen species; RNS, reactive nitrogen species; sGC, soluble guanylyl cyclase; SOD, superoxide dismutase; XD, xanthine dehydrogenase; XO, xanthine oxidase
Keywords: Reactive oxygen species, Reperfusion injury, Superoxide, Peroxynitrite
Highlights
► A short course on fundamentals in redox biology of oxyradicals. ► Discusses the good, bad and ugly side of ROS/RNS in biology and medicine. ► Discusses the pathophysiological role of ROS/RNS in drug therapy and clinical medicine.
This review is a first year medical student lecture taught during the Fall semester of 2012 at the Medical College of Wisconsin. I was asked by Dr. Nancy Dahms, the course director of M1 Biochemistry, to present a lecture on the medical relevance of active oxygen and oxidative stress. In the past, this lecture was taught by Dr. Owen Griffith. When I was asked to fill in his shoes (as if anyone could), I felt it was a great honor. I was also inspired by Dr. Barry Halliwell for emphasizing the importance of teaching free radical science to medical students [1]. I hope that this chapter will provide an increased awareness as to why understanding the basics of free radicals in disease mechanisms is so vital for providing better treatment and enhanced quality of life in clinical patients.
Introduction to reactive oxygen and nitrogen species (ROS/RNS)
A free radical is an atom or molecule with a single unpaired electron. Examples: Nitric oxide (•NO), superoxide (O2•–), hydroxyl radical (•OH), lipid peroxy radical (LOO•). Although molecular oxygen (O2) has two unpaired electrons in two different orbitals, it is not a free radical. Molecular oxygen, however, reacts rapidly with most other radicals, forming other free radicals that are more reactive and cause selective oxidation of lipid, protein, or DNA molecules [2].
Oxygen, or molecular oxygen, is vital for survival of all aerobic organisms. During aerobic metabolism in normal cells, 30–32 molecules of adenosine triphosphate (ATP) are generated from one molecule of oxygen. During this process, oxygen is reduced into water (4 electron reduction) (Fig. 1). Oxygen is an essential co-factor in many biological oxidations and drug metabolism catalyzed by monoxygenases or mixed function oxidases (cytochrome P450).
Fig. 1.
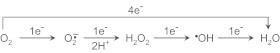
Sequential reduction of O2 to H2O.
In a perfect world, cells will use oxygen to produce ATP and water without any toxic byproducts or activated (also active) oxygen species. However, damage to mitochondria in pathophysiological conditions or during mitochondrial dysfunction, the electron-transport mechanism in the mitochondrial respiratory chain is impaired. This leads to reactive oxygen species formation (e.g., superoxide anion) from one-electron reduction of oxygen. Fig. 2 shows the molecular orbital diagrams of oxygen, superoxide, peroxide dianion, and the singlet oxygen. It is evident from the molecular orbital representation that not all active oxygen species is a free radical.
Fig. 2.
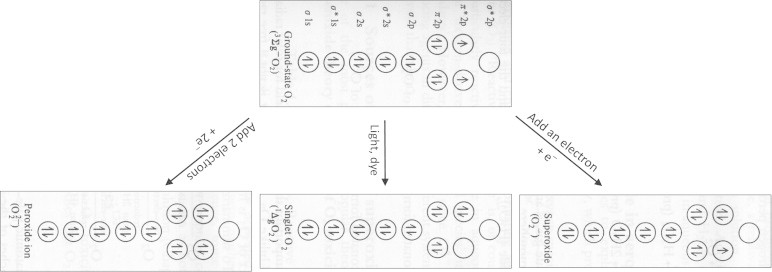
Molecular orbital representation of oxygen and active oxygens.
Superoxide is a free radical (species with a single electron) as is hydroxyl radical. Hydrogen peroxide is not a free radical, but is a precursor of free radicals. UV radiation causes the hemolytic cleavage of the oxygen–oxygen bond to form hydroxyl radicals (•OH). Redox metal ions (Fe2+ or Fe3+ or Cu+) react with hydrogen peroxide to generate hydroxyl radicals (the Fenton reaction). Singlet oxygen is not a free radical and is a photoexcited oxygen species. In terms of reactivity with biological constituents (nucleic acids, proteins, and lipids), hydroxyl radical is the most reactive, and the lifetime of this species in the presence of most biological constituents is extremely short (10–9 s). There is no selectivity for hydroxyl radical reactions—this species will react with most compounds at a nearly diffusion-controlled rate (1010 M–1 s–1).
Some antitumor drugs (e.g., doxorubicin) exert toxic side effects in cardiac myocytes through this mechanism [3]. One way to counteract the toxic side effect of this chemotherapeutic is through iron chelating molecules that tie up the iron ions and inhibit formation of hydroxyl radicals. Superoxide is much less reactive and much more selective. Its lifetime in biological systems is a few seconds. Superoxide rapidly reacts with another molecule of superoxide (self-dismutation reaction) to form hydrogen peroxide. Another important reaction in biology is the diffusion-controlled reaction between superoxide with nitric oxide (radical–radical reaction) to form a very potent oxidant and reactive nitrogen species, peroxynitrite (half-life, seconds) [4]. Hydrogen peroxide (H2O2) is relatively stable (half-life, months) when protected against light and trace metal contamination. H2O2 is, however, rapidly destroyed by antioxidant enzymes (catalase, glutathione peroxidase). Singlet oxygen reacts rapidly with histidine and cysteinyl groups present in proteins, unsaturated lipids and some nucleic acids [2]. Typically this species has a half-life of around 10 μs. Another immunologically relevant reactive oxygen species is hypochlorous acid (HOCl, or bleach). HOCl is formed from myeloperoxidase/H2O2-dependent oxidation of chloride anion. HOCl has intermediate reactivity-more reactive than H2O2 but considerably less reactive than hydroxyl radicals. Fig. 3 summarizes the reactions initiated by reactive oxygen and nitrogen species.
Fig. 3.
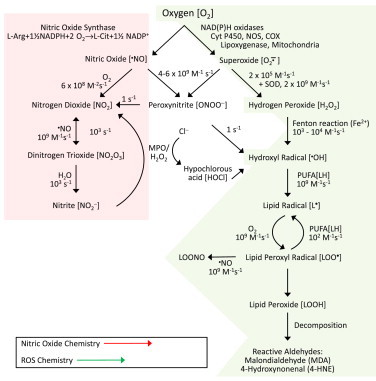
Reactive oxygen and nitrogen species: generation and reactions.
In contrast to normal cells, cancer cells avoid using the aerobic metabolism to produce ATP, and they utilize a rather inefficient pathway (anerobic glycolysis) to generate 2 molecules of ATP. Cancer cells are metabolically reprogrammed to make the switch from oxidative phosphorylation to glycolysis. Cancer cells are also hypoxic (less oxygen), which makes it harder to kill tumor cells more effectively during radiation and chemotherapy, that are based on selective generation of damaging reactive oxygen species. Some of the newer antitumor compounds force the cancer cells to use mitochondrial oxidative phosphorylation pathway to derive its source of energy (ATP). However, the mechanisms underlying this cytotoxic switch in cancer cells are not very clear.
Superoxide (the good side): respiratory burst or oxidative burst
Phagocytes (neutrophils, monocytes, eosinophils) kill invading pathogens and defend the host through generation of antimicrobial oxidants such as O2•– or O2•–-derived oxidants during the respiratory burst or enhanced oxygen consumption. Note: The respiratory burst can also be stimulated by opsonized microorganism (opsonized zymosan) and chemicals such as phorbol myristate acetate (PMA). Although O2•– itself is only weakly microbicidal, oxidants generated from O2•– are strongly microbicidal. The enzyme responsible for O2•– generation was shown to be a membrane-bound flavoprotein or the NADPH oxidase that uses NADPH as a co-factor. This enzyme is the cells come in contact with bacteria and other appropriate stimulus. As discussed earlier, the active NADPH oxidase assembly comprises of multiple cytosolic and plasma membrane components and catalyzes the one-electron reduction of oxygen to superoxide at the expense of NADPH [5].
The schematic illustration of the phagocytic process is shown in Fig. 4. The phagocyte finds a bacteria marked as foreign (i.e., coated by the complement or immune system) [2]. The phagocyte plasma membrane surrounds the bacteria and engulfs the microprobe into a phagocytic vacuole. Enzymes (NADPH oxidase) are selectively activated in the wall of vacuole, generating O2•– and H2O2 in the vacuolar lumen. Activation of NADPH oxidase requires translocation of several key cytosolic proteins (p44phox, p67phox, p40phox, and rac2) to the membrane. Although O2•– production is essential for bacterial cell killing, O2•– alone is not responsible for bacterial killing. H2O2 in the presence of released iron could form hydroxyl radical, a potent oxidant, via the Fenton mechanism (Fe2++H2O2→Fe3++•OH+OH−) or the Haber–Weiss mechanism (+O2). Hydroxyl radical is a potent antimicrobial oxidant [6].
Fig. 4.
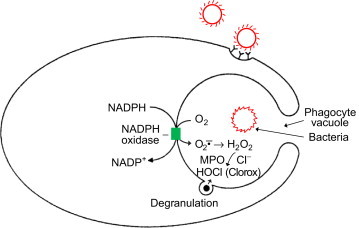
Schematic representation of a phagocyte engulfing a microbe into a phagocyte vacuole. The NADPH oxidase is selectively activated in the wall of the vacuole, generating superoxide and hydrogen peroxide in the vacuolar lumen. Enzymes are also released into the vacuole by degranulation of cytoplasmic granules.
The enzyme, myeloperoxidase (MPO), is released inside the phagocytic vacuole from granules and comprises 2–5% of total neutrophil protein. MPO is a heme-containing peroxidase that will oxidize halide anion (chloride, bromide, and iodide anions) in the presence of H2O2, forming the corresponding hypohalous acid. In the case of chloride anion, hypochlorous acid (HOCl) or Clorox (bleach) is formed. HOCl kills many bacteria and fungi in vitro. HOCl is very reactive and causes oxidation and chlorination of biological molecules. Although HOCl plays a role in bacterial killing by phagocytes, O2•– is more important than MPO in the overall bacterial killing during respiratory burst [7].
Chronic granulomatous disease (CGD) is a rare genetic disease of the immune system and is an inherited deficiency of NADPH oxidase [8]. CGD is caused by defects in one of the five subunits of phagocyte-derived NADPH oxidase including gp91phox, p22phox, p47phox, p67phox, and p40phox. Consequently, the NADPH oxidase is not fully functional in CGD patients, and the production of superoxide and other reactive intermediates is impaired in their phagocytic cells, leading to infections and other complications [2]. Neutrophils isolated from blood from CGD patients generate less O2•– and oxidative respiratory burst compared to healthy individuals. Neutrophil-derived O2•– and reactive oxidant formation may be used as a diagnostic indicator of the severity of the disease, and a strong predictor of the overall survival of CGD patients. Phagocytes of CGD patients lack the antimicrobial arsenal needed to “kill” the engulfed bacteria. CGD patients do not make sufficient O2•– and H2O2. As H2O2 is the substrate for the enzyme myeloperoxidase, neutrophils from CGD patients do not make hypochlorous acid or the bleach, a commonly used antibacterial agent.
Reperfusion injury—clinical implications (the bad side of O2•–)
Ischemia (or deprivation of oxygen) and regional ischemic episodes occur in several clinical situations due to severe restrictions in blood flow; for example, in heart tissues during myocardial infarction or in brain tissues during stroke. Clearly, the appropriate medical intervention is to restore oxygen immediately to tissues by removing the blockage in the artery (by administering clot dissolving drugs, tissue plasminogen activator (TPA), or use of balloon catheters). Although this treatment is mostly beneficial, resumption of blood flow (reoxygenation or reperfusion) in ischemic tissues has been shown to cause tissue damage or reperfusion injury, depending on the ischemic duration. This phenomenon is known as the “oxygen paradox”. It was proposed that reperfusion-derived free radicals were responsible for postischemic tissue injury [9]. The proposed biochemical mechanism for enhanced generation of superoxide during reperfusion is shown in Fig. 5.
Fig. 5.
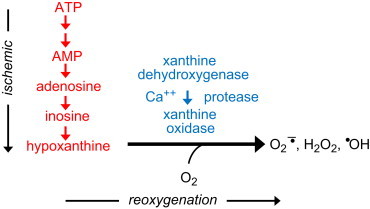
Proposed mechanism for ischemia-induced formation of reactive oxygen species. [Modified from: McCord, J. M. Oxygen-derived free radicals in postischemic tissue injury. New England Journal of Medicine312:159–163; 1985.]
Two major changes occur in ischemic or oxygen-deprived tissues: first, ischemic conditions promote the conversion of xanthine dehydrogenase (XD) to xanthine oxidase (XO). The enzyme XD is unable to transfer electrons to molecular oxygen, although it uses NADPH as an electron acceptor. The conversion of XD to XO involves either the oxidation of the sulfhydryl group or proteolysis triggered by the activation of a cytosolic protease as an adaptive mechanism for calcium transport in energy-starved cells. The molecule calmodulin also plays a role in proteolytic activation. The second major biochemical change that occurs in ischemic tissues is depletion of ATP to AMP, catabolism of AMP, and eventually accumulation of hypoxanthine, a purine substrate for the enzyme XO. Actually the buildup of hypoxanthine has been demonstrated in ischemic myocardium. As indicated earlier, the XO activity converts xanthine or hypoxanthine to uric acid, and molecular oxygen to superoxide and hydrogen peroxide. Ischemic conditions also promote the release of redox-active metals (e.g., iron) that can enable more potent oxidant (hydroxyl radical via the Fenton mechanism) formation. Thus, the biochemical machinery is ripe for increased oxygen-derived radical formation under ischemic conditions, and is facilitated by reperfusion that provides the remaining substrate, molecular oxygen [10].
Increased formation of oxygen-derived radicals is one of the factors of reperfusion-induced injury in many ischemic diseases of the heart, kidney, brain, liver, bowel, and in organ transplantation [10]. One way to protect against reperfusion-induced tissue damage is to use enzymatic scavengers of superoxide and hydrogen peroxide (SOD and CAT), iron chelators (deferoxamine), and inhibitors of the enzyme, XO (allopurinol, oxypurinol) [9].
Organ preservation and transplantation
Reperfusion injury is a potential complicating factor in the transplant surgery. Typically, once a donated organ (e.g., kidney) is removed from a cadaver, packed in ice, and maintained at low temperature in a preservation liquid, then transplanted and reperfused in the recipient, the transplanted organ can suffer oxygen radical-mediated reperfusion injury. This can lead to organ necrosis and organ failure. In preclinical studies, it has been shown that pretreatment with allopurinol or antioxidant enzymes improves organ function and graft survival [9].
The University of Wisconsin (UW) solution/Carolina rinse solution: strategies in mitigating storage/reperfusion injury
Not all cells are equally affected during storage/reperfusion process. For example, hepatocytes are very resistant to storage/reperfusion injury, even after many hours of cold hypoxia/anoxia. Multiple mechanisms and multiple cell types contribute to storage/reperfusion injury. The UW solution, introduced in the 1980s, is being used as a gold standard for organ preservation [11]. This solution contains multiple components: antioxidants (glutathione, alpha-tocopherol, ascorbic acid), apllopurinol, ATP precursors, amino acids, bioenergetics precursors and electrolytes.
In many respects, the Carolina rinse solution is similar in composition to the UW solution. This solution has been mostly used to mitigate storage/reperfusion injury to the liver. Different cells (endothelial cells and Kupffer cells) play a role in reperfusion injury after prolonged liver storage (Fig. 6).
Fig. 6.
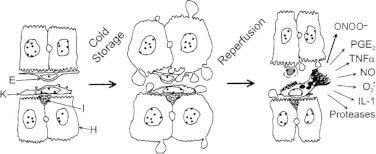
Reperfusion injury to liver cells. [Modified from: Lemasters, J. L.; Thurman, R.G. Reperfusion injury after liver preservation for transplantation. Annual Review of Pharmacology and Toxicology37:327–338; 1997.].
Activated Kupffer cells, upon reperfusion, release •NO, O2•–, and ONOO−, in addition to other inflammatory mediators (cytokines). Clearly, antioxidant strategies to improve organ preservation, graft survival, and organ function are a fruitful area of research investigation.
Singlet oxygen (the good side)
The singlet oxygen or the electronically excited oxygen differs from molecular oxygen in its electronic configuration. Whereas the oxygen that we breathe is in the triplet state with two unpaired electrons and is relatively unreactive with biological components, the singlet oxygen has no unpaired electron and both electrons occupy the same molecular orbital, leaving an empty orbital for reaction with other molecules. In contrast to molecular oxygen, the singlet oxygen reacts rapidly with most biological constituents and causes oxidative damage (e.g., conversion of an unsaturated lipid molecule to a lipid peroxide molecule) [2].
The singlet oxygen is formed from illuminating a photosensitizing dye (S) with visible light in the presence of oxygen [12]. The photosensitizing dye (e.g., Photofrin or merocyanine 540) absorbs the light energy in the visible region and activates the dye to form the excited singlet state (reaction 1) which undergoes intersystem crossing (IC) to the relatively long-lived triplet state dye (reaction 2), as shown below. The triplet state dye molecule interacts with molecular oxygen (transfers the excess energy to molecular oxygen), forming the singlet oxygen (reaction 3) which immediately reacts with the cellular target (membrane or protein) and causes oxidative damage (Fig. 7).
Fig. 7.

Dye-sensitized photoactivation of oxygen.
Photodynamic therapy (PDT)
PDT refers to the selective destruction of tumor c ells in the presence of a sensitizer (usually a dye molecule), visible light, and oxygen [12]. Selective cytotoxicity to tumor cells originates from selective binding to tumor cell membrane and accumulation/retention of the dye in tumor cells and/or from a localized light delivery and irradiation of tumor cells leading to singlet oxygen generation in tumor tissues. PDT is an anticancer therapy that results in localized tissue destruction due to focused light delivery and laser light. In one of the landmark clinical studies, investigators showed successful ablation of Barrett's esophagus with high-grade dysplasia using PDT [13].
Selective purging of cancer cells from bone marrow
Researchers at MCW discovered a novel PDT-based approach to selectively purge leukemic cells from bone marrow [14]. PDT was performed using a fluorescent dye, merocyanine 540 (MC540). MC540 selectively binds to the outer surface of the plasma membrane of leukemic cells which is responsible for the differential sensitivity of normal and neoplastic cells to MC540/PDT [14]. Simultaneous exposure to MC540 and visible light rapidly killed leukemic cells but spared the normal cells. Basic mechanistic studies revealed that singlet oxygen plays a major role in the MC540-mediated photodynamic killing of leukemic cells. MCW researchers were also involved in the MC540/PDT therapy (phase 1/11 clinical trials) for the purging of autologous bone marrow transplants in leukemia and lymphoma patients.
Singlet oxygen phototoxicity (the bad side)
Increased photosensitivity of the skin is the most common form of adverse side effects. The photosensitizing dye typically remains in the skin at low levels for a long period of time. Patients with photosensitive skin should be protected from sunlight for several months after treatment. The deleterious effects to normal cells during PDT result from singlet oxygen-mediated damaging effects.
Several disorders can result in excessive formation of singlet oxygen in the presence of light, including porphyria (defective heme biosynthesis). In cutaneous porphyrias, porphyrins accumulate in the skin. Exposure to light causes skin blistering and swelling mediated by singlet oxygen formation. Several anti-inflammatory drugs (e.g., benoxaprofen) have been discontinued due to potential phototoxicity arising from light-induced singlet oxygen formation.
Nitric oxide (the good side)
Depending upon their concentrations, not all free radicals are damaging and bad. For instance, •NO, a free radical gas, is an important intracellular messenger molecule that activates the soluble guanylyl cyclase (sGC) at nanomolar concentrations. Guanylyl cyclase is responsible for converting guanosine triphosphate (GTP) to cyclic GMP, which causes smooth muscle relaxation [15].
Prior to 1980, •NO, which is present in automobile exhaust and cigarette smoke, was largely considered as an environmental pollutant. This view of •NO changed in the 1980s when Robert Furchgott discovered that the endothelium released a labile factor, termed “endothelial-derived relaxing factor” (EDRF), which caused vascular relaxation when vascular smooth muscle cell preparations were stimulated with vasodilators (e.g., acetylcholine, histamine, and bradykinin) [16]. EDRF was also called “nitrovasodilator” because nitrovasodilators such as nitroglycerin, nitroprusside, other organic nitrites, cause vascular relaxation [17]. Subsequently, it was proposed that EDRF is •NO. For this discovery, a group of investigators (Ignarro, Furchgott, and Murad) received the Nobel Prize in Physiology/Medicine in 1998.
Clinical significance of •NO and cGMP signaling
Male impotence is a major, worldwide clinical problem. Abnormal vascular responsiveness leading to impaired relaxation of the smooth muscle of the corpus cavernosum was proposed as the underlying cause of impotence and erectile dysfunction [18]. •NO and cyclic GMP mediate the relaxation of the corpus cavernosum in response to stimulation by nonadrenergic, noncholinergic neurons [18,19]. This seminal finding paved the way for clinical treatment of erectile dysfunction with the drug, sildenafil (Viagra) that increase the steady-state levels of cyclic GMP in the corpus cavernosum vessels (Fig. 8). The use of this drug in patients who are taking organic nitrates (e.g., nitroglycerin) for angina may result in serious drug interactions causing hypotension and arrhythmias.
Fig. 8.
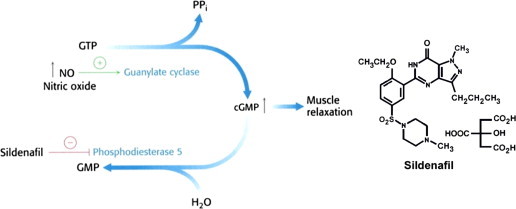
Sildenafil-induced smooth muscle cell relaxation.
Peroxynitrite (the ugly side of O2•– or the bad side of •NO?)
Peroxynitrite
The existence of the species peroxynitrite (ONOO−) has been demonstrated many years ago. Peroxynitrite anion is in equilibrium with the corresponding peroxynitrous acid (ONOOH). The pKa of this equilibrium is 6.8. Thus, under physiological conditions (pH=7.4), peroxynitrite exists mostly in the anionic form (80%) with about 20% in the protonated form. The anionic form, ONOO−, also reacts with the carbon dioxide molecule that is typically present in millimolar levels in cells. This reaction forms a very reactive adduct, nitrosoperoxycarbonate (ONOOCO2−) [20]. Both ONOO− and ONOOCO2− decays rapidly at physiological pHs forming other radicals (•OH, CO3–) that are more potent and oxidizing in nature. Peroxynitrite oxidizes protein thiols forming disulfide, nitrates tyrosyl groups in proteins forming nitrated proteins. One of the consequences of nitrating tyrosyl groups in protein is either a loss or gain of function in the activity (e.g., manganese superoxide dismutase; MnSOD). Nitration can interfere with signal transduction mechanism. Potentially, nitration decreases the pKa of the –OH group of the tyrosine from about 10 down to 7.5. As a result, the –OH group is ionized to –O−. This shift in the ionization of the –OH group present in the tyrosine group could impede phosphorylation and signaling mechanism. Peroxynitrite oxidizes unsaturated fatty acids in biological membranes to form nitrated fatty acids that show multiple and potent biological activities [21]. Nitrated derivatives of oleic, linolenic, and arachidonic acids were detected and characterized in human body fluids [22]. The reaction between peroxynitrite and DNA leads to multiple oxidized products from purine and pyrimidine bases. Some of the biomarkers of oxidative DNA damage (e.g., 8-hydroxy-2-deoxyguanosine (8-OHdG) and 8-nitroguanine) are formed from the reaction between peroxynitrite and DNA [23]. From the above, it is clear that formation of this species in cells would lead to cytotoxicity.
How is peroxynitrite formed in cells? Is it a viable intracellular reaction? It was shown that •NO and O2•– reacts at a nearly diffusion-controlled rate to form the peroxynitrite [4]. This is significant because the higher the value of the rate constant, the greater is the likelihood of the two species reacting to form the product. Thus, when these two species are formed in cells, despite the presence of radical scavengers and antioxidant enzymes, they will react because of the higher rate constant. This was first demonstrated in endothelial-derived relaxing factor (EDRF) measurements. Addition of SOD (enzyme that removes O2•– by catalyzing its dismutation reaction to H2O2) enhanced the relaxing mechanism by increasing the levels of •NO [24]. This reaction is also critical to the process of vasoconstriction and controlling the ratio of •NO and O2•– in hypertension.
What are the consequences of this reaction? Potentially, there are two consequences and both of which are bad. The •NO/O2•– reaction forms a more cytotoxic species. Second, it removes •NO required for normal vasodilation.
The enzymatic sources of •NO and ROS, and how they can be controlled
Enzymatic synthesis of •NO
Studies showed that •NO is produced from metabolism of the amino acid, l-arginine. The enzyme(s) catalyzing this metabolic pathway are known as nitric oxide synthases (•NO synthases, or NOS). NOS converts l-arginine to l-citrulline and •NO via a 5-electron oxidation of one of the guanidine nitrogens of l-arginine (Fig. 9). [25]. In the first two-electron oxidation step, N-hydroxy-l-arginine is formed as an enzyme-bound intermediate. In the second three-electron oxidation step, •NO is formed along with l-citrulline. The oxygen atom incorporated into •NO and l-citrulline originates from distinct molecules of oxygen during the catalytic turnover to generate •NO.
Fig. 9.
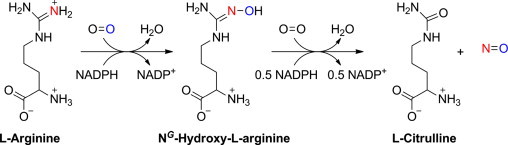
Enzymatic generation of •NO from l-arginine.
In addition to NADPH and oxygen, this reaction (NOS-catalyzed oxidation of l-arginine to •NO) also requires additional co-factors, calmodulin, calcium, and tetrahydrobiopterin (BH4).
Inhibitors of •NO biosynthesis
N-substituted analogs of l-arginine that inhibit the biosynthesis of •NO from l-arginine by all three NOS isoforms. These include N-methyl, N-nitro, and N-amino l-arginine (l-NAME, l-NMMA). Compounds, like 1400-W, inhibit only the iNOS isoform and not others. Similarly, there are nNOS-specific inhibitors as well. These agents are competitive inhibitors of the enzyme [26].
As mentioned earlier, BH4 is a key co-factor involved in the biosynthesis of •NO. Deficiency in intracellular BH4 levels caused by decreased biosynthesis of BH4 has been shown to inhibit •NO production. Thus, agents that modify BH4 synthesis and metabolism such as sepiapterin will affect endogenous •NO levels in cells and tissues.
There are three isoforms of NOS that have been characterized, purified, and cloned in chronological order: neuronal nitric oxide synthase (nNOS) or NOS-1, inducible nitric oxide synthase (iNOS) or NOS-2, and endothelial nitric oxide synthase (eNOS) or NOS-3. The three isoforms are encoded by three different genes on different chromosomes and share nearly 50–60% homology with each other and with the cytochrome P-450 enzymes.
NOS isoforms located in different cellular compartments generate •NO for different reasons: nNOS in neurons makes •NO for communication between nerve cells; iNOS in macrophages and smooth-muscle cells makes •NO in large quantities of •NO as part of its killing mechanism in response to lipopolysaccharide, cytokines, and glucocorticoids. Of course, if this form gets out of control, as occurs in septic shock and inflammatory disorders, severe consequences (death) follow; eNOS found in endothelium, brain, and heart makes •NO to relax blood vessels and maintain normal blood pressure.
Superoxide and hydrogen peroxide generation from NADPH oxidases
There are several enzymatic sources of superoxide, H2O2, and oxidants derived there from. Xanthine/xanthine oxidase reduces oxygen through a one-electron as well as a two-electron reduction mechanism, forming superoxide or H2O2 as a primary intermediate. During this process, xanthine is converted to uric acid. Allopurinol and oxypurinol inhibit formation of superoxide and H2O2 from xanthine oxidase. This enzyme is thought to play a major role in ischemia/reperfusion injury [27].
Other enzymes that have been proposed to generate reactive oxygen species are cytochrome P-450, cyclooxygenase, lipoxygenase, and mitochondria. However, these enzymes are not typical ROS generating systems, and their major property is not to generate O2•– and H2O2 as primary reactive species during their catalytic activity. For example, lipoxygenase oxidizes linoleic acid to generate biologically active lipid-derived products (HETEs) and cyclooxygenase forms prostaglandins from arachidonic acid. The primary function of mitochondria is to generate ATP. However, aberrant functioning of these systems in diseases or environmental toxicity generates ROS as a byproduct.
In contrast, NADPH oxidases (NOX) are a family of enzymes that generate ROS (either O2•– or H2O2) as the primary species during the catalytic metabolism of oxygen (Fig. 10). As indicated elsewhere, the phagocytic NOX use ROS to kill bacteria. In most inflammatory diseases and infections, enhanced ROS formation occurs predominantly from NOX activation.
Fig. 10.
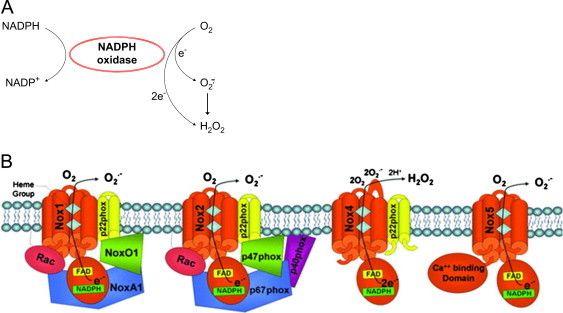
(A) Generation of reactive oxygen species from NADPH oxidases. (B) NOS structure and function. [Modified from: Streeter, J., et al. Cardiovascular Therapy; January 26 2012. http://dx.doi.org/10.1111/j.1755-5922.2011.00310.x. (Epub ahead of print)].
The NADPH oxidase is not a single enzyme, but it is a multi-enzyme complex. Several isoforms of NOX are expressed in mammalian cells. NADPH is the electron donor for all NOX isoforms (NOX1–5). Each of these isoforms contains a core catalytic subunit (NOX) and several regulatory subunits that are located in the cytosolic and membranous compartments (Fig. 10B). The various components are assembled at the plasma membrane in functionally active NADPH oxidase (or during activation) that transfers electrons from NADPH to oxygen producing O2•– or H2O2. Some NOXs form O2•– and others form H2O2. But their only known function is to generate ROS. Because of their role in inflammation, infection, and immune responses in various diseases, major pharmaceutical companies are actively involved in drug discovery targeting the different NOX isoforms.
Inhibition and interception of ROS and RNS
Aerobic organisms defend themselves against reactive oxygen and nitrogen species through enzymatic and non-enzymatic detoxication mechanisms. The enzymatic detoxification mechanisms involve antioxidant enzymes (superoxide dismutases, catalases, and peroxidases), small molecular-weight antioxidants (Vitamin E, Vitamin C, glutathione, ubiquinone, beta-carotene, etc.), and adaptive mechanism leading to antioxidant gene expression.
There are at least three types of superoxide dismutases (SODs): copper, zinc, SOD (cytosolic), manganese SOD (mitochondria), and EC-SOD (extracellular). SOD catalytically enhances the normal dismutation of superoxide (O2•–+O2•–+2H+→H2O2+O2). The rate constant for the normal dismutation is=105 M–1 s–1. In the presence of catalytic amount of SOD, the rate constant is increased to 109 M–1 s–1. In other words, the rate of this reaction is enhanced by 104-fold. The reaction mechanism of SOD (Enzyme-Cu2+) is shown in Scheme 1.
Scheme 1.
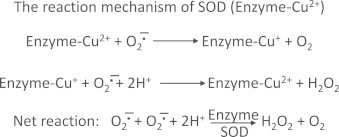
Superoxide reduces Cu2+ forming Cu++ and O2, and another superoxide molecule oxidizes Cu++ back to Cu2+, forming hydrogen peroxide. SOD is normally present at low micromolar concentrations in cells. This makes SOD-catalyzed dismutation of superoxide very feasible. By decreasing the intracellular steady state levels of s uperoxide, SOD could successfully inhibit the reaction between O2•– and •NO and the formation of a potent oxidant, ONOO−. In addition, SOD could prevent the release of iron ions from the iron–sulfur cluster enzymes, as superoxide is known to rapidly react with the iron–sulfur centers and release iron. Thus, SOD can also indirectly prevent the formation of hydroxyl radical from the Fenton reaction.
However, as shown earlier, SOD does not prevent the formation of H2O2. In some cases, SOD actually enhances H2O2 formation in cells. In contrast to O2•–, H2O2 is more stable and could freely diffuse to other compartments in cells. H2O2 is detoxified to water by catalases and peroxidases. The reaction between catalase (CAT) and H2O2 is shown in Scheme 2.
Scheme 2.
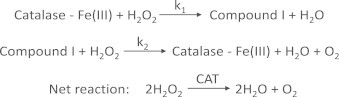
As shown, CAT reduces H2O2 to O2 and H2O. CAT is more important when H2O2 levels are low. At higher concentrations of H2O2, glutathione peroxidase enzyme (GPx) that is dependent on reduced glutathione is responsible for detoxifying H2O2. During this process, reduced glutathione (GSH) is oxidized to oxidized glutathione disulfide (GSSG) which is then reduced back to GSH by glutathione reductase using NADPH as a co-factor. The GPx enzyme is also responsible for detoxifying other lipid peroxides (LOOH) to the corresponding alcohol (LOH). The dynamics of the whole antioxidant enzyme machinery is shown in Fig. 11.
Fig. 11.
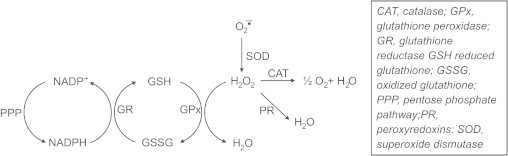
Enzymatic detoxification of hydrogen peroxide.
Phospholipid hydroperoxide-dependent glutathione peroxidase (PHGPx) detoxifies phospholipid hydroperoxides in the same way as GPx detoxifies lipid hydroperoxides. Phospholipid hydroperoxides that are located inside the membrane are not accessible to GPx. However, GPx and PLA2 (phospholipase A2, an enzyme that hydrolyzes phospholipid hydroperoxides and releases lipid hydroperoxides) are capable of detoxifying phospholipid hydroperoxides [28].
In addition to these enzymes, other mechanisms (peroxiredoxin, thioredoxin) are involved in detoxifying intracellular hydroperoxides. Peroxiredoxins are ubiquitous class of antioxidant or signaling proteins present at high concentrations (50–100 μM) in cytoplasm and mitochondria. Peroxiredoxins react very rapidly with H2O2 (k>107 M–1 s–1) [29].
In the presence of catalytic amounts of redox-active metals (copper and iron ions), aldehydes (MDA, 4-HNE) are formed as byproducts of lipid hydroperoxide decomposition. These products, when formed in excess, are biologically toxic as they will react with proteins and macromolecules and cause inactivation. However, as will be shown later in this section, these electrophiles are also able to induce antioxidant gene expression through activation of the Keap1–Nrf2 system. Aldehydes (MDA or 4-HNE) are detoxified by glutathione transferases which enables the conjugation of thiols (glutathione) forming the aldehyde–GSH conjugate [30]. Note: This type of detoxification reaction is very similar to the removal of N-acetylquinone amine by N-acetylcysteine.
The concentration of metal ions such as iron is regulated by ferritin and ceruloplasmin. Ferritin has numerous binding sites for iron. The cell will overexpress ferritin in order to protect itself from iron toxicity. Ferritin can indirectly inhibit formation of hydroxyl radicals and other toxic aldehydes.
Intracellular iron is tightly controlled by a very sophisticated iron-signaling mechanism. Under conditions of iron deficiency, cells acquire iron via increased synthesis of transferrin receptor that facilitates the uptake of iron-transferrin. During iron overload, TfR receptor synthesis is decreased and ferritin receptor synthesis is increased.
In addition to enzymatic mechanisms of protection again reactive oxygen species, there exist non-enzymatic small molecular weight antioxidants: ascorbic acid (also vitamin C), alpha-tocopherol (also vitamin E), reduced glutathione (present in millimolar levels in cells), beta-carotene (Fig. 12).
Fig. 12.
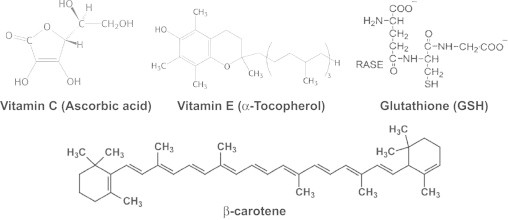
Structures of well-known antioxidants.
Antioxidants act in different ways. It is important to take into account the localization of antioxidants. These small molecular weight antioxidants are not able to deal with H2O2 as they will only react really slowly (if at all) with H2O2. However, vitamin E which is lipophilic (located in the biological membrane) will react with the lipid peroxyl radical produced during lipid peroxidation process. The vitamin E radical reacts with the lipid peroxyl radical and this reaction is a chain termination event. As a result, vitamin E is a very effective chain-breaking antioxidant that can stop lipid peroxidation in its tracks (so to speak). In contrast to vitamin E, vitamin C is water-soluble and will react rapidly with a variety of reactive oxygen species including superoxide and hydroxyl radical forming the ascorbate radical, which reacts with another ascorbate radical forming dehydroascorbate and ascobate. Ascorbate is, therefore, termed as a cytosolic antioxidant.
Although ascorbate is present in the cytosol, it can recycle the membrane-associated vitamin E radical back to vitamin E, thereby increasing the total antioxidant potential. The Pentose Phosphate Pathway keeps the ascorbate and vitamin E in the reduced form through the intracellular reductant, GSH (Fig. 13).
Fig. 13.
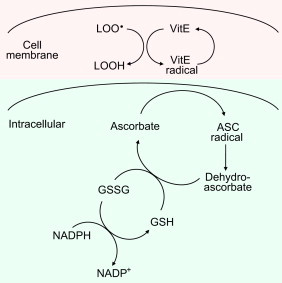
The cross-talk between lipophilic and hydrophilic antioxidants.
Oxidative stress, oxidative damage, and adaptation to oxidative stress
What is oxidative stress (OS)?
This is one of the most frequently used terms in free radical biology. OS refers to the imbalance between the prooxidant and antioxidant levels in favor of prooxidants in cells and tissues. This scenario leads to modification of lipids, proteins, and DNA. Such modifications or damage to these molecules are called oxidative damage. Prooxidant is a species that causes or promotes oxidation. Antioxidant is a molecule inhibits formation of prooxidants and inhibits oxidation. Antioxidants are usually present at a low concentration and yet inhibit oxidation of an oxidizable substrate.
Oxidative stress can result from diminished antioxidant levels or antioxidant enzymes or defects in antioxidant machinery expressing the antioxidant gene. Depletions of dietary antioxidants (e.g., vitamins E, C, and D, flavonoids and carotenoids) and micronutrients (e.g., iron, copper, zinc, selenium) that are needed for proper functioning of antioxidant enzymes (catalase, copper, zinc SOD, or glutathione peroxidase) results in the accumulation of prooxidants (free radicals or reactive oxygen species), leading to oxidative stress. Several diseases (e.g., kwashiorkor disease) associated with protein deficiency in children are caused by low levels of glutathione and enhanced iron (iron overload). Administering antioxidant therapy might be beneficial under these conditions.
Oxidative stress can also result from increased production of reactive oxygen species from exposure to toxins (e.g., paraquat or cigarette smoke) and activation of endogenous enzymes (e.g., NADPH oxidases) in chronic inflammatory conditions.
Oxidative damage
One of the consequences of oxidative stress is cell injury caused by oxidative damage. Targets of oxidative damage are DNA, lipid, and protein. Both purine (adenine and guanine) and pyrimidine (cytosine and thymine) react with hydroxyl radical to form specific products [2].
However, not all reactive species (O2•– and H2O2 or •NO) react with DNA bases or with the deoxyribose. •NO and O2•– together reacts with DNA bases. Hydroxy radical adds to the C-8 position of guanine and forms 8–hydroxyguanine (8-OH-guanine). Other DNA bases are also oxidized similarly by hydroxyl radical. A major consequence of oxidative damage to DNA bases is mutation caused by AT↔GC transition and by GC↔TA transversion. These mutations, if unrepaired, can lead to changes in protein gene expression.
Protein modifications caused by ROS/RNS include formation of carbonyls, dityrosine and nitrated and chlorinated tyrosines. Nitrated, chlorinated, and brominated tyrosines have been detected in high levels in diseased tissues in patients with inflammatory diseases.
Lipids (containing conjugated double bonds) undergo peroxidation in the presence of ROS/RNS and oxygen. Arachidonic acid with four double bonds is more easily oxidized than linolenic acid (three double bonds) and linoleic acid (two double bonds). Oleic acid with a single double bond is not peroxidized. During this chain oxidation reaction, several molecules of lipid (LH) are oxidized. In the initiation process, oxidants (hydroxyl radical) abstract the allylic hydrogen forming the carbon-centered lipid radical. This radical undergoes a molecular rearrangement to produce a more stable carbon-centered radical (L•) which rapidly reacts with oxygen to form a lipid peroxy radical (LOO•). In the propagation phase, the LOO• abstracts a hydrogen from another lipid molecule generating a new L•. (that continues the chain reaction) and lipid hydroperoxide (LOOH). This process will go on forever unless terminated by phenolic antioxidants (vitamin E or BHT). In the termination reaction, vitamin E or BHT donates a hydrogen atom to the LOO• species and forms a corresponding vitamin E or BHT radical that reacts with another LOO• forming non-radical products. These lipophilic phenolic antioxidants are known as the chain-breaking antioxidants. The rancidity of butter is due to the lipid peroxidation process, and the food additive BHT effectively terminates lipid peroxidation and keeps the butter fresh even when left outside at room temperature. Atherosclerosis, the formation of plaques in the arteries with time, is initiated from lipid peroxidation (the rancidity of the body fat) of low-density lipoprotein (LDL) (Fig. 14). The LOOH molecule is unstable and one usually detects isoprostanes or 4-HNE as specific end products of peroxidized lipid.
Fig. 14.
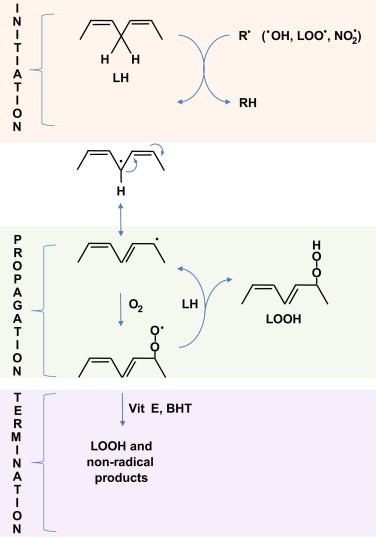
Lipid peroxidation: oxidation of unsaturated fatty acids to lipid hydroperoxide.
Adaptive response in cells exposed to mild to moderate oxidative stress.
As discussed in the previous section, cells cope with oxidants (O2•–, H2O2, LOOH) through several detoxification mechanisms afforded by endogenous antioxidant enzymes and small molecular-weight antioxidants. Paradoxically, this process (continued exposure to a mild dose of oxidants) actually results in increased syntheses of antioxidant enzymes. This is an intrinsic mechanism by which cells restore the oxidant/antioxidant balance. How does this happen? What makes these cells stronger? What is the signal transduction mechanism that is responsible for this paradoxical effect?
The three cellular components involved in this signaling mechanism at the transcriptional level are: Kelch-like ECH-associated protein 1 (Keap1), nuclear factor erythroid 2-related factor 2 (Nrf2), and antioxidant response elements (ARE). Research indicates that the Keap1-Nrf2-ARE signaling pathway is responsible for “turning on” the adaptive response against oxidant stress. The binding of Nrf2 to the DNA sequences present in ARE induce transcription of cytoprotective, antioxidant genes including SOD, GPx, CAT, gamma-GCS, GST. In addition, several other cytoprotective genes (heme oxygenase, NQO-1) are induced. As shown in Fig. 15, under normal conditions (reduced intracellular conditions), Nrf2 is stabilized through binding to Keap-1 in the cytoplasm. During enhanced ROS formation or exposure to electrophilic metabolites, the cysteine residues in Keap-1 are modified or oxidized causing the dissociation of Nrf2 and translocation to the nucleus and binding to the ARE. Depending upon the binding site present on the promoter region, the different antioxidant genes are induced.
Fig. 15.
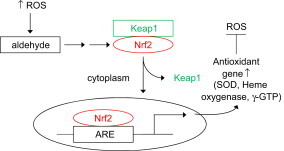
Activation of Keap1–Nrf2–ARE pathway by ROS.
Dietary antioxidants and exercise
Dietary antioxidants
Induction of the signaling pathway (Keap1/Nrf2/ARE) leading to overexpression of major inflammatory enzymes has been implicated in a variety of diseases as an adaptive mechanism to cope with oxidative stress. ARE inducers or activators are generally electrophilic. Electrophilic compounds typically contain a double bond conjugated to an electron-withdrawing carbonyl group. These compounds react readily with the sulfhydryl group present in the cysteinyl residues (this reaction is known as the Michael addition reaction) (Fig. 16). Several food components or phytochemicals (e.g., curcumin, cinnamon derivatives, diallyl sulfide, isothiocyanate) are known to activate the Keap1/Nrf2/ARE pathway and induce numerous cytoprotective genes involved in Phase 11 response. This includes induction of antioxidant and repair enzymes.
Fig. 16.
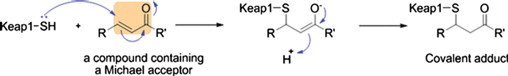
The proposed reaction mechanism of Michael addition reactions between Michael acceptors and cysteine sulfhydryl groups in Keap1. [From: Magesh, S., et al. Small molecule modulators of Keap1–Nrf2–ARE pathway as potential preventive and therapeutic agents. Medicinal Research Reviews32:687–726; 2012.].
Curcumin, or turmeric, a major component of Indian curry, has two phenolic functional groups and a beta-diketo group. This compound reacts readily with cysteine sulfhydryl group and activate Keap1-Nrf2-ARE pathway. The chemopreventive effect of green tea and broccoli is attributed to activation of Nrf2 (Fig. 17).
Fig. 17.

Botanical phenolics and brain health. [From: Mattson, M. P.; Cheng. A. Trends in Neurosciences29:632–639; 2006.]
All substances are poisons. “The right dose differentiates a poison from a remedy” (Paracelsus, 1493–1541). Are antioxidants always good? In a recent study published in the Proceedings of the National Academy of Sciences, researchers reported that antioxidants indeed prevented the health-promoting effects of physical exercise in humans. It is known that exercise promotes longevity, promotes expression of antioxidant defense (sod1/2, GPx1, catalase) and decreases type 2 diabetes mellitus and insulin resistance (mitohormesis) [31]. However, exercise also enhances mitochondrial formation of reactive oxygen species.
Daily ingestion of vitamin C and E totally abrogated the health-promoting effects of exercise, as physical exercise failed to promote insulin insensitivity and antioxidant gene expression by blocking exercise-dependent ROS formation in individuals supplemented with antioxidants. Clearly a little bit of exercise-induced oxidative stress is good to enhance the endogenous antioxidant defense that is longer lasting. I am reminded of the quote “everything in moderation, including moderation”.
Toxic side effects of commonly used drugs
Adverse side effects of drugs: role of reactive oxygen/nitrogen species.
The success of chemotherapy (treating cancer with drugs) is dependent upon the therapeutic window—the difference in cytotoxicity between the normal cells and the tumor cells. Several chemotherapeutic drugs (doxorubicin, cis-platin, bleomycin) that are being widely used in treating various cancers are beset with adverse side effects with prolonged use or even long after the cessation of chemotherapy in patients who are free of tumors. Understanding the molecular mechanism of adverse side effects in target organs of toxicity in these patients will greatly enhance the quality of life in cancer survivors.
Doxorubicin or Adriamycin
Doxorubicin (DOX) or Adriamycin, belongs to a group of anticancer drugs called anthracyclines. The chemical structure of all the anthracyclines is very similar and consists of a tetracycline ring (marked A–D).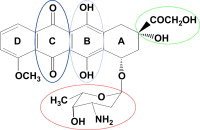 As shown in the structure of doxorubicin (DOX), there are four different functionally important regions that are color-coded: Quinone (dark blue), hydroquinone (light blue), alicyclic side chain (green), and the sugar group (red). The prototypical anthracycline, DOX, is currently being used, alone and in combination therapy, to treat a wide variety of cancers, including breast cancer, Hodgkin’s disease, leukemia, and testicular cancer.
As shown in the structure of doxorubicin (DOX), there are four different functionally important regions that are color-coded: Quinone (dark blue), hydroquinone (light blue), alicyclic side chain (green), and the sugar group (red). The prototypical anthracycline, DOX, is currently being used, alone and in combination therapy, to treat a wide variety of cancers, including breast cancer, Hodgkin’s disease, leukemia, and testicular cancer.
The clinical use of DOX in adult breast cancer patients, however, is associated with a dose-dependent increase in cardiotoxicity (arrhythmias, ventricular dysfunction, and cardiomyopathy or congestive heart failure) that is observed in patients decades after cessation of DOX chemotherapy. This problem also manifests in pediatric cancer patients. Children treated with DOX for acute lymphoblastic leukemia (ALL) developed heart problems years after completion of chemotherapy [32]. Estimates indicate that children who undergo DOX chemotherapy are nearly eight times more susceptible to develop heart problems later in life. Knowing the mechanism of injury to myocardial cells will lead to cardioprotective drug treatment post-chemotherapy.
Existing literature implicates the involvement of free radicals and reactive oxygen species in DOX cardiotoxicity. One-electron reduction of the quinone group in ring C of DOX results in the formation of a DOX semiquinone radical that undergoes redox-cycling in the presence of oxygen to form superoxide anion (Fig. 18). This is called a futile redox-cycling process (the substrate, DOX, is not consumed during the reaction). This reduction is catalyzed by several NADH-or NADPH-dependent enzymes such as cytochrome P450, mitochondrial dehydrogenase, or xanthine dehydrogenase (FH). Interaction between hydrogen peroxide and the redox-active iron that is released from the reaction between superoxide and the mitochondrial iron–sulfur protein (aconitase) results in the generation of hydroxyl radicals. DOX-induced generation of hydroxyl radicals are capable of oxidizing lipids, proteins, and nucleic acid in mitochondria, ultimately leading to lipid peroxidation, protein carbonyls, and DNA damage. A schematic representation of oxidative events initiated by DOX redox-cycling mechanism is shown in Fig. 18. Modifying the conformation of the –OH group present in the sugar moiety results in a less cardiotoxic form of DOX (epirubicin) that is metabolized faster.
Fig. 18.
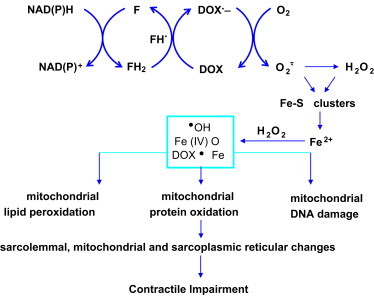
Free radical hypothesis of cardiotoxicity.
The target organ of DOX toxicity is the myocardium which is mitochondria-rich. One of the reasons why heart muscle is increasingly susceptible to oxidant-induced damage is due to the low levels of antioxidant enzymes (catalase, glutathione peroxidase) that can detoxify hydrogen peroxide and lipid peroxides. DOX also accumulates into mitochondria. Under these conditions, the antioxidant defenses can easily be overwhelmed, leading to oxidation of critical proteins involved in ATP generation in mitochondria. Recent research also suggests that DOX oxidative metabolism in mitochondria stimulates iron signaling, resulting in a “vicious” cycle of oxidant formation. Increased sensitivity of pediatric cancer patients to DOX-induced cardiotoxicity might be due to decreased antioxidant enzymes in the myocardium.
Several lines of research suggest that iron plays a major role in DOX-induced cardiotoxicity. Iron chelators have been used clinically to manage DOX toxicity in breast cancer patients. One of the frequently used drugs is ICRF-187, or dexrazoxane. ICRF-187 is a pro-drug but needs to be activated intracellularly to an active metabolite that has the iron-chelating ability. In both preclinical and clinical studies, pretreatment with dexrazoxane protects against cardiomyopathy. This drug also mitigates cardiac injury (as measured by troponin T levels) in children with ALL in response to DOX chemotherapy. Dexrazoxane does not compromise the efficacy of cancer treatment. This drug does not inhibit the DNA damage mediated by topoisomerase 11 present in cancer cells. Results from these studies indicate that DOX-induced myocardial injury may in part be related to iron-mediated oxidative damage. Emerging reports suggest that mitochondria-targeted antioxidants could effectively attenuate DOX-induced mitochondrial damage.
Cisplatin
Cisplatin (cis-diaminedichloroplatinum 11) is widely used as a first line of chemotherapy for treating a broad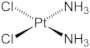 spectrum of tumors including testicular, cervical, and ovarian tumors. The clinical utility is, however, hampered due to its dose-dependent nephrotoxicity and ototoxicity (hearing loss due to damage to auditory cells). These adverse effects result in treatment with a lower dosage or termination of treatment. The primary cytotoxic effect is due its interaction and intercalation into DNA, leading to inter- and intra-strand cross links, defective DNA templates, impaired DNA repair, and inhibition of DNA replication in rapidly dividing tumor cells. Studies suggest that mitochondria are a major target of cisplatin. In addition to the first wave of oxidative damage, a secondary wave of reactive oxygen species formation occurs from proinflammatory cytokines from leukocyte infiltration induced by cisplatin and subsequent activation of NADPH oxidase activity. Increased expression of NOX enzymes responsible for sustained generation of O2•– and ONOO− was detected in the kidneys of cisplatin treated animals. Fig. 19 summarizes the sustained generation of reactive oxygen and nitrogen species in response to cisplatin-induced pro-inflammatory cytokines.
spectrum of tumors including testicular, cervical, and ovarian tumors. The clinical utility is, however, hampered due to its dose-dependent nephrotoxicity and ototoxicity (hearing loss due to damage to auditory cells). These adverse effects result in treatment with a lower dosage or termination of treatment. The primary cytotoxic effect is due its interaction and intercalation into DNA, leading to inter- and intra-strand cross links, defective DNA templates, impaired DNA repair, and inhibition of DNA replication in rapidly dividing tumor cells. Studies suggest that mitochondria are a major target of cisplatin. In addition to the first wave of oxidative damage, a secondary wave of reactive oxygen species formation occurs from proinflammatory cytokines from leukocyte infiltration induced by cisplatin and subsequent activation of NADPH oxidase activity. Increased expression of NOX enzymes responsible for sustained generation of O2•– and ONOO− was detected in the kidneys of cisplatin treated animals. Fig. 19 summarizes the sustained generation of reactive oxygen and nitrogen species in response to cisplatin-induced pro-inflammatory cytokines.
Fig. 19.
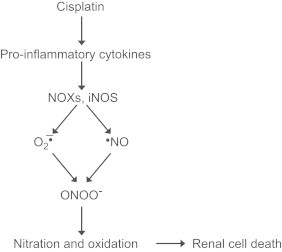
Molecular mechanism of cisplatin-induced inflammation.
Acetaminophen (or paracetamol, as known outside the US).
Acetaminophen is one of the most widely used analgesics worldwide. While safe at therapeutic doses, overdose or too-frequent high dose (supratherapeutic dose) leads to severe liver injury and even death. Acetaminophen poisoning (intentional overdose with therapeutic intent) results in nearly 500 deaths each year (in the US). Studies have shown that a majority of the drug is metabolized to nontoxic metabolites through conventional phase 2 metabolic pathways. However, a minor component (5%) of acetaminophen is metabolized by cytochrome P450 enzymes (CYP), especially CYP2E1 isoform, to the reactive, electrophilic intermediate or electrophile, N-acetyl-p-benzoquinone imine (NAPQI) [33]. NAPQI is very reactive and its toxicity in the liver results from its covalent binding to proteins (with cysteines) and nucleic acids. However, as long as sufficient glutathione is present in the liver, NAPQI is detoxified by glutathione (a strong nucleophile) forming cysteine and mercapturic acid conjugates. During this reaction, hepatic glutathione is depleted due to a rapid reaction between NAPQI and –SH moiety of GSH. This reaction is also catalyzed by glutathione transferase enzyme (GST) forming an acetaminophen–cysteine conjugate. In the absence of intracellular GSH or decreased GSH levels, NAPQI will indiscriminately react with proteins (present in the endoplasmic reticulum, the Ca2+ transport system, and/or mitochondria) and contribute to hepatic cell death. Note that alcoholics are more vulnerable to acetaminophen toxicity due to decreased GSH and elevated CYPE1 activity.
The only effective therapy that is currently available for acetaminophen toxicity is N-acetylcysteine therapy. N-acetylcysteine is more readily absorbed into hepatic tissues than cysteine. Once inside the cell, N-acetylcysteine is hydrolyzed to cysteine, a limiting substrate that restores glutathione synthesis and inhibits liver tissue injury [34]. In order to be therapeutically effective, N-acetylcysteine should be administered to acetaminophen-overdosed patients within 12–24 h following ingestion.
The importance of oxidative stress in acetaminophen toxicity remains controversial, although reports now indicate the activation of Kupffer cells and recruitment of neutrophils and monocytes (innate immune response) during the later phase of drug overdose. As a result of the inflammatory response, increased formation of •NO, O2•–, and ONOO− likely occurs during the later phase. Overall, acetaminophen overdose produces severe liver injury leading to cell necrosis via metabolic activation, glutathione depletion, alkylation of proteins, mitochondrial dysfunction, and formation of reactive oxygen and nitrogen species. Timely administration of N-acetylcysteine therapy protects against hepatotoxicity.
The pathopysiological role of ROS/RNS generation in pro-inflammatory diseases
Inflammatory diseases, oxidative conditions, decreased •NO
From the above discussion, it is evident that •NO plays a vital role in the maintenance of normal endothelial function. Under pathophysiological conditions promoting formation of reactive oxygen species, this situation dramatically changes, leading to a decreased •NO bioavailability. There are many examples in cardiovascular, neurodegenerative, and other chronic inflammatory diseases in which •NO levels are critically compromised. What is the basic chemical mechanism that is responsible for this acute shortage of •NO in the vasculature? One of the radical culprits that cause this problem is superoxide, as demonstrated in renovascular hypertension research (Fig. 20). In this disorder, the reduced arterial perfusion to one or both kidneys elevates circulating levels of angiotensin II due to increased production of renin. Angiotensin II activates membrane-bound NADPH oxidase that stimulates O2•– formation in the vessel wall in patients with hypertension [34].
Fig. 20.
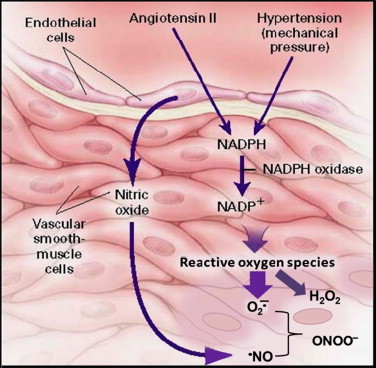
The role of angiotensin II and mechanical stress in the generation of reactive oxygen species in the vessel wall in patients with hypertension. [Modified from: Sowers, J. R. Hypertension, angiotensin II, and oxidative stress. New England Journal of Medicine346:1999–2001; 2002.].
Because of the diffusion-controlled reaction rate between •NO and O2•–, it is very likely that this reaction contributes to •NO depletion, thereby impairing endothelium-dependent vasodilation. Furthermore, an even more potent oxidant, peroxynitrite (ONOO−), is formed in this process. Peroxynitrite may further augment vascular injury by reacting with BH4, an essential co-factor involved in •NO biosynthesis by endothelial NOS and depleting BH4 in the vessel wall. This leads to a vicious cycle scenario in which •NO biosynthesis is ultimately inhibited by angiotensin-mediated O2•–. Note that published data suggest that this condition is corrected by renal angioplasty, leading to downregulation of the renin–angiotensin system (and inhibition of production of angiotensin II) or by administering ascorbic acid. Ascorbic acid will react with both O2•– and ONOO− but not with •NO. Consequently, the bioavailability of •NO is increased in the vessel wall in these patients.
The development of drugs that activate endogenous antioxidant enzymes for treating inflammatory diseases [chronic kidney disease (CKD) associated with type 2 diabetes] is currently underway. It has been shown that bardoxolone methyl, an orally active antioxidant drug, is able to decrease inflammation and oxidative stress associated with CKD. The mechanism of action appears to be related to activation of NrF2. Unfortunately, due to unexpected toxicity, clinical trials of this drug were suspended. Despite the setback, mitigating oxidative stress in inflammatory diseases may become a crucial factor for successful drug therapy. The question, “Doctor, can you tell me more about oxidants, antioxidants and free radicals,” is more realistic now than ever before.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Halliwell B. Tell me about free radicals, doctor: a review. Journal of the Royal Society of Medicine. 1989;82:747–752. doi: 10.1177/014107688908201216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halliwell B., Gutteridge J.M.C. 4th ed. Oxford University Press; Oxford: 2007. Free Radicals in Biology and Medicine. [Google Scholar]
- 3.Doroshow J.H. Doxorubicin-induced cardiac toxicity. New England Journal of Medicine. 1991;324:843–845. doi: 10.1056/NEJM199103213241210. [DOI] [PubMed] [Google Scholar]
- 4.Beckman J.S., Beckman T.W., Chen J., Marshall P.A., Freeman B.A. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proceedings of the National Academy of Science of USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babior B.M. NADPH oxidase. Current Opinion in Immunology. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Grant S.S., Kauffman B.B., Chand N.S., Hasely N., Hung D.T. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proceedings of the National Academy of Science of USA. 2012;109:12147–12152. doi: 10.1073/pnas.1203735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss S.J. Tissue destruction by neutrophils. New England Journal of Medicine. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 8.Kuhns D.B., Alvord W.G., Heller T., Feld J.J., Pike K.M., Marciano B.E. Residual NADPH oxidase and survival in chronic granulomatous disease. New England Journal of Medicine. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCord J.M. Oxygen-derived free radicals in postischemic tissue injury. New England Journal of Medicine. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 10.Odeh M. The role of reperfusion-induced injury in the pathogenesis of the crush syndrome. New England Journal of Medicine. 1991;324:1417–1422. doi: 10.1056/NEJM199105163242007. [DOI] [PubMed] [Google Scholar]
- 11.Uhlmann D., Armann B., Ludwig S., Escher E., Pietsch U.C., Tannapfel A. Comparison of Celsior and UW solution in experimental pancreas. Journal of Surgical Research. 2002;105:173–180. doi: 10.1006/jsre.2002.6408. [DOI] [PubMed] [Google Scholar]
- 12.Kessel D. Adventures in photodynamic therapy: 1976–2008. Journal of Porphyrins and Phthalocyanines. 2008;12:877–880. doi: 10.1142/s1088424608000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishioka N.S., Lauwers G.Y. Case 10-2006: a 66-year-old woman with Barrett’s esophagus with high-grade dysplasia. New England Journal of Medicine. 2006;354:1403–1409. doi: 10.1056/NEJMcpc059007. [DOI] [PubMed] [Google Scholar]
- 14.Sieber F., Stuart R.K., Rowley S.D., Sharkis S.J., Sensenbrenner L.L. Dye-mediated photolysis of normal and neoplastic hematopoietic cells. Leukemia Research. 1987;11:43–49. doi: 10.1016/0145-2126(87)90104-4. [DOI] [PubMed] [Google Scholar]
- 15.Murad F. Nitric oxide and cyclic GMP in cell signaling and drug development. New England Journal of Medicine. 2006;355:2003–2011. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 16.Furchgott R.F. A research trail over half a century. Annual Review of Pharmacology and Toxicology. 1995;35:1–27. doi: 10.1146/annurev.pa.35.040195.000245. [DOI] [PubMed] [Google Scholar]
- 17.Ignarro L. Endothelium-derived nitric oxide: pharmacology and relationships to the actions of organic nitrate esters. Pharmaceutical Research. 1989;6:1–9. doi: 10.1023/a:1015926119947. [DOI] [PubMed] [Google Scholar]
- 18.Rajfer J., Aronson W.J., Bush P.A., Dorey F.J., Ignarro L.J. Nitric oxide as a mediator of relaxation of the corpus cavernosum in response to nonadrenogenic, noncholinergic neurotransmission. New England Journal of Medicine. 1992;326:90–94. doi: 10.1056/NEJM199201093260203. [DOI] [PubMed] [Google Scholar]
- 19.Bush P.A., Aronson W.J., Buga G.M., Rajfer J., Ignarro L.J. Nitric oxide is a potent relaxant of human and rabbit corpus cavernosum. Journal of Urology. 1992;147:1650–1655. doi: 10.1016/s0022-5347(17)37671-1. [DOI] [PubMed] [Google Scholar]
- 20.Pryor W.A., Jin X., Squadrito G.L. One- and two-electron oxidations of methionine by peroxynitrite. Proceedings of the National Academy of Sciences of USA. 1994;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trostchansky A.; Bonilla L.; Gonzalez-Perilli L.; Rubbo; H. Nitro-fatty acids: formation, redox signaling, and therapeutic potential. Antioxidants and Redox Signalling 2012 Dec 20 [Epub ahead of print] [DOI] [PubMed]
- 22.Schopfer F.J., Cipolina C., Freeman B.A. Formation and signaling actions of electrophilic lipids. Chemical Review. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohshima H., Sawa T., Akaike T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: formation, occurrence, and implications in inflammation and carcinogenesis. Antioxidants and Redox Signalling. 2006;8:1033–1045. doi: 10.1089/ars.2006.8.1033. [DOI] [PubMed] [Google Scholar]
- 24.Gryglewski R.J., Palmer R.M., Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 25.White K.A., Marletta M.A. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry. 1992;28:6627–6631. doi: 10.1021/bi00144a001. [DOI] [PubMed] [Google Scholar]
- 26.Babu B.R., Griffith O.W. Design of isoform-selective inhibitors of nitric oxide synthase. Current Opinion in Chemical Biology. 1998;2:491–500. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 27.Grisham M.B., Granger D.N. Metabolic sources of reactive oxygen metabolites during oxidant stress and ischemia with reperfusion. Clinics in Chest Medicine. 1989;10:71–81. [PubMed] [Google Scholar]
- 28.Thomas J.P., Girotti A.W. Reactivity of photochemically-generated lipid hydroperoxides in cell membranes with glutathione peroxidase. Photochemistry and Photobiology. 1989;49:153–156. doi: 10.1111/j.1751-1097.1989.tb04089.x. [DOI] [PubMed] [Google Scholar]
- 29.Peskin A.V., Low F.M., Paton L.N., Maghzai G.J., Hampton M.B., Winterbourn C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. Journal of Biological Chemistry. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 30.Zimniak P. Relationship of electrophilic stress to aging. Free Radical Biology and Medicine. 2011;51:1087–1105. doi: 10.1016/j.freeradbiomed.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ristow M., Zarse K., Oberbach A., Klöting N., Birringer M., Kiehntopf M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences of USA. 2009;106:8665–8680. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipshultz S.E., Rifai N., Dalton V.M., Levy D.E., Silverman L.B., Lipsitz S.R. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. New England Journal of Medicine. 2004;351:145–153. doi: 10.1056/NEJMoa035153. [DOI] [PubMed] [Google Scholar]
- 33.Heard K.J. Acetylcysteine for acetaminophen poisoning. New England Journal of Medicine. 2008;359:285–292. doi: 10.1056/NEJMct0708278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sowers J.R. Hypertension, angiotensin II, and oxidative stress. New England Journal of Medicine. 2002;346:1999–2001. doi: 10.1056/NEJMe020054. [DOI] [PubMed] [Google Scholar]