

Abstract
Nearly 30 years have passed since the discovery of xanthine oxidoreductase (XOR) as a critical source of reactive species in ischemia/reperfusion injury. Since then, numerous inflammatory disease processes have been associated with elevated XOR activity and allied reactive species formation solidifying the ideology that enhancement of XOR activity equates to negative clinical outcomes. However, recent evidence may shatter this paradigm by describing a nitrate/nitrite reductase capacity for XOR whereby XOR may be considered a crucial source of beneficial •NO under ischemic/hypoxic/acidic conditions; settings similar to those that limit the functional capacity of nitric oxide synthase. Herein, we review XOR-catalyzed reactive species generation and identify key microenvironmental factors whose interplay impacts the identity of the reactive species (oxidants vs. •NO) produced. In doing so, we redefine existing dogma and shed new light on an enzyme that has weathered the evolutionary process not as gadfly but a crucial component in the maintenance of homeostasis.
Abbreviations: GAGs, glycosaminoglycans; H2O2, hydrogen peroxide; I/R, ischemia/reperfusion; •NO, nitric oxide; NOS, nitric oxide synthase; O2•−, superoxide; ROS, reactive oxygen species; XDH, xanthine dehydrogenase; XO, xanthine oxidase; XOR, xanthine oxidoreductase)
Keywords: Free radicals, Hypoxia, Inflammation, Nitric oxide, Nitrite, Oxygen tension, Xanthine oxidoreductase
Highlights
-
•
Inflammation-induced elevation of XO has long been associated with negative outcomes.
-
•
Yet, XO-derived reactive species generation is poorly understood leading to misconceptions.
-
•
For example, H2O2 and not O2•− is the major reactive product of XO.
-
•
And, recent reports demonstrate beneficial •NO production by XO and nitrite.
-
•
As such, a detailed reevaluation of XO-catalyzed reactive species generation is crucial.
Introduction
Xanthine oxidoreductase (XOR) is a molybdoflavin enzyme that catalyzes the terminal two reactions in purine degradation in primates; oxidation of hypoxanthine to xanthine and the subsequent oxidation of xanthine to uric acid. XOR is a homodimer of ~300 kD with each subunit consisting of four redox centers: a molybdenum cofactor (Mo-co), one FAD site and two Fe/S clusters, Fig. 1. The Mo-co is the site of purine oxidation while NAD+ and O2 reduction occur at the FAD. The two Fe/S clusters provide the conduit for electron flux between the Mo-co and the FAD [1–3]. The enzyme is transcribed as a single gene product, xanthine dehydrogenase (XDH) where substrate-derived electrons reduce NAD+ to NADH, Fig. 1A. However, during inflammatory conditions, oxidation of key cysteine residues (535 and 992) and/or limited proteolysis converts XDH to xanthine oxidase (XO) [4]. In the oxidase form, affinity for NAD+ at the FAD is greatly decreased while affinity for oxygen is significantly enhanced resulting in univalent and divalent electron transfer to O2 generating O2•− and hydrogen peroxide (H2O2), respectively, Fig. 1B [5]. This capacity to reduce O2 led to XOR being identified as the first source of biological O2•− formation and subsequently as a significant source of reactive species mediating ischemia/reperfusion injury [6,7]. As the redox field progressed, several additional enzymatic and non-enzymatic sources of free radicals and reactive species have been identified yet, to date, XOR remains the most pharmacologically targetable thus incentivizing extensive exploration of inhibition strategies to address disease processes where elevated rates of reactive species formation are contributory.
Fig. 1.
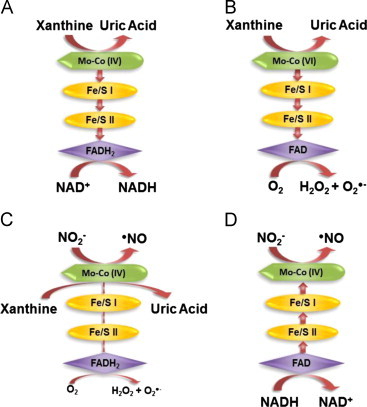
XOR-Catalyzed Reactions. (A) For XDH, xanthine is oxidized to uric acid and electrons transferred via 2 Fe/S centers to the FAD where NAD+ is reduced to NADH. (B) For XO, xanthine is oxidized to uric acid and electrons are transferred to the FAD where O2 is reduced to O2•− and H2O2. Under normal O2 tension and pH the Mo-co would reside more often in the oxidized +6 (VI) valence as electrons are rapidly transferred to O2 at the FAD. (C) Nitrite (NO2−) undergoes a 1 electron reduction to •NO at the Mo-cofactor of XO (electrons are donated directly to Mo by xanthine). (D) NO2− is reduced to •NO at the of XO (electrons are supplied by NADH and transferred retrograde reducing the Mo). Under low O2 tensions and pH the Mo-co would reside more often in the reduced +4 (IV) valence as electrons are more slowly transferred to O2. This decrease in electron flux from the Mo-co to the FAD is depicted in (C) as diminished arrows whereas in (D) NADH-mediated electron donation at the FAD is out-competing O2-mediated electron withdrawal and thus the arrows are reversed indicating flux from the FAD to the Mo-co.
Oxidant formation
Most reports refer to XO as a source of O2•− and assume H2O2 formation is a result of spontaneous dismutation of O2•−. This premise is completely invalid as attainment of 100% O2•− generation requires XO turnover at pH 10.0 in an environment of 100% O2 [8]. However, under room air and pH 7.4, XO transfers over 72% of its substrate-derived electrons to O2 divalently to generate H2O2 and thus 28% to O2•− formation. This observation is critically important as it clearly demonstrates that, under conditions approaching those encountered in vivo, H2O2 is the major reactive product of XO-catalyzed O2 reduction [8,9]. The prime determinate of the relative quantities of O2•− and H2O2 generated by XO is O2 tension. For example, at pH 7.4 and 10% O2 XO generates ~26% O2•− and thus ~74% H2O2 whereas at 1% O2, XO forms ~90% H2O2 and only ~10% O2•−, Fig. 2 [9]. In addition to O2 tension, pH and purine concentration also play a significant role in divalent versus univalent electron transfer to O2. The reaction of hypoxanthine/xanthine at the Mo-co of XO is based-catalyzed with a pH optimum of 8.9 and a Km=~6.5 µM. Under normal physiologic conditions, hypoxanthine + xanthine levels in humans are ~1–3 µM; however, under hypoxic/inflammatory conditions these levels have been reported as high as 50–100 µM while pH concomitantly drops below 7.0 [10–12]. When this occurs, total purine (hypoxanthine+xanthine) concentration is well above the Km and thus will not significantly impact either rates of electron deposition at the Mo-co or resultant transfer to the FAD. However, acidic pH will significantly retard purine–Mo-co reaction thereby reducing the electron flux rate which favors divalent transfer to O2 to generate H2O2. Therefore, under ischemic and/or hypoxic conditions, where both O2 levels and pH are reduced, H2O2 formation is favored suggesting that XO activity may be influential in numerous signaling cascades where H2O2 has been noted to participate. However, this hypoxia-mediated proclivity for H2O2 production cannot overshadow the fact that rates of O2•− formation by XO under these same conditions are sufficient to mediate alterations in vascular function by reducing •NO bioavailability via direct reaction (•NO + O2•−→ONOO−) [13–15].
Fig. 2.
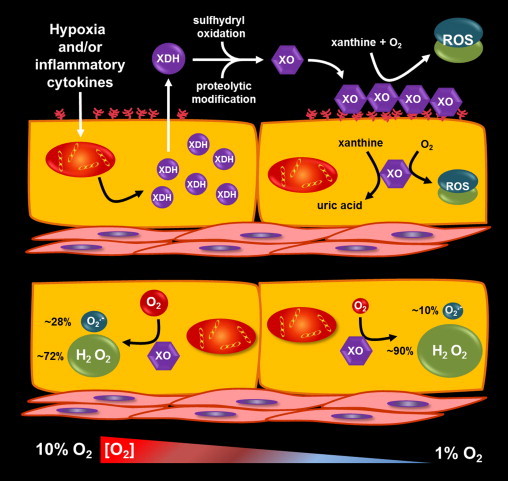
Hypoxic/inflammatory induction of XOR and vascular consequences. (Top) Inflammatory cytokines and/or hypoxia induce XDH transcription and resultant protein expression. In vascular endothelial cells XDH is exported to the circulation where it is rapidly converted to XO by plasma proteases. However, cellular export is not requisite for XDH conversion to XO as enhanced oxidative stress within the endothelium can induce oxidation of critical cysteine residues that mediate reversible conversion to XO. Once in the circulation, negatively charged glycosaminoglycans (GAGs) on the luminal surface of the endothelium bind and sequester XO by high affinity (Kd=6 nM) interaction with pockets of cationic amino acids on the surface of the enzyme. This sequestration amplifies local XO levels creating a vascular milieu whereby, in the presence of hypoxanthine and/or xanthine, enhanced rates of O2•− and H2O2 formation ensue. (Bottom) A key determinate regulating the relative amounts of O2•− and H2O2 generated by XO is the concentration of molecular O2. Shown is a cartoon representing the change in relative percentages of O2•− and H2O2 formed by XO at 10% O2 (~130 µM O2) compared to 1% O2 (~13 µM O2). This range of O2 tension is critically important as it represents from well above to 50% below the Km-O2 at the FAD-cofactor of 27 µM or ~2% O2. As the O2 tension drops below this Km value the FAD-cofactor assumes more time in the fully reduced FADH2 state where, upon reaction with O2, divalent electron transfer is preferred. This process assumes constant electron from the Mo-co (e.g. [hypoxanthine+xanthine] above the 6.5 µM Km at the Mo-co) which would be expected under conditions similar to those encountered in the lumen of an ischemic/hypoxic vessel. In addition, it is critical to note that XO–GAG association as well as acidic pH serves to further favor H2O2 formation. Taken together, moderate to severe hypoxia induces XDH expression, export and conversion to XO that is subsequently captured by GAGs in an environment the primed for catalyzing the formation of H2O2 as well as a little O2•−.
While the post-translational conversion of XDH to XO has become synonymous with conversion from a source of reducing equivalents to a source of reactive oxygen species (ROS), it is important to recognize that under certain circumstances XDH effectively reduces O2 to generate ROS. Although NAD+ is the preferred electron acceptor for XDH, when levels of this substrate are low XDH will utilize O2 [16]. These conditions include hypoxia either localized, regional or systemic where O2-dependent alterations in cellular respiration lead to decreased mitochondrial NADH oxidation and thus significant diminution of NAD+ levels [17]. This being said, care should be taken not to exclusively associate XDH with the form of XOR that does not produce ROS.
XO-endothelial interaction
In humans, XOR is ubiquitously expressed with the liver and intestines displaying the highest specific activity [18]. Hypoxia as well as inflammatory cytokines (TNF-α, IL-1β, IFN-γ), induce XDH expression in tissues and vascular endothelial cells where it is released to the circulation, Fig. 2 [18,19]. Circulating XDH is rapidly (<1 min) converted to XO where it avidly binds to negatively charged glycosaminoglycans (GAGs) on the apical surface of vascular endothelial cells [20,21]. This XO–endothelium interaction is exemplified in animal models and clinical studies of cardiovascular disease where intravenous administration of heparin results in a substantive increase in plasma XO activity, suggesting heparin-mediated mobilization of XO from vascular endothelial GAGs [21–23]. While XO exhibits a net negative charge at physiological pH, pockets of cationic amino acid motifs on the surface of the protein result in high affinity for GAGs (Kd=6 nM) [21,24,25]. Binding to and sequestration of XO on GAGs: (1) amplifies local XO concentration and subsequent ROS generation; (2) alters XO kinetic properties further shifting oxidant formation from O2•− to H2O2 and (3) confers significant resistance to inhibition from the pyrazalopyrimidine-based inhibitors, allo/oxypurinol [26]. For example, when compared to XO in free in solution, XO–GAG association decreases substrate binding affinity and thus: (1) increases the Km for xanthine over 3-fold (6.5→21.2 µM); (2) reduces O2•− production by 34% favoring H2O2 formation and (3) induces a 5-fold increase in the Ki for allo/oxypurinol (85→451 nM) [26]. Taken together, inflammation-mediated up-regulation of XDH, export to the circulation, rapid conversion to XO and sequestration by the endothelium coalesce to generate a vascular milieu favoring increased rates of reactive species generation that can participate in mediating the loss of homeostasis, Fig. 2. This deleterious action of XO has been noted in various reports of vascular and cardiopulmonary diseases including heart failure, chronic obstructive pulmonary disease (COPD), pulmonary hypertension, sickle cell disease and Type I and II diabetes [14,27–30].
XOR knockouts and inhibition strategies
For an enzyme whose activity was described in 1889 followed by it being named xanthine oxidase in 1901 and first purified in 1939, surprisingly little detail is known regarding its regulation and subsequent interplay with biomolecular pathways when compared to other enzymes with much more recent history [31]. Potential reasons for this discrepancy in understanding include: (1) lethality of global XDH knockouts; (2) absence of reports utilizing tissue-specific conditional knockouts; (3) side-effects resulting from pharmacological knockdown with tungsten supplementation; (4) promiscuity of the active site resulting in ambiguity regarding both substrate identity and inhibition by compounds designed to specifically target other molybdopterin enzymes and (5) resistance to inhibition by allo/oxypurinol conferred by binding to vascular endothelial GAGs. Attempts to establish homozygous knockouts of XDH in mice have resulted in the death of pups before 30 days of age due to kidney fibrosis and failure attributed to excessive hypoxanthine deposition [32,33]. Similar effects were obtained with heterozygous XDH knockouts where both nutrient absorption and kidney failure resulted in death in a similar timeframe as XDH−/−. These unfortunate side-effects have relegated investigators to utilize allo/oxypurinol-based inhibition or global XOR knockdown with dietary tungsten (W) supplementation for proof-of-principle experimentation. Dietary supplementation with sodium tungstate (NaW) results in replacement of the active site Mo with W producing an enzyme that is inactive with respect to hypo/xanthine oxidation to uric acid. However, it is important to note that W-mediated inactivation of the Mo-co does not affect the capacity of the FAD in XOR to be reduced by NADH and subsequently react with and reduce O2 to produce O2•− and H2O2. In addition, treatment with NaW also inactivates other members of the molybdopterin family including aldehyde oxidase, sulfite oxidase and mitochondrial amidoxime reducing component 1 (MARC1) which can lead to significant ambiguity regarding interpretation of results. On the other hand, inhibition of XOR with allo/oxypurinol is also not optimal as: (1) allopurinol can mediate effects on other purine catabolic pathways including those resulting in alteration of adenosine levels [34]; (2) reaction of allopurinol with XO induces enzyme turnover resulting in O2•− and H2O2 formation [35] and (3) plasma allo/oxypurinol concentrations (100–400 µM) well above those tolerated clinically (30–90 µM) are incapable of fully inhibiting XO when it is sequestered by vascular GAGs [26,35]. As a result of these limitations we have recently identified febuxostat (Uloric) to be more optimal for exploring contributions of XOR both in vivo and tissue culture. For example, febuxostat concentrations (25–50 nM) well below the reported plasma Cmax (15 µM) for the clinic demonstrate over 3 orders of magnitude greater potency than allopurinol (Ki=0.9 nM vs. 1.6 µM), are not affected by XOR-vascular GAG association and do not alter other purine catabolism pathways [34,35]. In toto, these findings clearly demonstrate the potential benefit of using febuxostat to interrogate XOR-dependence in various experimental models.
XOR-catalyzed •NO production
For decades, the dogma in the field has been as described above; specifically that inflammation/hypoxia-induced enhancement of XO activity equates to elevated rates of XO-derived ROS generation, propagation/exacerbation of the disease process and ultimately poor clinical outcomes. This correlation has been substantiated in several disease models where XO inhibition leads to a reduction in symptoms and measurable restoration of function. However, recent reports have posed a bold challenge to the standing paradigm by demonstrating a nitrate/nitrite (NO2−) reductase function for XOR (1e− reduction of NO2− to •NO) suggesting XOR to be a source of beneficial •NO under these same hypoxic/inflammatory conditions. In essence, these observations directly countervail a substantive body of literature indicating XO inhibition to be beneficial and as such affirm the need to more closely interrogate XOR-catalyzed reactions and potential factors that alter product formation. For example, reduction of NO2− to •NO is indeed catalyzed by purified XO under anoxic conditions when electrons are supplied by either xanthine or NADH [36–38]. Nitrite reduction occurs at the reduced Mo-co (Mo-co IV) and electrons driving this reaction can be supplied directly by xanthine (Fig. 1C) or indirectly by NADH via electron donation at the FAD with subsequent retrograde flow to the Mo-co, Fig. 1D [39]. At this point, it is critical to note that work with the purified enzyme has revealed two issues requiring resolution before the biological relevance of XOR-derived •NO can be substantiated. First, the NO2− reductase activity of XOR is inhibited by O2 which results from oxidation of the Mo-co mediated by electron withdrawal from the FAD [40]. Second, the affinity for NO2− at the Mo-co of XOR is 3 orders of magnitude less than for xanthine (Km-NO2−=2.5 mM vs. Km-xanthine=6.5 µM) [38]. Despite these formidable issues, several reports demonstrate significant reduction in or ablation of salutary outcomes attributable to NO2− treatment upon inhibition of XOR activity with allo/oxypurinol affirming the need for more vigorous investigation to fully elucidate this reductive process. For example, systemic inhibition of XOR activity has diminished protective effects of NO2− treatment in models of intimal hyperplasia [41], lung injury [42,43], myocardial infarction [44], pulmonary hypertension [45] and ischemia/reperfusion (I/R)-induced damage [46–49]. It is also important to note that plasma levels of NO2− are reported to be enhanced in an XOR-dependent manner by treatment with nitrate (NO3−) where XOR serves first as a NO3− reductase (NO3−+1e−→NO2−) and ultimately a NO2− reductase (NO2−+1e−→•NO). This XO-catalyzed process was described over 50 years ago [50] and recently expanded to in vivo models [51]. In these experiments treatment of germ-free mice (void of bacterial NO3− reductases) with NO3− resulted in elevation of plasma NO2− levels that were not observed when mice received co-treatment with allopurinol and thus are consistent with previous biochemical reports demonstrating NO3− reductase activity for XOR [52]. In the aggregate, there is a new body of evidence suggesting a protective role for XOR under hypoxic and inflammatory conditions in the presence of elevated levels of NO2−, summarized in Fig. 3. However, several key issues remain unclear regarding the microenvironmental conditions necessary for operative and biologically relevant nitrite reductase activity of XOR in vivo and were recently extensively reviewed [53].
Fig. 3.
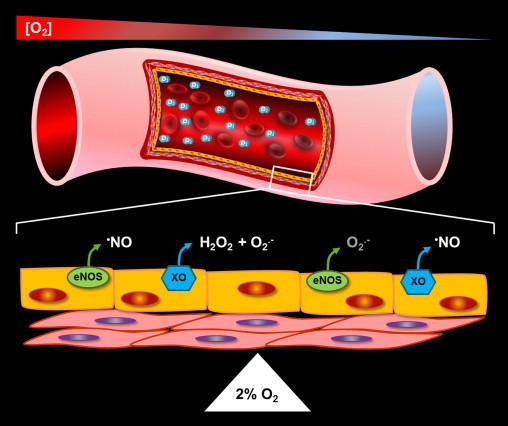
Hypoxic conversion of XOR from oxidant to •NO production. Hypoxia mediates the alteration of microenvironmental factors that coalesce to both facilitate the conversion of XO from oxidant to •NO production and diminish the capacity of eNOS to catalyze •NO formation as well as enhance its potential to uncouple and produce O2•−. These factors include: (1) acidic pH; (2) elevation of NADH levels; (3) oxidation of biopterin and, of course; (4) diminution of O2 tension. Nitrite reduction at the Mo-co of XO is acid catalyzed with a pH optimum ~6–6.5 while xanthine oxidation the Mo-co is base catalyzed with a pH optimum=8.9. Therefore, lower pH confers a reduction in affinity for xanthine while increasing affinity for NO2−. Thus, lower pH results in an environmental setting more favorable for NO2− to compete with xanthine for the Mo-co. This shift in affinity away from xanthine and toward NO2− is further augmented by reduction in O2 tension to values below the Km-O2 at the FAD (27 µM or ~2% O2). Once this occurs, electron withdrawal from the FAD slows resulting in the Mo-co assuming a more reduced state (Mo-co IV, see Fig. 1C and D) which is crucial for two reasons: (1) NO2− reduction requires a reduced Mo-co and (2) xanthine oxidation requires an oxidized Mo-co. Therefore, O2 tensions at or below 2% further assist the ability of NO2− to compete with xanthine for reaction at the Mo-co. As seen in Fig. 1D, hypoxia-mediated elevation of NADH levels can also further augment the potential for NO2− reduction at the Mo-co by competing with O2 for reaction at the FAD. In this case, NADH-FAD reaction results in reduction of the FAD to FADH2 inducing Mo-co reduction by retrograde electron flux as well as inhibition of O2-mediated electron withdrawal. On the other hand, this same inflammatory setting negatively impacts •NO formation by eNOS. For example, as O2 tensions fall below 2% (27 µM): (1) O2 becomes limiting as a substrate for eNOS-catalyzed •NO production where the Km-O2 for eNOS=23 µM and (2) acid pH coupled with elevated levels of oxidants drive eNOS uncoupling and the propensity for eNOS-mediated O2•− generation (depicted above the cell on the right in small font). Taken together, diminution of O2 tension, acidic pH, elevation of NADH levels, and oxidation of biopterin converge to generate an environment whereby the burden for •NO production shifts from eNOS to XOR. Furthermore, the critical O2 concentration where this shift or “switch” is triggered is assumed to be near 2% where the Km-O2 values for both XO and eNOS collide (depicted by a pivot point in the cartoon). However, it is crucial to note that if this process is to be of biological relevance then: (1) NO2− and/or NO3− levels must be significantly elevated by dietary or pharmacologic supplementation and (2) the proposed interplay between the components of these concerted reactions must be vigorously pursued and validated.
Although XOR has been studied for 114 years, it is clear from the information provided herein that we have only begun to understand the complexity regarding the interplay between crucial microenvironmental factors and the identity/generation of XOR-derived reactive products as well as their impact on cellular signaling both in normal and pathophysiology. Suffice it to say the long-standing dogma identifying XDH as a housekeeping enzyme and XO as a mediator of negative clinical outcomes is beginning to crumble as we uncover new roles for XOR in the network of adaptive responses that serve to maintain homeostasis.
Acknowledgments
This work was supported by AHA National Scientist Development Grant 10SDG3560005 and University of Pittsburgh, Department of Anesthesiology Development Grant (EEK).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Enroth C., Eger B.T., Okamoto K., Nishino T., Nishino T., Pai E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proceedings of the National Academy of Sciences USA. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishino T., Okamoto K. The role of the [2Fe–2s] cluster centers in xanthine oxidoreductase. Journal of Inorganic Biochemistry. 2000;82:43–49. doi: 10.1016/s0162-0134(00)00165-3. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki T., Okamoto K., Nishino T., Mizushima J., Hori H. Sequence motif-specific assignment of two [2Fe–2S] clusters in rat xanthine oxidoreductase studied by site-directed mutagenesis. Journal of Biochemistry. 2000;127:771–778. doi: 10.1093/oxfordjournals.jbchem.a022669. [DOI] [PubMed] [Google Scholar]
- 4.Kuwabara Y., Nishino T., Okamoto K., Matsumura T., Eger B.T., Pai E.F., Nishino T. Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proceedings of the National Academy of Sciences USA. 2003;100:8170–8175. doi: 10.1073/pnas.1431485100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris C.M., Massey V. The oxidative half-reaction of xanthine dehydrogenase with NAD; reaction kinetics and steady-state mechanism. Journal of Biological Chemistry. 1997;272:28335–28341. doi: 10.1074/jbc.272.45.28335. [DOI] [PubMed] [Google Scholar]
- 6.Granger D.N., Rutili G., McCord J.M. Superoxide radicals in feline intestinal ischemia. Gastroenterology. 1981;81:22–29. [PubMed] [Google Scholar]
- 7.McCord J.M., Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) Journal of Biological Chemistry. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 8.Fridovich I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. Journal of Biological Chemistry. 1970;245:4053–4057. [PubMed] [Google Scholar]
- 9.Kelley E.E., Khoo N.K., Hundley N.J., Malik U.Z., Freeman B.A., Tarpey M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radical Biology & Medicine. 2010;48:493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lartigue-Mattei C., Chabard J.L., Bargnoux H., Petit J., Berger J.A., Ristori J.M., Bussiere J.L., Catilina P., Catilina M.J. Plasma and blood assay of xanthine and hypoxanthine by gas chromatography–mass spectrometry: physiological variations in humans. Journal of Chromatography. 1990;529:93–101. doi: 10.1016/s0378-4347(00)83810-4. [DOI] [PubMed] [Google Scholar]
- 11.Pesonen E.J., Linder N., Raivio K.O., Sarnesto A., Lapatto R., Hockerstedt K., Makisalo H., Andersson S. Circulating xanthine oxidase and neutrophil activation during human liver transplantation. Gastroenterology. 1998;114:1009–1015. doi: 10.1016/s0016-5085(98)70321-x. [DOI] [PubMed] [Google Scholar]
- 12.Quinlan G.J., Lamb N.J., Tilley R., Evans T.W., Gutteridge J.M. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. American Journal of Respiratory and Critical Care Medicine. 1997;155:479–484. doi: 10.1164/ajrccm.155.2.9032182. [DOI] [PubMed] [Google Scholar]
- 13.Aslan M., Freeman B.A. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease—mechanisms and consequences. Cellular and Molecular Biology (Noisy.-le-grand) 2004;50:95–105. [PubMed] [Google Scholar]
- 14.Aslan M., Ryan T.M., Adler B., Townes T.M., Parks D.A., Thompson J.A., Tousson A., Gladwin M.T., Patel R.P., Tarpey M.M., Batinic-Haberle I., White C.R., Freeman B.A. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proceedings of the National Academy of Sciences USA. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White C.R., Darley-Usmar V., Berrington W.R., McAdams M., Gore J.Z., Thompson J.A., Parks D.A., Tarpey M.M., Freeman B.A. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proceedings of the National Academy of Sciences USA. 1996;93:8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris C.M., Massey V. The reaction of reduced xanthine dehydrogenase with molecular oxygen. Reaction kinetics and measurement of superoxide radical. Journal of Biological Chemistry. 1997;272:8370–8379. doi: 10.1074/jbc.272.13.8370. [DOI] [PubMed] [Google Scholar]
- 17.Zhou L., Stanley W.C., Saidel G.M., Yu X., Cabrera M.E. Regulation of lactate production at the onset of ischaemia is independent of mitochondrial NADH/NAD+: insights from in silico studies. Journal of Physiology. 2005;569:925–937. doi: 10.1113/jphysiol.2005.093146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radical Biology & Medicine. 2002;33:774–797. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 19.Kelley E.E., Hock T., Khoo N.K., Richardson G.R., Johnson K.K., Powell P.C., Giles G.I., Agarwal A., Lancaster J.R., Jr., Tarpey M.M. Moderate hypoxia induces xanthine oxidoreductase activity in arterial endothelial cells. Free Radical Biology & Medicine. 2006;40:952–959. doi: 10.1016/j.freeradbiomed.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Parks D.A., Williams T.K., Beckman J.S. Conversion of xanthine dehydrogenase to oxidase in ischemic rat intestine: a reevaluation. American Journal of Physiology. 1988;254:G768–G774. doi: 10.1152/ajpgi.1988.254.5.G768. [DOI] [PubMed] [Google Scholar]
- 21.Houston M., Estevez A., Chumley P., Aslan M., Marklund S., Parks D.A., Freeman B.A. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. Journal of Biological Chemistry. 1999;274:4985–4994. doi: 10.1074/jbc.274.8.4985. [DOI] [PubMed] [Google Scholar]
- 22.Granell S., Gironella M., Bulbena O., Panes J., Mauri M., Sabater L., Aparisi L., Gelpi E., Closa D. Heparin mobilizes xanthine oxidase and induces lung inflammation in acute pancreatitis. Critical Care Medicine. 2003;31:525–530. doi: 10.1097/01.CCM.0000049948.64660.06. [DOI] [PubMed] [Google Scholar]
- 23.Landmesser U., Spiekermann S., Dikalov S., Tatge H., Wilke R., Kohler C., Harrison D.G., Hornig B., Drexler H. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation. 2002;106:3073–3078. doi: 10.1161/01.cir.0000041431.57222.af. [DOI] [PubMed] [Google Scholar]
- 24.Fukushima T., Adachi T., Hirano K. The heparin-binding site of human xanthine oxidase. Biological & Pharmaceutical Bulletin. 1995;18:156–158. doi: 10.1248/bpb.18.156. [DOI] [PubMed] [Google Scholar]
- 25.Adachi T., Fukushima T., Usami Y., Hirano K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochemical Journal. 1993;289:523–527. doi: 10.1042/bj2890523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley E.E., Trostchansky A., Rubbo H., Freeman B.A., Radi R., Tarpey M.M. Binding of xanthine oxidase to glycosaminoglycans limits inhibition by oxypurinol. Journal of Biological Chemistry. 2004;279:37231–37234. doi: 10.1074/jbc.M402077200. [DOI] [PubMed] [Google Scholar]
- 27.Butler R., Morris A.D., Belch J.J., Hill A., Struthers A.D. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. doi: 10.1161/01.hyp.35.3.746. [DOI] [PubMed] [Google Scholar]
- 28.Desco M.C., Asensi M., Marquez R., Martinez-Valls J., Vento M., Pallardo F.V., Sastre J., Vina J. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–1124. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 29.Farquharson C.A., Butler R., Hill A., Belch J.J., Struthers A.D. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106:221–226. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- 30.Heunks L.M.A., Vina J., van Herwaarden C.L.A., Folgering H.T.M., Gimeno A., Dekhuijzen P.N.R. Xanthine oxidase is involved in exercise-induced oxidative stress in chronic obstructive pulmonary disease. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology. 1999;277:R1697–R1704. doi: 10.1152/ajpregu.1999.277.6.R1697. [DOI] [PubMed] [Google Scholar]
- 31.Ball E.G. Xanthine oxidase: purification and properties. Journal of Biological Chemistry. 1939;128:51–67. [Google Scholar]
- 32.Cheung K.J., Tzameli I., Pissios P., Rovira I., Gavrilova O., Ohtsubo T., Chen Z., Finkel T., Flier J.S., Friedman J.M. Xanthine oxidoreductase is a regulator of adipogenesis and PPARgamma activity. Cell Metabolism. 2007;5:115–128. doi: 10.1016/j.cmet.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Ohtsubo T., Rovira I.I., Starost M.F., Liu C., Finkel T. Xanthine oxidoreductase is an endogenous regulator of Cyclooxygenase-2. Circulation Research. 2004;95:1118–1124. doi: 10.1161/01.RES.0000149571.96304.36. [DOI] [PubMed] [Google Scholar]
- 34.Takano Y., Hase-Aoki K., Horiuchi H., Zhao L., Kasahara Y., Kondo S., Becker M.A. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sciences. 2005;76:1835–1847. doi: 10.1016/j.lfs.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 35.Malik U.Z., Hundley N.J., Romero G., Radi R., Freeman B.A., Tarpey M.M., Kelley E.E. Febuxostat inhibition of endothelial-bound XO: implications for targeting vascular ROS production. Free Radical Biology & Medicine. 2011;51:179–184. doi: 10.1016/j.freeradbiomed.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Godber B.L.J., Doel J.J., Sapkota G.P., Blake D.R., Stevens C.R., Eisenthal R., Harrison R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. Journal of Biological Chemistry. 2000;275:7757–7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 37.Millar T.M., Stevens C.R., Benjamin N., Eisenthal R., Harrison R., Blake D.R. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Letters. 1998;427:225–228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 38.Li H., Samouilov A., Liu X., Zweier J.L. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide gereration in anoxic tissues. Journal of Biological Chemistry. 2001;276:24482–24489. doi: 10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- 39.Maia L.B., Moura J.J. Nitrite reduction by xanthine oxidase family enzymes: a new class of nitrite reductases. Journal of Biological Inorganic Chemistry. 2011;16:443–460. doi: 10.1007/s00775-010-0741-z. [DOI] [PubMed] [Google Scholar]
- 40.Li H., Samouilov A., Liu X., Zweier J.L. Characterization of the effects of oxygen on xanthine oxidase-mediated nitric oxide formation. Journal of Biological Chemistry. 2004;279:16939–16946. doi: 10.1074/jbc.M314336200. [DOI] [PubMed] [Google Scholar]
- 41.Alef M.J., Vallabhaneni R., Carchman E., Morris S.M., Jr., Shiva S., Wang Y., Kelley E.E., Tarpey M.M., Gladwin M.T., Tzeng E., Zuckerbraun B.S. Nitrite-generated NO circumvents dysregulated arginine/NOS signaling to protect against intimal hyperplasia in Sprague–Dawley rats. Journal of Clinical Investigation. 2011;121:1646–1656. doi: 10.1172/JCI44079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samal A.A., Honavar J., Brandon A., Bradley K.M., Doran S., Liu Y., Dunaway C., Steele C., Postlethwait E.M., Squadrito G.L., Fanucchi M.V., Matalon S., Patel R.P. Administration of nitrite after chlorine gas exposure prevents lung injury: effect of administration modality. Free Radical Biology & Medicine. 2012;53:1431–1439. doi: 10.1016/j.freeradbiomed.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pickerodt P.A., Emery M.J., Zarndt R., Martin W., Francis R.C., Boemke W., Swenson E.R. Sodium nitrite mitigates ventilator-induced lung injury in rats. Anesthesiology. 2012;117:592–601. doi: 10.1097/ALN.0b013e3182655f80. [DOI] [PubMed] [Google Scholar]
- 44.Baker J.E., Su J., Fu X., Hsu A., Gross G.J., Tweddell J.S., Hogg N. Nitrite confers protection against myocardial infarction: role of xanthine oxidoreductase, NADPH oxidase and K(ATP) channels. Journal of Molecular and Cellular Cardiology. 2007;43:437–444. doi: 10.1016/j.yjmcc.2007.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuckerbraun B.S., Shiva S., Ifedigbo E., Mathier M.A., Mollen K.P., Rao J., Bauer P.M., Choi J.J., Curtis E., Choi A.M., Gladwin M.T. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation. 2010;121:98–109. doi: 10.1161/CIRCULATIONAHA.109.891077. [DOI] [PubMed] [Google Scholar]
- 46.Sugimoto R., Okamoto T., Nakao A., Zhan J., Wang Y., Kohmoto J., Tokita D., Farver C.F., Tarpey M.M., Billiar T.R., Gladwin M.T., McCurry K.R. Nitrite reduces acute lung injury and improves survival in a rat lung transplantation model. American Journal of Transplantation. 2012;12:2938–2948. doi: 10.1111/j.1600-6143.2012.04169.x. [DOI] [PubMed] [Google Scholar]
- 47.Tripatara P., Patel N.S., Webb A., Rathod K., Lecomte F.M., Mazzon E., Cuzzocrea S., Yaqoob M.M., Ahluwalia A., Thiemermann C. Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. Journal of the American Society of Nephrology. 2007;18:570–580. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 48.Webb A., Bond R., McLean P., Uppal R., Benjamin N., Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia–reperfusion damage. Proceedings of the National Academy of Sciences USA. 2004;101:13683–13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu P., Liu F., Yao Z., Wang C.Y., Chen D.D., Tian Y., Zhang J.H., Wu Y.H. Nitrite-derived nitric oxide by xanthine oxidoreductase protects the liver against ischemia–reperfusion injury. Hepatobiliary & Pancreatic Diseases International. 2005;4:350–355. [PubMed] [Google Scholar]
- 50.Huang L., Borniquel S., Lundberg J.O. Enhanced xanthine oxidoreductase expression and tissue nitrate reduction in germ free mice. Nitric Oxide. 2010;22:191–195. doi: 10.1016/j.niox.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 51.Fridovich I. and Handler P., Xanthine oxidase. V. Differential inhibition of the reduction of various electron acceptors., J Biol Chem., 237:916–21, 1962. [PubMed]
- 52.Millar T.M., Stevens C.R., Benjamin N., Eisenthal R., Harrison R., Blake D.R. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Letters. 1998;427:225–228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 53.Cantu-Medellin N., Kelley E.E. Xanthine oxidoreductase-catalyzed reduction of nitrite to nitric oxide: insights regarding where, when and how. Nitric Oxide. 2013 doi: 10.1016/j.niox.2013.02.081. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]