

Abstract
Mitochondrial radical production is important in redox signaling, aging and disease, but the relative contributions of different production sites are poorly understood. We analyzed the rates of superoxide/H2O2 production from different defined sites in rat skeletal muscle mitochondria oxidizing a variety of conventional substrates in the absence of added inhibitors: succinate; glycerol 3-phosphate; palmitoylcarnitine plus carnitine; or glutamate plus malate. In all cases, the sum of the estimated rates accounted fully for the measured overall rates. There were two striking results. First, the overall rates differed by an order of magnitude between substrates. Second, the relative contribution of each site was very different with different substrates. During succinate oxidation, most of the superoxide production was from the site of quinone reduction in complex I (site IQ), with small contributions from the flavin site in complex I (site IF) and the quinol oxidation site in complex III (site IIIQo). However, with glutamate plus malate as substrate, site IQ made little or no contribution, and production was shared between site IF, site IIIQo and 2-oxoglutarate dehydrogenase. With palmitoylcarnitine as substrate, the flavin site in complex II (site IIF) was a major contributor (together with sites IF and IIIQo), and with glycerol 3-phosphate as substrate, five different sites all contributed, including glycerol 3-phosphate dehydrogenase. Thus, the relative and absolute contributions of specific sites to the production of reactive oxygen species in isolated mitochondria depend very strongly on the substrates being oxidized, and the same is likely true in cells and in vivo.
Abbreviations: IF, flavin site of complex I; IQ, quinone-binding site of complex I; IIF, flavin site of complex II; IIIQo, quinol oxidation site of complex III; CDNB, 1-chloro-2,4-dinitrobenzene; Eh, redox potential; ETF, electron transferring flavoprotein; ETF:QOR, ETF:ubiquinone oxidoreductase; mGPDH, mitochondrial glycerol 3-phosphate dehydrogenase; OGDH, 2-oxoglutarate dehydrogenase; PDH, pyruvate dehydrogenase; Q, ubiquinone; QH2, ubiquinol; ROS, reactive oxygen species
Keywords: Superoxide, Hydrogen peroxide, Respiratory complexes, NADH, Ubiquinone, Cytochrome b
Graphical abstract
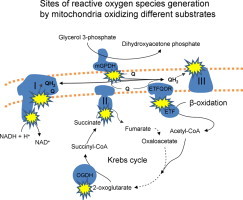
Highlights
-
•
Mitochondria oxidizing four substrate combinations make superoxide/H2O2 from six defined sites.
-
•
The absolute rates of mitochondrial superoxide/H2O2 depend on the substrate oxidized.
-
•
The relative contributions of specific sites of superoxide/H2O2 vary greatly between substrates.
Introduction
An increasing number of hypotheses propose that production of mitochondrial reactive oxygen species (ROS) plays a crucial role in different areas of physiology and pathology [1–4]. Despite this, we know very little about which mitochondrial sites in the electron transport chain and associated metabolic enzymes are responsible for physiological or pathological ROS production under native conditions (i.e. in the absence of added inhibitors).
It is well established that isolated mitochondria can produce hydrogen peroxide (H2O2) in vitro [5,6]. Since the earliest observations by Chance and colleagues [7], this field has expanded considerably and many characteristics of mitochondrial superoxide/H2O2 production have been revealed. ROS are produced by the leak of electrons from donor redox centers to molecular oxygen. The mitochondrial electron transport chain (Fig. 1) consists of several complexes containing multiple redox centers that normally facilitate transfer of electrons to their final acceptor, molecular oxygen, which is reduced by four electrons to water at complex IV. Premature single electron reduction of molecular oxygen earlier in the chain forms the superoxide radical (O2•−) and divalent reduction forms H2O2. Superoxide dismutase-2 in the matrix converts superoxide to H2O2, which can escape and be assayed in the surrounding medium. The general term ‘ROS’ can refer to several different species, but in this context we use it to refer only to superoxide or H2O2.
Fig. 1.
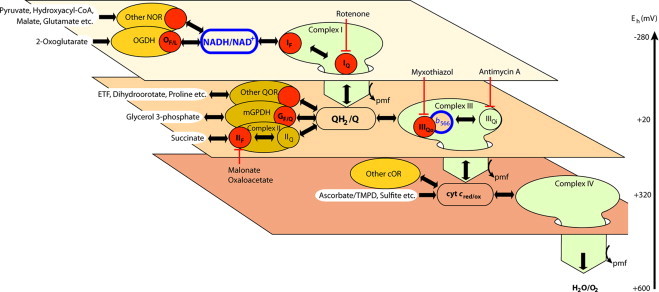
Isopotential groups, electron flow, and endogenous reporters of superoxide/H2O2 production at different sites in mitochondria. The three planes represent different isopotential groups of redox centers, each operating at about the same redox potential (Eh): NADH/NAD+ at Eh∼−280 mV, QH2/Q at Eh∼+20 mV, and cytochrome c at Eh∼+320 mV [31]. The normal flow of electrons from substrate dehydrogenases through NADH to oxygen is indicated by the large green arrows dropping down through the isopotential planes. The enzymes that feed electrons into each isopotential group are represented as ovals, and relevant inhibitors are drawn with blunted arrows. Electrons from NAD-linked substrates enter the NADH/NAD+ pool at Eh∼−280 mV through appropriate NAD oxidoreductases (NOR) including 2-oxoglutarate dehydrogenase (OGDH) and malate dehydrogenase (MDH) and flow into complex I (site IF). They then drop down via site IQ to QH2/Q at Eh∼+20 mV in the next isopotential pool, providing the energy to pump protons and generate protonmotive force (pmf). Q oxidoreductases (QOR) including complex II and mitochondrial glycerol 3-phosphate dehydrogenase (mGPDH) can also pass electrons into the Q pool. Electrons flow from QH2 through complex III to cytochrome c at Eh∼+320 mV in the next isopotential pool, again pumping protons and generating pmf. Ascorbate plus N,N,N′,N′-tetramethyl-phenylenediamine (TMPD) and cytochrome c oxidoreductases including sulfite oxidase can also pass electrons to cytochrome c. Finally, electrons flow through complex IV to oxygen at Eh∼+600 mV, generating pmf. The redox state of NADH (outlined in blue), measured as NAD(P)H using autofluorescence, reports the redox state of the first isopotential group. The redox state of cytochrome b566 (outlined in blue), measured using absorbance spectroscopy, reports the redox state of the second isopotential group [21]. Red dots are sites of superoxide/H2O2 production. Named sites are the flavin/lipoate of 2-oxoglutarate dehydrogenase (OF/L), the complex I flavin and Q-binding sites (IF and IQ, respectively), the flavin site of complex II (IIF), the flavin/quinone site of mGPDH (GF/Q), and the outer quinol-binding site of complex III (IIIQo). Other sites, both known and unknown, are indicated by unlabeled red dots. These include less well-described sites such as pyruvate dehydrogenase, the ETF/ETF:QOR system, proline dehydrogenase, and dihydroorotate dehydrogenase.
Using inhibitors to manipulate the redox states of particular sites and prevent superoxide generation from others, at least ten different sites of superoxide/H2O2 production in the electron transport chain and associated enzymes (Krebs cycle, β-oxidation etc.) have been identified in mammalian mitochondria (Fig. 1). Each of these sites has been at least partially characterized. The sites that are often invoked as the most important mitochondrial superoxide producers are in respiratory complexes I and III [5,6]. In complex I there are two sites: the flavin in the NADH-oxidizing site (site IF) and the ubiquinone-reducing site (site IQ) [8]. In complex III, the superoxide is thought to arise from the quinol oxidizing site (site IIIQo) [9–11]. However, other sites of superoxide/H2O2 production have also been defined, including 2-oxoglutarate dehydrogenase (OGDH) [12,13]; pyruvate dehydrogenase (PDH) [14]; complex II (site IIF) [15]; and glycerol 3-phosphate dehydrogenase (mGPDH) [16]. In addition, there are suggestions that other less well-described sites may also be involved in H2O2 production: the electron transferring flavoprotein/ETF:Q oxidoreductase (ETF/ETF:QOR) system of fatty acid β-oxidation [17,18]; proline dehydrogenase [19]; and dihydroorotate dehydrogenase [16,20].
Despite our understanding of the superoxide/H2O2-producing capacities of mitochondrial enzymes in vitro, we know very little about the native ROS-producing behavior of mitochondria in vitro or in situ. This is because the standard way to implicate a specific ROS-producing site in a particular phenotype is to inhibit or genetically modify the site, and observe the change in ROS signal or in the downstream phenotype. However, this approach is fundamentally flawed, because blocking a site of electron transport will invariably interrupt normal electron flow and alter the redox states of other sites in the electron flow pathway, which can dramatically alter their rates of ROS production, leading to unreliable or incorrect conclusions. This raises the question: how can the individual contributions from a complex suite of superoxide/H2O2-producing sites be assessed within intact mitochondria under native conditions? To address this question, we developed a novel method of estimating the rates of superoxide generation from two specific sites (IF and IIIQo) by determining the dependence of superoxide production from each site (defined using inhibitors) on the redox state of its electron donor (reported by the redox states of NAD(P)H and cytochrome b566, respectively), then measuring the redox state of the reporter under native conditions in the absence of added inhibitors to predict the contribution of the reported site to overall H2O2 production [21].
In the present study, we extend this approach of using endogenous reporters under native conditions to encompass many more superoxide/H2O2-producing sites and a greater variety of substrates. We determine the contributions of each site to overall H2O2 production by isolated skeletal muscle mitochondria oxidizing four different substrate combinations in the absence of inhibitors: (a) succinate, (b) glycerol 3-phosphate, (c) palmitoylcarnitine plus carnitine, and (d) glutamate plus malate. The results show that the absolute and relative contribution of each site differs greatly with different substrates.
Materials and methods
Animals, mitochondrial preparation, and reagents
Female Wister rats (Harlan Laboratories), age 5–8 weeks, were fed chow ad libitum and given free access to water. Mitochondria from hind limb skeletal muscle were isolated at 4 °C in Chappell–Perry buffer (CP1; 100 mM KCl, 50 mM Tris, 2 mM EGTA, pH 7.1 at 25 °C) by standard procedures [22]. The animal protocol was approved by the Buck Institute Animal Care and Use Committee, in accordance with IACUC standards. All reagents were from Sigma except Amplex UltraRed, which was from Invitrogen.
Superoxide/H2O2 production
Rates of superoxide/H2O2 production were measured collectively as rates of H2O2 production, as two superoxide molecules are dismutated by endogenous or exogenous superoxide dismutase to yield one H2O2. H2O2 was detected using the horseradish peroxidase and Amplex UltraRed detection system [22]. Mitochondria (0.3 mg protein ml−1) were suspended under non-phosphorylating conditions in medium at 37 °C containing 120 mM KCl, 5 mM Hepes, 5 mM K2HPO4, 2.5 mM MgCl2, 1 mM EGTA, and 0.3% (w/v) bovine serum albumin (pH 7.0 at 37 °C), together with 5 U ml−1 horseradish peroxidase, 25 U ml−1 superoxide dismutase, 50 µM Amplex UltraRed and, except for experiments with palmitoylcarnitine, 1 µg ml−1 oligomycin. Reactions were monitored fluorometrically in a Shimadzu RF5301-PC or Varian Cary Eclipse spectrofluorometer (λexcitation=560 nm, λemission=590 nm) with constant stirring, and calibrated with known amounts of H2O2 [22].
To correct for losses of H2O2 caused by peroxidase activity in the matrix and give a better estimate of superoxide/H2O2 production rates, all H2O2 production rates were mathematically corrected to the rates that would have been observed in these mitochondria after pre-treatment with 1-chloro-2,4-dinitrobenzene (CDNB) to deplete glutathione and decrease glutathione peroxidase and peroxiredoxin activity, as described in Refs. [21,23], using an empirical equation
| (1) |
(rates in pmol H2O2 min−1 mg protein−1).
NAD(P)H redox state
Experiments were performed using 0.3 mg mitochondrial protein ml−1 at 37 °C in parallel with measurements of H2O2 production and cytochrome b566 redox state in the same medium with the same additions. The reduction state of endogenous NAD(P)H was determined by autofluorescence (most of the autofluorescence signal is from NADH bound in the matrix, and NADPH hardly changes in our experiments, but for full disclosure we use “NAD(P)H”) [21,23] using a Shimadzu RF5301-PC or Varian Cary Eclipse spectrofluorometer at λexcitation=365 nm, λemission=450 nm. NAD(P)H was assumed to be 0% reduced after 5 min without added substrate and 100% reduced with 5 mM malate plus 5 mM glutamate and 4 µM rotenone. Intermediate values were determined as % reduced NAD(P)H relative to the 0% and 100% values.
Cytochrome b566 redox state
Experiments were performed at 1.5 mg mitochondrial protein ml−1 in parallel with measurements of H2O2 production and NAD(P)H redox state in the same medium. The reduction state of endogenous cytochrome b566 was measured with constant stirring at 37 °C in an Olis DW-2 dual wavelength spectrophotometer as the absorbance difference at 566–575 nm [11,21]. Cytochrome b566 was assumed to be 0% reduced after 5 min without added substrate and 100% reduced with saturating substrates plus antimycin A. Intermediate values were determined as % reduced b566 relative to the 0% and 100% values.
Definition of sites and calibration of endogenous reporters
Site IF was defined as the site producing superoxide (measured as H2O2) in the presence of malate to reduce NAD to NADH, and rotenone to inhibit reoxidation of complex I by the Q-pool. Any H2O2 arising from reverse flow from the NADH pool into NAD oxidoreductases such as OGDH or PDH will appear in the analysis as a component of site IF (Fig. 2a). To decrease the contributions of forward electron flow at OGDH and PDH to H2O2 production assigned to site IF, we added 1.5 mM aspartate to remove endogenous 2-oxoglutarate by transamination and 2.5 mM ATP to decrease carbon flows at various points in the Krebs cycle, particularly succinate thiokinase. This change to our previous protocol [18,21] strongly decreased our earlier estimates of the rates from site IF. The rate of superoxide/H2O2 production from site IF defined by inhibitors in this way was measured as a function of the redox state of NAD(P)H at different malate concentrations between 0.02 mM and 5 mM (Fig. 2b). The data were arbitrarily fitted by non-linear regression to a single exponential, to give the parameter values in the following equation:
| (2) |
where is the rate of H2O2 production. This equation was used to predict the rate of superoxide/H2O2 production from site IF at any observed NAD(P)H redox state in the absence of inhibitors in subsequent experiments.
Fig. 2.
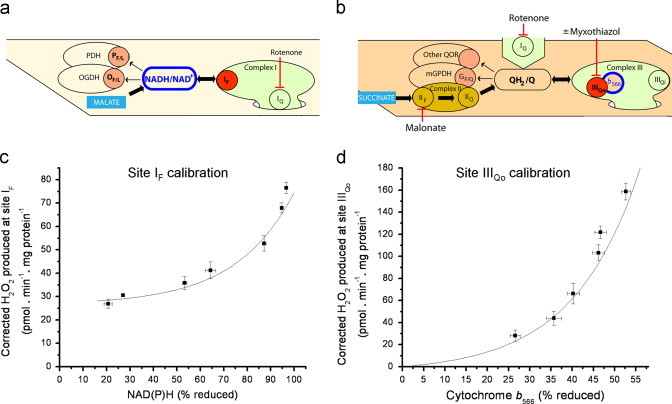
Calibration of endogenous reporters in isolated rat skeletal muscle mitochondria. (a) Definition of site IF (red dot) using malate as substrate in the presence of rotenone to inhibit site IQ and cause reduction of upstream sites and oxidation of all downstream sites. The definition includes any small contribution from backflow of electrons from NADH into other sites, e.g. OGDH and PDH, shown in pink. PF/L denotes the flavin/lipoate of pyruvate dehydrogenase. (b) Dependence of superoxide production (measured as extramitochondrial appearance of H2O2) from site IF on the redox state of NAD(P)H (measured by autofluorescence). Malate was titrated from 20 μM−5 mM in the presence of 4 μM rotenone, 1.5 mM aspartate and 2.5 mM ATP. (c) Definition of site IIIQo (red dot) using succinate as substrate, rotenone to inhibit site IQ, and sufficient succinate+malonate (5 mM) to suppress site IIF. The rate of superoxide/H2O2 production from site IIIQo was defined as the rate that was inhibited by 2 µM myxothiazol (after correction for changes at site IF using the calibration curve in (b)). The definition includes any small contribution from backflow of electrons from QH2 into other sites, e.g. mGPDH or other Q-oxidoreductases, shown in pink. (d) Dependence of myxothiazol-sensitive superoxide production from site IIIQo on the redox state of cytochrome b566 (measured by the absorbance spectroscopy). Electron input was titrated by adding different ratios of succinate:malonate (sum=5 mM) in the presence of 4 µM rotenone. Data are means±SEM (n≥4) recalculated from [21] using the new calibration in (b). For curve-fitting see Materials and methods.
Superoxide production by site IIIQo was measured as H2O2 production in the presence of succinate to reduce the Q pool, malonate to keep total succinate plus malonate at 5 mM and inhibit superoxide/H2O2 formation from site IIF without fully inhibiting succinate oxidation [15], and rotenone to inhibit superoxide formation from site IQ (Fig. 2c). Site IIIQo was defined as the component of the observed H2O2 production under these conditions that was sensitive to myxothiazol (a specific inhibitor of site IIIQo) after correction for the small difference in rates from site IF before and after myxothiazol addition (calculated from parallel measurements of NAD(P)H and application of the calibration curve in Fig. 2b). The rate of superoxide/H2O2 production from site IIIQo defined by inhibitors in this way was measured as a function of the redox state of cytochrome b566 at different succinate: malonate ratios ranging from 75% to 100% succinate at 5% increments (total dicarboxylate concentration 5 mM) (Fig. 2d). The data were arbitrarily fitted in the same way as for site IF to give the parameter values using the following equation:
| (3) |
This equation was used to predict the rate of superoxide/H2O2 production from site IIIQo at any observed cytochrome b566 redox state in the absence of inhibitors in subsequent experiments.
See Ref. [21] for more extensive discussion and descriptions.
Statistics
When using the calibration curves in Fig. 2 to calculate rates of H2O2 production at a given reduction state of the reporter, the error in the measurements during calibration was taken into account. This error was calculated by standard methods of error propagation through all the steps, as detailed in [21].
The significance of differences between reported and experimentally measured rates of H2O2 production in each experimental condition was tested using Welch's t-test. Because error propagation was used to capture uncertainty in the calibration curves, individual data-points could not be used for statistical analysis. Instead we used the traits describing the population of data (mean, SEM based on error propagation and number of observations) to calculate if differences were significant (p<0.05).
Results and discussion
The aim of the present study was to determine the contributions of different sites of superoxide/H2O2 production to the total observed H2O2 generation by mitochondria during oxidation of different substrates in the absence of inhibitors. Electrons leak from the respiratory chain to generate superoxide or H2O2 at two different redox potentials (Eh): at the isopotential group of redox carriers around the NADH/NAD+ pool at Eh∼−280 mV and at the isopotential group of redox carriers around the QH2/Q pool at Eh∼+20 mV (Fig. 1). At each isopotential group, an important determinant of the rate of superoxide or H2O2 production is the redox state: a more reduced carrier generally leaks electrons to oxygen at a faster rate. We exploited this relationship to create assays of the rate of superoxide/H2O2 production from different sites, either by defining the site precisely with inhibitors and determining its dependence on the redox state of the appropriate pool as described in Materials and methods (Fig. 2), or by inhibiting the site and determining the change in superoxide/H2O2 production after correction for secondary changes in the redox states of the two pools. Specifically, to predict rates from each site with different respiratory substrates in the absence of inhibitors, we measured the redox states of the endogenous reporters NAD(P)H and cytochrome b566 as proxies of the redox states of the two isopotential groups, and determined the rates of superoxide/H2O2 production from sites IF and IIIQo, respectively, using the calibration curves in Fig. 2. Assessing the contribution of other sites was a little more complicated, so we first provide an example of how the endogenous reporter calibration curves and the careful use of inhibitors can be used to quantify the sites of H2O2 production during oxidation of the substrate succinate.
Sites of superoxide/H2O2 production during oxidation of succinate: a worked example
When succinate is oxidized in the absence of rotenone and other electron transport chain inhibitors, electrons flow through complex II into the Q pool. Next, there are two options (Fig. 3a). The electrons can flow forward to complex III and thermodynamically downhill to more oxidized isopotential groups at cytochrome c and H2O/O2, pumping protons at complexes III and IV and generating protonmotive force. Alternatively, they can flow in reverse to complex I, driven thermodynamically uphill to the more negative isopotential group by reversal of the proton pumps in complex I driven by the protonmotive force generated by proton pumping at complexes III and IV. In the process, a number of possible sites of superoxide/H2O2 production may be engaged, particularly sites IIF, IIIQo, IQ and IF (Fig. 3a). However, it is generally agreed that the primary mechanism of H2O2 production during succinate oxidation is reverse electron transport into complex I, because this H2O2 production is very sensitive to rotenone, the classic Q-site inhibitor of complex I [24,25]. What is not usually discussed is that when an inhibitor such as rotenone is added there are subsequent shifts in electron distribution that invariably change the rates of superoxide/H2O2 production by sites both upstream and downstream of the inhibition site [21]. Therefore, the decrease in rate observed after the addition of an inhibitor is not an accurate indication of the rate from its target site before inhibition. The endogenous reporter method described here circumvents this problem: by monitoring the changes in the NADH isopotential group (through NAD(P)H) and the Q isopotential group (through cytochrome b566), the changes in electron distribution after inhibitors are added can be quantified and corrected for.
Fig. 3.
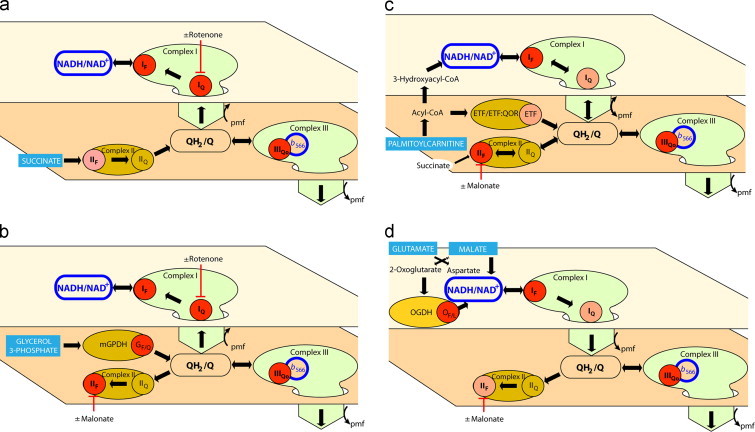
Electron flows and sites of superoxide/H2O2 production during oxidation of different substrates by rat skeletal muscle mitochondria. Electron flows and postulated (red and pink dots) and observed (red dots) sites of superoxide/H2O2 production during oxidation of (a) succinate, (b) glycerol 3-phosphate, (c) palmitoylcarnitine plus carnitine and (d) glutamate plus malate.
In this worked example, we quantified the sites of H2O2 production during succinate oxidation by measuring the redox states of the endogenous reporters, and the changes in these redox states after addition of rotenone. From this information, we could predict the contribution of each site to the total H2O2 production observed during succinate oxidation. To begin, we measured the rate of H2O2 production during succinate oxidation in the absence of rotenone (891 pmol H2O2 min−1 mg protein−1) (Fig. 4a). In parallel, in different cuvettes, we measured the reduction states of the reporters. We observed that NAD(P)H was 89% reduced and cytochrome b566 was 46% reduced (Fig. 4b, Table 1). From this information, we could predict using the equations describing the calibration curves in Fig. 2 that site IF was producing 59 pmol H2O2 min−1 mg protein−1 (7% of the total), and site IIIQo was producing 91 pmol H2O2 min−1 mg protein−1 (10%) (Fig. 4c, Table 1), with the remaining 741 pmol H2O2 min−1 mg protein−1 coming from other sites.
Fig. 4.
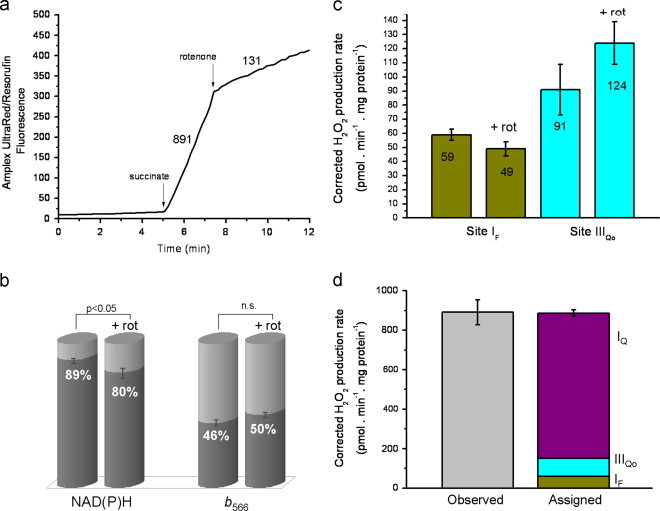
Sites of superoxide/H2O2 production during succinate oxidation: a worked example. (a) Representative trace of Amplex UltraRed/resorufin fluorescence during oxidation of 5 mM succinate by rat skeletal muscle mitochondria before and after addition of 4 µM rotenone. Numbers by the traces represent average calibrated rates in pmol H2O2 min−1 mg protein−1. (b) Reduction states of the endogenous reporters NAD(P)H and cytochrome b566 before and after rotenone addition. The difference in reduction state before and after rotenone was significant for NAD(P)H (p=0.01), but not for cytochrome b566 (p=0.11) by Student's t-test. (c) Rates of superoxide/H2O2 production from sites IF and IIIQo before and after rotenone addition, predicted from (b) using the calibration curves in Fig. 2. Results are means±SEM (n=4). To account for error in both the measurements and the calibration curves, a Welch's t-test was used. The difference in rates before and after rotenone did not fall within the 95% confidence interval (p=0.09 for site IF and 0.15 for IIIQo). (d) Observed total rate of H2O2 production during succinate oxidation (before addition of rotenone) and assigned rates of superoxide/H2O2 production from sites IF, IIIQo and IQ during succinate oxidation. Results are means±SEM (n=4); the error bars on the sum column show the combined propagated errors in the total sum value. There was no significant difference between the observed rate and the sum of the assigned rates (Welch's t-test; p<0.05).
Table 1.
Reduction levels of reporters and corresponding calculated rates of H2O2 production from site IF and site IIIQo in the presence of different substrates and inhibitors. With each substrate, the redox states of NAD(P)H and cytochrome b566 were measured in parallel with the measurements of overall rates of H2O2 production reported in Fig. 5. Eqs. (2) and (3) describing the calibration curves in Fig. 2 were used to calculate the rates of superoxide/H2O2 production from site IF and site IIIQo from these redox measurements. Concentrations of substrates and inhibitors are given in Fig. 5. All rates are in pmol H2O2 min−1 mg protein−1. Data are means±SEM (n≥4). Error values on the calculated rates of H2O2 production represent the propagated error as described in Materials and methods and Ref. [21].
| Substrates and inhibitors |
Redox state of site IFreporter and calculated rate of superoxide/H2O2production by site IF |
Redox state of site IIIQoreporter and calculated rate of superoxide/H2O2production by site IIIQo |
||
|---|---|---|---|---|
| Reduced NAD(P)H (%) | Reported rate of H2O2 production | Reduced cytochrome b566 (%) | Reported rate of H2O2 production | |
| Succinate | 89±1 | 59±4 | 46±2 | 91±18 |
| Succinate+rotenone | 80±2 | 49±4 | 50±1 | 124±15 |
| Glycerol 3-phosphate | 59±3 | 34±6 | 29±2 | 27±7 |
| Glycerol 3-phosphate+malonate | 59±3 | 34±6 | 29±2 | 27±7 |
| Glycerol 3-phosphate+malonate+rotenone | 39±2 | 31±5 | 31±3 | 33±9 |
| Palmitoylcarnitine+carnitine | 80±1 | 49±4 | 42±3 | 72±18 |
| Palmitoylcarnitine+carnitine+malonate | 79±1 | 48±4 | 44±2 | 80±17 |
| Glutamate+malate | 85±7 | 54±9 | 38±5 | 54±18 |
To identify and quantify these other sites, we added rotenone to inhibit site IQ, and measured the changes in H2O2 production and the changes in the reduction state of the reporters (Figs. 4a and b, Table 1). After rotenone addition, the overall rate of H2O2 production dropped to 131 pmol H2O2 min−1 mg protein−1, NAD(P)H became significantly more oxidized and cytochrome b566 appeared to become slightly more reduced (Fig. 4 a and b, Table 1). Again, using the calibration curves in Fig. 2 we determined that site IF now appeared to produce slightly less superoxide/H2O2 (49 pmol H2O2 min−1 mg protein−1) and site IIIQo appeared to produce more (124 pmol H2O2 min−1 mg protein−1) than before the addition of rotenone (Fig. 4c, Table 1). When rotenone was added, not only was reverse electron flow abolished, leading to partial oxidation of NAD(P)H as the electron supply from succinate through complex I failed, but there appeared to be a small increase in the reduction level of cytochrome b566 as all flow was diverted through complex III. The total change in superoxide/H2O2 production at site IF was calculated by subtracting the rate before rotenone addition (59 pmol H2O2 min−1 mg protein−1) from the rate after rotenone addition (49 pmol H2O2 min−1 mg protein−1) to give a final change in rate from site IF of −10 pmol H2O2 min−1 mg protein−1. The equivalent calculation performed for site IIIQo gave a total change of +33 pmol H2O2 min−1 mg protein−1. The rotenone-sensitive rate (759 pmol H2O2 min−1 mg protein−1) was then corrected for the changes in superoxide/H2O2 production from sites IF and IIIQo to give a final adjusted rate of 736 pmol H2O2 min−1 mg protein−1, all assigned to site IQ (it was not from site IF, as has been suggested [26], because site IF superoxide production was fully corrected for using NAD(P)H as the endogenous reporter). This painted a final picture (Fig. 4d) showing that during succinate oxidation, site IQ contributed 83% of the observed rate of H2O2 production while site IF accounted for 7% and site IIIQo accounted for 10%. Since there was no unaccounted-for rate of H2O2 production, either before (891 pmol H2O2 min−1 mg protein−1 observed versus 886 pmol H2O2 min−1 mg protein−1 assigned, N.S.) or after addition of rotenone (131 pmol H2O2 min−1 mg protein−1 observed versus 171 pmol H2O2 min−1 mg protein−1 assigned, N.S.), no other site made a substantial contribution to the total observed rate. The reason site IIF did not contribute is that the conventional succinate concentration used (5 mM) was sufficiently high to effectively abolish production of superoxide/H2O2 by site IIF [15].
There are no surprises in these conclusions (although they are more accurate than simply assigning the rotenone-sensitive signal to complex I): site IQ is the dominant superoxide/H2O2 producer when succinate is oxidized in the absence of rotenone. However, this example illustrates clearly that during oxidation of a single substrate, superoxide/H2O2 are produced from multiple sites (Fig. 4d). It also clearly illustrates that site-specific inhibitors not only affect superoxide/H2O2 production at the site that they inhibit, but also (through changes in electron distribution) change superoxide/H2O2 production at other sites.
The contributions of specific sites to superoxide/H2O2 production during oxidation of different substrates
The approach described above as a worked example using succinate as substrate was used to define the contributions of specific sites to superoxide/H2O2 production during oxidation of three additional important conventional substrates: glycerol 3-phosphate, palmitoylcarnitine plus carnitine, and glutamate plus malate. In each condition, the reduction state of the reporters was measured in parallel with H2O2 production. The changes in reduction state of the reporters after addition of inhibitors of complex I or complex II were measured and used to correct for consequent changes in electron distribution and superoxide/H2O2 production at other sites. Table 1 details the observed redox states of the reporters with each substrate before and after addition of the inhibitors rotenone or malonate.
Fig. 5 shows the central result of the present paper: the contributions of each of the individual sites of superoxide/H2O2 production to overall H2O2 generation by isolated skeletal muscle mitochondria with each of the four substrate combinations under native conditions in the absence of added inhibitors. The first striking result is that the overall rates of H2O2 generation were very different with different substrates, as previously observed by [27] and others [28–30]. The second, crucial, observation is that the relative contribution of each site was very different with different substrates. Thus, the relative and absolute contributions of specific sites to the production of reactive oxygen species in isolated mitochondria depend very strongly on the substrates being oxidized.
Fig. 5.
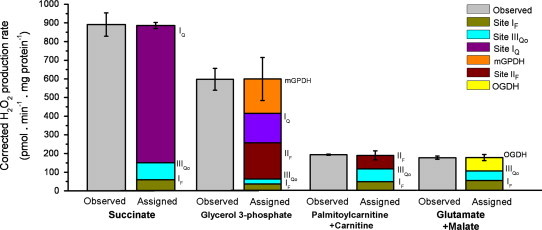
Native rates of superoxide/H2O2 production by mitochondria oxidizing different substrates. Observed total rate of H2O2 production (gray bars) and sum of assigned rates of superoxide/H2O2 production from different sites (colored stacked bars) in the presence of different substrates as indicated: 5 mM succinate; 27 mM disodium rac-α/β-glycerol phosphate (25% active optical isomer sn-glycerol 3-phosphate); 15 µM palmitoyl-L-carnitine plus 2 mM L-carnitine; and 5 mM L-glutamate plus 5 mM L-malate. With each substrate, the reduction states of NAD(P)H and cytochrome b566 were measured in parallel with H2O2 production and the calibration curves in Fig. 2 were used to predict the rates of production from sites IF and IIIQo. With succinate, 4 µM rotenone was subsequently added to allow calculation of the rate from site IQ as described in the text (data from Fig. 4d). With glycerol 3-phosphate, 1 mM malonate and 4 µM rotenone were subsequently added to allow calculation of the rates from sites IIF and IQ, respectively. The rate assigned to mGPDH was calculated by difference. With palmitoylcarnitine plus carnitine, 1 mM malonate was subsequently added to allow calculation of the rate from site IIF (data recalculated from [18]). With glutamate plus malate, the rate assigned to OGDH was calculated by difference (data recalculated from [21]). Results are means±SEM (n=4–6); the error bars on the sum columns show the combined propagated errors in the total sum value. There was no significant difference between observed and assigned rates with succinate or with palmitoylcarnitine plus carnitine (Welch's t-test; p<0.05).
To determine the contributions of individual sites to overall H2O2 generation with each substrate, the rates of H2O2 production at site IF and IIIQo were first estimated from the redox states of NAD(P)H and cytochrome b566 using the calibration curves in Fig. 2. With succinate and glycerol 3-phosphate, the contribution of site IQ was then estimated from the decrease in the rate of H2O2 production after addition of rotenone followed by correction for changes in the rate from site IF (calculated from the redox state of NAD(P)H before and after rotenone addition using the calibration curve in Fig. 2a) and from site IIIQo (calculated from the redox state of cytochrome b566 before and after rotenone addition using the calibration curve in Fig. 2c). With glycerol 3-phosphate, palmitoylcarnitine plus carnitine, and glutamate plus malate, the contribution of site IIF was estimated from the decrease in rate of H2O2 production after addition of malonate followed by correction for changes in the rate from site IF (calculated from the redox state of NAD(P)H before and after malonate addition) and from site IIIQo (calculated from the redox state of cytochrome b566 before and after malonate addition). The contributions of mGPDH (with glycerol 3-phosphate as substrate) and OGDH (with glutamate plus malate as substrate) were estimated as the difference between the sum of the rates estimated for the other sites and the observed total rate.
Sites of superoxide/H2O2 production during oxidation of glycerol 3-phosphate
Glycerol 3-phosphate oxidation by mGPDH results directly in Q pool reduction (Fig. 3b). From the Q pool, electrons may flow both forward (to complex III and beyond, generating protonmotive force) and in reverse (to complex I and complex II). In the process, a number of possible sites of superoxide/H2O2 production may be engaged, particularly mGPDH and sites IIF, IIIQo, IQ and IF (Fig. 3b). Protonmotive force is required for reverse electron flow to complex I [25], but not for reversal into complex II [16]. Complex II has been shown to generate superoxide in both the forward and reverse reactions [15].
With glycerol 3-phosphate as substrate, the total observed rate of H2O2 production was 622 pmol H2O2 min−1 mg protein−1. Sites IF (6%) and IIIQo (4%) made only modest contributions to this total rate (Fig. 5). The contributions of sites IQ (33%) and IIF (26%) (after correction for changes in sites IF and IIIQo following the addition of inhibitors) were more substantial. The absolute rates from sites IF, IIIQo and IQ were lower with glycerol 3-phosphate than with succinate, because the two isopotential pools were less reduced (Table 1), presumably because succinate was a better substrate than glycerol 3-phosphate in this experiment, which had sub-optimal glycerol 3-phosphate and calcium concentrations. After these assignments, 192 pmol H2O2 min−1 mg protein−1 (31% of the total) remained unaccounted for. mGPDH itself is known to generate ROS (predominantly superoxide), which are released to both the matrix and the intermembrane space [16]. In this case, we assume that the remaining observed rates of H2O2 production must have arisen from mGPDH, since all other known sites were already accounted for, and we assign this site by difference (31%). It is unlikely that any of the unassigned H2O2 generation in this condition was from uncharacterized sites, since such sites would have to be engaged during oxidation of glycerol 3-phosphate but not during oxidation of succinate (where there was no unassigned H2O2 production, Fig. 5), despite the less extensive reduction of the two isopotential groups with glycerol 3-phosphate as substrate (Table 1).
Sites of superoxide/H2O2 production during oxidation of palmitoylcarnitine plus carnitine
Carnitine enhances oxidation of palmitoylcarnitine by removing inhibitory acetyl-CoA in the form of acetylcarnitine and by promoting entry of palmitoylcarnitine into the mitochondrial matrix [18]. When palmitoylcarnitine is metabolized by the β-oxidation pathway, electrons enter the respiratory chain at two sites: from acyl-CoA dehydrogenase through the electron transferring flavoprotein (ETF) and ETF:ubiquinone oxidoreductase (ETF:QOR) to the Q-pool, and from 3-hydroxyacyl-CoA dehydrogenase to NADH (Fig. 3c). Oxidation of the end product, acetyl-CoA, in the citric acid cycle leads to further electron input through the NAD-linked dehydrogenases and through complex II during oxidation of succinate. Electron entry by more than one route suggests that fatty acid oxidation may generate superoxide/H2O2 from several different sites, particularly ETF/ETF:QOR and sites IIIQo, IQ and IF (Fig. 3c). Site IIF may also contribute by reverse flow from the Q pool, or by forward flow from succinate. Importantly, under this condition the acetyl-CoA generated by β-oxidation will tend to deplete inhibitory oxaloacetate through the action of citrate synthase, making site IIF more prone to generate superoxide/H2O2 [18].
With palmitoylcarnitine plus carnitine as substrate, the total observed rate of H2O2 production was 199 pmol H2O2 min−1 mg protein−1. From the redox states of the reporters and the corrected effects of malonate on H2O2 production, we were able to account for the entirety of this observed H2O2 production. There were approximately equal contributions from sites IIF (38%) and IIIQo (36%), with site IF producing slightly less (25%) (Fig. 5). The absence of unaccounted-for H2O2 production indicated that neither site IQ nor the ETF/ETF:QOR system generated substantial superoxide/H2O2. Indeed, the ETF/ETF:QOR system does not generate measureable superoxide/H2O2 except under very specific conditions and in the presence of respiratory chain inhibitors [18].
Sites of superoxide/H2O2 production during oxidation of glutamate plus malate
In the final condition, the substrate pair glutamate plus malate was used to generate NADH (Fig. 3d). During oxidation of this substrate combination, malate is oxidized to oxaloacetate by malate dehydrogenase and NADH is generated. Glutamate is used to transaminate the oxaloacetate to form aspartate and 2-oxoglutarate (and some glutamate may be oxidized to 2-oxoglutarate by glutamate dehydrogenase). The aspartate is exchanged for glutamate on the glutamate–aspartate antiporter, and much of the 2-oxoglutarate is exchanged for malate on the oxoglutarate–malate antiporter (although some may be oxidized by 2-oxoglutarate dehydrogenase). In this way, removal of oxaloacetate maximizes NADH generation by malate dehydrogenase [31].
With glutamate plus malate as substrate, the total observed rate of H2O2 production was 182 pmol H2O2 min−1 mg protein−1. Under this condition, sites IF and IIIQo contributed equally (30% each) [21] (Fig. 5). Malonate did not significantly inhibit the rate (3% inhibition±5%), so site IIF was not a significant contributor, presumably because it was inhibited by oxaloacetate or malate under these conditions [15]. A significant proportion (41%) of the total H2O2 production was unassigned, indicating that another site also contributed under this condition. We assume this was from forward flow through OGDH (Fig. 5) because some 2-oxoglutarate was likely formed during transamination of glutamate, but we cannot exclude a contribution from site IQ or uncharacterized other sites in this complex metabolic condition. In principle the contribution of site IQ could be assessed by the decrease in rate observed following addition of rotenone after correction for secondary changes in other defined sites, but definitive conclusions could not be drawn because of the relatively small rates of unassigned H2O2 production, large changes in redox states of the reporters following addition of rotenone, relatively large errors involved, and possible changes in other uncharacterized sites following addition of rotenone.
Advantages and limitations of the approach used
The great strength of the approach we have used here to measure the contributions of different sites to overall mitochondrial H2O2 production [21] is that, unlike all previous approaches, it reports rates under native conditions in the absence of added inhibitors. This gives it great potential for future studies with physiological substrate mixes in mitochondria and in intact cells and organisms. Its main limitation is the assumption that the calibration curves measured in the presence of inhibitors and particular substrates apply under native conditions using other substrates. The good agreement between calculated and measured total rates (Fig. 5) supports the validity of this assumption.
Potential pathological and physiological implications
The results obtained using the four different substrates described in Fig. 5 provide a very clear illustration of the remarkably different H2O2-producing profiles that can be attained by isolated muscle mitochondria oxidizing conventional substrates. When taken as a whole, two results are striking. (i) The native rates differed greatly between substrates. This implies that mitochondrial superoxide/H2O2 production rates in vivo likely depend critically on the substrate being oxidized, so physiological or pathological changes in substrate may be very important determinants of rates of radical production, even at the same overall rate of oxygen consumption. (ii) The contribution of each site differed markedly between substrates. With succinate, site IQ dominated, with relatively small contributions from sites IF and IIIQo. However, with palmitoylcarnitine plus carnitine, site IIF was an important contributor, and with glycerol 3-phosphate, five sites contributed, including site IIF and mGPDH. Thus, which sites contribute to superoxide and H2O2 production in mitochondria, in cells, and in vivo under both physiological and pathological conditions likely depends critically on the substrates being utilized.
Notably, the sites are known to differ markedly in the topology of superoxide production [6,16,27,32,33]. Essentially all superoxide/H2O2 from sites IF, IQ and IIF is directed to the matrix, but about half the superoxide from site IIIQo and mGPDH appears in the intermembrane space. Thus, the strength of mitochondrial superoxide signaling in the cytosol (and also the amount of oxidative damage caused by superoxide and H2O2 in the matrix) will differ substantially between substrates, even at identical total rates of mitochondrial superoxide/H2O2 production.
Acknowledgments
Supported by National Institutes of Health grants P01 AG025901, PL1 AG032118, R01 AG03354 and TL1 AG032116 (M.D.B., C.L.Q.) and The Ellison Medical Foundation, grant AG-SS-2288-09 (M.D.B., I.V.P.). Fellowship support was from The Glenn Foundation (I.V.P) and The Carlsberg Foundation (M.H-M).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Powers S.K., Duarte J., Kavazis A.N., Talbert E.E. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Experimental Physiology. 2010;95:1–9. doi: 10.1113/expphysiol.2009.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ralph S.J., Neuzil J. Mitochondria as targets for cancer therapy. Molecular Nutrition and Food Research. 2009;53:9–28. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]
- 3.Sundaresan M., Yu Z.X., Ferrans V.J., Irani K., Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 4.Witte M.E., Geurts J.J., de Vries H.E., van der Valk P., van Horssen J. Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion. 2010;10:411–418. doi: 10.1016/j.mito.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 5.Murphy M.P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brand M.D. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochemical Journal. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Treberg J.R., Quinlan C.L., Brand M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) Journal of Biological Chemistry. 2011;286:27103–27110. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kramer D.M., Roberts A.G., Muller F., Cape J.L., Bowman M.K. Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes. Methods in Enzymology. 2004;382:21–45. doi: 10.1016/S0076-6879(04)82002-0. [DOI] [PubMed] [Google Scholar]
- 10.Muller F.L., Roberts A.G., Bowman M.K., Kramer D.M. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry. 2003;42:6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 11.Quinlan C.L., Gerencser A.A., Treberg J.R., Brand M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. Journal of Biological Chemistry. 2011;286:31361–31372. doi: 10.1074/jbc.M111.267898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunik V.I., Sievers C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. European Journal of Biochemistry. 2002;269:5004–5015. doi: 10.1046/j.1432-1033.2002.03204.x. [DOI] [PubMed] [Google Scholar]
- 13.Tretter L., Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. Journal of Neuroscience. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starkov A.A., Fiskum G., Chinopoulos C., Lorenzo B.J., Browne S.E., Patel M.S., Beal M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. Journal of Neuroscience. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan C.L., Orr A.L., Perevoshchikova I.V., Treberg J.R., Ackrell B.A., Brand M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. Journal of Biological Chemistry. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orr A.L., Quinlan C.L., Perevoshchikova I.V., Brand M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. Journal of Biological Chemistry. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seifert E.L., Estey C., Xuan J.Y., Harper M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 Emission during long-chain fatty acid oxidation. Journal of Biological Chemistry. 2010;285:5748–5758. doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.I.V. Perevoshchikova, C.L. Quinlan, A.L. Orr, M.D., Brand. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle. Free Radical Biology and Medicine 61, 2013, 298–309. [DOI] [PMC free article] [PubMed]
- 19.White T.A., Krishnan N., Becker D.F., Tanner J.J. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. Journal of Biological Chemistry. 2007;282:14316–14327. doi: 10.1074/jbc.M700912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forman J.H., Kennedy J. Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. Journal of Biological Chemistry. 1975;250:4322–4326. [PubMed] [Google Scholar]
- 21.Quinlan C.L., Treberg J.R., Perevoshchikova I.V., Orr A.L., Brand M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radical Biology and Medicine. 2012;53:1807–1817. doi: 10.1016/j.freeradbiomed.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Affourtit C., Quinlan C.L., Brand M.D. Measurement of proton leak and electron leak in isolated mitochondria. Methods in Molecular Biology. 2012;810:165–182. doi: 10.1007/978-1-61779-382-0_11. [DOI] [PubMed] [Google Scholar]
- 23.Treberg J.R., Quinlan C.L., Brand M.D. Hydrogen peroxide efflux from muscle mitochondria underestimates matrix superoxide production—a correction using glutathione depletion. FEBS Journal. 2010;277:2766–2778. doi: 10.1111/j.1742-4658.2010.07693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansford R.G., Hogue B.A., Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. Journal of Bioenergetics and Biomembrane. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 25.Lambert A.J., Brand M.D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochemical Journal. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proceedings of the National Academy of Sciences of the USA. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.St-Pierre J., Buckingham J.A., Roebuck S.J., Brand M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. Journal of Biological Chemistry. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 28.Muller F.L., Liu Y., Abdul-Ghani M.A., Lustgarten M.S., Bhattacharya A., Jang Y.C., Van Remmen H. High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochemical Journal. 2008;409:491–499. doi: 10.1042/BJ20071162. [DOI] [PubMed] [Google Scholar]
- 29.Zoccarato F., Cavallini L., Alexandre A. Succinate is the controller of O2−/H2O2 release at mitochondrial complex I: negative modulation by malate, positive by cyanide. Journal of Bioenergetics and Biomembranes. 2009;41:387–393. doi: 10.1007/s10863-009-9238-2. [DOI] [PubMed] [Google Scholar]
- 30.Zoccarato F., Cavallini L., Bortolami S., Alexandre A. Succinate modulation of H2O2 release at NADH:ubiquinone oxidoreductase (Complex I) in brain mitochondria. Biochemical Journal. 2007;406:125–129. doi: 10.1042/BJ20070215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicholls D.G., Ferguson S.J. Academic Press; London: 2002. Bioenergetics 3. [Google Scholar]
- 32.Miwa S., Brand M.D. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochimica Biophysica Acta. 2005;1709:214–219. doi: 10.1016/j.bbabio.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Muller F.L., Liu Y., Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. Journal of Biological Chemistry. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]