

Abstract
The Nuclear factor-2 erythroid related factor-2 (Nrf2) is a redox regulated transcription factor involved in the regulation of antioxidant defence systems. It drives the production of endogenous antioxidant defences and detoxifying enzymes. Nuclear factor-kappa light chain enhancer of B cells (NF-κB) is a transcription factor, involved in proinflammatory cytokine production, in addition to its immunological function. Both Nrf2 and NF-κB regulation are co-ordinated in order to maintain redox homeostasis in healthy cells. However, during pathological conditions this regulation is perturbed offering an opportunity for therapeutic intervention. Diabetic neuropathy is a condition, in which change in expression pattern of Nrf2 and NF-κB has been reported. This review aims to focus on the role of the Nrf2 and NF-κB in diabetic neuropathy and summarizes the therapeutic outcomes of various pharmacological modulators targeted at the Nrf2–NF-κB axis in diabetic neuropathy.
Keywords: Nrf2, NF-κB, Diabetic neuropathy
Highlights
-
•
Hyperglycaemia-induced changes in NF-κB and Nrf2 activity of Nrf2–NF-κB axis contributes to the pathophysiology of diabetic neuropathy.
-
•
Crosstalk between the Nrf2 and NF-κB pathways can induce neuronal damage via oxidative stress and neuroinflammation.
-
•
Simultaneous targeting of Nrf2 and NF-κB may be beneficial in diabetic neuropathy.
Introduction
Diabetes is one of the most debilitating conditions in patients affecting a substantial proportion of the world's population. Diabetes can predispose an individual to metabolic, cardiovascular disturbances and obesity, and these pathologies are accompanied by vascular complications [1]. Hyperglycaemia-induced damage to the endothelial cells results in micro-vascular complications of the diabetes such as diabetic neuropathy, nephropathy and retinopathy and macro-vascular complications such as cardiomyopathy [2]. Diabetic neuropathy remains the most severe form of complication affecting 40–50% of people with both types of diabetes. The clinical features of diabetic neuropathy range from sensory deficit to allodynia and hyperalgesia. Diabetic neuropathy arises from the long term effects of hyperglycaemia induced damage to peripheral nervous tissue as well as the vasa nervorum [3].
The current knowledge of pathophysiological mechanisms of hyperglycaemia-induced diabetic neuropathy is substantial and recent advances made in field could lead to the development of some novel therapeutic strategies targeted at advance glycation end products (AGE), sorbitol accumulation, protein kinase C (PKC) activation and hexosamine pathway. The axis of pathophysiological factors responsible for diabetes and diabetic neuropathy converge at two of the most extensively studied pathways, oxidative–nitrosative stress and neuroinflammation (Fig. 1). Molecular studies have revealed the involvement of transcriptional regulators such as Nrf2-Keap1 and the NF-κB inflammatory cascade in the pathophysiology of many diseases [4].
Fig. 1.
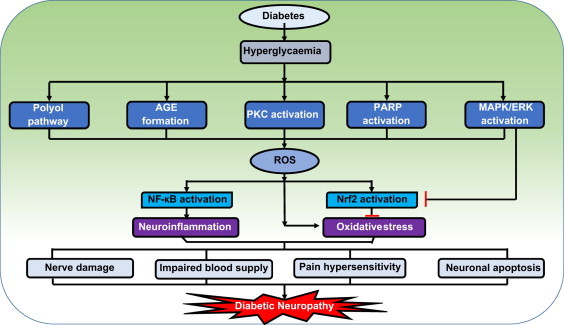
Possible factors in the pathophysiology of diabetic neuropathy. Hyperglycaemia induces excess formation of sorbitol through polyol pathway, advanced glycation end products (AGE), mitochondrial dysfunction and causes mitogen activated protein kinases (MAPK), poly ADP ribosyl polymerase (PARP) and protein kinase C (PKC) hyper-activation [7]. All these pathways can contribute to nitrosative/oxidative stress in neuronal cells and endothelial cells of the vasa nervorum through depletion of endogenous antioxidant defences and excess generation of reactive oxygen species (ROS). The resulting oxidative stress leads to the activation of redox regulated transcription factors such as nuclear factor erythroid 2 related factor-2 (Nrf2), nuclear factor kappa light chain enhancer of B cells (NF-κB). Although Nrf2 is transiently activated by oxidative stress, the hyperglycaemic stress induced extracellular related kinase (ERK) activation restrain continued Nrf2 activation [8]. A decline in Nrf2 activity and a persistent increase in NF-κB activity can lead to neuroinflammation and increased nitrosative–oxidative stress. These further lead to cumulative damage to peripheral nerve fibres, impaired blood supply to neuronal tissue [9], release of algogens like bradykinins and prostaglandins, which cause hypersensitivity to pain and hence, result in the development of neuropathic pain [10]. Oxidative and nitrosative stress can also lead to massive DNA damage, which is a strong stimulator of PARP, hence causes neuronal apoptosis. All these events will culminate in the development of diabetic neuropathy (DN).
NF-κB has been shown to respond to the cellular redox status since a reducing environment prevents its activation whereas oxidative/nitrosative stress promotes phosphorylation and degradation of IκB [5]. Nrf2 increases intracellular GSH levels and GSH-dependent enzymes favouring a reducing environment thereby inhibiting NF-κB. Li et al. demonstrated that Nrf2-deficient mice exhibit greater induction of pro-inflammatory genes regulated by NF-κB such as interleukins, TNF-α, iNOS and COX-2 pointing towards the fact that Nrf2 deficiency enhances NF-κB-mediated pro-inflammatory reactions [6]. Soares et al. showed that HO-1 inhibited the TNF-α dependent activation of NF-κB in endothelial cells. It has been postulated that HO-1 induced by the Nrf2-EpRE interaction inhibits the NF-κB dependent transcriptional apparatus. Inhibition of NF-κB downstream of IκB phosphorylation/degradation and nuclear translocation has been hypothesized to be the site of action of HO-1 [11]. These data further support the concept that the Nrf2 directed increase in the expression of HO-1 is one of the hubs for cross-talk between Nrf2 and NF-κB (Figs. 2 and 3).
Fig. 2.
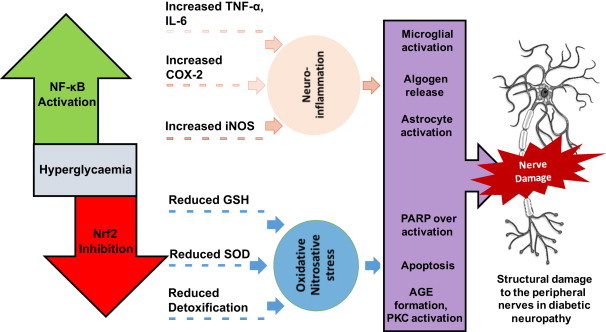
Role of NF-κB and Nrf2 in diabetic neuropathy. Hyperglycaemia induced imbalance in Nrf2–NF-κB regulation can contribute to the pathogenesis of diabetic neuropathy. Enhanced NF-κB activity during the hyperglycaemic state is associated with excess production of proinflammatory cytokines such as IL-6, TNF-α, COX-2 and iNOS. These proteins and enzymes are prerequisite mediators for the initiation and amplification of inflammatory processes in neuronal cells [9]. Reduced Nrf2 activity results in impaired antioxidant defence and is characterized by decline in superoxide dismutase (SOD), catalase and glutathione (GSH) levels. Additionally, it decreases the production of detoxifying enzymes such as, haem-oxygenase-1 (HO-I) and NADPH quinone oxidoreductase (NQO1), leading to nitrosative and oxidative stress [14]. Neuroinflammation due to elevated NF-κB can also activate microglia and astrocytes, which further augments the release of proinflammatory mediators, thus develops a vicious cycle of inflammation. Release of algogenic mediators, such as prostaglandins, bradykinins and chemokines due to neuroinflammation, can sensitise the nerve fibres to painful stimulus and end up in sensorimotor alterations [10]. NF-κB mediated neuroinflammation can also result in endoneurial hypoxia due to decreased blood supply to the nervous tissue and ganglion [9]. This neuronal hypoxic condition leads to dysfunction of mitochondrial electron transport chain (etc), decreased efficiency of mitochondria, and results in increased ROS production. Nitrosative and oxidative stress generated in the nerve cells can also lead to increased AGE formation and protein kinase C (PKC) activation [7] and increased peroxynitrite mediated PARP over-activation and apoptosis. The manifestations of neuroinflammation and oxidative stress can cumulatively cause the structural damage which can lead to the functional, sensorimotor and biochemical deficits which are characteristic of diabetic neuropathy.
Fig. 3.
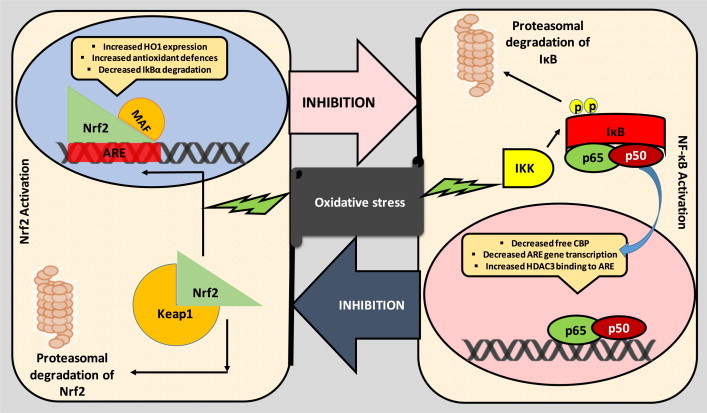
Crosstalk between the Nrf2 and NF-κB pathways. Oxidative stress mediated Nrf2 activation can lead to the production of antioxidant enzymes and cytoprotective enzymes such as SOD, catalase, GSH, haem-oxygenase-1 (HO-1) and NADPH quinone oxidoreductase (NQO1) and increased synthesis of GSH, NADPH and multidrug transporters. Nrf2 bind to the antioxidant response element (ARE) on DNA and maintains redox balance in body [14]. Normally Nrf2 is bound to cytosolic repressor Kelch-like ECH-associated protein 1 (Keap1) and labelled for polyubiquitination mediated proteasomal degradation. However, during oxidative insult, the sulfhydryl groups on Keap1 are oxidised, causing the alteration in confirmation of Keap1, and hence releases Nrf2, which then binds with ARE elements of the genome along with small Maf proteins [14,15]. NF-κB is another redox regulated transcription factor, involved in inflammation, immune function, cellular growth and apoptosis [16]. p65 is a Rel protein with transactivation efficiency whereas its partner p50 does not possess transcriptional activity. Oxidative stress can cause the activation of IκB kinase (IKK). Activation of IKK causes phosphorylation of inhibitor of NF-κB, IκB and hence targets the later for polyubiquitination mediated proteasomal degradation, results in release of NF-κB, which then migrates into the nucleus, binds with the κ region of genome. With the help of other coactivators and histone acetyl transferases (HAT), NF-κB causes the transcription of proinflammatory mediators such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2), interleukin-1 (IL-1), intracellular adhesion molecule (ICAM) and inducible nitric oxide synthase (iNOS) [16]. Further these two pathways are proposed to inhibit each other at their transcription level via protein–protein interactions or through secondary messenger effects. Nrf2 pathway inhibits the activation of NF-κB pathway by increasing antioxidant defences and HO-1 expression, which efficiently neutralizes ROS and detoxify toxic chemicals and hence, reduces ROS mediated NF-κB activation [11]. Nrf2 pathway also inhibits NF-κB mediated transcription by preventing the degradation of IκB-α. Similarly, NF-κB mediated transcription reduces the Nrf2 activation by reducing the ARE gene transcription, decreases free CREB binding protein (CBP) by competing with Nrf2 for CH1-KIX domain of CBP [12]. NF-κB also enhances the recruitment of histone deacetylase3 (HDAC3) to the ARE region by binding to Mafk and hence interferes with the transcriptional facilitation of Nrf2 [15].
Recent studies have shown that NF-κB suppresses the transcriptional activity of Nrf2. Liu et al. demonstrated that NF-κB p65 subunit repressed the beneficial effects of Nrf2 by promoting the localisation of transcription repressors, histone deacetylases with Nrf2/ARE and sequestering coactivators like CREB binding protein (CBP) [12]. Cells over-expressing NF-κB showed lesser expression of HO-1 which further confirms that NF-κB activation can act as a repressor of Nrf2 transcriptional activity. In a recent study, Yu et al. found that the N-terminal region of p65 subunit of NF-κB was physically associated with Keap1, and thus provide an additional mechanism for Nrf2–ARE inhibition. It was also suggested that NF-κB not only interacted with cytosolic Keap1 but also promoted nuclear translocation of Keap1 [13].
Previous studies with agents like curcumin [17], melatonin [18], resveratrol [19] and sulphoraphane [20] have reported beneficial effects in ameliorating various functional (motor nerve conduction velocity and nerve blood flow), sensorimotor (thermal and mechanical hyperalgesia) and biochemical deficits in experimental diabetic neuropathy (Fig. 4). These agents also suppressed the increased activity and levels of NF-κB and associated proteins and hence protected against neuroinflammation in diabetic neuropathy. As expected, treatment with these agents increased the levels of Nrf2 and HO-1 which further modulating the redox regulation of pro-inflammatory signalling pathways. Additional studies to find any common co-activators or co-repressors shared by these transcription factors and co-regulation by upstream and downstream signalling in these cascades will enable a better appreciation of the crosstalk between these two transcription factors in diabetic neuropathy.
Fig. 4.
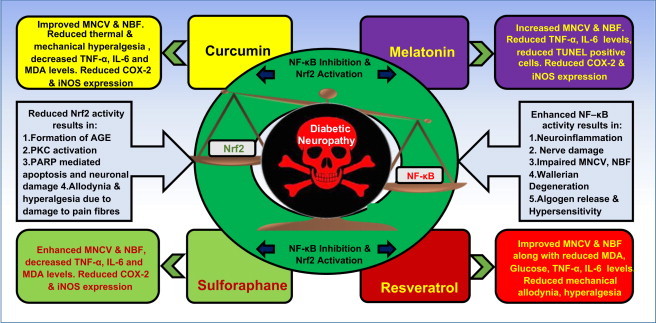
Imbalance of Nrf2–NF-κB in diabetic neuropathy and effects of simultaneous-targeting Nrf2–NF-kB pathway using pharmacological agents. Hyperglycaemia induced disturbances in Nrf2–NF-κB axis can contribute to the development of several diabetic vascular complications such as neuropathy, nephropathy and retinopathy. Enhanced NF-κB activity in diabetic neuropathy leads to the development of neuroinflammation, production of proinflammatory cytokines, nerve damage, impaired motor nerve conduction velocity (MNCV) due to demyelination, impaired neuronal blood flow (NBF) and production of algogenic mediators and hence causes pain hypersensitivity. Reduced Nrf2 activity is associated with enhanced oxidative stress in the neurons, which leads to the activation of PARP mediated neuronal apoptosis, AGE formation, PKC activation and allodynia and hyperalgesia due to the damage to the sensory fibres. However, this disturbed balance can be modulated pharmacologically to attenuate various deficits in diabetic neuropathy. The pharmacological modulation of NF-κB–Nrf2 axis by some pharmacological agents like Curcumin [17], Melatonin [18], Resveratrol[19] and Sulforaphane [20] produced beneficial effect by inhibiting NF-κB and activating Nrf2. The experimental outcomes of the studies of above mentioned compounds in streptozotocin (STZ) induced diabetic neuropathy model are manifested in the form of improved MNCV and NBF, decreased lipid peroxidation, IL-6, TNF-α levels, decreased expression of COX-2 and iNOS and reduced apoptosis.
In summary, Nrf2 and NF-κB individually affect many signalling cascades to maintain a redox homeostasis; additionally they interact with each other to further modulate level of key redox modulators in health and disease. Studies with specific agents that might regulate the crosstalk between the two central pleiotropic transcription factors, Nrf2 and NF-κB, may be one of the prospective strategies that might aid in finding newer therapeutic choices for prevention and treatment of diabetic neuropathy.
Acknowledgement
Authors would like to acknowledge Department of Pharmaceuticals, Ministry of Chemical and fertilizers, Government of India for their support.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Forbes J.M., Cooper M.E. Mechanisms of diabetic complications. Physiological reviews. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 2.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 3.Edwards J.L., Vincent A.M., Cheng H.T., Feldman E.L. Diabetic neuropathy: mechanisms to management. Pharmacology & Therapeutics. 2008;120:1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Negi G., Kumar A., Sharma S.S. Adopting Nrf2 and NF-kB from cancer: is there any role of the duo in diabetes? Nature Precedings. 2011 [Google Scholar]
- 5.Banning A., Brigelius-Flohe R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxidants & Redox Signaling. 2005;7:889–899. doi: 10.1089/ars.2005.7.889. [DOI] [PubMed] [Google Scholar]
- 6.Li W., Khor T.O., Xu C., Shen G., Jeong W.S., Yu S., Kong A.N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochemical Pharmacology. 2008;76:1485–1489. doi: 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlee M. The pathobiology of diabetic complications a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 8.Tan Y., Ichikawa T., Li J., Si Q., Yang H., Chen X., Goldblatt C.S., Meyer C.J., Li X., Cai L. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cameron N.E., Cotter M.A. Pro-inflammatory mechanism in diabetic neuropathy: focus on the nuclear factor kappa B pathway. Current Drug Targets. 2008;9:60–67. doi: 10.2174/138945008783431718. [DOI] [PubMed] [Google Scholar]
- 10.Scholz J., Woolf C.J. The neuropathic pain triad: neurons, immune cells and glia. Nature Neuroscience. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 11.Soares M.P., Seldon M.P., Gregoire I.P., Vassilevskaia T., Berberat P.O., Yu J., Tsui T.Y., Bach F.H. Haem oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. Journal of Immunology. 2004;172:3553–3563. doi: 10.4049/jimmunol.172.6.3553. [DOI] [PubMed] [Google Scholar]
- 12.Liu G.H., Qu J., Shen X. NF-kappaB/p65 antagonizes Nrf2–ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochimica Biophysica Acta. 2008;1783:713–727. doi: 10.1016/j.bbamcr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Yu M., Li H., Liu Q., Liu F., Tang L., Li C., Yuan Y., Zhan Y., Xu W., Li W., Chen H., Ge C., Wang J., Yang X. Nuclear factor p65 interacts with Keap1 to repress the Nrf2–ARE pathway. Cell Signaling. 2011;23:883–892. doi: 10.1016/j.cellsig.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 14.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.-L. The Keap1–Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biology. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakabayashi N., Slocum S.L., Skoko J.J., Shin S., Kensler T.W. When NRF2 talks, who's listening? Antioxidants & Redox Signaling. 2010;13:1649–1663. doi: 10.1089/ars.2010.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karin M., Yamamoto Y., Wang Q.M. The IKK NF-kappa B system: a treasure trove for drug development. Nature Reviews Drug Discovery. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 17.Joshi R.P., Negi G., Kumar A., Pawar Y.B., Munjal B., Bansal A.K., Sharma S.S. SNEDDS curcumin formulation leads to enhanced protection from pain and functional deficits associated with diabetic neuropathy: an insight into its mechanism for neuroprotection. Nanomedicine. 2013;9:776–785. doi: 10.1016/j.nano.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Negi G., Kumar A., Sharma S.S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-κB and Nrf2 cascades. Journal of Pineal Research. 2011;50:124–131. doi: 10.1111/j.1600-079X.2010.00821.x. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A., Sharma S.S. NF-kappaB inhibitory action of resveratrol: a probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochemical and Biophysical Research Communications. 2010;394:360–365. doi: 10.1016/j.bbrc.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Negi G., Kumar A., Sharma S.S. Nrf2 and NF-B modulation by sulforaphane counteracts multiple manifestations of diabetic neuropathy in rats and high glucose-induced changes. Current Neurovascular Research. 2011;8:294–304. doi: 10.2174/156720211798120972. [DOI] [PubMed] [Google Scholar]