

Abstract
Superoxide (O2•−) contributes to the development of cardiovascular disease. Generation of O2•− occurs in both the intracellular and extracellular compartments. We hypothesized that the gene transfer of cytosolic superoxide dismutase (SOD1) or extracellular SOD (SOD3) to blood vessels would differentially protect against O2•−-mediated endothelial-dependent dysfunction. Aortic ring segments from New Zealand rabbits were incubated with adenovirus (Ad) containing the gene for Escherichia coli β-galactosidase, SOD1, or SOD3. Activity assays confirmed functional overexpression of both SOD3 and SOD1 isoforms in aorta 24 h following gene transfer. Histochemical staining for β-galactosidase showed gene transfer occurred in the endothelium and adventitia. Next, vessels were prepared for measurement of isometric tension in Kreb's buffer containing xanthine. After precontraction with phenylephrine, xanthine oxidase impaired relaxation to the endothelium-dependent dilator acetylcholine (ACh, max relaxation 33±4% with XO vs. 64±3% without XO, p<0.05), whereas relaxation to the endothelium-independent dilator sodium nitroprusside was unaffected. In the presence of XO, maximal relaxation to ACh was improved in vessels incubated with AdSOD3 (55±2%, p<0.05 vs. control) but not AdSOD1 (34±4%). We conclude that adenoviral-mediated gene transfer of SOD3, but not SOD1, protects the aorta from xanthine/XO-mediated endothelial dysfunction. These data provide important insight into the location and enzymatic source of O2•− production in vascular disease.
Keywords: Superoxide dismutase, Oxygen free radicals, Gene transfer, Endothelial dysfunction
Graphical abstract
Highlights
-
•
Xanthine oxidase (XO)-derived O2•− inhibits endothelium-dependent relaxation.
-
•
Extracellular SOD alleviates XO-mediated vasomotor dysfunction.
-
•
Increased expression of cytosolic SOD fails to protect from XO-mediated dysfunction.
-
•
To maintain •NO bioavailability, SOD must localize to the site of O2•− production.
Introduction
Reactive oxygen species (ROS) have an important role in mediating vascular disease in hypertension, diabetes mellitus, and atherosclerosis, as well as in acute conditions such as hypoxia-reoxygenation. One clinically-relevant enzymatic source of ROS is xanthine oxidase (XO). Circulating levels of XO are increased with hypercholesterolemia [1], [2]. Moreover, activity of XO is elevated in human atherosclerotic lesions [3], [4], [5]. Infusion of a XO inhibitor in humans with cardiovascular disease improves vascular function [6]. The pathophysiologic effects of ROS causing vascular dysfunction can occur through multiple mechanisms. Superoxide anion (O2•−) reacts with endothelium-derived nitric oxide (•NO) via a radical-radical reaction at a diffusion-limited rate to generate peroxynitrite (ONOO−), a potent oxidant and mediator of vascular tissue injury [7]. Inactivation of •NO impairs the ability of vessels to relax normally [8]. Peroxynitrite can oxidize lipoproteins and damage lipid membranes. Oxidation of LDL in the vessel wall by O2•− can alter signal transduction and cause cytotoxicity [9]. Excess levels of O2•− increase platelet aggregation [10], and the adhesion and migration of monocytes [11]. Cell proliferation and cell death are also influenced by the cellular pro-oxidant state [12]. By activating metalloproteinases, O2•− can also modulate matrix degradation [13].
Perhaps the most important cellular mechanism by which to protect against endothelial dysfunction involves the activity of superoxide dismutases (SODs) in the vessel wall [14]. In cardiovascular disease, O2•−-generating systems can result in both the intracellular and extracellular generation of O2•−. Of the two copper–zinc containing SODs, one resides in the cytoplasm (SOD1) and the second is extracellular and has an affinity for cell-surface heparin sulfate proteoglycans (SOD3) [15]. Adenoviral delivery of SOD3 improves endothelial-dependent relaxation in multiple disease states [16], [17], [18], [19]. In contrast, gene transfer of SOD1 restores acetylcholine-dependent vasodilation response in diabetic rabbits [20], whereas arterial relaxation in either hyperlipidemic rabbits or rabbits treated with Ang-II is not improved [21], [22]. A limitation of these studies is that O2•− localization may be intracellular and/or extracellular.
In this study, we compared the roles of intracellular and extracellular SOD in protecting against extracellular oxidant-induced endothelial dysfunction. Our data demonstrate that adenoviral transfer of SOD3 but not SOD1 improves vasomotor function following exposure to xanthine/XO. We conclude that, to maintain bioavailability of •NO, SOD must be localized to the site of O2•− production rather than •NO production, suggesting that the reaction of •NO with O2•− occurs outside the cell.
Methods
Animals and tissue preparation
All protocols were approved by the Animal Care and Use Committee at the University of Iowa. New Zealand white rabbits (n=17, either sex, 2.5–3.0 kg) were sacrificed with an overdose of pentobarbital and the thoracic aorta immediately dissected and placed into ice cold Kreb's bicarbonate buffer of the following composition of (in mM) 118.0 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 25.0 NaHCO3, 1.2 KH2PO4, 5.5 glucose, 0.0025 EDTA. The aorta was trimmed free of fat and loosely adhering connective tissue, and cut into approximately 2.5 mm wide ring segments. Vessel segments were incubated for two hours at 37 °C in 1.0 ml of Dulbecco's Modified Eagle Medium (DMEM) containing 0.1 ml of either replication deficient adenovirus (5×109 pfu/ml) or phosphate buffered saline as described below. After two hours, the vessel segments were transferred to DMEM containing 10% bovine serum and placed in an incubator at 37 °C for 24 h.
Adenoviral-mediated gene transfer
Adenovirus (Ad) with CMV promoters used for this study were obtained from the University of Iowa Gene Vector Core Facility and include Adβgal for expression of the histochemical marker β-galactosidase, AdSOD3 [21] for expression of the human extracellular isoform of SOD, or AdSOD1 [23] for expression of the human copper–zinc cytoplasmic isoform of SOD.
β-galactosidase activity assay
Rings were rinsed in phosphate buffered saline and fixed in 2% paraformaldehyde for 10 min. Segments were then stained in 1 mg/ml of X-gal for 4 h at 37 °C. Tissue was further fixed in 2% paraformaldehyde, imbedded in paraffin, and sectioned for histologic analysis.
SOD activity assay
SOD activity was measured by the method of NADPH oxidation described by Paoletti and Mocali [24]. Because this method relies on the oxidation of NADPH, it is less prone than other SOD assays to interference by nonspecific reduction from cellular components. Care was taken to insure specimens were free of blood. Aorta from four different rabbits were combined for each intervention (control, Adβgal, AdSOD3, and AdSOD1) and homogenized with a Dounce homogenizer in 50 mM KPO4 buffer (pH 7.4, 4 °C) containing anti-proteolytics (PMSF 1.0 mM, leupeptin 10 µg/ml, aprotinin 10 µg/ml, and pepstatin A 10 µg/ml, Sigma). The homogenate was sonicated (five second burst times five) and centrifuged at 3000g for ten minutes at 4 °C. Protein concentrations were determined by the Lowry assay. The assay was performed in triplicate and values averaged.
Isometric force measurement
Vasomotor reactivity of rabbit aorta was measured by determination of isometric force, 24 h after virus incubation. Ring segments were mounted in organ baths containing 10 ml of Kreb's buffer containing xanthine (10−4 M) and catalase (500 U/ml) at 37 °C and continuously gassed with 20% O2, 5% CO2, and 75% N2. Rings were stretched to an optimal resting tension of 6 g as determined by repeated administration of KCl (75 mM). Vessels were equilibrated for 30 min and then constricted twice with KCl (75 mM). After the segments were washed, phenylephrine was added to achieve a tension of 50–100% of the maximal contraction to KCl. Xanthine oxidase (XO, Sigma) was added to the buffer for generation of O2•− immediately prior to performing a concentration response to the endothelium-dependent vasodilator acetylcholine (ACh, 10−9–10−5 M) or the endothelium-independent dilator sodium nitroprusside (SNP, 10−9–10−5 M). Although in some vessels the addition of XO produced an increase in tension, this increase was not significant.
Based on our findings that 5 U/L of XO moderately impaired relaxation to ACh without affecting the response to SNP, protocols examining vasomotor responses following gene transfer were performed with 5 U/L of XO. Only one concentration-dose response curve was performed in each vessel. In this way, abnormalities in relaxation could not be attributed to prior exposure to xanthine/XO.
Data analysis
Results are expressed as mean±standard error. For the ring studies, data were obtained in duplicate for each intervention (control, Adβgal, AdSOD3, and AdSOD1) and averaged such that n is equal to the number of animals studied. Relaxations are the percent change from the precontracted tension. Dilator responses are compared among groups using a two factor repeated measures ANOVA with a Bonferroni correction for multiple comparisons. A non-linear curve fit (3 parameter with a Hill slope of 1.0) was used to determine the maximal and EC50 relaxation (GraphPad Prism for Windows). Statistical significance was accepted if the null hypothesis was rejected at p<0.05.
Results and discussion
Effect of xanthine and xanthine oxidase on relaxation of aorta
We adapted a previously described model whereby incubation of rabbit aortic ring segments in the presence of xanthine/XO impairs endothelial-dependent relaxation [2]. After contraction with phenylephrine (mean 75±4%), increasing concentrations of the endothelium-dependent dilator ACh produced a dose-dependent relaxation (Fig. 1A). Addition of XO immediately prior to ACh impaired relaxation. Our protocol for oxidant production was designed such that generation of O2•− occurred simultaneously with ACh-induced release of •NO. The maximal relaxation at 10−5 M ACh was 88±4% for control, 61±7% for 1 U/L XO, 50±8% for 5 U/L XO, and 32±5% for 10 U/L XO (p<0.05 for each XO dose vs. control). These results confirmed that oxidants derived from XO mediate vascular dysfunction in a concentration-dependent manner [2].
Fig. 1.
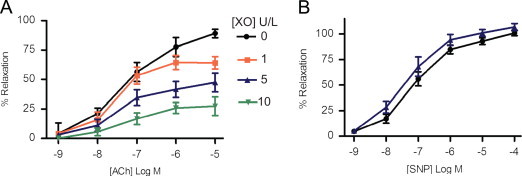
Xanthine oxidase-derived O2•− impairs endothelial-dependent relaxation of rabbit aorta. (A) Dose-dependent relaxation to endothelium-dependent dilator acetylcholine (ACh) in the absence or presence of increasing concentrations of XO as indicated. (B) Dose-dependent relaxation to endothelium-independent dilator sodium nitroprusside (SNP) in the absence (black line) or presence (blue line) of 5 U/L XO. (n=8–12) ⁎p<0.05 vs. control (no XO). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To confirm that the observed impairment in dilation was endothelium specific, we examined the relaxation of rabbit aorta to SNP. Increasing concentrations of SNP produced a dose-dependent relaxation that was not affected by XO (Fig. 1B, relaxation at 10−5 M SNP is 100±5% for control and 97±4% for 5 U/L XO, p=NS, n=5). Based on these data, 5 U/L of XO was used in subsequent protocols to produce free radical-mediated impairment in relaxation.
Gene transfer of SOD to rabbit aorta increases SOD expression and activity
Following gene transfer of β-galactosidase into vessel segments, histochemical staining demonstrated β-galactosidase in the endothelium and adventitia, indicative of efficient gene transfer (Fig. 2A). Others have provided evidence that SOD3 and SOD1 properly bind to the membrane and localize to the cytosol, respectively, when expressed in vivo using these adenoviral constructs [17], [25], [26]. To confirm gene transfer of SOD3 and SOD1 produced functional antioxidant protein in the vessel segments, SOD activity was determined by measuring NADPH oxidation. Total SOD activity in vessels after transduction of SOD was approximately 4–6 times higher than in non-transduced vessels (Fig. 2B).
Fig. 2.
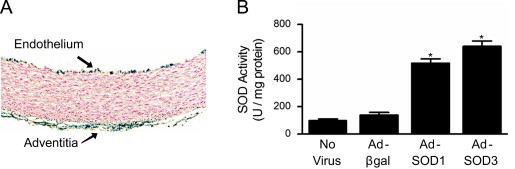
Adenoviral-mediated gene transfer of SOD3 or SOD1 increases total SOD activity in rabbit aorta. (A) β-galactosidase activity as determined by X-gal staining of aortic segments exposed to Adβgal. Blue staining denotes β-galactosidase activity, which limited to the endothelium and adventitial layers. (B) SOD activity in rabbit aorta one day after adenoviral-mediated gene transfer of SOD1 or SOD3. Values are the mean of three assays performed on combined homogenate from four aortae. ⁎p<0.05 vs. Adβgal.
Effects of gene transfer of SOD on superoxide mediated vasomotor dysfunction
We next examined the ability of gene transfer of SOD in aorta to protect against oxidant-mediated abnormal relaxation. Following gene transfer of β-galactosidase, the concentration-dependent relaxation to ACh was impaired in the presence of XO (Fig. 3A). Overexpression of SOD3 in the presence of XO restored relaxation to control levels observed in the absence of XO. The maximal relaxation of control vessels was markedly reduced by XO, whereas gene transfer of SOD3 restored relaxation to ACh (Fig. 3B). There was no difference in the EC50 between groups (Fig. 3C). Bovine SOD added exogenously to the circulating buffer prior to addition of XO improved relaxation to ACh as compared to aorta in XO without SOD (Fig. 3A, C). This concentration of exogenous SOD was sufficient to inhibit O2•− accumulation by xanthine/XO detected by lucigenin-enhanced chemiluminescence (Fig. 3D). In contrast to overexpression of SOD3, ACh-induced relaxation of vessels overexpressing SOD1 was unchanged from control vessels after XO (Fig. 4). As in Fig. 3, bovine SOD in the presence of XO restored relaxation to ACh (Fig. 4). In all groups, relaxation to the endothelium-independent dilator SNP was similar (Fig. 5). These data indicate that extracellular, but not cytosolic, SOD improved endothelium-dependent dilation during generation of O2•− by xanthine and XO.
Fig. 3.
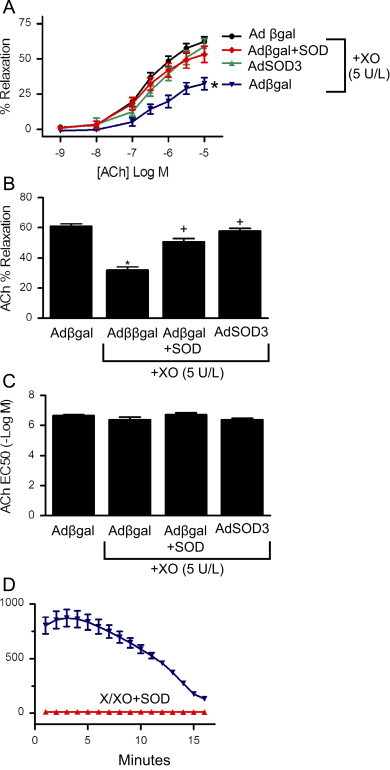
Gene transfer of SOD3 protects aorta against XO-mediated vasomotor dysfunction. Following gene transfer of SOD3, (A) dose-dependent relaxation to ACh in the absence or presence of XO was determined. Gene transfer of β-galactosidase serves as negative control and exogenously applied bovine SOD (500 U/ml) serves as positive control. (n=6–10) ⁎p<0.05 vs Adβgal without XO. (B) Maximal relaxation to ACh based on non-linear curve fit of data in (A). ⁎p<0.05 vs Adβgal without XO; +p<0.05 Adβgal with XO. (C) EC50 relaxation to ACh based on non-linear curve fit of data in (A). (D) Lucigenin-enhanced chemiluminescence of xanthine/XO in the presence or absence of bovine SOD (n=3); p<0.05.
Fig. 4.
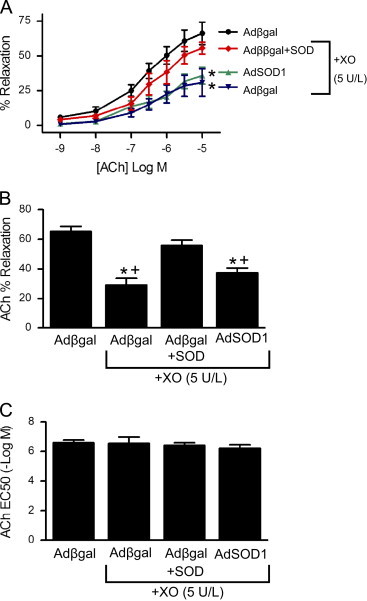
Gene transfer of SOD1 does not protect against XO-mediated vasomotor dysfunction. Following gene transfer of SOD1, (A) dose-dependent relaxation to ACh in the absence or presence of XO was determined as described for Fig. 3. ⁎p<0.05 vs Adβgal without XO. (B) Maximal relaxation and (C) EC50 to ACh based on non-linear curve fit of data in (A). ⁎p<0.05 vs Adβgal without XO;+p<0.05 Adβgal+SOD+XO.
Fig. 5.
Relaxation to SNP is preserved in the presence of xanthine-XO. Relaxation response to 10−5 M SNP following gene transfer and treatment with XO. Gene transfer of β-galactosidase serves as negative control and exogenously applied bovine SOD (500 U/ml) serves as positive control. (n=6–10).
In aorta incubated in xanthine/XO, gene transfer of SOD3 only partially restored relaxation to ACh. Although greater vascular levels of SOD, through increased transgene expression, may have further improved ACh-induced dilation, this is unlikely since vessels incubated in AdSOD3 relaxed similarly to vessels incubated in exogenous SOD (500 U/ml). These observations suggest that the inability of SOD to completely restore dilator responses of vessels in xanthine/XO results from non-superoxide mediated mechanisms. This hypothesis is consistent with studies showing hydrogen peroxide contributes to vascular injury caused by xanthine/XO [27].
The observation that gene transfer of SOD3 but not SOD1 improved relaxation of aorta during generation of O2•− can be explained by the cellular compartmentalization of the antioxidants. Despite relatively similar SOD activity following gene transfer of the two SOD isoforms, SOD1 is localized to the cytosol and therefore has limited access to O2•− generated in the extracellular space [15]. In contrast, since SOD3 is primarily bound to extracellular proteoglycans, it has greater access to O2•− generated by xanthine/XO. A limitation of this interpretation is based on the concentration of XO at the endothelial surface via its reversible interaction with glycosaminoglycans [28]. In this way, SOD3 may displace XO and reduce its surface concentration, thereby limiting endothelial dysfunction independent of its antioxidant effect. In addition to XO, multiple other cellular enzyme systems have been implicated in vascular disease, including NADPH oxidases, uncoupled endothelial nitric oxide synthase (eNOS), mitochondrial respiratory chain, and arachidonic acid metabolizing enzymes. Our model specifically examines the ability of SOD1 vs. SOD3 in restoring endothelial function in the presence of extracellular XO and do not necessarily address the contribution of these other enzymes that generate intracellular O2•−.
Conclusion
In summary, our data demonstrate that adenoviral mediated gene transfer of either SOD3 or SOD1 to rabbit aorta increases antioxidant activity, but only SOD3 protects against xanthine/XO-induced endothelial dysfunction. Selective overexpression of SOD to intracellular or extracellular compartments provides important insights into the location and enzymatic source of O2•− production in vascular disease. Our data are particularly interesting when considering that both XO and SOD3 can be internalized into the cell via endocytosis [29], [30]. The lack of protection with SOD1 provides strong evidence that the interaction of •NO with O2•− occurs either outside of the cell, or potentially in an endocytic vesicle. XO is implicated as the source of O2•− in the vascular dysfunction associated with ischemia/reperfusion, diabetes, and atherosclerosis [31], [32], [33], denoting the relevance of our studies with xanthine/XO. Whereas oxypurinol and allopurinol improve vascular relaxation in humans [6], this strategy of inhibiting XO-derived O2•− is most effective in patients that are hyperuricemic [34]. By contrast, SOD3 is protective in the pathogenesis of ischemia/reperfusion injury, atherosclerosis, and hypertension [15]. Thus, our data suggest that selective targeting of extracellular O2•−, i.e. with SOD3, may be broadly applicable to multiple cardiovascular disease settings.
Sources of funding
This work was supported by an American Heart Association Grant-In-Aid, NIH Mentored Clinical Scientist Development Award (HLO3669), and Office of Research and Development, Department of Veterans Affairs 1BX001729 to FJM. The funding sources had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Acknowledgments
We thank the Gene Transfer Vector Core at the University of Iowa, supported in part by a trust from the Carver Foundation, for developing and providing adenoviral constructs, and Dr. James Crapo for providing reagents.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.White C.R., Parks D.A., Patel R.P., Shelton J., Tarpey M.M., Freeman B.A., Darley-Usmar V.M. L-Arginine inhibits xanthine oxidase-dependent endothelial dysfunction in hypercholesterolemia. FEBS Letters. 2004;561:94–98. doi: 10.1016/S0014-5793(04)00137-1. [DOI] [PubMed] [Google Scholar]
- 2.White C.R., Darley-Usmar V., Berrington W.R., McAdams M., Gore J.Z., Thompson J.A., Parks D.A., Tarpey M.M., Freeman B.A. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proceedings of the National Academy of Sciences of USA. 1996;93:8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swain J., Gutteridge J.M. Prooxidant iron and copper, with ferroxidase and xanthine oxidase activities in human atherosclerotic material. FEBS Letters. 1995;368:513–515. doi: 10.1016/0014-5793(95)00726-p. [DOI] [PubMed] [Google Scholar]
- 4.Spiekermann S., Landmesser U., Dikalov S., Bredt M., Gamez G., Tatge H., Reepschlager N., Hornig B., Drexler H., Harrison D.G. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation. 2003;107:1383–1389. doi: 10.1161/01.cir.0000056762.69302.46. [DOI] [PubMed] [Google Scholar]
- 5.Guzik T.J., Sadowski J., Guzik B., Jopek A., Kapelak B., Przybylowski P., Wierzbicki K., Korbut R., Harrison D.G., Channon K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:333–339. doi: 10.1161/01.ATV.0000196651.64776.51. [DOI] [PubMed] [Google Scholar]
- 6.Higgins P., Dawson J., Lees K.R., McArthur K., Quinn T.J., Walters M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: a systematic review and meta-analysis. Cardiovascular Therapy. 2012;30:217–226. doi: 10.1111/j.1755-5922.2011.00277.x. [DOI] [PubMed] [Google Scholar]
- 7.Gryglewsli R.J., Palmer R.M.J., Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 8.White C.R., Brock T.A., Chang L., Crapo J., Briscoe P., Ku D., Bradley W.A., Gianturco S.H., Gore J., Freeman B.A., Tarpey M.M. Superoxide and peroxynitrite in atherosclerosis. Proceedings of the National Academy of Sciences of USA. 1994;91:1044–1048. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zmijewski J.W., Landar A., Watanabe N., Dickinson D.A., Noguchi N., Darley-Usmar V.M. Cell signalling by oxidized lipids and the role of reactive oxygen species in the endothelium. Biochemical Society Transactions. 2005;33:1385–1389. doi: 10.1042/BST20051385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikeda H., Koga Y., Oda T., Kuwano K., Nakayama H., Ueno T., Toshima H., Michael L.H., Entman M.L. Free oxygen radicals contribute to platelet aggregation and cyclic flow variations in stenosed and endothelium-injured canine coronary arteries. Journal of the American College og Cardiology. 1994;24:1749–1756. doi: 10.1016/0735-1097(94)90183-x. [DOI] [PubMed] [Google Scholar]
- 11.Rattan V., Sultana C., Shen Y., Kalra V.K. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. American Journal of Physiology. 1997;273:E453–E461. doi: 10.1152/ajpendo.1997.273.3.E453. [DOI] [PubMed] [Google Scholar]
- 12.Li P.F., Dietz R., von Harsdorf R. Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells. Circulation. 1997;96:3602–3609. doi: 10.1161/01.cir.96.10.3602. [DOI] [PubMed] [Google Scholar]
- 13.Rajagopalan S., Kurz S., Munzel T., Tarpey M., Freeman B.A., Griendling K.K., Harrison D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. Journal of Clinical Investigations. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heistad D.D. Oxidative stress and vascular disease: 2005 Duff lecture. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:689–695. doi: 10.1161/01.ATV.0000203525.62147.28. [DOI] [PubMed] [Google Scholar]
- 15.Fukai T., Folz R.J., Landmesser U., Harrison D.G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovascular Research. 2002;55:239–249. doi: 10.1016/s0008-6363(02)00328-0. [DOI] [PubMed] [Google Scholar]
- 16.Lund D.D., Gunnett C.A., Chu Y., Brooks R.M., Faraci F.M., Heistad D.D. Gene transfer of extracellular superoxide dismutase improves relaxation of aorta after treatment with endotoxin. American Journal of Physiology—Heart and Circulatory Physiology. 2004;287:H805–811. doi: 10.1152/ajpheart.00907.2003. [DOI] [PubMed] [Google Scholar]
- 17.Chu Y., Iida S., Lund D.D., Weiss R.M., DiBona G.F., Watanabe Y., Faraci F.M., Heistad D.D. Gene transfer of extracellular superoxide dismutase reduces arterial pressure in spontaneously hypertensive rats: role of heparin-binding domain. Circulatory Research. 2003;92:461–468. doi: 10.1161/01.RES.0000057755.02845.F9. [DOI] [PubMed] [Google Scholar]
- 18.Levonen A.L., Vahakangas E., Koponen J.K., Yla-Herttuala S. Antioxidant gene therapy for cardiovascular disease: current status and future perspectives. Circulation. 2008;117:2142–2150. doi: 10.1161/CIRCULATIONAHA.107.718585. [DOI] [PubMed] [Google Scholar]
- 19.Qin Z., Reszka K.J., Fukai T., Weintraub N.L. Extracellular superoxide dismutase (ecSOD) in vascular biology: an update on exogenous gene transfer and endogenous regulators of ecSOD. Translational Research. 2008;151:68–78. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanetti M., Sato J., Katusic Z.S., O'Brien T. Gene transfer of superoxide dismutase isoforms reverses endothelial dysfunction in diabetic rabbit aorta. American Journal of Physiology—Heart and Circulatory Physiology. 2001;280:H2516–2523. doi: 10.1152/ajpheart.2001.280.6.H2516. [DOI] [PubMed] [Google Scholar]
- 21.Miller F.J., Jr., Gutterman D.D., Rios C.D., Heistad D.D., Davidson B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circulatory Research. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 22.Nakane H., Miller F.J., Jr., Faraci F.M., Toyoda K., Heistad D.D. Gene transfer of endothelial nitric oxide synthase reduces angiotensin II-induced endothelial dysfunction. Hypertension. 2000;35:595–601. doi: 10.1161/01.hyp.35.2.595. [DOI] [PubMed] [Google Scholar]
- 23.Zwacka R.M., Epperly M.W., Greenberger J.L., Engelhardt J.F. Redox gene therapy protects human IB-3 lung epithelial cells against ionizing radiation-induced apoptosis. Human Gene Therapy. 1998;9:1381–1386. doi: 10.1089/hum.1998.9.9-1381. [DOI] [PubMed] [Google Scholar]
- 24.Paoletti F., Mocali A. Determination of superoxide dismutase activity by purely chemical system based on NAD(P)H oxidation. Methods in Enzymology. 1990;186:209–220. doi: 10.1016/0076-6879(90)86110-h. [DOI] [PubMed] [Google Scholar]
- 25.Chu Y., Alwahdani A., Iida S., Lund D.D., Faraci F.M., Heistad D.D. Vascular effects of the human extracellular superoxide dismutase R213G variant. Circulation. 2005;112:1047–1053. doi: 10.1161/CIRCULATIONAHA.104.531251. [DOI] [PubMed] [Google Scholar]
- 26.Weydert C.J., Cullen J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nature Protocols. 2010;5:51–66. doi: 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowell F.J., Hamilton C.A., McMurray J., Reid J.L. Effects of a xanthine oxidase/hypoxanthine free radical and reactive oxygen species generating system on endothelial function in New Zealand white rabbit aortic rings. Journal of Cardiovascular Pharmacology. 1993;22:792–979. doi: 10.1097/00005344-199312000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Tan S., Yokoyama Y., Dickens E., Cash T.G., Freeman B.A., Parks D.A. Xanthine oxidase activity in the circulation of rats following hemorrhagic shock. Free Radical Biology and Medicine. 1993;15:407–414. doi: 10.1016/0891-5849(93)90040-2. [DOI] [PubMed] [Google Scholar]
- 29.Houston M., Estevez A., Chumley P., Aslan M., Marklund S., Parks D.A., Freeman B.A. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. Journal of Biological Chemistry. 1999;274:4985–4994. doi: 10.1074/jbc.274.8.4985. [DOI] [PubMed] [Google Scholar]
- 30.Chu Y., Piper R., Richardson S., Watanabe Y., Patel P., Heistad D.D. Endocytosis of extracellular superoxide dismutase into endothelial cells: role of the heparin-binding domain. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:1985–1990. doi: 10.1161/01.ATV.0000234921.88489.5c. [DOI] [PubMed] [Google Scholar]
- 31.Terada L.S., Guidot D.M., Leff J.A., Willingham I.R., Hanley M.E., Piermattei D., Repine J.E. Hypoxia injures endothelial cells by increasing endogenous xanthine oxidase activity. Proceedings of the National Academy of Sciences of USA. 1992;89:3362–3366. doi: 10.1073/pnas.89.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee B.E., Toledo A.H., Anaya-Prado R., Roach R.R., Toledo-Pereyra L.H. Allopurinol xanthine oxidase, and cardiac ischemia. Journal of Investigative Medicine. 2009;57:902–909. doi: 10.2310/JIM.0b013e3181bca50c. [DOI] [PubMed] [Google Scholar]
- 33.Gunnett C.A., Heistad D.D., Faraci F.M. Interleukin-10 protects nitric oxide-dependent relaxation during diabetes: role of superoxide. Diabetes. 2002;51:1931–1937. doi: 10.2337/diabetes.51.6.1931. [DOI] [PubMed] [Google Scholar]
- 34.Doehner W., Landmesser U. Xanthine oxidase and uric acid in cardiovascular disease: clinical impact and therapeutic options. Seminars in Nephrology. 2011;31:433–440. doi: 10.1016/j.semnephrol.2011.08.007. [DOI] [PubMed] [Google Scholar]