

Abstract
4-hydroxynonenal (HNE) is a lipid hydroperoxide end product formed from the oxidation of n-6 polyunsaturated fatty acids. The relative abundance of HNE within the vasculature is dependent not only on the rate of lipid peroxidation and HNE synthesis but also on the removal of HNE adducts by phase II metabolic pathways such as glutathione-S-transferases. Depending on its relative concentration, HNE can induce a range of hormetic effects in vascular endothelial and smooth muscle cells, including kinase activation, proliferation, induction of phase II enzymes and in high doses inactivation of enzymatic processes and apoptosis. HNE also plays an important role in the pathogenesis of vascular diseases such as atherosclerosis, diabetes, neurodegenerative disorders and in utero diseases such as pre-eclampsia. This review examines the known production, metabolism and consequences of HNE synthesis within vascular endothelial and smooth muscle cells, highlighting alterations in mitochondrial and endoplasmic reticulum function and their association with various vascular pathologies.
Abbreviations: Akt, Protein kinase B; AR, Aldose reductase; ARE, Antioxidant response element; AP-1, Activator protein-1; ATF6, Activating transcription factor 6; BH4, Tetrahydrobiopterin; BAEC, Bovine aortic endothelial cells; BLMVEC, Bovine lung microvascular vein endothelial cells; BPAEC, Bovine pulmonary arterial endothelial cells; BTB, Broad complex Tramtrack and Bric–brac domain; CHOP, C/EBP-homologous protein; CREB, cAMP response element-binding protein; EGFR, Epidermal growth factor receptor; elF2α, Eukaryotic translation initiation factor 2α; Elk1, ETS domain-containing protein; eNOS, Endothelial nitric oxide synthase; EpRE, Electrophile response element; ER, Endoplasmic reticulum; ERAD, Endoplasmic reticulum assisted degradation; ERK1/2, Extracellular signal-regulated kinase 1/2; FAK, Focal adhesion kinase; FAP, Familial amyloidotic polyneuropathy; GCLC, Glutamate cysteine ligase catalytic subunit; GCLM, Glutamate cysteine ligase modifier subunit; GS-DHN, Glutathionyl-1,4 dihydroxynonene; GSH, Glutathione; GS-HNE, HNE-conjugates; GST, Glutathione-S-transferase; GTPCH, Guanosine triphosphate cyclohydrolase I; HASMC, Human aortic smooth muscle cells; HCSMC, Human coronary smooth muscle cells; HERP, Homocysteine inducible ER protein; HMEC, Human microvascular endothelial cells; HO-1, Heme oxygenase-1; HNE, 4-hydroxynonenal; Hsp-70/72/90, Heat shock proteins-70/ -72/ -90; HUVEC, Human umbilical vein endothelial cells; iNOS, Inducible nitric oxide synthase; IRE1, Inositol requiring enzyme 1 IRE1; IVR, Central intervening region; JNK, c-jun N-terminal kinase; Keap1, Kelch-like ECH-associated protein 1; MASMC, Mouse aortic smooth muscle cells; MEK1/2, Mitogen activated protein kinase kinase 1/2; MMP-1/2, Matrix metalloproteinase-1/ -2; MPEC, Mouse pancreatic islet endothelial cells; NAC, N-acetylcysteine; NFκB, Nuclear factor kappa B; NO, Nitric oxide; NQO1, NAD(P)H quinone oxidoreductase; Nrf2, Nuclear factor-E2-related factor 2; oxLDL, Oxidized low density lipoprotein; PCEC, Porcine cerebral endothelial cells; PDGF, Platelet-derived growth factor; PDI, Protein disulfide isomerases; PERK, Protein kinase-like endoplasmic reticulum kinase; 15d-PGJ2, 15-deoxy-Delta (12,14) prostaglandin-J2; PKC, Protein kinase C; PUFAs, Polyunsaturated fatty acids; RASMC, Rat aortic smooth muscle cells; ROS, Reactive oxygen species; RVSMC, Rat vascular smooth muscle cells; SMC, Smooth muscle cell; tBHP, Tert-butylhydroperoxide; TKR, Tyrosine kinase receptor; UPR, Unfolded protein response; xCT, cystine/glutamate amino acid transporter
Keywords: 4-hydroxynonenal, Vascular biology, Endothelial cells, Vascular smooth muscle cells, Redox signaling, Nrf2
Highlights
-
•
HNE is a lipid peroxidation endproduct regulating vascular redox signaling.
-
•
HNE detoxification is tightly regulated in vascular and other cell types.
-
•
Elevated HNE levels are associated with various vascular diseases.
HNE production in the vasculature
The oxidation and subsequent β-cleavage of n-6 polyunsaturated fatty acids (PUFAs) by H+ abstracting agents (e.g. hydroxyl radicals, OH•) yields a variety of lipid hydroperoxide products [1]. Of these, the alkenal 4-hydroxy-2-trans-nonenal (HNE) is considered one of the most abundant and bioactive species [1,2] and has been studied extensively both in the context of physiological as well as pathological vascular events. HNE can form adducts with DNA [3,4] but is also highly reactive with phospholipids (containing PUFAs such as linoleic and arachidonic acid) and nucleophilic amino acids (e.g cysteine, histidine and lysine residues, [5]), making lipid membranes particularly vulnerable to HNE modification. Furthermore, HNE Michael additions can induce further cross-linking and/or other HNE conjugates, with roles in physiology and pathophysiology [6]. In addition to the plasma membrane, specific organelles, such as the mitochondria and endoplasmic reticulum (ER), are highly susceptible to HNE-induced damage, with propagating lipid peroxidation ultimately resulting in organelle dysfunction and loss of redox homeostasis Fig. 1.
Fig. 1.
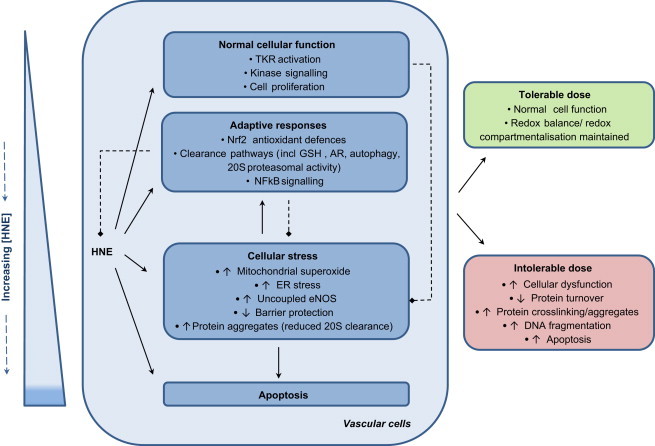
Hormetic modulation of vascular cell function in response to HNE. Whilst low physiological levels of HNE can directly target specific cellular pathways such as tyrosine kinase receptor (TKR) activation and downstream kinase signaling, exerting positive effects on cell function such as cell proliferation, progressively increasing concentrations of HNE can be detrimental. Adaptive responses in phase II metabolism largely through activation of the redox-sensitive Nrf2 serve to neutralize HNE protein adduction and have a potential role in preventing off-target effects of HNE, prolonged organelle stress and loss of function. Chronic exposure to pathological concentrations of HNE found in disease states or which may accumulate in discrete cellular regions or organelles are associated with cellular stress and dysfunction, characterized by loss of function of enzymatic systems, cellular damage and activation of pro-inflammatory and ultimately pro-apoptotic signaling.
It is estimated that free HNE concentrations in the plasma of healthy individuals range between 0.3 and 0.7 µM [7]. HNE levels increase significantly in plasma and tissues during aging [8] and in diseases associated with oxidative stress such as atherosclerosis and diabetes [9]. As a lipid peroxidation derived product, HNE is highly associated with the generation of reactive oxygen species (ROS), and hence levels of HNE and HNE conjugates in plasma, tissues and urine are frequently used as markers of oxidative stress. In disease states, levels of HNE within the low micromolar range (∼1–20 µM) have been reported, although conceivably far higher concentrations (∼100 µM) may be achieved in discreet areas such as the plasma membrane [1]. In vitro incubation of vascular cells with linoleic and arachidonic acid directly increases HNE formation [10], with HNE also known to accumulate indirectly within vessels via macrophage infiltration. Cholesterol consumption reportedly increases HNE synthesis [11], with HNE inducing low-density lipoprotein oxidation and increased uptake by macrophages [12], and HNE accumulation within atherosclerotic plaques [13].
Metabolism of HNE in vascular cells
The half-life of HNE is relatively short within cells, with HNE rapidly removed by phase II reactions, allowing for excretion of water soluble conjugates [14]. HNE modified proteins can also be removed by autophagic and proteasomal degradation pathways [15–17]. Although HNE can form Michael adducts directly with the abundant cellular antioxidant glutathione (GSH), HNE conjugation is accelerated by enzymatic systems [14]. HNE conjugation is catalyzed by enzymes including glutathione-S-transferases (GST), alcohol dehydrogenases and aldehyde dehydrogenases, leading to the formation of glutathionyl HNE (GS-HNE), 1,4-dihydroxynonene and 4-hydroxy-2-nonenoic acid, respectively. Among them, GST mediated conjugation of HNE to glutathione (GSH) accounts for approximately 60% of the total degradation in vascular cells [18,19]. The GST isoform GST-4 displays a high selectivity for α,β unsaturated aldehydes and overexpression of GST-4 has been shown to protect endothelial cells from HNE-mediated apoptosis [20]. In human endothelial and vascular smooth muscle cells (SMCs), the primary HNE-glutathione conjugate can be further converted to glutathionyl-1,4 dihydroxynonene (GS-DHN) by aldose reductase (AR, AKR1B1), a member of the aldo-keto reductase superfamily and the rate-limiting enzyme involved in metabolizing glucose via the polyol pathway [18]. These metabolites may then be exported for secondary degradation [21].
In hepatocytes, the half-life of 100 μM HNE is only 5 min [22]. Srivastava et al. reported that in human endothelial cells incubated with 5 μM HNE, 95% of the compound was metabolized after 30 min [18]. Following addition of 25 μM HNE to bovine aortic endothelial cells (BAEC), intracellular HNE levels peak at 4 h and decline by 50% after 6 h [23]. In contrast, vascular smooth muscle cells may be more resistant to HNE modification. Hill et al. reported that exposure of primary rat aortic SMCs to HNE concentrations below 50 µM fails to induce widespread cellular accumulation of HNE protein adducts, with adducts only apparent after at least 30 min incubation with 50 µM HNE [15]. Thus, clearance is faster in SMCs than endothelial cells, with complete removal of HNE adducts in SMC treated with 50 µM HNE occurring within ∼8 h [15].
The rate of HNE metabolism is critically dependent on cellular levels of GSH and relevant enzyme activity, reflecting the antioxidant capacity of different cells [24,25]. SMCs are known to induce rapid synthesis of intracellular GSH from cysteine or extracellular GSH [26], and basal GSH levels are markedly higher in SMCs compared with endothelial cells [27]. Furthermore, SMCs exposed to HNE promote the induction of aldose reductase (AR) activity and expression, increasing GS-HNE metabolite clearance and promoting cell survival [28,29]. Collectively, the enhanced induction of these clearance pathways within SMCs may account for the apparent increased sensitivity of endothelial cells to HNE.
Downstream effects of HNE production on cellular function
Although the majority of HNE is rapidly removed from cells, approximately 1–8% of HNE may remain bound to functional amino acid groups including cysteine, histidine and lysine and thereby continue to exert cellular effects [19]. Among them, cysteine residues display the highest reactivity with HNE at their C-3 electrophilic center [30]. Effects of HNE on endothelial and smooth muscle cells are summarized in Tables 1 and 2, respectively. For a more comprehensive overview of the biochemistry and general cell signaling of lipid peroxidation products we refer readers to existing reviews [14,31,32]. In biological systems, lipid peroxidation products have been referred to as ‘second messengers’ of free radicals, as they are more stable and diffuse easily to affect distant targets enabling participation in a diverse number of cellular events. Cellular effects of HNE may evoke both damaging and hormetic cytoprotective actions. Whilst high concentrations of HNE can inhibit cellular processes inducing widespread cellular damage ultimately resulting in apoptosis [1,33], a range of relatively low concentrations of HNE produced in response to mild oxidative challenge appear to modulate normal physiological processes including upregulation of endogenous antioxidant defense pathways [34,35].
Table 1.
Effects of HNE and 15d-PGJ2 on endothelial cells.
| Cell type | Treatment | Conc.⁎ (μmol/L) | Time | Main findings | References |
|---|---|---|---|---|---|
| RTK and MAPK activation | |||||
| HEC | HNE | 0.1 | 3 h | HNE (0.1 µM) and oxLDL (200 µg/ml)↑EGFR activation | [36] |
| oxLDL | [49] | ||||
| BLMVEC | HNE | 10–100 | 2 h | ↑Phosphorylation of ERK, JNK, p38 MAPK | |
| MPEC | HNE | 20 | 0–2 h | ↑p∼JNK within 30 min,↔p∼p38, p∼ERK1/2 | [20] |
| BLMVEC | HNE | 25 | 30 min | ↑MAPK activation | [88] |
| BPAEC | HNE | 50 | 30 min | ↑Phospholipase D activation via protein tyrosine phosphorylation | [37,141] |
| HUVEC | 15d-PGJ2 | 10 | 2 h | ↑Phosphorylation of MAPK JNK (2 h) | [142] |
| Transcriptional activity | |||||
| HUVEC | HNE | 5 | 12 h | ↑HO-1 and NQO1 via Nrf2, protects HUVEC from tBHP attack | [55] |
| HUVEC | HNE | 1–10 | 12 h | ↔NFκB activation, ↓IL-8 and ICAM-1 production in a concentration dependent manner indicating ↓inflammatory responses | [10] |
| HUVEC | 15d-PGJ2 | 2.5 | 2–8 h | ↑Nrf2 nuclear accumulation, ↑HO-1, ↑NQO1, ↑adducts with Keap1 at cysteines residues in IVR region, which may mediate shear induced Nrf2 activation | [143] |
| BAEC | 15d-PGJ2 | 2 | 16 h | ↑GSH, HO-1, ↑Keap1 adduct formation | [144] |
| HUVEC | 15d-PGJ2 | 5 | 24 h | ↑GSH, ↑GCLC, ↑GCLM, ↑resistance to oxidative stress which relies on de novo GSH synthesis, ARE responsible for GCLC induction, Cys273 and Cys278 of Keap1 conjugated by 15d-PGJ2 | [52,145] |
| Oxidative stress and protein adduct clearance | |||||
| BAEC | HNE | 5 | 1 h | Modifies thioredoxin-1 at cys-73 and ↓activity, ↑ROS, ↓GSH | [90] |
| HAEC | HNE | 5 | 24 h | Protects HAEC from 6-hydroxydopamine induced cell death | |
| BAEC | HNE | 25 | 4 h | Proposed ↑proteasomal degradation of GTPCH and Hsp90 | [23] |
| Cellular dysfunction | |||||
| BLMVEC | HNE | 10–100 | 2 h | ↑Endothelial permeability; ↑Michael adducts formation, actin fiber remodeling via phosphorylation of ERK, JNK, p38 MAPK | [49] |
| BLMVEC | HNE | 25 | 30 min | Induces actin rearrangement, ↓GSH, ↑cell adhesion and surface integrins; ↓tyrosine phosphorylation of FAK,↑MAPK activation, ↑Michael adducts with ↑focal adhesion & adherens junctional proteins, reversed by pretreatment NAC | [88] |
| BAEC | HNE | 5 | 1 h | ↑Monocyte adhesion | [90] |
| BAEC | HNE | 10 | 5 d | ↑Membrane phospholipid perturbation, ↑prostacyclin, ↑monocyte migration | [146] |
| BAEC | HNE | 10 | 6–8 h | ↓Junctional communication | [87] |
| BAEC | HNE | 25 | 4 h | ↑ROS, ↓NO, GSH, ↑apoptosis involving caspase-3 activation which can be attenuated by BH4 supplementation. ↓BH4 by ↓GTPCH leading to ↑eNOS uncoupling, ↓HSP90 leading to ↓eNOS phosphorylation, proposed ↑proteasomal activity and proteasomal degradation of BH4 and HSP 90 | [23] |
| Mitochondrial function | |||||
| BAEC | HNE | 5–10 | 4–16 h | ↑GSH and HO-1 at 16 h, ↑ROS from mitochondria after 4 h | [71] |
| HUVEC | 15d-PGJ2 | 2.5 | 24 h | ↑GSH, ↑complex I activity in cell extracts, dependent on de novo protein synthesis in response to ROS but independent of GSH depletion | [94] |
| BAEC | 15d-PGJ2 | 10 | 16 h | ↑HO-1, ↓mitochondrial membrane potential ↑Nrf2 nuclear accumulation, blocked IBTP, a mitochondrial-targeted thiol reactive compound. | [53] |
| ER stress | |||||
| HUVEC | HNE | 25 | 30 min – 2 h | ↑Protein adduction (HSP90, HSP70,PDI), ↑xBP-1 splicing, ↑PERK, p∼eIF2α, and ATF6 translocation; ↑Grp78 and HERP, ↑ICAM-1, cell adhesion, TNF-α, IL-6, and IL-8; activation of endothelial cells mediated by ↑ER stress, noting depletion of GSH cannot ↑ER stress | [107] |
| HMEC | HNE | 20 | 24 h | ↑PERK and ATF6; ↑p∼IRE1 and p∼eIF2α; ↑ER stress, which is prevented by pretreatment with NAC | [108] |
| HMEC | HNE/oxLDL | 0–25 (HNE)/ 200 µg/ml oxLDL | 14–18 h | Forms adducts with PDI,↓PDI activity and↓cell viability. ↑CHOP and xBP1s mRNA as indicators of ↑ER stress. Loss of PDI activity and reduced viability prevented by NAC. | [110] |
| Apoptosis | |||||
| HUVEC | HNE | 10–50 | 3 h | ↑Cell death and ↓growth capacity but this effect is affected by serum in the medium | [147] |
| HUVEC | HNE | 10 | 12 h | ↑Apoptosis, ↓protein synthesis | [10] |
| PCEC | HNE | 1–50 | 3 h | ↑Chromosomal aberrations and micronuclei formation | [113] |
| BAEC | HNE | 25 | 4 h | ↑ROS, ↓NO,↓GSH, ↑apoptosis involving caspase-3 activation which can be attenuated by BH4 supplementation. | [23] |
| ↓BH4 resulting from ↓GTPCH and ↓HSP90 levels leads to ↑eNOS uncoupling and ↓eNOS phosphorylation. Proposed ↑proteasomal activity towards GTPCH and HSP 90 underlies impaired NO production | |||||
| MPEC | HNE | 40 | 8 h | ↑p∼JNK leading to ↑p53 and Bax expression and↑apoptosis. Abolished by GST4 overexpression | [20] |
| HUVEC | 15d-PGJ2 | 10 | 2–16 h | ↑ROS (1 h), ↑phosphorylation of MAPK and JNK (2 h), ↑p53 expression and phosphorylation (8 h), ↑caspase associated apoptosis | [142] |
Cell type abbreviations: MPEC, mouse pancreatic islet endothelial cells; HEC human endothelial cell line CRL-1998; BAEC, bovine aortic endothelial cells; BLMVEC, Bovine lung microvascular vein endothelial cells; BPAEC, bovine pulmonary arterial endothelial cells; HMEC, human microvascular endothelial cells; HUVEC, human umbilical vein endothelial cells; PCEC, porcine cerebral endothelial cells.
Table 2.
Effects of HNE on vascular smooth muscle cells.
| Cell type | [HNE] μmol/L | Time | Findings | References |
|---|---|---|---|---|
| RTK, MAPK and PI3K/Akt activation | ||||
| MASMC | 0.1 | 10–90 min | In young but not aged animals↑p∼ERK1/2,↔p∼p38, p∼Jnk | [44] |
| RASMC | 1 | 5 min | ↑p∼ERK1/2 | [43] |
| HCSMC | 10 | 10 min | ↑p∼PDGFR, ↑p∼ERK1/2 | [40] |
| MASMC | 1 | 5–120 min | ↑p∼ERK1/2 that was maximal within 60 min,↑p∼p38 maximal within 30 min and p∼JNK reduced after 30 min of HNE treatment | [42] |
| RASMC | 1 | 5–120 min | Dose-dependent↑Akt activity | [41] |
| RASMC | 1 | 4 h | ↑PKC activation, abolished by AR inhibition | [45] |
| Proliferation | ||||
| RASMC | 0.1–1 | 12–24 h | Dose-dependent↑MMP-2 activity, mRNA and protein expression, which was dependent on Akt activation | [41] |
| MASMC | 1 | 24 h | ↑MMP-2 expression and activity which was dependent on HNE-induced ERK activation | [42] |
| MASMC | 0.1–10 | 24 h | 0.1 µM HNE ↑cell density in cells from young but not aged mice,↑ERK1/2 activation and downstream cyclin D1 mRNA expression. Higher doses 1–10 µM HNE↓cell density | [44] |
| 0.1 µM HNE ↑cell survival in cells from young but not aged mice. Aged cells showed↑ROS generation in response to HNE, with NAC↑survival in aged cell populations | ||||
| RASMC | 0.1–10 | 24 h | HNE and HNE conjugates (e.g. GS-HNE) <1 µM↑cell proliferation, with higher doses↓cell density. Aldose reductase activity required for cell survival/proliferation. Inhibition of HNE conjugate removal by RLIP76 transporter↑cell growth | [45] |
| HCSMC | 2–20 | 24 h | Dose-dependent ↑MMP-1 requiring HNE-induced PDGFR and downstream ERK1/2 activation | [40] |
| RASMC | 1 | 48–72 h | ↑Cell proliferation, attenuated by growth factor PDGF receptor autoantibodies | [43] |
| RASMC | 2.5 | 48 h | ↑Proliferation | [74] |
| HASMC | 2.5 | 72 h | ↑Cell proliferation, dependent on aldose reductase activity | [29] |
| Transcriptional activity | ||||
| HASMC | 1 | 0.5–6 h | 30 min following HNE exposure↑IκBα phosphorylation without inducing IƙB degradation. ↑NFκB DNA binding was also increased from 30 min of HNE stimulation | [72] |
| RASMC | 1 | 4 h | ↑NFκB and AP-1 activation | [45] |
| RASMC | 1 | 12 h | ↑NFκB activity but no change in AP-1, c-jun or CREB activity. NFƙB activation was dependent on mitochondria derived superoxide mediated Akt activation which and proceeds MMP-2 expression | [41] |
| RVSMC | 0–5 | 24 h | Dose-dependent ↓NFκB activation, with 50 µM HNE shown to prevent LPS/IFN induced proteasomal IκBα degradation. Accordingly, HNE dose-dependently (0–5 µM) ↓LPS/IFN induced nitrite production, with doses >12.5 µM↓iNOS expression | [73] |
| RASMC | 1–10 | 3 h | ↑Elk1, c-jun, CHOP and AP-1 activity, ↑c-jun and c-fos mRNA | [74] |
| RASMC | 2.5 | 1–2 h | ↑c-fos and c-jun expression, ↑AP-1 DNA binding | [43] |
| MASMC | 20 | 0–5 h | ↑Nrf2 nuclear accumulation and downstream HO-1, Prx1 and A170 mRNA, absent in cells derived from Nrf2 knockout mice | [54] |
| Phase II metabolism | ||||
| HASMC | 0.05 | 30 min | ↑Aldose reductase metabolized GSH conjugates | [29] |
| 2.5 | 8–12 h | ↑Aldose reductase mRNA (8 h) and protein (12 h) expression | ||
| RASMC | 1 | 5 min | ↓Cellular thiol content | [43] |
| RVSMC | 5–10 | 0–24 h | ↑Aldose reductase mRNA (7 h), protein (12 h) and activity (12–24 h) | [28] |
| RASMC | 0.1–10 | 24 h | HNE and HNE conjugates (e.g. GS-HNE) <1 µM↑cell proliferation, with higher doses↓cell density. Aldose reductase activity required for cell survival/proliferation. Inhibition of HNE conjugate removal by RLIP76 transporter↑cell growth | [45] |
| Oxidative stress and protein adduct clearance | ||||
| HASMC | 1 | 0–12 h | ↑8-isoprostane induced following 9 h HNE treatment | [72] |
| RASMC | 50 | 30 min | ↑Autophagy but not proteasomal HNE degradation | [15] |
| RVSMC | 50 | 4 h | ↓LPS/IFN induced proteasomal IƙBα degradation | [73] |
| RBASMC | 0–3000 | 4.5 h | ↓Recognition and clearance of LDL with increasing [HNE] | [12] |
| Cellular dysfunction | ||||
| PBMS | 10–100 | 0 min | ↓Carbachol induced relaxation | [148] |
| RVSMC | 0–5 | 24 h | HNE dose-dependently (0–5 µM) ↓LPS/IFN induced nitrite production, with doses >12.5 µM↓iNOS expression | [73] |
| Mitochondria | ||||
| RASMC | 1–30 | 10–60 min | HNE dose-dependently↑ROS production measured by DCF fluorescence with inhibitors of mitochondria abolishing HNE-induced superoxide generation | [96] |
| RASMC | 1 | 30 min | ↑Mitochondria derived superoxide | [41] |
| RASMC | 20 | 20–180 min | ↓Oxygen consumption rate (OCR),↔extracellular acidification rate (ECAR) | [97] |
| Apoptosis | ||||
| RASMC | 1–30 | 24 h | HNE >10 µM↑apoptosis, with 30 µM HNE also increasing necrosis | [44,96] |
| RVSMC | 5–15 | 6 h | HNE dose-dependently↑apoptosis, with AR inhibition↑HNE-induced apoptosis | [28] |
| HASMC | 0–100 | 6 h | 100 µM HNE induced DNA fragmentation | [72] |
Cell type abbreviations: HCSMC, human coronary smooth muscle cells; HASMC, Human aortic smooth muscle cells; RVMC, rat vascular smooth muscle cells; RASMC, rat aortic smooth muscle cells; RBASMC, rabbit aortic smooth muscle cells MASMC, mouse aortic smooth muscle cells; PBMS, pig bladder muscle strips.
Targeted signaling induced by HNE within the vasculature
Instead of a mere by-product of oxidative stress, both reactive oxygen species and resulting lipid hydroperoxide products (e.g. HNE) are capable of modifying nucleophilic residues and are now recognized as signaling agents participating in a wide array of cellular pathways. Key pathways targeted by HNE in vascular cells include tyrosine kinase receptor (TKR) activation and modulation of transcriptional activity.
Enzyme and receptor targets of HNE signaling
A classical signaling pathway known to be induced by HNE is the activation of TKRs, with limited data also supporting their activation in vascular endothelial and SMCs. HNE (0.1 µM) is reported in a human endothelial cell line to modify and induce autophosphorylation of the epidermal growth factor receptor (EGFR) which is associated with activation of intrinsic tyrosine kinase activity and implicated in cell proliferation/differentiation [36]. The formation of HNE protein adducts with sulfhydryl groups, lysine and histidine is a probable mechanism accounting for the activation of TKRs triggering pleiotropic regulatory pathways. In bovine aortic endothelial cells, Natarajan et al. also demonstrated protein tyrosine phosphorylation is responsible for the activation of phospholipase D (PLD) by HNE, although the concentration of HNE (50 µM) in their treatments may be somewhat higher than reported pathophysiological levels [37]. Activation of PLD is recognized as a trigger to generate the intracellular lipid messenger phosphatidic acid and the latter is associated with diverse cellular functions including reorganization of the cytoskeleton, intracellular membrane transport and cell signaling which is reviewed elsewhere [38].
Interestingly, modulation of intracellular thiol levels, either by supplementing with a thiol rich compound (N-acetyl-L-cysteine) or treatment with a GSH chelating compound (L-buthionine-(S,R)-sulfoximine, BSO), attenuates or enhances PLD activation by HNE respectively, implicating a potential competitive role of intracellular reactive free thiols in modulating PLD activation [39]. These findings highlight the importance of basal redox status in determining the various physiological or pathological consequences of HNE exposure. In SMCs relatively low concentrations of HNE (1–2.5 µM) also lead to platelet-derived growth factor (PDGF) receptor activation, preceding metalloproteinase-1 and extracellular signal-regulated kinase 1/2 (ERK1/2) activation [40]. HNE activation of extracellular matrix degrading matrix metalloproteinases-1 and -2 [40–42] is essential for SMC proliferation [43], as is the activation of ERK1/2 [43,44] which occurs within 5 min of HNE stimulation. Although in SMCs, ERK1/2 appears to be the principal kinase pathway activated by physiological concentrations (0.1–1 µM) of HNE, presumably indirectly via activation of upstream mitogen-activated protein kinase kinase 1/2 (MEK 1/2), several other kinase pathways are HNE sensitive. In SMCs, HNE (1 µM) promotes rapid protein kinase B (Akt) [41], p38 MAPK [42] as well more latent protein kinase C (PKC) [45] activity, whereas c-jun N-terminal kinase (JNK) signaling appears to be suppressed [42,44].
The downstream effects of Akt, p38, PKC and JNK activation in SMC function in response to low concentrations of HNE are not well characterized, and the predominant activation of ERK is somewhat surprising given the well characterized induction of cysteine rich PKC in non-vascular cell types [46–48]. As highlighted in Table 2, studies assessing kinase activation in SMC following challenge with pathological concentrations of HNE (∼10 µM) are limited. In contrast, in vitro studies with endothelial cells have largely used rather high HNE concentrations (>10 µM) to demonstrate upregulation of JNK, ERK1/2 and/or p38 [20,49]. Interestingly, both HNE and its conjugates (e.g. GS-HNE) appear to be bioactive and participate in cell signaling [45]. Furthermore, based on proliferative responses of SMCs to a range of HNE concentrations, it is notable that low ‘signaling’ doses (∼1 µM) of HNE elicit proliferative or ‘pro-survival’ responses in SMCs, whereas stimulation with higher and pathological levels of HNE (∼10 µM) cause apoptosis in both endothelial and SMCs (see Apoptosis Section).
Transcriptional targets of HNE
HNE can also induce the activation of a number of transcription factors. Due to its integral role as a mediator of oxidative stress, the best described role of HNE mediated transcriptional regulation is its activation of nuclear factor-E2-related factor 2 (Nrf2) antioxidant defenses. Biphasic effects of HNE are characterized by an initial decrease in GSH levels over 1–2 h and a later rebound upregulation of GSH (12–24 h) and other antioxidant enzymes, including heme oxygenase 1 (HO-1), NAD(P)H:quinone oxidoreductase (NQO1), glutamate cysteine ligase catalytic subunit (GCLC), glutamate cysteine ligase modifier subunit (GCLM) and the cystine/glutamate amino acid transporter (xCT), all of which enhance cellular defenses against further oxidative stress [34,50]. These cytoprotective enzymes possess an antioxidant response element (ARE)/electrophile response element (EpRE) in their promoter region which binds Nrf2, a key transcription factor regulating redox homeostasis [34,51]. Lipid peroxidation derivatives significantly increase nuclear accumulation of Nrf2 [35,52–54] and knockdown of Nrf2 with siRNA almost completely abolishes the adaptive upregulation of GSH by HNE [55], highlighting the importance of this transcriptional pathway in HNE triggered cellular responses.
Nrf2 is a member of the CNC (‘cap n collar’) family of transcription factors which possesses a b-Zip binding motif [56]. Nrf2 deficient mice are more susceptible to vascular damage due to diminished glutathione levels and an impaired compensatory induction of GSH synthesis (Chan & Kwong, 2000), highlighting a fundamental role for Nrf2 in antioxidant defenses against oxidative stress. Activation of Nrf2 is mainly regulated by its cytosolic inhibitor Kelch-Like ECH-Associated Protein 1 (Keap1), which under basal conditions targets Nrf2 for ubiquitin-dependent proteasomal degradation [57–59]. Keap1 is a cysteine rich protein that serves as a key redox sensor [60]. Modification of cysteine residues on Keap1 by electrophiles, particularly Cys 151 in the BTB (Broad complex Tramtrack, and Bric–brac) domain and Cys 273 and Cys 288 in the central intervening region (IVR) domain, suppress Keap1 mediated degradation of Nrf2 [61,62]. Although the exact residues modified by HNE have yet to be determined, Michael addition of HNE to Keap1 has been demonstrated [52]. When the level of Nrf2 exceeds the sequestration capacity of Keap1, excess Nrf2 can accumulate in the nucleus where it initiates transcription [56].
In addition to the genes mentioned above, the Nrf2 inducible enzymes aldo–keto reductase (AR) [63,64] and GSTs including GST4 [65,66] are critical in neutralizing HNE in vascular cells. Increased levels of GSH can detoxify HNE conjugated proteins such as protein disulfide isomerases (PDIs), heat shock protein 72 (Hsp72) and restore their function [67]. Interestingly, knockdown of Nrf2 results in increased HNE accumulation in tissues of aged (>24 months) mice [68], and in aged humans elevated HNE levels correlate with diminished Nrf2 activity [30,69]. However, the mechanisms underlying loss of Nrf2 function during aging remain to be elucidated. Because activation of cytoprotective pathways could be enhanced by HNE treatment, moderate doses of HNE may thus exert beneficial effects on cell survival, overcoming its initial detrimental damage. In SMCs, HNE challenge increases AR activity, mRNA and protein expression [28,29], which is associated with increased cell survival [45]
Moreover, Chen et al. reported that pre-treatment of human arterial endothelial cells with 5 μM HNE for 24 h significantly improved cell survival following oxidative stress induced by 6-hydroxydopamine [70]. Consistent with these findings, 5 μM HNE markedly reduced tert-butyl hydroperoxide-induced cytotoxicity in human umbilical vein endothelial cells, which was accompanied by increased expression of HO-1 and NQO1 and activated Nrf2/ARE signaling [55]. In addition, pretreatment of 15-deoxy-Delta (12,14) prostaglandin J2 (15d-PGJ2), another peroxidation product with a highly reactive electrophilic center that shares common functions with HNE, also enhanced cell resistance to oxidative stress via a similar mechanism [71], indicating hormetic actions of HNE in endothelial cells.
Surprisingly, whilst the transcriptional effects of HNE in endothelial cells seem to be predominantly confined to Nrf2, in SMCs the effects of HNE appear to be more diverse. In SMCs relatively low concentrations of HNE (∼1 µM) also promote IκBα phosphorylation, leading to rapid (30 min) nuclear factor kappa B (NFκB) DNA binding, with NFκB activation maintained for at least 12 h and promoting SMC proliferation [41,45,72]. In contrast, higher concentrations of HNE (5–50 µM) inhibit IκB-α degradation via the proteasome [73], consistent with reduced proliferation observed in response to higher concentrations of HNE [44,45]. In contrast to SMCs, HNE (10 µM) fails to modulate NFκB signaling in endothelial cells [10]. Furthermore, other transcription factors and transcriptional activators including activator protein-1 (AP-1), cAMP response element-binding protein (CREB) and ETS domain-containing protein (Elk1) are acutely (1–4 h), albeit transiently, activated in SMCs in response to HNE [43,45,74] but similar reports for endothelial cells are to our knowledge lacking. Whether HNE directly activates redox sensitive transcription factors such as AP-1, or whether this occurs as a consequence of perturbations within the cell redox environment remains to be established.
Restoration and clearance pathways
In addition to inducing the expression of HNE-responsive GSTs (e.g. GST4) and aldo–keto reductases (e.g AR) to metabolize HNE (see Section 2), export of GS-HNE conjugates in many cell types predominantly via the non-ABC transporter RLIP76 is also known to modulate HNE-induced cell signaling [75]. Chemical or antibody based inhibition of RLIP76 has been shown to prevent GS-DHN removal and to potentiate low dose HNE-induced SMC proliferation [45]. These findings highlight a specific signaling rather than cytotoxic role for low physiological concentrations of HNE and its conjugates in the vasculature, a finding confirmed in many other cell types [76]. The export of HNE conjugates by RLIP76 can be saturated, with purified RLIP76 shown to have a Km ∼2.5 µM for GS-HNE conjugates [77]. Exposure to high levels of HNE, exceeding the capacity of RLIP76, may be a critical factor mediating the hormetic actions of HNE. Indeed, inhibition of RLIP76 in vitro has been shown to induce HNE accumulation and activation of JNK mediated apoptosis [78]. In RLIP76−/− mice, HNE and GS-HNE metabolites accumulate in tissues [79] and, whilst the activity of RLIP76 in aging has yet to be determined, increased RLIP76 autoantibodies have been found in other vascular diseases and are implicated in metabolic syndrome [78,80].
Failure to induce HNE clearance can lead to the accumulation of HNE protein adducts, which below a certain threshold can be removed via proteasomal or autophagic degradation pathways to restore redox balance. In the cytosol, the proteasome largely consists of a 20S catalytic core, capped by regulatory subunits 19S (PA700) and/or 11S (PA28) subunits [81]. The 20S core is capped at either end by a 19S subunit, collectively termed the 26S proteasome, and has been shown to be key for normal turnover of cellular proteins in an ubiquitin-dependent manner. Oxidized proteins are a poor substrate for the 26S proteasome, but detachment of the 19S regulatory subunits allows oxidized proteins to be degraded by the 20S proteasome independent of ubiquitin conjugation [81]. It is generally accepted that whilst mild HNE-induced protein oxidation stimulates 20S proteasomal clearance, extensively oxidized and cross-linked proteins are more resistant to proteasome degradation leading to the accumulation of oxidatively modified and cytotoxic protein aggregates [17]. The proteasome itself may also be a target for HNE adduction, with HNE at concentrations as low as 10 µM shown to directly bind specific subunits within the 20S proteasomal catalytic core inhibiting their activity [82] and potentially exacerbating HNE-modified protein accumulation.
In endothelial cells, Vieira et al. reported that exposure to 50–200 µg/ml oxLDL induced protein adduct formation which was replicated by 1 µM HNE treatment [83]. Protein oxidation coinsided with elevated proteolysis peaking within 3–5 h and was inhibited by the proteasomal inhibitors LLnL and PSI and partly dependent on the previous ubiquitination of substrates [83]. Whitsett et al. also proposed that increased proteasomal degradation in response to HNE (25 µM) underlies loss of guanosine triphosphate cyclohydrolase I (GTPCH) in bovine aortic endothelial cells [23]. In SMCs, there is limited information on the effects of HNE on proteasomal activity and autophagy, however proteasomal activity appears limited following exposure to pathological concentrations of HNE [15], perhaps due to extensive protein crosslinking. Studies by Hill et al., demonstrate that treatment of rat aortic SMCs with 50 µM HNE induces widespread protein-adduction, with inhibitors of autophagy (3-MA) but not the proteasome (lactacystin) further increasing the presence of adduct, suggestive of an active autophagic response in SMCs to remove HNE-adducts [15]. In other studies, HNE (50 µM) was shown to prevent IκB proteasomal degradation in response to LPS/IFN treatment, with similar results obtained using the proteasomal inhibitor MG115 [73]. Collectively, these findings support an increase in HNE clearance pathways as a necessary step to prevent widespread and aberrant HNE modifications, ultimately resulting in cellular dysfunction and activation of pro-apoptotic signaling pathways.
More recently, in addition to increasing phase II metabolism, a role for Nrf2 has been implicated in the induction of proteasomal genes in response to oxidative stress. Hydrogen peroxide and a range of dietary Nrf2 inducers have been shown to increase the expression of 20S proteasomal and immunoproteasomal subunits, resulting in a rebound increase in oxidized protein degradation capacity [51,84]. Although, as reviewed elsewhere [51], not all Nrf2 inducers have the capacity to modulate proteasomal gene expression, it is likely given the known role of HNE in modulating proteasomal activity that HNE may indeed be able to do so. Studies monitoring the effects HNE on proteasomal activity in normal and Nrf2 deficient cells have yet to be conducted to confirm this, but the effects of HNE on proteasomal/autophagic clearance remains a potential factor influencing the transition from physiological to pathological HNE-induced cellular events.
Aberrant HNE modifications and misguided targets of lipid peroxidation
Whilst relatively low doses of HNE can orchestrate cell signaling events, higher concentrations of HNE appear to modify a further set of target proteins, inhibiting or dysregulating previously functional cellular processes and organelle functions. In particular, endothelial cells, which form the primary vascular interface for potentially oxidized circulating components, appear to be highly susceptible to HNE induced damage [10,85]. HNE can exert a range of pathophysiological effects, including interfering with the synthesis and release of vasoactive mediators, breakdown of the endothelial barrier function and inducing a pro-inflammatory phenotype within the vessel wall.
Loss of vasoprotection
Although not directly reactive with nitric oxide (NO), HNE is able to reduce bioavailability of the key vasoactive agent NO via modulation of nitric oxide synthase (NOS) activity. In BAEC treated with HNE, Whitsett et al. showed an elevation of peroxynitrite and uncoupled endothelial nitric oxide synthase (eNOS) with reduced tetrahydrobiopterin (BH4) levels and eNOS phosphorylation, resulting from a loss of guanosine triphosphate cyclohydrolase I (GTPCH) and heat shock protein 90 (HSP90) [23]. Reduced GTPCH and HSP90 induced by HNE can be reversed by proteasomal inhibition, suggesting accelerated proteasomal degradation of these proteins following HNE adduction [23,86]. However, as no additional studies were conducted to assess whether these proteins are adducted by HNE or whether HNE is modulating proteasomal activity, the mechanism mediating these effects remains uncertain. In SMCs, HNE also inhibits the production of nitrite from NO through modulating gene expression, decreasing expression of inducible nitric oxide synthase (iNOS) following inhibition of NFκB activation [73].
Elevated plasma levels of HNE also damage endothelial barrier function [10,87] due to impaired cell–cell communication and inhibition of membrane associated enzymes [85]. Treatment of endothelial cells with HNE increases Michael additions to adhesion and adherens junction proteins, reduces surface integrin expression and phosphorylation of focal adhesion kinase (FAK) and redistributes tight junction proteins, leading to impaired focal adhesion and intercellular gap formation [88]. Intracellular F-actin arrangement may also be remodeled by HNE, contributing to gap formation and barrier dysfunction [49]. Notably, all these changes can be inhibited by pre-treatment of cells with the thiol protectant N-acetylcysteine, indicating an underlying redox sensitive mechanism [49,88]. It has been suggested that sulfhydryl groups (e.g. Cys374 on actin regulating polymerization) are potential targets of HNE addition [89]. Moreover, lipid peroxidation products induce inflammatory responses and increase the adhesion of monocytes to endothelial cells [88,90], with HNE also reported to modify LDL leading to increased uptake and reduced clearance of oxLDL from SMCs [12]. As discussed in Section 6, these processes may be active contributors towards the development of vascular diseases.
Organelle specific effects—mitochondrial dysfunction
Mitochondria are a major energy source in cells and generate reactive oxygen radicals (e.g. superoxide which can be rapidly dismutated to hydrogen peroxide) via the electron respiratory chain, and thereby not only contribute to cell signaling but also to the development of disease [91]. Mitochondrial membranes are rich in protein thiols which make them potential targets for lipid peroxidation products [92]. Similar to other non-vascular cell types, reviewed elsewhere [31], HNE is reported to increase cellular generation of reactive oxygen species in both endothelial and SMCs, with superoxide generated by the mitochondria the predominant source. Landar et al. reported that in BAECs, HNE (5 µM) evokes an increase in mitochondrial reactive oxygen species generation measured by DCF fluorescence [71]. However, in our studies, acute treatment with HNE (20 µM) fails to induce superoxide generation in human umbilical vein endothelial cells measured by lucigenin and L-012 chemiluminescence. Although no studies have yet determined the effects of mitochondrial inhibitors in endothelial cells in response to HNE evoked superoxide generation, rotenone, a complex I inhibitor, can inhibit 15-deoxy-Delta (12,14) prostaglandin-J2 (15d-PGJ2) induced mitochondrial ROS generation [93]. Similarly, in BAEC treated with 15d-PGJ2 for an extended time period, mitochondrial membrane potential is reduced [53] with elevated complex I activity [94], perhaps suggestive of mitochondrial uncoupling providing a feedback mechanism in mitochondria to combat sustained HNE stress [95]. In SMCs, HNE has been shown to dose-dependently increase ROS generation, with inhibition of mitochondrial complex I preventing HNE-induced superoxide generation [44,96]. Interestingly, exposure of SMCs to relatively high (20 µM) HNE concentrations fails to induce any change in O2 consumption [97], indicating that rather than exerting a direct effect on the respiratory chain HNE may be modulating clearance of reactive oxygen species by GSH or other antioxidant pathways.
Although yet to be investigated, it is worthwhile highlighting the role that mitochondria may play in HNE induced activation of the Nrf2 defense pathway. Using immunofluorescence, Lo et al. have shown that cytosolic Keap1 is closely associated with mitochondria [98]. Modulation of the mitochondrial redox status in endothelial cells has been proposed as a necessary step in the activation of Nrf2 signaling by lipid peroxidation products [53] and shear stress [99], although a specific role for mitochondria in HNE-induced Nrf2 activation in endothelial or SMCs has yet to be established. Addition of a mitochondrial targeted thiol reactive compound 4-iodobutyl-triphenylphosphonium abolishes hemin and 15d-PGJ2 induced Nrf2 signaling [53] without interfering with Nrf2-Keap1 dissociation, indicating an intermediate role for mitochondrial thiols. Interestingly, using modified 15d-PGJ2 specifically targeted to mitochondria, Diers et al. found that the modified compound caused profound mitochondrial reactive oxygen species generation and apoptosis, with minimal induction of HO-1 or adaptive increases in GSH. These findings suggest that both cytosolic and mitochondrial modifications induced by lipid peroxidation products are required for activating Nrf2 [100]. Moreover, modulation of redox regulating enzymes rather than altered ROS generation in mitochondira may be responsible for the activation of Nrf2/ARE signaling, as overexpression of mitochondrial thiol reductive enzyme thioredoxin 2 (Trx2) can abrogate the induction of this pathway [101]. Although HNE-induced modification of Trx2 has yet to be determined, inhibition of thioredoxin 1 by HNE conjugation has been previously reported [90] in aortic endothelial cells, and may thus serve as a mechanism for the activation of Nrf2 by HNE.
Organelle specific effects - endoplasmic reticulum (ER) stress
ER stress and the subsequent unfolded protein response (UPR) are emerging as key mechanisms determining the fate of cells and may contribute to the development of atherosclerosis, diabetes and neurodegenerative disorders. Oxidative stress (e.g from mitochondria), lipid peroxides such as HNE and elevation of intracellular Ca2+ are known activators of ER stress [102]. In eukaryotic cells, the ER regulates chaperone-assisted protein folding, which requires tight regulation of its internal redox environment to prevent mis-directed disulfide bond formation and accumulation of misfolded proteins [103]. ER stress, induced by excessive accumulation of misfolded proteins, results in activation of the UPR to (i) transiently reduce ER load and induce chaperones to enhance correct folding capacity, (ii) degrade misfolded proteins via proteasomal and autophagy ER assisted degradation (ERAD) pathways and (iii) induce antioxidant responses to restore redox homeostasis and reduce ER stress [102,104]. If a balanced folding capacity cannot be achieved, induction of pro-apoptotic signaling (e.g. C/EBP-homologous protein, CHOP transcription) triggers programmed cell death. The UPR is activated by the disassociation of Grp78 (BiP) from ER transmembrane UPR effectors and its association with accumulated misfolded proteins, allowing activation of transmembrane ER effectors [105] inositol requiring enzyme 1 (IRE1), RNA-dependent protein kinase-like endoplasmic reticulum kinase (PERK) and activating transcription factor 6 (ATF6), and down-stream signaling cascades including the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and xBP-1 splicing [102,106].
Similar to mitochondria, ER proteins are also thiol abundant and targets of lipid peroxidation products [107]. Whilst not affecting intracellular Ca2+, incubation of endothelial cells with 20 μM HNE for 24 h significantly elevates phosphorylation of IRE1 and eIF2α, which is effectively inhibited by pre-treatment with NAC, indicating increased ER stress by HNE is thiol dependent [107,108]. Moreover, depleting GSH alone with tert-butylhydroperoxide (tBHP) cannot trigger ER stress [107], implicating a direct modification of ER proteins by HNE. Liquid chromatography–mass spectrometry analysis has shown that chaperones including Grp78, protein disulfide isomerases (PDIs), and heat shock proteins including HSP90 and HSP70 are predominantly conjugated by HNE (25 μM, 30 min) in human umbilical vein endothelial cells [107]. It is presently unclear whether HNE Michael addition via lysine residues to Grp78 is alone sufficient to trigger UPR activation, but it is likely that HNE adduction to PDI may trigger activation. PDI is a key ER-resident enzyme catalyzing the formation and rearrangement of disulfide bonds, and thus determines protein unfolding capacity in ER. Activity of PDI can be inhibited by HNE in a concentration dependent manner due to cysteine modifications [67], leading to accumulation of unfolded proteins and consequential ER stress [109,110]. Fortunately, PDI modification is reversible by physiological concentrations of GSH [67] allowing for eventual resolution of ER stress.
Secondary to ER stress, activation of the UPR by HNE is evidenced by the upregulation of ER regulators and chaperones (e.g. Grp78 and homocysteine inducible ER protein (HERP)) to enhance overall protein folding capacity [107]. It worth noting that Nrf2 has been identified as a direct downstream signaling target activated by PERK phosphorylation [111], suggesting that HNE-induced Nrf2 activation may also occur secondary to ER stress as well as a consequence of cytosolic or potentially mitochondrial oxidation. Depletion of Nrf2 renders cells more susceptible to ER stress, and Nrf2 activation in response to ER stress activators has been shown to be critical for cell survival [109,111]. These findings highlight that, in addition to enhancing protein folding capacity, restoration of redox homeostasis through activation of Nrf2 mediated induction of GSH related genes, phase II enzymes, antioxidant enzymes and potentially 20S proteasomal gene expression and oxidized protein clearance, is necessary for cellular recovery from HNE stress. Together these Nrf2-mediated responses prevent continued ER stress and activation of apoptotic signaling cascades. Unfortunately, information concerning ER stress in SMCs is extremely limited and to our knowledge there are no reports on whether HNE is capable of inducing ER stress in SMCs. However, induction of ER stress in SMCs by non-lipid peroxides has been reviewed [112] and, as highlighted in this review, a causative relationship between ER stress and apoptosis is lacking in SMCs. Notably, ER stress has important implications for diseases such as atherosclerosis and diabetes in which it has actively been assigned a role in disease pathogenesis.
Apoptosis
Failure to remove HNE adducts following exposure to pathological concentrations of HNE results in cellular dysfunction and oxidative damage in organelles such as mitochondria and the ER in vascular cells. In human endothelial cells, pathological concentrations of HNE increase caspase activation and induce apoptosis [23]. Similarly in SMCs, HNE has been shown to increase mitochondrial generation of reactive oxygen radicals and induce apoptosis [96], which was augmented by AR inhibition and reduced HNE clearance [28]. DNA adduction and subsequent fragmentation has also been reported to occur following HNE treatment of both endothelial and SMCs [72,113].
In most studies, pathological concentrations of HNE are required to initiate apoptosis, with the hormetic effects of HNE highlighted in the proliferation responses of SMCs. Whilst low physiological levels of HNE (0.1–1 µM) dose-dependently increase protein synthesis and SMC proliferation, exposure to higher concentrations >1 µM progressively inhibit protein synthesis and induce cell apoptosis. It has recently been suggested that detoxification of HNE through GSH phase II reactions may represent an important buffering system within cells to limit and create specificity in lipid peroxide signaling by preventing aberrant HNE modifications [31]. Indeed, the capacity of cells to remove HNE and HNE-damaged proteins (via GSTS, AR, RLIP76, proteasome, autophagy, UPR etc.) clearly plays a critical role in determining cell fate. Moreover, in vascular diseases in which cells may be exposed chronically to elevated levels of HNE, it could be argued that HNE-induced apoptosis can occur not only as a result of accumulative damage but as a loss of normal cell signaling (e.g NO production, TKR activation and downstream kinase signaling) inducing progressive endothelial dysfunction ultimately resulting in programmed cell death.
Role of HNE in vascular diseases
Lipid peroxide induced damage is widely thought to be an active contributor to a number of vascular diseases, including, atherosclerosis, neurodegenerative disorders, diabetes and pregnancy diseases such as pre-eclampsia and gestational diabetes. Unlike reactive oxygen species, lipid peroxides are generally more stable allowing them to diffuse through membranes and affect distant organelles or tissues including fetal tissues following transplacental transfer [1,114,115]. Indeed, HNE-modified oxLDL accumulates in atherosclerotic plaques [13,116], with HNE adducts and associated peroxides also reported pancreatic beta cell islets [117], neuronal tissue [118] and in circulating cord blood [119]. Notably, and as highlighted below, whilst HNE accumulation in many vascular disease models is well documented, evidence directly linking HNE to cellular dysfunction is in most cases associative rather than causal, underlining the need for further in vitro studies to assess the specific effects of HNE on cellular organelles.
In utero diseases
Lipid peroxides including HNE have been extensively implicated in the pathogenesis of in utero diseases such as pre-eclampsia through the induction of pro-inflammatory signaling cascades [120] in the maternal as well as fetal vasculature. Moreover, as reviewed elsewhere [119,120], enhanced HNE formation appears to correlate with alterations in fatty acid metabolism and co-insides with impairments in antioxidant defenses, with these changes ultimately resulting in systemic vascular dysfunction. Commonly, central to the role of HNE in disease is the dysregulation of organelles such as mitochondria and the ER. Whilst PE appears to have a vast inflammatory component associated with its etiology, markers of ER stress and mitochondrial dysfunction have been detected. Activation of UPR effector pathways is clearly evidenced in the placentas of PE pregnancies [121], although no causative link has yet been made to suggest that ER stress occurs secondary to lipid peroxidation. Furthermore, ER stress remains to be assessed in the fetal vasculature, although markers of mitochondrial dysfunction are found in fetal endothelial cells [122] and are well documented in the placenta to co-inside with markers of oxidative stress [123].
Atherosclerosis
In atherosclerosis and cardiac disorders, there are indications that HNE-induced mitochondrial dysfunction and ER stress may play a key role in disease pathogenesis through the modulation of vascular cells. In spontaneously hypertensive rats, HNE adducts to mitochondrial NADP+-isocitrate dehydrogenase and reduced enzymatic activity have been reported and shown to enhance cardiac hypertrophy [124]. Furthermore, in vitro oxLDL has been shown to lead to endothelial HNE accumulation and the induction of ER stress through the inhibition of PDI activity, consequent downstream effector signaling (e.g xBP-1 splicing) and endothelial caspase activation and apoptosis [110]. In vivo, endothelial ER stress in vessel regions associated with low or disturbed shear stress have also been reported [125,126] and even to precede loss of Nrf2 [126]. ER stress appears to have a negative effect on endothelial cell stability [125], which may in part account for plaque susceptibility to rupture through ER stress-mediated apoptosis, promoting loss of the endothelial layer covering the plaque. Without direct confirmation that HNE can induce ER stress within SMCs, it is more difficult to assess whether this vascular cell type can also be destabilized during atherosclerosis as a consequence of oxLDL/HNE accumulation.
Diabetes
ER stress has also been implicated in diabetic pathologies, including in beta cell death [102,127] and vascular dysfunction in diabetic retinopathy [128]. Whilst there is little information regarding the potential role of lipid peroxides in inducing ER stress within pancreatic beta cells, ER stress markers have been found in the retinas of human diabetic patients [128] and may be as a consequence of oxidative stress or elevated HNE production. In vitro human retinal capillary pericytes treated with oxLDL or HNE exhibit markers of ER stress and mitochondrial dysfunction [128], with ER stress also found in human retinal Muller cells following oxLDL or HNE exposure [129]. Athough not well characterized in diabetes, it is likely that the vascular endothelium may also be affected, since treatment of healthy human endothelial cells with HNE induces ER stress [107]. Similarly, mitochondrial dysfunction and the resulting elevation of reactive oxygen species generation has been implicated in the pathogenesis of diabetes [50], and there is evidence to support HNE-induced mitochondrial dysfunction in vascular diabetic pathologies. In diabetic rat hearts HNE adducts formed with complex II subunit SDHA have been shown to result in reduced mitochondrial respiration, with insulin administration able to prevent HNE adduction and loss of mitochondrial function [130]. Conversely, in diabetic skeletal muscle biopsies reduced mitochondrial respiration has been detected with the absence of HNE accumulation in tissue, suggesting that in diabetes mitochondrial dysfunction may be induced independently from lipid peroxide generation [131].
Neurodegerative diseases
In neurodegenerative diseases associated with amyloid, α-Synuclein or transthyretin protein deposition such as Alzheimer’s disease, Parkinson’s disease or familial amyloidotic polyneuropathy (FAP), indirect associations have been made between HNE-induced oxidative stress and downstream ER stress/UPR activation or mitochondrial dysfunction. HNE colocalises with Lewy body protein accumulations present within brain tissues, such as in Parkinsons disease [118], indirectly implicating HNE in the pathogenesis of these diseases. In addition, HNE has been shown to directly form Michael additions with proteins such as α-Synuclein implicated in familial Parkinson’s [132], however the oligomers formed, whilst toxic, are not fibrilar implying that HNE alone cannot account of the formation of Lewy bodies. In Parkinson’s patients, the accumulation of HNE [118] and markers of ER stress such as the upregulation of UPR target HERP have been reported in Lewy bodies present within the substantia nigra [133]. Whilst not providing a definite link, in retinal Muller glial cells HNE has been shown to induce ER stress in vitro [129] in agreement with the possibility that in the brain HNE-induced ER stress may be a cause rather than consequence of neuronal injury. In FAP, use of antioxidant scavengers was able to reduce HNE accumulation and markers of ER stress in stomach tissue and reduce transthyretin accumulation [134], suggestive of an active role of redox regulation in transthyretin deposition. Conversely, transthyretin has been shown to directly activate ER stress, though whether this is through HNE accumulation remains unknown [135]. Furthermore, following neuronal injury such as in stroke, HNE concentrations are increased in plasma and positively correlate with homocysteine concentrations [136]. Whilst not directly implicating HNE in the pathogenesis of stroke, spontaneously hypertensive stroke prone rats exhibit higher circulating levels of HNE [136] and HNE has also been shown form Michael additions to various adhesion molecules inducing breakdown of endothelial barrier function, which may have important implications for blood-brain barrier integrity [88]. Consistent with a role for HNE in inducing mitochondrial dysfunction, Sompol et al. have reported that in an animal model of Alzheimer’s disease, HNE accumulation is associated with reduced mitochondrial membrane potential in neuronal cells [137]. As overexpression of MnSOD or use of a SOD mimetic was able to partially restore mitochondrial function, it is likely that HNE modulates mitochondrial superoxide output either directly by adduction to the respiratory chain or indirectly through inhibition of antioxidant proteins such as Trx2.
Concluding remarks and future research prospectives
Bioactive lipid peroxides such as HNE are increasingly becoming recognized as exerting hormetic actions within the vasculature (Fig. 1). Low physiological levels of HNE appear to positively modulate the cell cycle and proliferation of vascular cells [44] and, by inducing non-toxic protein oxidation, can induce long-term cytoprotection though the activation of Nrf2 antioxidant defenses protecting against later oxidant insults [55]. Indeed, oxLDL, containing 0.5 µM free HNE, and 5 µM HNE alone have been shown in human endothelial cells to upregulate a variety of Nrf2-dependent genes with a similar transcriptional profile to laminar shear stress [138], arguably the ultimate vasoprotective stimulus exerted in the vasculature. In endothelial cells, there is limited information concerning the effects of physiological concentrations of HNE (<1 µM) on endothelial function, which may have important implications for regulation of basal transcriptional activity and thus merits further investigation. Progressively higher concentrations of HNE, extending into the pathological range, are well documented to exert opposing effects. At high doses, HNE induces widespread oxidation of cellular proteins [1], particularly those located within cholesterol rich membranes or in organelles with a high thiol content such as the mitochondrial or the ER [71,107]. Modification of such proteins can not only induce a pro-inflammatory phenotype or disrupt endothelial barrier function [88], but can also precipitate further oxidative stress by disrupting protein folding and potentially mitochondrial function, which if not counteracted by Nrf2 antioxidant defenses ultimately results in apoptosis [10,71,107]. Further studies using both physiological and pathological HNE concentrations will enhance our understanding of the extent and effects of HNE adduction to mitochondrial or ER associated proteins in vascular endothelial and SMCs, and will have important implications for targeting endogenous antioxidant defense pathways to prevent or limit the progression of vascular diseases.
Levels of HNE within tissues are dependent not only on the rate of fatty acid oxidation and thus HNE production, but also on its removal by phase II metabolic pathways. Key to the removal of HNE is its conjugation to the abundant thiol GSH, which in vascular cells is primarily mediated via GST-4 [20]. Whilst GSH conjugation is essential for removal of HNE, importantly some HNE conjugates such as GS-DHN produced by AR are also bioactive and may perhaps help to prolong the biological actions of short-lived HNE following low-dose HNE stimulation [45]. Interestingly, whilst modulation of some HNE degradation pathways such as the HNE-conjugate transporter RLIP76 appear to have a clear role in disease pathologies such as in atherosclerotic lesions or metabolic syndrome [78,80], others such as AR are less clearly defined. Thus, when considering redox modulating therapies, there is a need to consider both the hormetic effects of HNE in vascular pathologies and other potential off-target effects following HNE exposure.
Whilst studies clearly demonstrate short-term inhibition of AR is able to upregulate low dose HNE proliferative effects within vascular cells, AR activity is also critical in promoting cell survival following aberrant HNE modifications in response to higher HNE doses [45]. Moreover, in diseases such as diabetic retinopathy associated with HNE accumulation [139], AR activity is reportedly higher [140], presumably in part to increase HNE clearance and combat oxidative stress. Whilst elevated AR activity may be of benefit in removing HNE conjugates, increased AR activity also increases glucose metabolism to sorbitol, resulting in osmotic stress and cytotoxicity [140]. In such instances where it is clear that dampening AR activity may be of overall benefit in terms of restoring redox homeostasis via relieving glucose-induced cytotoxicity, increasing HNE degradation by other means may provide an alternative strategy to alleviate HNE-induced oxidative injury.
In conclusion, HNE is capable of modulating both survival and pro-apoptotic pathways within vascular endothelial and SMCs. Aging or disease pathologies associated with HNE accumulation may be due to both elevated HNE synthesis and/or decline in phase II metabolic pathways required to remove HNE, inducing aberrant HNE protein oxidation and resulting in vascular dysfunction. Whilst HNE can form widespread protein adducts, specific organelles including the plasma membrane, mitochondria and ER clearly more susceptible to HNE modification, and thus the convergence of HNE concentration and activation of clearance pathways within these organelles may be the critical determinant as to whether cells can induce adaptive antioxidant defenses via Nrf2 to counteract HNE modifications or ultimately undergo apoptosis. Targeting of endogenous antioxidant defenses such as Nrf2-linked genes to remove HNE adducts may be of therapeutic benefit, but considerations concerning the effects on parallel metabolic pathways should considered when pursuing this strategy.
Acknowledgments
We gratefully acknowledge the support of Heart Research UK, British Heart Foundation, The Henry Smith Charity and The Great Britain Sasakawa Foundation.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology and Medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Benedetti A., Comporti M., Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochimica et Biophysica Acta. 1980;620:281–296. doi: 10.1016/0005-2760(80)90209-x. [DOI] [PubMed] [Google Scholar]
- 3.Chung F.L., Nath R.G., Ocando J., Nishikawa A., Zhang L. Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: detection and potential sources. Cancer Research. 2000;60:1507–1511. [PubMed] [Google Scholar]
- 4.Nair J., De Flora S., Izzotti A., Bartsch H. Lipid peroxidation-derived etheno-DNA adducts in human atherosclerotic lesions. Mutation Research. 2007;621:95–105. doi: 10.1016/j.mrfmmm.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Doorn J.A., Petersen D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chemico-Biological Interactions. 2003;143–144:93–100. doi: 10.1016/s0009-2797(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 6.Uchida K., Stadtman E.R. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. Journal of Biological Chemistry. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 7.Selley M.L., Bartlett M.R., McGuiness J.A., Hapel A.J., Ardlie N.G. Determination of the lipid peroxidation product trans-4-hydroxy-2-nonenal in biological samples by high-performance liquid chromatography and combined capillary column gas chromatography-negative-ion chemical ionisation mass spectrometry. Journal of Chromatography. 1989;488:329–340. doi: 10.1016/s0378-4347(00)82957-6. [DOI] [PubMed] [Google Scholar]
- 8.Gil L., Siems W., Mazurek B., Gross J., Schroeder P., Voss P., Grune T. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radical Research. 2006;40:495–505. doi: 10.1080/10715760600592962. [DOI] [PubMed] [Google Scholar]
- 9.Selley M.L. Determination of the lipid peroxidation product (E)-4-hydroxy-2-nonenal in clinical samples by gas chromatography--negative-ion chemical ionisation mass spectrometry of the O-pentafluorobenzyl oxime. Journal of Chromatography B: Biomedical Sciences and Applications. 1997;691:263–268. doi: 10.1016/s0378-4347(96)00446-x. [DOI] [PubMed] [Google Scholar]
- 10.Herbst U., Toborek M., Kaiser S., Mattson M.P., Hennig B. 4-Hydroxynonenal induces dysfunction and apoptosis of cultured endothelial cells. Journal of Cellular Physiology. 1999;181:295–303. doi: 10.1002/(SICI)1097-4652(199911)181:2<295::AID-JCP11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 11.Ong W.Y., Jenner A.M., Pan N., Ong C.N., Halliwell B. Elevated oxidative stress, iron accumulation around microvessels and increased 4-hydroxynonenal immunostaining in zone 1 of the liver acinus in hypercholesterolemic rabbits. Free Radical Research. 2009;43:241–249. doi: 10.1080/10715760802691455. [DOI] [PubMed] [Google Scholar]
- 12.Hoff H.F., O’Neil J., Chisolm G.M., 3rd, Cole T.B., Quehenberger O., Esterbauer H., Jurgens G. Modification of low density lipoprotein with 4-hydroxynonenal induces uptake by macrophages. Arteriosclerosis. 1989;9:538–549. doi: 10.1161/01.atv.9.4.538. [DOI] [PubMed] [Google Scholar]
- 13.Salomon R.G., Kaur K., Podrez E., Hoff H.F., Krushinsky A.V., Sayre L.M. HNE-derived 2-pentylpyrroles are generated during oxidation of LDL, are more prevalent in blood plasma from patients with renal disease or atherosclerosis, and are present in atherosclerotic plaques. Chemical Research in Toxicology. 2000;13:557–564. doi: 10.1021/tx000007u. [DOI] [PubMed] [Google Scholar]
- 14.Gueraud F., Atalay M., Bresgen N., Cipak A., Eckl P.M., Huc L., Jouanin I., Siems W., Uchida K. Chemistry and biochemistry of lipid peroxidation products. Free Radical Research. 2010;44:1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- 15.Hill B.G., Haberzettl P., Ahmed Y., Srivastava S., Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochemical Journal. 2008;410:525–534. doi: 10.1042/BJ20071063. [DOI] [PubMed] [Google Scholar]
- 16.Botzen D., Grune T. Degradation of HNE-modified proteins--possible role of ubiquitin. Redox Report. 2007;12:63–67. doi: 10.1179/135100007X162130. [DOI] [PubMed] [Google Scholar]
- 17.Grune T., Davies K.J. The proteasomal system and HNE-modified proteins. Molecular Aspects of Medicine. 2003;24:195–204. doi: 10.1016/s0098-2997(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 18.Srivastava S., Liu S.Q., Conklin D.J., Zacarias A., Srivastava S.K., Bhatnagar A. Involvement of aldose reductase in the metabolism of atherogenic aldehydes. Chemico-Biological Interactions. 2001;130-132:563–571. doi: 10.1016/s0009-2797(00)00299-4. [DOI] [PubMed] [Google Scholar]
- 19.Siems W., Grune T. Intracellular metabolism of 4-hydroxynonenal. Molecular Aspects of Medicine. 2003;24:167–175. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y., Yang Y., Trent M.B., He N., Lick S.D., Zimniak P., Awasthi Y.C., Boor P.J. Glutathione-S-transferase A4-4 modulates oxidative stress in endothelium: possible role in human atherosclerosis. Atherosclerosis. 2004;173:211–221. doi: 10.1016/j.atherosclerosis.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 21.Tjalkens R.B., Cook L.W., Petersen D.R. Formation and export of the glutathione conjugate of 4-hydroxy-2, 3-E-nonenal (4-HNE) in hepatoma cells. Archives of Biochemistry and Biophysics. 1999;361:113–119. doi: 10.1006/abbi.1998.0946. [DOI] [PubMed] [Google Scholar]
- 22.Siems W.G., Zollner H., Grune T., Esterbauer H. Metabolic fate of 4-hydroxynonenal in hepatocytes: 1,4-dihydroxynonene is not the main product. Journal of Lipid Research. 1997;38:612–622. [PubMed] [Google Scholar]
- 23.Whitsett J., Picklo M.J., Sr, Vasquez-Vivar J. 4-Hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2340–2347. doi: 10.1161/ATVBAHA.107.153742. [DOI] [PubMed] [Google Scholar]
- 24.Poot M., Verkerk A., Koster J.F., Esterbauer H., Jongkind J.F. Influence of cumene hydroperoxide and 4-hydroxynonenal on the glutathione metabolism during in vitro ageing of human skin fibroblasts. European Journal of Biochemistry. 1987;162:287–291. doi: 10.1111/j.1432-1033.1987.tb10598.x. [DOI] [PubMed] [Google Scholar]
- 25.Romero F.J., Romero M.J., Bosch-Morell F., Martinez M.C., Medina P., Lluch S. 4-hydroxynonenal-induced relaxation of human mesenteric arteries. Free Radical Biology and Medicine. 1997;23:521–523. doi: 10.1016/s0891-5849(97)00119-6. [DOI] [PubMed] [Google Scholar]
- 26.Voskoboinik I., Soderholm K., Cotgreave I.A. Ascorbate and glutathione homeostasis in vascular smooth muscle cells: cooperation with endothelial cells. American Journal of Physiology. 1998;275:C1031–1039. doi: 10.1152/ajpcell.1998.275.4.C1031. [DOI] [PubMed] [Google Scholar]
- 27.Buckley B.J., Whorton A.R. Adaptive responses to peroxynitrite: increased glutathione levels and cystine uptake in vascular cells. American Journal of Physiology—Cell Physiology. 2000;279:C1168–1176. doi: 10.1152/ajpcell.2000.279.4.C1168. [DOI] [PubMed] [Google Scholar]
- 28.Spycher S., Tabataba-Vakili S., O’Donnell V.B., Palomba L., Azzi A. 4-hydroxy-2,3-trans-nonenal induces transcription and expression of aldose reductase. Biochemical and Biophysical Research Communications. 1996;226:512–516. doi: 10.1006/bbrc.1996.1386. [DOI] [PubMed] [Google Scholar]
- 29.Ruef J., Liu S.Q., Bode C., Tocchi M., Srivastava S., Runge M.S., Bhatnagar A. Involvement of aldose reductase in vascular smooth muscle cell growth and lesion formation after arterial injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:1745–1752. doi: 10.1161/01.atv.20.7.1745. [DOI] [PubMed] [Google Scholar]
- 30.Poli G., Schaur R.J., Siems W.G., Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Medical Care Research and Review. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 31.Higdon A., Diers A.R., Oh J.Y., Landar A., Darley-Usmar V.M. Cell signaling by reactive lipid species: new concepts and molecular mechanisms. Biochemical Journal. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forman H.J., Fukuto J.M., Miller T., Zhang H., Rinna A., Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Archives of Biochemistry and Biophysics. 2008;477:183–195. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. American Journal of Clinical Nutrition. 1993;57:779S–785S. doi: 10.1093/ajcn/57.5.779S. [DOI] [PubMed] [Google Scholar]
- 34.Ishii T., Itoh K., Takahashi S., Sato H., Yanagawa T., Katoh Y., Bannai S., Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. Journal of Biological Chemistry. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 35.Ishii T., Itoh K., Ruiz E., Leake D.S., Unoki H., Yamamoto M., Mann G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circulation Research. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 36.Suc I., Meilhac O., Lajoie-Mazenc I., Vandaele J., Jurgens G., Salvayre R., Negre-Salvayre A. Activation of EGF receptor by oxidized LDL. FASEB Journal. 1998;12:665–671. doi: 10.1096/fasebj.12.9.665. [DOI] [PubMed] [Google Scholar]
- 37.Natarajan V., Scribner W.M., Vepa S. Phosphatase inhibitors potentiate 4-hydroxynonenal-induced phospholipase D activation in vascular endothelial cells. American Journal of Respiratory Cell and Molecular Biology. 1997;17:251–259. doi: 10.1165/ajrcmb.17.2.2623. [DOI] [PubMed] [Google Scholar]
- 38.McDermott M., Wakelam M.J., Morris A.J., Phospholipase D. Biochemistry and Cell Biology. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- 39.Parinandi N.L., Scribner W.M., Vepa S., Shi S., Natarajan V. Phospholipase D activation in endothelial cells is redox sensitive. Antioxidants and Redox Signaling. 1999;1:193–210. doi: 10.1089/ars.1999.1.2-193. [DOI] [PubMed] [Google Scholar]
- 40.Akiba S., Kumazawa S., Yamaguchi H., Hontani N., Matsumoto T., Ikeda T., Oka M., Sato T. Acceleration of matrix metalloproteinase-1 production and activation of platelet-derived growth factor receptor beta in human coronary smooth muscle cells by oxidized LDL and 4-hydroxynonenal. Biochimica et Biophysica Acta. 2006;1763:797–804. doi: 10.1016/j.bbamcr.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Lee S.J., Seo K.W., Yun M.R., Bae S.S., Lee W.S., Hong K.W., Kim C.D. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-kappaB signaling pathways. Free Radical Biology and Medicine. 2008;45:1487–1492. doi: 10.1016/j.freeradbiomed.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 42.Seo K.W., Lee S.J., Kim C.E., Yun M.R., Park H.M., Yun J.W., Bae S.S., Kim C.D. Participation of 5-lipoxygenase-derived LTB(4) in 4-hydroxynonenal-enhanced MMP-2 production in vascular smooth muscle cells. Atherosclerosis. 2010;208:56–61. doi: 10.1016/j.atherosclerosis.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 43.Ruef J., Rao G.N., Li F., Bode C., Patterson C., Bhatnagar A., Runge M.S. Induction of rat aortic smooth muscle cell growth by the lipid peroxidation product 4-hydroxy-2-nonenal. Circulation. 1998;97:1071–1078. doi: 10.1161/01.cir.97.11.1071. [DOI] [PubMed] [Google Scholar]
- 44.Lee T.J., Lee J.T., Moon S.K., Kim C.H., Park J.W., Kwon T.K. Age-related differential growth rate and response to 4-hydroxynonenal in mouse aortic smooth muscle cells. International Journal of Molecular Medicine. 2006;17:29–35. [PubMed] [Google Scholar]
- 45.Ramana K.V., Bhatnagar A., Srivastava S., Yadav U.C., Awasthi S., Awasthi Y.C., Srivastava S.K. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. Journal of Biological Chemistry. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 46.Paola D., Domenicotti C., Nitti M., Vitali A., Borghi R., Cottalasso D., Zaccheo D., Odetti P., Strocchi P., Marinari U.M., Tabaton M., Pronzato M.A. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochemical and Biophysical Research Communications. 2000;268:642–646. doi: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- 47.Marinari U.M., Nitti M., Pronzato M.A., Domenicotti C. Role of PKC-dependent pathways in HNE-induced cell protein transport and secretion. Molecular Aspects of Medicine. 2003;24:205–211. doi: 10.1016/s0098-2997(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 48.Gopalakrishna R., Jaken S. Protein kinase C signaling and oxidative stress. Free Radical Biology and Medicine. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 49.Usatyuk P.V., Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. Journal of Biological Chemistry. 2004;279:11789–11797. doi: 10.1074/jbc.M311184200. [DOI] [PubMed] [Google Scholar]
- 50.Cheng X., Siow R.C., Mann G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxidants and Redox Signaling. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- 51.Chapple S.J., Siow R.C., Mann G.E. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. International Journal of Biochemistry and Cell Biology. 2012;44:1315–1320. doi: 10.1016/j.biocel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 52.Levonen A.L., Landar A., Ramachandran A., Ceaser E.K., Dickinson D.A., Zanoni G., Morrow J.D., rley-Usmar V.M. Cellular mechanisms of redox cell signaling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochemical Journal. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ricart K.C., Bolisetty S., Johnson M.S., Perez J., Agarwal A., Murphy M.P., Landar A. The permissive role of mitochondria in the induction of haem oxygenase-1 in endothelial cells. Biochemical Journal. 2009;419:427–436. doi: 10.1042/BJ20081350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itoh K., Mochizuki M., Ishii Y., Ishii T., Shibata T., Kawamoto Y., Kelly V., Sekizawa K., Uchida K., Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2) Molecular and Cellular Biology. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishikado A., Nishio Y., Morino K., Ugi S., Kondo H., Makino T., Kashiwagi A., Maegawa H. Low concentration of 4-hydroxy hexenal increases heme oxygenase-1 expression through activation of Nrf2 and antioxidative activity in vascular endothelial cells. Biochemical and Biophysical Research Communications. 2010;402:99–104. doi: 10.1016/j.bbrc.2010.09.124. [DOI] [PubMed] [Google Scholar]
- 56.Motohashi H., Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends in Molecular Medicine. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 57.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J.D., Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes and Development. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang M.I., Kobayashi A., Wakabayashi N., Kim S.G., Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proceedings of the National Academy of Sciences of U.S.A. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McMahon M., Itoh K., Yamamoto M., Hayes J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. Journal of Biological Chemistry. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 60.Sekhar K.R., Rachakonda G., Freeman M.L. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicology and Applied Pharmacology. 2010;244:21–26. doi: 10.1016/j.taap.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dinkova-Kostova A.T., Holtzclaw W.D., Cole R.N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proceedings of the National Academy of Sciences of U.S.A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamamoto T., Suzuki T., Kobayashi A., Wakabayashi J., Maher J., Motohashi H., Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Molecular and Cellular Biology. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jung K.A., Choi B.H., Nam C.W., Song M., Kim S.T., Lee J.Y., Kwak M.K. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicological Letters. 2013;218:39–49. doi: 10.1016/j.toxlet.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 64.Macleod A.K., McMahon M., Plummer S.M., Higgins L.G., Penning T.M., Igarashi K., Hayes J.D. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis. 2009 doi: 10.1093/carcin/bgp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanduyn N., Settivari R., Wong G., Nass R. SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicological Sciences. 2010;118:613–624. doi: 10.1093/toxsci/kfq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chanas S.A., Jiang Q., McMahon M., McWalter G.K., McLellan L.I., Elcombe C.R., Henderson C.J., Wolf C.R., Moffat G.J., Itoh K., Yamamoto M., Hayes J.D. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochemical Journal. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carbone D.L., Doorn J.A., Kiebler Z., Petersen D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chemical Research in Toxicology. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 68.Miller C.J., Gounder S.S., Kannan S., Goutam K., Muthusamy V.R., Firpo M.A., Symons J.D., Paine R., III, Hoidal J.R., Rajasekaran N.S. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochimica et Biophysica Acta. 2012;1822:1038–1050. doi: 10.1016/j.bbadis.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 69.Suh J.H., Shenvi S.V., Dixon B.M., Liu H., Jaiswal A.K., Liu R.M., Hagen T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proceedingsof the National Academy of Sciences of U.S.A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen Z.H., Yoshida Y., Saito Y., Noguchi N., Niki E. Adaptive response induced by lipid peroxidation products in cell cultures. FEBS Letters. 2006;580:479–483. doi: 10.1016/j.febslet.2005.12.045. [DOI] [PubMed] [Google Scholar]
- 71.Landar A., Zmijewski J.W., Dickinson D.A., Le G.C., Johnson M.S., Milne G.L., Zanoni G., Vidari G., Morrow J.D., rley-Usmar V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. American Journal of Physiology—Heart and Circulatory Physiology. 2006;290:H1777–H1787. doi: 10.1152/ajpheart.01087.2005. [DOI] [PubMed] [Google Scholar]
- 72.Ruef J., Moser M., Bode C., Kubler W., Runge M.S. 4-hydroxynonenal induces apoptosis, NF-kappaB-activation and formation of 8-isoprostane in vascular smooth muscle cells. Basic Research in Cardiology. 2001;96:143–150. doi: 10.1007/s003950170064. [DOI] [PubMed] [Google Scholar]
- 73.Hattori Y., Hattori S., Kasai K. 4-hydroxynonenal prevents NO production in vascular smooth muscle cells by inhibiting nuclear factor-kappaB-dependent transcriptional activation of inducible NO synthase. Arteriosclerosis Thrombosis and Vascular Biology. 2001;21:1179–1183. doi: 10.1161/hq0701.092135. [DOI] [PubMed] [Google Scholar]
- 74.Kakishita H., Hattori Y. Vascular smooth muscle cell activation and growth by 4-hydroxynonenal. Life Science. 2001;69:689–697. doi: 10.1016/s0024-3205(01)01166-3. [DOI] [PubMed] [Google Scholar]
- 75.Awasthi Y.C., Sharma R., Cheng J.Z., Yang Y., Sharma A., Singhal S.S., Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Molecular Aspects of Medicine. 2003;24:219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 76.Sharma R., Yang Y., Sharma A., Dwivedi S., Popov V.L., Boor P.J., Singhal S.S., Awasthi S., Awasthi Y.C. Mechanisms and physiological significance of the transport of the glutathione conjugate of 4-hydroxynonenal in human lens epithelial cells. Investigative Ophthalmology and Visual Science. 2003;44:3438–3449. doi: 10.1167/iovs.03-0051. [DOI] [PubMed] [Google Scholar]
- 77.Sharma R., Sharma A., Yang Y., Awasthi S., Singhal S.S., Zimniak P., Awasthi Y.C. Functional reconstitution of Ral-binding GTPase activating protein, RLIP76, in proteoliposomes catalyzing ATP-dependent transport of glutathione conjugate of 4-hydroxynonenal. Acta Biochimica Polonica. 2002;49:693–701. [PubMed] [Google Scholar]
- 78.Margutti P., Matarrese P., Conti F., Colasanti T., Delunardo F., Capozzi A., Garofalo T., Profumo E., Rigano R., Siracusano A., Alessandri C., Salvati B., Valesini G., Malorni W., Sorice M., Ortona E. Autoantibodies to the C-terminal subunit of RLIP76 induce oxidative stress and endothelial cell apoptosis in immune-mediated vascular diseases and atherosclerosis. Blood. 2008;111:4559–4570. doi: 10.1182/blood-2007-05-092825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Singhal J., Singhal S.S., Yadav S., Suzuki S., Warnke M.M., Yacoub A., Dent P., Bae S., Sharma R., Awasthi Y.C., Armstrong D.W., Awasthi S. RLIP76 in defense of radiation poisoning. International Journal of Radiation Oncology, Biology, Physics. 2008;72:553–561. doi: 10.1016/j.ijrobp.2008.06.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singhal J., Nagaprashantha L., Vatsyayan R., Awasthi S., Singhal S.S. RLIP76, a glutathione-conjugate transporter, plays a major role in the pathogenesis of metabolic syndrome. PLoS One. 2011;6:e24688. doi: 10.1371/journal.pone.0024688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davies K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 82.Farout L., Mary J., Vinh J., Szweda L.I., Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Archives of Biochemistry and Biophysics. 2006;453:135–142. doi: 10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 83.Vieira O., Escargueil-Blanc I., Jurgens G., Borner C., Almeida L., Salvayre R., Negre-Salvayre A. Oxidized LDLs alter the activity of the ubiquitin-proteasome pathway: potential role in oxidized LDL-induced apoptosis. FASEB Journal. 2000;14:532–542. doi: 10.1096/fasebj.14.3.532. [DOI] [PubMed] [Google Scholar]
- 84.Pickering A.M., Linder R.A., Zhang H., Forman H.J., Davies K.J. Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. Journal of Biological Chemistry. 2012;287:10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uchida K., Toyokuni S., Nishikawa K., Kawakishi S., Oda H., Hiai H., Stadtman E.R. Michael addition-type 4-hydroxy-2-nonenal adducts in modified low-density lipoproteins: markers for atherosclerosis. Biochemistry. 1994;33:12487–12494. doi: 10.1021/bi00207a016. [DOI] [PubMed] [Google Scholar]
- 86.Carbone D.L., Doorn J.A., Petersen D.R. 4-Hydroxynonenal regulates 26S proteasomal degradation of alcohol dehydrogenase. Free Radical Biology and Medicine. 2004;37:1430–1439. doi: 10.1016/j.freeradbiomed.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 87.Radu A., Moldovan N. 4-Hydroxynonenal reduces junctional communication between endothelial cells in culture. Experimental Cell Research. 1991;196:121–126. doi: 10.1016/0014-4827(91)90463-5. [DOI] [PubMed] [Google Scholar]
- 88.Usatyuk P.V., Parinandi N.L., Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. Journal of Biological Chemistry. 2006;281:35554–35566. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- 89.Usatyuk P.V., Natarajan V. Hydroxyalkenals and oxidized phospholipids modulation of endothelial cytoskeleton, focal adhesion and adherens junction proteins in regulating endothelial barrier function. Microvascular Research. 2012;83:45–55. doi: 10.1016/j.mvr.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Go Y.M., Halvey P.J., Hansen J.M., Reed M., Pohl J., Jones D.P. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. American Journal of Pathology. 2007;171:1670–1681. doi: 10.2353/ajpath.2007.070218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Quintero M., Colombo S.L., Godfrey A., Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proceedings of the National Academy of Sciences of USA. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin T.K., Hughes G., Muratovska A., Blaikie F.H., Brookes P.S., Darley-Usmar V., Smith R.A., Murphy M.P. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. Journal of Biological Chemistry. 2002;277:17048–17056. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- 93.Pignatelli M., Sanchez-Rodriguez J., Santos A., Perez-Castillo A. 15-deoxy-Delta-12,14-prostaglandin J2 induces programmed cell death of breast cancer cells by a pleiotropic mechanism. Carcinogenesis. 2005;26:81–92. doi: 10.1093/carcin/bgh308. [DOI] [PubMed] [Google Scholar]
- 94.Ceaser E.K., Ramachandran A., Levonen A.L., rley-Usmar V.M. Oxidized low-density lipoprotein and 15-deoxy-delta 12,14-PGJ2 increase mitochondrial complex I activity in endothelial cells. American Journal of Physiology—Heart and Circular Physiology. 2003;285:H2298–H2308. doi: 10.1152/ajpheart.00508.2003. [DOI] [PubMed] [Google Scholar]
- 95.Echtay K.S., Esteves T.C., Pakay J.L., Jekabsons M.B., Lambert A.J., Portero-Otin M., Pamplona R., Vidal-Puig A.J., Wang S., Roebuck S.J., Brand M.D. A signaling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO Journal. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee J.Y., Jung G.Y., Heo H.J., Yun M.R., Park J.Y., Bae S.S., Hong K.W., Lee W.S., Kim C.D. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicology Letters. 2006;166:212–221. doi: 10.1016/j.toxlet.2006.07.305. [DOI] [PubMed] [Google Scholar]
- 97.Sansbury B.E., Jones S.P., Riggs D.W., Darley-Usmar V.M., Hill B.G. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chemico-Biological Interactions. 2011;191:288–295. doi: 10.1016/j.cbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lo S.C., Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Experimental Cell Research. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han Z., Varadharaj S., Giedt R.J., Zweier J.L., Szeto H.H., Alevriadou B.R. Mitochondria-derived reactive oxygen species mediate heme oxygenase-1 expression in sheared endothelial cells. Journal of Pharmacology and Experimental Therapeutics. 2009;329:94–101. doi: 10.1124/jpet.108.145557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Diers A.R., Higdon A.N., Ricart K.C., Johnson M.S., Agarwal A., Kalyanaraman B., Landar A., Darley-Usmar V.M. Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signaling mechanisms. Biochemical Journal. 2010;426:31–41. doi: 10.1042/BJ20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Imhoff B.R., Hansen J.M. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochemical Journal. 2009;424:491–500. doi: 10.1042/BJ20091286. [DOI] [PubMed] [Google Scholar]
- 102.Scheuner D., Kaufman R.J. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocrine reviews. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Higa A., Chevet E. Redox signaling loops in the unfolded protein response. Cellular Signaling. 2012;24:1548–1555. doi: 10.1016/j.cellsig.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 104.Fujita E., Kouroku Y., Isoai A., Kumagai H., Misutani A., Matsuda C., Hayashi Y.K., Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Human Molecular Genetics. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 105.Bertolotti A., Zhang Y., Hendershot L.M., Harding H.P., Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 106.Marciniak S.J., Ron D. Endoplasmic reticulum stress signaling in disease. Physiological Reviews. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 107.Vladykovskaya E., Sithu S.D., Haberzettl P., Wickramasinghe N.S., Merchant M.L., Hill B.G., McCracken J., Agarwal A., Dougherty S., Gordon S.A., Schuschke D.A., Barski O.A., O’Toole T., D’Souza S.E., Bhatnagar A., Srivastava S. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. Journal of Biological Chemistry. 2012;287:11398–11409. doi: 10.1074/jbc.M111.320416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sanson M., Auge N., Vindis C., Muller C., Bando Y., Thiers J.C., Marachet M.A., Zarkovic K., Sawa Y., Salvayre R., Negre-Salvayre A. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circulation Research. 2009;104:328–336. doi: 10.1161/CIRCRESAHA.108.183749. [DOI] [PubMed] [Google Scholar]
- 109.Cullinan S.B., Diehl J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. Journal of Biological Chemistry. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 110.Muller C., Bandemer J., Vindis C., Camare C., Mucher E., Gueraud F., Larroque-Cardoso P., Bernis C., Auge N., Salvayre R., Negre-Salvayre A. Protein disulfide isomerase modification and inhibition contribute to ER stress and apoptosis induced by oxidized low density lipoproteins. Antioxidants and Redox Signaling. 2013;18:731–742. doi: 10.1089/ars.2012.4577. [DOI] [PubMed] [Google Scholar]
- 111.Cullinan S.B., Zhang D., Hannink M., Arvisais E., Kaufman R.J., Diehl J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and Cellular Biology. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scull C.M., Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arteriosclerosis Thrombosis and Vascular Biology. 2011;31:2792–2797. doi: 10.1161/ATVBAHA.111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karlhuber G.M., Bauer H.C., Eckl P.M. Cytotoxic and genotoxic effects of 4-hydroxynonenal in cerebral endothelial cells. Mutation Research. 1997;381:209–216. doi: 10.1016/s0027-5107(97)00170-x. [DOI] [PubMed] [Google Scholar]
- 114.Braekke K., Harsem N.K., Staff A.C. Oxidative stress and antioxidant status in fetal circulation in preeclampsia. Pediatric Research. 2006;60:560–564. doi: 10.1203/01.pdr.0000242299.01219.6a. [DOI] [PubMed] [Google Scholar]
- 115.Gveric-Ahmetasevic S., Sunjic S.B., Skala H., Andrisic L., Stroser M., Zarkovic K., Skrablin S., Tatzber F., Cipak A., Jaganjac M., Waeg G., Gveric T., Zarkovic N. Oxidative stress in small-for-gestational age (SGA) term newborns and their mothers. Free Radical Research. 2009;43:376–384. doi: 10.1080/10715760902783285. [DOI] [PubMed] [Google Scholar]
- 116.Leonarduzzi G., Chiarpotto E., Biasi F., Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Molecular Nutrition and Food Research. 2005;49:1044–1049. doi: 10.1002/mnfr.200500090. [DOI] [PubMed] [Google Scholar]
- 117.Sakuraba H., Mizukami H., Yagihashi N., Wada R., Hanyu C., Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 118.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Molecular Aspects of Medicine. 2003;24:293–303. doi: 10.1016/s0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 119.Chamy V.M., Lepe J., Catalan A., Retamal D., Escobar J.A., Madrid E.M. Oxidative stress is closely related to clinical severity of pre-eclampsia. Biological Research. 2006;39:229–236. doi: 10.4067/s0716-97602006000200005. [DOI] [PubMed] [Google Scholar]
- 120.Negre-Salvayre A., Auge N., Ayala V., Basaga H., Boada J., Brenke R., Chapple S., Cohen G., Feher J., Grune T., Lengyel G., Mann G.E., Pamplona R., Poli G., Portero-Otin M., Riahi Y., Salvayre R., Sasson S., Serrano J., Shamni O., Siems W., Siow R.C., Wiswedel I., Zarkovic K., Zarkovic N. Pathological aspects of lipid peroxidation. Free Radical Research. 2010;44:1125–1171. doi: 10.3109/10715762.2010.498478. [DOI] [PubMed] [Google Scholar]
- 121.Lian I.A., Loset M., Mundal S.B., Fenstad M.H., Johnson M.P., Eide I.P., Bjorge L., Freed K.A., Moses E.K., Austgulen R. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta. 2011;32:823–829. doi: 10.1016/j.placenta.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Illsinger S., Janzen N., Sander S., Schmidt K.H., Bednarczyk J., Mallunat L., Bode J., Hagebolling F., Hoy L., Lucke T., Hass R., Das A.M. Preeclampsia and HELLP syndrome: impaired mitochondrial function in umbilical endothelial cells. Reproductive Sciences. 2010;17:219–226. doi: 10.1177/1933719109351597. [DOI] [PubMed] [Google Scholar]
- 123.Padmini E., Lavanya S., Uthra V. Preeclamptic placental stress and over expression of mitochondrial HSP70. Clinical Chemistry and Laboratory Medicine. 2009;47:1073–1080. doi: 10.1515/CCLM.2009.247. [DOI] [PubMed] [Google Scholar]
- 124.Benderdour M., Charron G., DeBlois D., Comte B., Des Rosiers C. Cardiac mitochondrial NADP+-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: an event that precedes hypertrophy development. Journal of Biological Chemistry. 2003;278:45154–45159. doi: 10.1074/jbc.M306285200. [DOI] [PubMed] [Google Scholar]
- 125.Zeng L., Zampetaki A., Margariti A., Pepe A.E., Alam S., Martin D., Xiao Q., Wang W., Jin Z.G., Cockerill G., Mori K., Li Y.S., Hu Y., Chien S., Xu Q. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proceedings of the National Academy of Sciences of USA. 2009;106:8326–8331. doi: 10.1073/pnas.0903197106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Civelek M., Manduchi E., Riley R.J., Stoeckert C.J., Jr, Davies P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circulation Research. 2009;105:453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Song B., Scheuner D., Ron D., Pennathur S., Kaufman R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. Journal of Clinical Investigation. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fu D., Wu M., Zhang J., Du M., Yang S., Hammad S.M., Wilson K., Chen J., Lyons T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia. 2012;55:3128–3140. doi: 10.1007/s00125-012-2692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu M., Yang S., Elliott M.H., Fu D., Wilson K., Zhang J., Du M., Chen J., Lyons T. Oxidative and endoplasmic reticulum stresses mediate apoptosis induced by modified LDL in human retinal Muller cells. Investigative Ophthalmology and Visual Science. 2012;53:4595–4604. doi: 10.1167/iovs.12-9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lashin O.M., Szweda P.A., Szweda L.I., Romani A.M. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radical Biology and Medicine. 2006;40:886–896. doi: 10.1016/j.freeradbiomed.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 131.Mogensen M., Sahlin K., Fernstrom M., Glintborg D., Vind B.F., Beck-Nielsen H., Hojlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 132.Qin Z., Hu D., Han S., Reaney S.H., Di Monte D.A., Fink A.L. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. Journal of Biological Chemistry. 2007;282:5862–5870. doi: 10.1074/jbc.M608126200. [DOI] [PubMed] [Google Scholar]
- 133.Slodzinski H., Moran L.B., Michael G.J., Wang B., Novoselov S., Cheetham M.E., Pearce R.K., Graeber M.B. Homocysteine-induced endoplasmic reticulum protein (herp) is up-regulated in parkinsonian substantia nigra and present in the core of Lewy bodies. Clinical Neuropathology. 2009;28:333–343. [PubMed] [Google Scholar]
- 134.Macedo B., Magalhaes J., Batista A.R., Saraiva M.J. Carvedilol treatment reduces transthyretin deposition in a familial amyloidotic polyneuropathy mouse model. Pharmacological Research. 2010;62:514–522. doi: 10.1016/j.phrs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 135.Teixeira P.F., Cerca F., Santos S.D., Saraiva M.J. Endoplasmic reticulum stress associated with extracellular aggregates. Evidence from transthyretin deposition in familial amyloid polyneuropathy. Journal of Biological Chemistry. 2006;281:21998–22003. doi: 10.1074/jbc.M602302200. [DOI] [PubMed] [Google Scholar]
- 136.Lee W.C., Wong H.Y., Chai Y.Y., Shi C.W., Amino N., Kikuchi S., Huang S.H. Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker? Biochemical and Biophysical Research Communications. 2012;425:842–847. doi: 10.1016/j.bbrc.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 137.Sompol P., Ittarat W., Tangpong J., Chen Y., Doubinskaia I., Batinic-Haberle I., Abdul H.M., Butterfield D.A., St, Clair D.K. A neuronal model of Alzheimer’s disease: an insight into the mechanisms of oxidative stress-mediated mitochondrial injury. Neuroscience. 2008;153:120–130. doi: 10.1016/j.neuroscience.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Noguchi N. Role of oxidative stress in adaptive responses in special reference to atherogenesis. Journal of Clinical Biochemistry and Nutrition. 2008;43:131–138. doi: 10.3164/jcbn.2008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Polak M., Zagorski Z. Lipid peroxidation in diabetic retinopathy. Annales Universitatis Mariae Curie-Sklodowska. Sectio D: Medicina. 2004;59:434–437. [PubMed] [Google Scholar]
- 140.Srivastava S.K., Ramana K.V., Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocrine Reviews. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- 141.Natarajan V., Scribner W.M., Taher M.M. 4-Hydroxynonenal, a metabolite of lipid peroxidation, activates phospholipase D in vascular endothelial cells. Free Radical Biology and Medicine. 1993;15:365–375. doi: 10.1016/0891-5849(93)90036-t. [DOI] [PubMed] [Google Scholar]
- 142.Ho T.C., Chen S.L., Yang Y.C., Chen C.Y., Feng F.P., Hsieh J.W., Cheng H.C., Tsao Y.P. 15-deoxy-Delta(12,14)-prostaglandin J2 induces vascular endothelial cell apoptosis through the sequential activation of MAPKS and p53. Journal of Biological Chemistry. 2008;283:30273–30288. doi: 10.1074/jbc.M804196200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hosoya T., Maruyama A., Kang M.I., Kawatani Y., Shibata T., Uchida K., Warabi E., Noguchi N., Itoh K., Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. Journal of Biological Chemistry. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 144.Oh J.Y., Giles N., Landar A., Darley-Usmar V. Accumulation of 15-deoxy-delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochemical Journal. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Levonen A.L., Dickinson D.A., Moellering D.R., Mulcahy R.T., Forman H.J., Darley-Usmar V.M. Biphasic effects of 15-deoxy-delta(12,14)-prostaglandin J(2) on glutathione induction and apoptosis in human endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21:1846–1851. doi: 10.1161/hq1101.098488. [DOI] [PubMed] [Google Scholar]
- 146.Moldovan N.I., Lupu F., Moldovan L., Simionescu N. 4-Hydroxynonenal induces membrane perturbations and inhibition of basal prostacyclin production in endothelial cells, and migration of monocytes. Cell Biology International. 1994;18:985–992. doi: 10.1006/cbir.1994.1020. [DOI] [PubMed] [Google Scholar]
- 147.Kaneko T., Kaji K., Matsuo M. Cytotoxicities of a linoleic acid hydroperoxide and its related aliphatic aldehydes toward cultured human umbilical vein endothelial cells. Chemico-Biological Interactions. 1988;67:295–304. doi: 10.1016/0009-2797(88)90065-8. [DOI] [PubMed] [Google Scholar]
- 148.de Jongh R., Haenen G.R., van Koeveringe G.A., Dambros M., van Kerrebroeck P.E. Lipid peroxidation product 4-hydroxynonenal contributes to bladder smooth muscle damage. Urology. 2008;71:974–978. doi: 10.1016/j.urology.2007.11.014. [DOI] [PubMed] [Google Scholar]