

Abstract
Cystic Fibrosis (CF) is a frequent and lethal autosomal recessive disease, caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Before the discovery of the CFTR gene, several hypotheses attempted to explain the etiology of this disease, including the possible role of a chloride channel, diverse alterations in mitochondrial functions, the overexpression of the lysosomal enzyme α-glucosidase and a deficiency in the cytosolic enzyme glucose 6-phosphate dehydrogenase. Because of the diverse mitochondrial changes found, some authors proposed that the affected gene should codify for a mitochondrial protein. Later, the CFTR cloning and the demonstration of its chloride channel activity turned the mitochondrial, lysosomal and cytosolic hypotheses obsolete. However, in recent years, using new approaches, several investigators reported similar or new alterations of mitochondrial functions in Cystic Fibrosis, thus rediscovering a possible role of mitochondria in this disease. Here, we review these CFTR-driven mitochondrial defects, including differential gene expression, alterations in oxidative phosphorylation, calcium homeostasis, oxidative stress, apoptosis and innate immune response, which might explain some characteristics of the complex CF phenotype and reveals potential new targets for therapy.
Keywords: Cystic Fibrosis, CFTR, Mitochondrial complex I, Reactive oxygen species, Apoptosis, Innate immunity
Highlights
► Earlier findings on mitochondrial abnormalities in CF were erroneously disregarded. ► Recent findings demonstrate affected mitochondrial functions in CF. ► These findings include the discovery of CFTR-dependent genes, some of which are mitochondrial. ► CFTR modulation on calcium, ROS, Complex I, SODs, GSH and autophagy are discussed. ► A profound influence on apoptosis, inflammation and innate immunity might exist.
Introduction
Cystic Fibrosis (CF) is one of the most severe and frequent hereditary diseases [1]. It is the main cause of chronic lung damage and exocrine pancreatic insufficiency in the first three decades of life [2]. Other organs and all exocrine glands are also affected to a greater or lesser extent [3].
Lowe and May have described CF as a genetic recessive disorder back in 1949 [4,5]. However, the affected gene was not discovered until 1989, when the team led by Lap-Chee Tsui [6] cloned the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator) and found a deletion of three base pairs in both copies of the gene [6–8]. The deletion causes the loss of a phenylalanine residue at position 508 of the CFTR protein (ΔF508) [6]. Experiments of expression then demonstrated that CFTR was a chloride channel [9–16], as it was suspected for decades [17–32] and intuitively known for many centuries [1,33,34].
Today, more than 1900 mutations have been described for the CFTR gene (Cystic Fibrosis Mutation Database; www.genet.sickkids.on.ca), although the ΔF508 mutation is the most abundant in CF patients, with an occurrence of approximately 66 % [35,36]. It is noteworthy that a clear relationship between genotype and phenotype could not be demonstrated in CF [2]. This is because the different degrees and high variability of the parameters studied in CF reflect the interaction of diverse factors, including genetic and environmental influences, infectious events and differences in treatments [37].
CFTR is an integral membrane glycoprotein frequently located in the apical membrane of the epithelial cells; it is also present in non-epithelial cells from blood, brain, heart, liver, kidney and other tissues [38–42]. In addition to chloride, the CFTR channel is able to transport bicarbonate [43] and glutathione [44]. On the other hand, the possible role of CFTR as an ATP channel has been controversial [45–54].
The CFTR channel belongs to the superfamily of ABC (ATP Binding Cassette) transporter proteins. The protein includes two nucleotide-binding domains (NBD1 and NBD2) that hydrolyze ATP to regulate the channel activity [55]. In addition, an intracytoplasmic regulatory domain (domain R) is activated by protein kinase A (PKA) phosphorylation to control the opening of the channel [16]. Thus, the chloride transport activity of CFTR is a cyclic AMP-regulated process, a feature that differentiates CFTR from the other chloride channels. The kinases PKC [56] and SRC also regulate the CFTR channel activity [57]. Actually, all molecules or pathways capable of modulating cAMP levels contribute to CFTR activation, including epinephrine, isoproterenol, carbachol, phosphodiesterase inhibitors [58] and G proteins, among other factors [59]. The CFTR protein also forms a macromolecular complex with several proteins, interacting either directly or indirectly [60,61]. Some interactions involve PDZ domain-containing proteins [60,62,63].
Different factors regulate CFTR expression. Cell differentiation in Caco-2 cells induces CFTR up-regulation; the same effect was not observed in T84 cells [64]. In addition, cAMP up-regulates CFTR mRNA levels [65] through a variant cAMP response element (CRE) present at position -48 to -41 of the CFTR promoter [66]. On the other hand, INF-γ [67], TNF-α [67] and progesterone [68] are able to down-modulate CFTR expression. IL-1β modulates CFTR expression of T84 cells in a biphasic way [69]. The CFTR mRNA levels are up-regulated when the cytokine concentration is near 1 ng/ml (≈6 fM) and down-modulated at higher concentrations. This biphasic response involves at least two different mechanisms: NF-κB accounts for the stimulation of CFTR mRNA levels at low concentrations of IL-1β (around and below 1 ng/ml) [70] whereas AP-1 appears to be involved in the down-modulation observed at higher concentrations of this cytokine (2–5 ng/ml and higher) [70,71]. Interestingly, IL-1β has been found in sputum from CF children at high concentrations, between 2.8 and 32 ng/ml [72]. At these concentrations, according to the results obtained in T84 cells [69], the CFTR should be strongly down-modulated. Other modulators of CFTR include protein kinase C (PKC), protein tyrosine kinases (PTKs) and phospholipase C (PLC) [69], as well as steroid hormones [73–76], nitric oxide [77–80], hyperosmolarity [81], vasoactive intestinal peptide (VIP) [82,83], c-Src tyrosine kinase (SRC) [57], with-no-lysine [K] 4 kinase (WNK4) [84], casein kinase 2 (CK2) [85] and spleen tyrosine kinase (SYK) [85].
In addition to its CFTR-chloride transport activity, CFTR indirectly regulates the expression of different CFTR-dependent genes, such as SRC and MUC1 [86], MT-ND4 [87], CISD1 [88], RANTES [89], and other genes involved in inflammation or metabolic functions [90–93]. It is noteworthy that two of these genes, MT-ND4 and CISD1, encode for mitochondrial proteins having a reduced gene expression in CF cells [87,88,94–107]. Since ND4 (encoded by mitochondrial MT-ND4) is a key subunit for the activity of mitochondrial Complex I (mtCx-I), the early work of Burton L. Shapiro and colleagues [108–113] regarding possible mitochondrial failures in CF was rethought. Subsequent work of our laboratory demonstrated that indeed the activity of mtCx-I was reduced in CFTR defective cells [87,94,96,97,99]. Later, the mtCx-I failure and other alterations found in the pioneer work of Shapiro and colleagues, including glutathione and calcium metabolism [112–122], were recently confirmed by other laboratories. Here we review the earlier work and the recent findings on CFTR-induced mitochondrial alterations, and their possible pathophysiological consequences.
Initial findings involving mitochondrial alterations in CF
Irena Antonowicz, from the laboratory of Harry Shwachman at Harvard, studied several mitochondrial and lysosomal enzymes in cultures of lymphoid cells obtained from CF patients [123]. They found only alterations in the lysosomal α-glucosidase (which degrades glycogen) without any changes detected in the three mitochondrial enzymes tested: succinate dehydrogenase (succinate-coenzyme Q reductase (SQR) or respiratory Complex II), glutamate dehydrogenase (GLDH), and malate dehydrogenase (MDH). From these results, the authors concluded that CF might be a lysosomal disorder [123] (unfortunately the authors did not measure NADH dehydrogenase). On the other hand, Congdon and Littlewood found a glucose 6-phosphate dehydrogenase (G6PD or G6PDH) deficiency in CF [124], although this observation was not further explored.
During the decade before the cloning of CFTR, Shapiro and Feigal reported the existence of different mitochondrial abnormalities in CF [108–113,115,117,125]. First, in 1979, based on previous work from other laboratories [126–130], they found alterations on calcium uptake and oxygen consumption in mitochondria isolated from fibroblasts of CF patients [113]. In the same year, they found alterations in the optimal pH of mitochondrial Complex I (mtCx-I; NADH dehydrogenase, NADH:ubiquinone reductase (H(+)-translocating), EC 1.6.5.3) and suggested that the mutant gene responsible for CF might be expressed within the mtCx-I [111]. Later, these same authors found that the increased calcium uptake by mitochondria was associated with an altered respiratory system activity [117] and found a reduced Km of mitochondrial NADH dehydrogenase in whole cell homogenates of cultured skin fibroblasts [110]. After these initial works, several studies described mitochondrial changes in CF [108,109,115,131–133], until the CFTR was cloned and found to be a chloride channel [6,7,134]. Following this important finding, the hypotheses of possible mitochondrial, lysosomal or cytosolic alterations in CF were totally disregarded. Future work mostly focused on the involvement of CFTR as a chloride channel. Few explored other areas. Among them, Picci et al. reported differences in the 2D electrophoretic patterns of mitochondrial proteins in CF [135]. Also, de Meer et al., found higher intracellular pH in CF patients in vivo, during workload [136]. More recently, Day et al. [137] found decreased glutathione levels in epithelial lining fluid of CFTR knockout mice, the last finding also in agreement with the early work of B.L. Shapiro and colleagues, regarding glutathione [119–122]. Now, by using different approaches, several authors are reporting different results in agreement with the pioneer work of Shapiro and colleagues, reinforcing the idea that mitochondrial dysfunctions are indeed present in CF, although as an indirect consequence of CFTR signaling mechanisms, yet undefined.
Differential expression of genes in Cystic Fibrosis
Back in 1994, as an approach to explain the diverse phenotypic characteristics of CF, we hypothesized that perhaps the CFTR activity could indirectly affect the expression of several genes. This hypothesis was tested by applying the method of differential display [138]. First, it was necessary to find a way to avoid the frequent false positive/negative signals of this method. Initially we use T84 colon carcinoma cells treated with the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), a drug that down-modulates CFTR mRNA levels [139]. Since it was difficult to determine which effects were due to TPA or CFTR, we later changed the model system by using instead CFDE cells (derived from a CF patient) and the same cells ectopically expressing wt-CFTR (CFDE/6RepCFTR cells). The hypothesis was correct, and we found several CFTR-dependent genes [86,140,141]. Some spots of the differential display corresponded to unknown genes. One spot overexpressed in CFDE cells (CF cells) was studied in detail; its cDNA sequence corresponded to the SRC/c-Src tyrosine kinase. Thus, the mRNA and protein expression of SRC/c-Src was found increased in CFDE cells (CF cells). Since SRC/c-Src was known to regulate several mucins, we tested the expression of MUC1, and found that this mucin was also upregulated in CFDE cells, under SRC/c-Src modulation. These results suggested that SRC/c-Src might constitute a bridge between the CFTR failure and mucin overexpression, at least in the case of MUC1 and CFDE cells [86]. Interestingly, the effects occurred as a primary failure of CFDE cells, in the absence of any bacterial infection.
Almost in parallel, by using microarrays, Srivastava [90,91,93], Galvin [92], Eidelman [142], Pollard [143] and colleagues found several differentially expressed genes in CF cells. In addition, Ichikawa et al. [144] studied the differential expression of genes in the cell line A549, derived from lung pneumocytes, exposed to Pseudomonas aeruginosa, thus analyzing differential gene expression in response to bacterial–host interaction. Interestingly, Xu et al. found differential expression of 54 RNAs, corresponding to genes that influence gene transcription, inflammation, intracellular trafficking, signal transduction, and ion transport, including the CEBPδ transcription factor and IL-1β [145,146], both modulators of CFTR transcription [69,70,147]. More recently, Ogilvie et al. have studied the differential expression of genes in CF by using the Illumina HumanRef-8 Expression BeadChips [148]. They conclude that CF and non-CF nasal and bronchial epithelium are transcriptionally distinct. Also, the CF nasal epithelium, with 15 differentially expressed genes, was not a good surrogate for the lung epithelium that has 863 differentially expressed genes. In addition, these cells showed a distinct pattern of enriched pathways, being inflammation pathways predominant in bronchial cells, whereas the pathways controlling amino acid metabolism were predominant in nasal epithelial cells [148]. Lately, antibody microarrays were used instead of DNA microarrays to identify differentially expressed proteins as possible serum biomarkers for CF [93,149].
After characterizing SRC/c-Src and MUC1 as CFTR-dependent genes, we decided to study two additional spots of the differential display, which contrary to SRC/c-Src and MUC1 were down-modulated in CFDE cells. Surprisingly, both spots resulted in genes encoding for mitochondrial proteins: CISD1 (a nuclear DNA-encoded gene) [88,98,101,103,105,107,150–152] and MT-ND4 (a mitochondrial DNA-encoded gene) [87,100,102,104,106,141,153–155]. These results are discussed with more detail in the next two sections.
CISD1 expression in CF cells
As mentioned above, a differential display (DD) obtained in 2002 from CFDE cells (CF cells) and CFDE/6RepeCFTR (CF corrected cells) showed a cDNA spot that contrary to SRC had a reduced expression in CF cells. It was isolated, cloned and sequenced from a cDNA DD spot of 477 bp derived from CFDE cells. Initially, we called this gene CFTR-RG2 (CFTR-regulated Gene 2), since it was the second CFTR-dependent cDNA isolated and sequenced in our laboratory [150]. Then, in 2004, to assign a name related to its possible function, it was called KLPx, since we found sequence similarities to kinesin motor proteins [107]. Then we realize that the kinesin motor region was absent and therefore we preferred to call it ZCD1, owing to the presence of a Zn finger-like motif, the CDGSH motif [105], defined in a previously annotated sequence obtained by Zhao M. et al. from hematopoietic stem/progenitor cells (AF220049; unpublished). The mRNA corresponding to the ZCD1 sequence from CFDE cells was finally annotated in GenBank as “Homo sapiens zinc finger CDGSH-type domain 1 (ZCD1) mRNA” (accession number AY960578 and AAY32336 for the deduced protein).
Previously, in 1999, Zhao et al. from the Chinese National Human Genome Center at Shanghai had annotated a 636 bp cDNA sequence obtained from hematopoietic stem/progenitor cells as “Homo sapiens uncharacterized hematopoietic stem/progenitor cells protein MDS029” (AF220049, unpublished). They also annotated the deduced protein sequence (AAY32336), defining the region 55.93 as “ZnF_CDGSH” a “CDGSH-type zinc finger”, using the conserved domain database CDD [156]. They also mentioned the SMART signaling domain [157] smart00704, assigning an unknown function to it. Since these results were unpublished and further details are not available, it is not clear who actually defined the CDGSH-type zinc finger, which corresponds today to a superfamily of proteins (CDD cl02748: zf-CDGSH superfamily). In addition, Strausberg et al. [158] annotated the CISD1 sequence as C10orf70 (which stand for “chromosome 10 open reading frame 70”) during the generation and analysis of more than 15,000 full-length human cDNA sequences (ORFs) from the human genome, which was sequenced one year before [159].
In parallel, in studies linked to diabetes published in 2003, Colca et al. [161] identified a pioglitazone binding protein from bovine brain and rat liver mitochondria. Pioglitazone, an antidiabetic drug, also binds to the nuclear peroxisome proliferator-activated receptor gamma (PPAR-γ), which is an important target for antidiabetic drugs [160]. The sequence was obtained by using nanospray LC–mass spectroscopy (MS/MS) and N-terminal sequencing confirmed the sequence; it was named mitoNEET [161].
Finally, when the crystal structure was determined by other laboratories [162–164], instead of the predicted Zn2+, a [2Fe2S] cluster was found within the ZCD1 molecule [162–166]. In consequence, the official HUGO symbol (www.genenames.org) was finally agreed as “CISD1” (Z was eliminated and “IS” was added, which stands for Iron Sulfur binding motif). It is noteworthy that recent evidences suggest that Zn2+ can also bind to CISD1 and even may replace the [2Fe2S] cluster in E. Coli cultures [167]. In consequence, Zn2+ might also bind to CISD1 in mammalian cells in addition to [2Fe2S]. A direct measurement of Fe2+ and Zn2+ content of CISD1 isolated from mammalian cells or tissues might indicate in which proportion these ions bind to CISD1 and under which circumstances.
Although the exact function of this protein is unknown yet, it has been shown that mitoNEET/CISD1 is a mitochondrial specific binding protein receptor for pioglitazone [161] and similar drugs [168]. Co-immunoprecipitation assays suggested that mitoNEET/CISD1 was associated with proteins that belong to the mitochondrial complex I (mtCx-I), among other mitochondrial proteins [161]. The protein was also proposed to be a modulator of the oxidative capacity of cells [166], a sensor of the intracellular redox state [164,169,170], and a mediator for the transference of a [2Fe–2S] cluster to apoproteins [169,171]. We found that an eGFP-CISD1 chimera was mainly located in mitochondria after 24 h of transfection, suggesting that its primary location and function was indeed mitochondrial [88]. However, the mechanisms involved in all these possible functions, the targets of these functions, and their relevance for the mitochondrial and cellular functioning, remain to be established. The mechanisms by which CFTR controls CISD1 expression and down-modulation in CF cells [88], and the possible consequences for the cellular pathophysiology, are still unknown.
MT-DN4 expression in CF cells
The second gene found reduced in CF cells, MT-ND4, encodes for ND4, one of the seven subunits (ND1 to ND7) of the mitochondrial Complex I (mtCx-I) [172]. The mammalian mtCx-I is a holoenzyme comprising 45 subunits that constitute a complex of approximately one MDa. This complex is the entry point of electrons for the oxidative phosphorylation system (OXPHOS system) [173]. ND4 subunit constitutes a fundamental component for the assembly and correct activity of mtCx-I [172,174–180]. Thus, a MT-ND4 mutation in Leber's Hereditary Optic Neuropathy disease (LHON) determines a low efficiency in the NADH oxidation due to a mtCx-I failure [172,179,181]. The reduced expression of MT-ND4 found in CF cells suggested that the activity of mtCx-I might be reduced in CF cells, and, as it will be discussed below, we later found that the activity of these mitochondrial complexes was indeed reduced in CF cells or in cells with impaired CFTR activity (inhibitors, iRNA, etc.).
Mitochondrial complex I and Cystic Fibrosis
As mentioned in the previous section, we found a downregulation of MT-ND4 in CF, a gene encoding for ND4, a mtCx-I subunit essential for its assembly and activity [100]. The CFTR-mediated down-modulation of MT-ND4 found in CF cells was in agreement with the early findings of Shapiro et al., showing that the activity of mtCx-I was affected in CF cells [108,110,111]. The results obtained and the previous work by other laboratories prompted as to measure the mtCx-I activity in CF cells. Consistent with the earlier observations, we found a decreased mtCx-I activity in cells derived from trachea of CF patients (CFDE and IB3-1 cells) when compared to the same cells ectopically expressing wild type CFTR (wt-CFTR) (CFDE/6RepCFR and S9, respectively) [87,94,97,99]. This was also found in colon carcinoma cells (T84 and Caco-2), which express high levels of endogenous wt-CFTR, treated either with pharmacological inhibitors of CFTR activity or RNAi [87,94,97,99]. The decreased activity of mtCx-I found in CF cells might be a consequence of the MT-ND4 down-regulation induced by CFTR mutations or inhibition [100]. In agreement with these findings, Kelly-Aubert et al. have recently reported a decrease in mtCx-I activity in CF cells as compared to cells rescued by ectopic expression of wt-CFTR, and also by using a CFTR-knockout mice [182]. These authors suggested that the observed reduction in the specific activity of mtCx-I was caused by oxidation because of the oxidative stress originated in reduced glutathione levels [182]. Altogether, these observations suggest that the mtCx-I activity is altered in CF. The possible mechanisms involved in regulation of mtCx-I by CFTR (CFTR→mtCx-I) are unknown yet.
Altered Ca2+ homeostasis in mitochondria
Recently, Antigny et al. have reviewed the changes occurring in calcium homeostasis and calcium signaling in CF [183]. Donnell et al., back in 1961, were perhaps the first to show alterations of calcium homeostasis in CF [184]. They found evidences of calcium deposition and signs of reparative fibrosis in the bowel of CF patients. Then, Marmar et al. showed altered calcium in parotid gland secretions [185] and Blomfield et al., reported hypersecretion of zymogen granules in the submandibular saliva, with elevated calcium concentrations [186]. Since these initial reports, many investigators found alterations in calcium secretion and homeostasis in CF. Later, Shapiro and Lam [114], based on previous observations from different laboratories [126–130], found increased intracellular calcium concentration ([Ca2+]i) in fibroblasts derived from CF patients. Feigal et al. also found an increased mitochondrial Ca2+ uptake attributed to alterations in oxidative phosphorylation [115,117].
More recently, in contrast to the earlier observations of Feigal and Shapiro, Antigny et al. have found a decreased mitochondrial Ca2+ uptake in CF airway epithelial cells (ΔF508 homozygous) [187]. The reason for this discrepancy is unknown. In addition, they observed a fragmentation of mitochondria and a decrease of the mitochondrial membrane potential (ΔΨmit) in CF cells as compared to control cells. These changes in mitochondrial Ca2+ homeostasis were linked to a decreased mitochondrial membrane potential (ΔΨmit), which in turn occurs owing to changes in the oxidative phosphorylation [187].
Among several intracellular pathways activated by calcium, it has been reported that increased [Ca2+]i may lead to activation of NF-κB (“Nuclear Factor Kappa B”) and its proinflammatory effects [188,189]. Thus, the rise in [Ca2+]i observed in CF cells may induce or at least contribute to the increased NF-κB activation seen in these cells. As discussed below, the increased oxidative stress in CF also contribute to activation of NF-κB.
Diverse processes affected in CF and related to mitochondrial pathways
Oxidative stress
Reactive oxygen species (ROS) levels are increased in airway diseases, including CF [182,190–197]. CF cells also show higher sensibility to oxidative stress caused by environmental factors, such as ozone [198] and air pollution [199], or infections [200]. The origin and consequences of high ROS levels are not fully understood. Cleeter et al. suggest that a defect in mtCx-I may lead to an increased ROS yield, which in turn further affect mtCx-I activity [201]. On the other hand, Esposito et al. have shown that inhibition of the OXPHOS system in Ant1(tm2Mgr) (-/-) mice, lacking the heart/muscle isoform of the adenine nucleotide translocation protein Ant, increase ROS levels. Interestingly, in the same work, as a compensation response, increased levels of manganese superoxide dismutase (Mn-SOD or SOD2) were also observed [202]. They concluded that, if the antioxidant defenses are not enough to deal with high ROS levels, then increased mtDNA damage might occur. Similar results were obtained by Lian and Godley in human RPE cells [203]. The increased mtDNA damage may in turn further reduce the OXPHOS system activity, producing a vicious cycle that results in additional ROS generation [204,205].
Autophagy
Autophagy is a natural process by which cells degrade or recycle damaged or unneeded proteins, organelles, and pathogens. The mechanisms of autophagy and their relationship with disease are becoming of high interest [206]. Luciani et al. showed that the CFTR failure in CF induces autophagy inhibition, since defective CFTR upregulates ROS and tissue transglutaminase (TG2), which in turn drive the cross-linking of beclin 1 [195]. This process favors the accumulation of beclin 1 in aggresomes, resulting in a decreased degradation of these juxtanuclear inclusion bodies that appear as a response to misfolded proteins [207]. Rescuing cells from autophagy through beclin 1 overexpression, cystamine or antioxidants, results in improved CFTR transport and reduced ROS formation and inflammation [195,208]. Abdulrahman et al. observed similar results rescuing autophagy by using the immunosuppressant drug rapamycin on CFTR ΔF508 mouse macrophages [209]. Thus, the excessive intracellular accumulation of defective CFTR observed in CF cells seem to be a consequence of autophagy inhibition and aggresome accumulation [195,208]. Targeting autophagy might be a useful strategy for CF therapy [210].
Glutathione
The higher sensitivity to oxidative stress in CF might be associated with several factors that affect the redox balance and the susceptibility to high ROS levels. One of these factors is the ratio between reduced and oxidized glutathione. Reduced glutathione is the most abundant antioxidant inside cells and the ratio between its reduced and oxidized form reflects the redox state and health of cells. A deficient extracellular glutathione (eGSH) transport that cause an imbalance between reduced glutathione and oxidized glutathione (GSH/GSSG) outside cells has been linked to the low CFTR activity found in CF [44,211–213]. This deficiency in eGSH transport might decrease the antioxidant protection of the extracellular space against oxidative stress. In addition, lower mitochondrial GSH (mGSH) levels were found in CFTR-knockout mice (CFTR −/−), and in tracheal cells from CF patients, accompanied with increased ROS production [193]. Recently, other authors have confirmed a decreased mGSH levels in CF that occurs in the absence of a defect in the transport of GSH through mitochondria [182]. Furthermore, these authors reported a decrease in the mtCx-I activity of CF cells and CFTR-knockout mice that was reverted to control values by treating cells with GSH monoethyl ester (GSH-EE) [182]. GSH-EE is a membrane permeable analog of GSH proved to be effective to increase mGSH levels in several cellular models [214–216]. This reactive, as well as other antioxidant compounds that allow increased mGSH levels, might be potentially useful for CF therapy [182].
Peroxiredoxin
In addition to GSH levels, other components of the antioxidant defense mechanism are affected in CF. It is noteworthy that the enzyme peroxiredoxin 6 (Prdx6, EC 1.11.1.15), which plays an important role in the defense against oxidative damage in lung [217–219], has a decreased expression and activity in CFTR-knockout mice [217]. This enzyme uses GSH as electron donor to reduce hydrogen peroxide (H2O2), fatty acid hydroperoxides and phospholipid hydroperoxides [220]. Although the mechanism responsible for decreased Prdx6 expression in CF is unknown, these results are in agreement with the general idea of a redox imbalance and higher susceptibility to oxidative damage in CF.
Superoxide Dismutases (SODs)
Another factor that affects the redox balance is the level and activity of superoxide dismutases (SOD, EC 1.15.1.1). SOD enzymes are key components of the cellular defense system against the production of the free radical superoxide (O2−), by dismutation of O2− in oxygen (O2) and hydrogen peroxide (H2O2). A decreased protein expression of Cu/Zn-SOD (SOD1, cytosolic localization) and Mn-SOD (SOD2, mitochondrial localization) was found in pancreatic and tracheal cells derived from CF patients, without changes in their activities [192]. By the contrary, a decrease in the activity of extracellular SOD (EC-SOD/SOD3) was observed in CF cells, without changes in its protein expression level [192]. These results are in agreement with previous work in which a decrease in the SOD activities was found in plasma of CF patients [221]. They are also in agreement with a decrease in the Cu/Zn-SOD activity of blood cells (mononuclear cells and polymorphonuclear cells) from CF patients [222,223]. The diminished extracellular SOD activity, together with the diminished eGSH, could produce a misbalance in the extracellular redox state, affecting the structure and function of many membrane and extracellular proteins.
Possible pathophysiological consequences
Apoptosis
Except for one work [224], different investigators have found increased apoptosis in CF [191,192,198,199,225–228]. Although several studies suggest that apoptosis in CF is a consequence of the recurrent bacterial infections [225,226,228], others suggest that apoptosis in CF is a primary defect that occurs even in the absence of bacterial infections [191,192,227,229]. In infected human conjunctiva epithelial Chang cells, apoptosis mediated by mitochondrial alterations was induced after infection with P. aeruginosa. These alterations include mitochondrial depolarization, enhanced ROS production, release of cytochrome c, and activation of c-Jun N-terminal kinases (JNKs) [200]. On the other hand, in JME cells (CF ΔF508CFTR nasal epithelial cells), the homoserine lactone C12 rapidly activates apoptosis. However, no differences were seen between JME cells and CFTR corrected JME cells after C12 treatment, suggesting that CFTR is not an important modulator of C12-induced apoptosis [228]. It should be pointed-out, however, that corrected JME cells ectopically express wt-CFTR under the control of a foreign promoter, thus possible feedbacks on its own promoter are missing.
Other studies suggest that the increased apoptosis in CF might be related to a decrease in the antioxidant protection system, which may contribute to self-perpetuate the inflammatory process characteristic of CF [191,192,199]. In particular, Rottner et al. reported a marked sensitivity of CF cells to start apoptosis linked to activation of the NF-κB pathway, suggesting that the secretion of proinflammatory cytokines from these cells promote apoptosis [227]. Recently, a link between the high level of the oxidative stress and apoptosis has been attributed to a decrease in the antioxidant defenses, caused by reduced activity and expression of superoxide dismutase (SOD) enzymes [192]. Hence, a tendency to increased apoptosis mediated by the mitochondrial pathway seems to occur in CF cells, probably exacerbated upon bacterial infections.
Chronic inflammation
Chronic lung inflammation is an important characteristic of the CF phenotype [230,231]. Yet, it is difficult to define and distinguish primary defects due to the CFTR failure from secondary defects that occur after bacterial infections. It is even more difficult to determine their relative weight in defining the CF phenotype or its clinical manifestations.
In early studies, several cytokines were found to be elevated in sputum obtained from CF patients, including IRAP, IL-1α, IL-1β, IL-6, and TNF-α [232,233]. In addition, in neutrophils from CF patients, IL-8 was found at high levels and its receptors down-modulated [234]. After these initial studies, several reports suggest elevated concentrations of cytokines, chemokines (small cytokines) and other inflammatory mediators in CF patients [72,235–250].
The role of Interleukin-1, neutrophil elastase and lipopolysaccharide to regulate inflammation in CF has been reviewed by Carroll et al. [251]. Studying possible modulators of CFTR expression, we found that IL-1β was able to modulate CFTR expression in a biphasic way. At low concentrations (<1 ng/ml) IL-1β up-regulates CFTR mRNA levels [69] through NF-κB [70,71]. However, at concentrations over 2.5 ng/ml (between 2.3 and 32 ng/ml were found in CF children [72]), the IL1-β effect was the contrary, inducing a down-regulation of CFTR expression. The mechanism involved in this second phase was not fully defined yet and might involve the AP-1 transcription factor [71], as occurs with TNF-α, which down-modulates CFTR mRNA by increasing its degradation through the AP1 pathway [252–255]. These results suggest that CF children, having high concentrations of IL-1β and TNF-α, should have a strong down-modulation of CFTR, which together with the low levels of ΔF508-CFTR able to reach the cell membranes in CF cells, might constitute a negative loop with profound consequences for the disease. In addition, CFTR negatively regulates NF-κB-mediated innate immune response [256], establishing an additional loop between CFTR and NF-κB (CFTR--|NF-κB→CFTR). In addition to its effects on CFTR, IL-1β reduces the mtCx-I activity in human chondrocyte cells [257] and its mRNA was found elevated in cells with impaired CFTR activity [95,258,259]. Therefore, IL-1β might be an important piece of the CFTR→→mitochondrial signaling.
Following these initial studies, additional cytokines were found increased in CF, including IL-8 [259–265] and IL-17 [266–270]. The increased concentrations of different cytokines might be responsible for the abundant neutrophil recruitment observed in CF and drive the activation of transcription factors such as the nuclear factor-κB (NF-κB) [271,272] and the activator protein 1 (AP-1) [273], increasing in turn the inflammatory response with the consequent tissue damage. Now, numerous studies support the concept that alterations in the inflammatory process are present in CF [247,264,274–278].
Innate immunity
A central question is whether the mitochondrial defects found in CF are strong enough to influence innate immunity and, in the long-term, favor lung bacterial infections. Initial works in models of viral infection connected mitochondria, apoptosis and innate immunity [279–282]. Then, death receptors (DDs) have been recognized as modulators of immune and inflammatory responses [283]. Also, it has been shown that exacerbated inflammation might impair the function and structure of mitochondria [284]. More recently, studies on pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and cytosolic Nod-like receptors (NLRs), have become important players in the innate immune and inflammatory responses [285,286]. These studies led to the characterization of the “inflammasome”, a macromolecular complex that uses NLRs as scaffold proteins, which recruits and activates inflammatory caspases, essential mediators of inflammation and cell death responses. The activation of caspases directly connects to the mitochondrial apoptotic pathways [285]. Mitochondria, in turn, play a very important role in modulation of the cell death pathway during defense against bacterial infections [287]. In addition, the process of autophagy [288–290] and the production of ROS [291,292] became new players in defining innate immunity. Even more, mitochondria by itself, once released from damaged cells, can trigger an acute inflammatory response [293]. Thus, mitochondria emerges as a fundamental piece of the innate immune response [294–297], modulating the inflammasome-mediated production of proinflammatory cytokines [298]. Dysfunctional mitochondria generate ROS and induce the NLRP3 inflammasome activation [299,300]. On the other hand, autophagy negatively regulates the NLRP3 inflammasome [299]. In consequence, the role of autophagy [195,208,209,301–305] and the inflammasome [303,306,307] both have increased relevance in CF research.
Concluding remarks
Fig. 1 summarizes the mitochondrial abnormalities found in CF cells or tissues. Among them, differential expression of genes or proteins, alterations on calcium homeostasis, membrane potential, increased ROS, and reduced Complex I activity, all appear to occur in mitochondria due to the CFTR failure. These mitochondrial effects in turn induce changes in the ratio of reduced/oxidized glutathione, trigger apoptotic events, and produce inflammatory responses that affect innate immunity. These alterations might influence the phenotype or clinical manifestations of CF and have profound pathophysiological consequences.
Fig. 1.
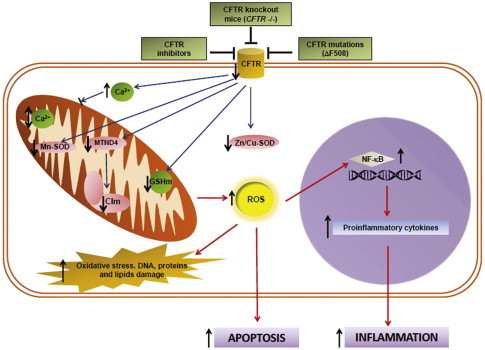
Representative scheme for mitochondrial alterations in CF. The effects of a CFTR failure on ROS production, apoptosis and inflammation are shown, together with the main relationships between mitochondrial activity and antioxidant defense system in CF. CFTR inhibition or mutation leads to mitochondrial alterations that produce an imbalance in the antioxidant defense system and an impairment of the OXPHOS system, inducing an excessive ROS production by mitochondria. The noxious effects of ROS include DNA, protein and lipids damage, apoptosis and inflammation, the latter due to ROS-induced NF-κB. Also, the CFTR failure induces an alteration in calcium homeostasis for the mitochondria. All these effects suggest an important role of the mitochondria in defining at least some of the phenotypic characteristics of CF. The vertical black arrows indicate the net effect of a CFTR failure. The red connectors illustrate positive interactions and the black ones ( _| ) inhibition. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
A complete understanding of these interactions, their mechanisms of action and the relative importance of each pathway, will help to better define the CF phenotype and clinical manifestations, and to find possible new targets for CF therapy. Thus, the role of CFTR in regulating mitochondrial functions, in particular de OXPHOS pathway, and the role of mitochondria in modulating the inflammatory process, and their possible effects on innate immunity, are issues of increasing interest in CF research.
Acknowledgments
This work was supported by the National Agency for the Promotion of Science and Technology (ANPCYT, grant PICT 2007-00628 to TASC) and the National Research Council of Argentina (CONICET, grant PIP 11220080 102551, 2009–2011 to TASC) and research fellowships from CONICET to AGV, and Pontifical Catholic University of Argentina (grant to TASC and fellowships to AGV).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Angel Gabriel Valdivieso, Email: agvaldivieso@gmail.com, angel_valdivieso@uca.edu.ar.
Tomás A. Santa-Coloma, Email: tsantacoloma@gmail.com, tomas_santacoloma@uca.edu.ar.
References
- 1.Quinton P.M. Cystic fibrosis: lessons from the sweat gland. Physiology (Bethesda) 2007;22:212–225. doi: 10.1152/physiol.00041.2006. [DOI] [PubMed] [Google Scholar]
- 2.Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67:117–133. doi: 10.1159/000029497. [DOI] [PubMed] [Google Scholar]
- 3.Heijerman H. Infection and inflammation in cystic fibrosis: a short review. Journal of Cystic Fibrosis. 2005;4(Suppl. 2):3–5. doi: 10.1016/j.jcf.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Lowe C.U., May C.D., Reed S.C. Fibrosis of the pancreas in infants and children; a statistical study of clinical and hereditary features. American Journal of Diseases of Children. 1949;78:349–374. doi: 10.1001/archpedi.1949.02030050362008. [DOI] [PubMed] [Google Scholar]
- 5.May C.D., Lowe C.U. Fibrosis of the pancreas in infants and children; an illustrated review of certain clinical features with special emphasis on the pulmonary and cardiac aspects. Journal of Pediatrics. 1949;34:663–687. doi: 10.1016/s0022-3476(49)80333-7. [DOI] [PubMed] [Google Scholar]
- 6.Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L., Drumm M.L., Iannuzzi M.C., Collins F.S., Tsui L.C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 7.Tsui L.C., Rommens J.M., Burns J., Zengerling S., Riordan J.R., Carlock L.R., Grzeschik K.H., Buchwald M. Progress towards cloning the cystic fibrosis gene. Philosophical Transactions of the ROYAL Society of London Series B, Biological Sciences. 1988;319:263–273. doi: 10.1098/rstb.1988.0048. [DOI] [PubMed] [Google Scholar]
- 8.Rommens J.M., Iannuzzi M.C., Kerem B., Drumm M.L., Melmer G., Dean M., Rozmahel R., Cole J.L., Kennedy D., Hidaka N., Zsiga M., Buchwald M., Riordan J.R., Tsui L.C., Collins F.S. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 9.Drumm M.L., Pope H.A., Cliff W.H., Rommens J.M., Marvin S.A., Tsui L.C., Collins F.S., Frizzell R.A., Wilson J.M. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990;62:1227–1233. doi: 10.1016/0092-8674(90)90398-x. [DOI] [PubMed] [Google Scholar]
- 10.Gregory R.J., Cheng S.H., Rich D.P., Marshall J., Paul S., Hehir K., Ostedgaard L., Klinger K.W., Welsh M.J., Smith A.E. Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature. 1990;347:382–386. doi: 10.1038/347382a0. [DOI] [PubMed] [Google Scholar]
- 11.Jefferson D.M., Valentich J.D., Marini F.C., Grubman S.A., Iannuzzi M.C., Dorkin H.L., Li M., Klinger K.W., Welsh M.J. Expression of normal and cystic fibrosis phenotypes by continuous airway epithelial cell lines. American Journal of Physiology. 1990;259:L496–505. doi: 10.1152/ajplung.1990.259.6.L496. [DOI] [PubMed] [Google Scholar]
- 12.Rich D.P., Anderson M.P., Gregory R.J., Cheng S.H., Paul S., Jefferson D.M., McCann J.D., Klinger K.W., Smith A.E., Welsh M.J. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature. 1990;347:358–363. doi: 10.1038/347358a0. [DOI] [PubMed] [Google Scholar]
- 13.Anderson M.P., Gregory R.J., Thompson S., Souza D.W., Paul S., Mulligan R.C., Smith A.E., Welsh M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 14.Anderson M.P., Rich D.P., Gregory R.J., Smith A.E., Welsh M.J. Generation of cAMP-activated chloride currents by expression of CFTR. Science. 1991;251:679–682. doi: 10.1126/science.1704151. [DOI] [PubMed] [Google Scholar]
- 15.Bear C.E., Duguay F., Naismith A.L., Kartner N., Hanrahan J.W., Riordan J.R. Cl− channel activity in Xenopus oocytes expressing the cystic fibrosis gene. Journal of Biological Chemistry. 1991;266:19142–19145. [PubMed] [Google Scholar]
- 16.Sheppard D.N., Welsh M.J. Structure and function of the CFTR chloride channel. Physiological Reviews. 1999;79:S23–45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 17.Gahm N., Shwachman H. Studies in cystic fibrosis of the pancreas; a simple test for the detection of excessive chloride on the skin. New England Journal of Medicine. 1956;255:999–1001. doi: 10.1056/NEJM195611222552107. [DOI] [PubMed] [Google Scholar]
- 18.Barbero G.J., Sibinga M.S. The electrolyte abnormality in cystic fibrosis. Pediatric Clinics of North America. 1964;11:983–1001. doi: 10.1016/s0031-3955(16)31633-9. [DOI] [PubMed] [Google Scholar]
- 19.Hansen L., Buechele M., Koroshec J., Warwick W.J. Sweat chloride assay for cystic fibrosis. Using pilocarpine iontophoresis stimulation, filter paper collection and cotlove chloridometer analysis. Minnesota Medicine. 1967;50:1191–1196. [PubMed] [Google Scholar]
- 20.McCombs M.L. Research in cystic fibrosis: a review. Texas Reports on Biology and Medicine. 1973;31:615–629. [PubMed] [Google Scholar]
- 21.Steinrud J., Winkel S., Flensborg E.W. Screening for cystic fibrosis with chloride electrode. An investigation of sweat chloride with chloride electrode orion 417 in normal persons and in patients with cystic fibrosis. Danish Medical Bulletin. 1974;21:251–255. [PubMed] [Google Scholar]
- 22.Bowman B.H., Barnett D.R. Recent advances in cystic fibrosis research. Birth Defects Original Article Series. 1976;12:197–212. [PubMed] [Google Scholar]
- 23.Quinton P.M. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. doi: 10.1038/301421a0. [DOI] [PubMed] [Google Scholar]
- 24.Boucher R.C., Ross D.W., Knowles M.R., Gatzy J.T., Parker J.C. Cl− permeabilities in red blood cells and peripheral blood lymphocytes from cystic fibrosis and control subjects. Pediatric Research. 1984;18:1336–1339. doi: 10.1203/00006450-198412000-00024. [DOI] [PubMed] [Google Scholar]
- 25.Widdicombe J.H., Welsh M.J., Finkbeiner W.E. Cystic fibrosis decreases the apical membrane chloride permeability of monolayers cultured from cells of tracheal epithelium. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:6167–6171. doi: 10.1073/pnas.82.18.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinton P.M. Missing Cl− conductance in cystic fibrosis. American Journal of Physiology. 1986;251:C649–652. doi: 10.1152/ajpcell.1986.251.4.C649. [DOI] [PubMed] [Google Scholar]
- 27.Illsley N.P., Verkman A.S. Membrane chloride transport measured using a chloride-sensitive fluorescent probe. Biochemistry. 1987;26:1215–1219. doi: 10.1021/bi00379a002. [DOI] [PubMed] [Google Scholar]
- 28.Widdicombe J.H. Altered chloride transport of tracheal epithelium in cystic fibrosis. Progress in Clinical and Biological Research. 1987;254:115–126. [PubMed] [Google Scholar]
- 29.Berschneider H.M., Knowles M.R., Azizkhan R.G., Boucher R.C., Tobey N.A., Orlando R.C., Powell D.W. Altered intestinal chloride transport in cystic fibrosis. FASEB Journal. 1988;2:2625–2629. doi: 10.1096/fasebj.2.10.2838365. [DOI] [PubMed] [Google Scholar]
- 30.Boucher R.C., Cotton C.U., Gatzy J.T., Knowles M.R., Yankaskas J.R. Evidence for reduced Cl− and increased Na+ permeability in cystic fibrosis human primary cell cultures. Journal of Physiology. 1988;405:77–103. doi: 10.1113/jphysiol.1988.sp017322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J.H., Schulman H., Gardner P. A cAMP-regulated chloride channel in lymphocytes that is affected in cystic fibrosis. Science. 1989;243:657–660. doi: 10.1126/science.2464852. [DOI] [PubMed] [Google Scholar]
- 32.Boyd C.A. Function of cystic fibrosis gene product. Lancet. 1990;336:938. doi: 10.1016/0140-6736(90)92305-2. [DOI] [PubMed] [Google Scholar]
- 33.Busch R. On the history of cystic fibrosis. Nordisk Medicinhistorisk Arsbok. 1991:95–98. [PubMed] [Google Scholar]
- 34.Quinton P.M. Physiological basis of cystic fibrosis: a historical perspective. Physiological Reviews. 1999;79:S3–S22. doi: 10.1152/physrev.1999.79.1.S3. [DOI] [PubMed] [Google Scholar]
- 35.Kerem E., Corey M., Kerem B.S., Rommens J., Markiewicz D., Levison H., Tsui L.C., Durie P. The relation between genotype and phenotype in cystic fibrosis—analysis of the most common mutation (delta F508) New England Journal of Medicine. 1990;323:1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 36.Mateu E., Calafell F., Ramos M.D., Casals T., Bertranpetit J. Can a place of origin of the main cystic fibrosis mutations be identified? American Journal of Human Genetics. 2002;70:257–264. doi: 10.1086/338243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noone P.G., Knowles M.R. ‘CFTR-opathies’: disease phenotypes associated with cystic fibrosis transmembrane regulator gene mutations. Respiratory Research. 2001;2:328–332. doi: 10.1186/rr82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lange T., Jungmann P., Haberle J., Falk S., Duebbers A., Bruns R., Ebner A., Hinterdorfer P., Oberleithner H., Schillers H. Reduced number of CFTR molecules in erythrocyte plasma membrane of cystic fibrosis patients. Molecular Membrane Biology. 2006;23:317–323. doi: 10.1080/09687860600738304. [DOI] [PubMed] [Google Scholar]
- 39.Mulberg A.E., Wiedner E.B., Bao X., Marshall J., Jefferson D.M., Altschuler S.M. Cystic fibrosis transmembrane conductance regulator protein expression in brain. Neuroreport. 1994;5:1684–1688. doi: 10.1097/00001756-199408150-00035. [DOI] [PubMed] [Google Scholar]
- 40.Levesque P.C., Hart P.J., Hume J.R., Kenyon J.L., Horowitz B. Expression of cystic fibrosis transmembrane regulator Cl− channels in heart. Circulation Research. 1992;71:1002–1007. doi: 10.1161/01.res.71.4.1002. [DOI] [PubMed] [Google Scholar]
- 41.Horowitz B., Tsung S.S., Hart P., Levesque P.C., Hume J.R. Alternative splicing of CFTR Cl− channels in heart. American Journal of Physiology. 1993;264:H2214–2220. doi: 10.1152/ajpheart.1993.264.6.H2214. [DOI] [PubMed] [Google Scholar]
- 42.Tizzano E.F., Chitayat D., Buchwald M. Cell-specific localization of CFTR mRNA shows developmentally regulated expression in human fetal tissues. Human Molecular Genetics. 1993;2:219–224. doi: 10.1093/hmg/2.3.219. [DOI] [PubMed] [Google Scholar]
- 43.Chan H.C., Shi Q.X., Zhou C.X., Wang X.F., Xu W.M., Chen W.Y., Chen A.J., Ni Y., Yuan Y.Y. Critical role of CFTR in uterine bicarbonate secretion and the fertilizing capacity of sperm. Molecular and Cellular Endocrinology. 2006;250:106–113. doi: 10.1016/j.mce.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 44.Kogan I., Ramjeesingh M., Li C., Kidd J.F., Wang Y., Leslie E.M., Cole S.P., Bear C.E. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO Journal. 2003;22:1981–1989. doi: 10.1093/emboj/cdg194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reisin I.L., Prat A.G., Abraham E.H., Amara J.F., Gregory R.J., Ausiello D.A., Cantiello H.F. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. Journal of Biological Chemistry. 1994;269:20584–20591. [PubMed] [Google Scholar]
- 46.Li C., Ramjeesingh M., Bear C.E. Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel. Journal of Biological Chemistry. 1996;271:11623–11626. doi: 10.1074/jbc.271.20.11623. [DOI] [PubMed] [Google Scholar]
- 47.Prat A.G., Reisin I.L., Ausiello D.A., Cantiello H.F. Cellular ATP release by the cystic fibrosis transmembrane conductance regulator. American Journal of Physiology. 1996;270:C538–545. doi: 10.1152/ajpcell.1996.270.2.C538. [DOI] [PubMed] [Google Scholar]
- 48.Cantiello H.F. Nucleotide transport through the cystic fibrosis transmembrane conductance regulator. Bioscience Reports. 1997;17:147–171. doi: 10.1023/a:1027381412574. [DOI] [PubMed] [Google Scholar]
- 49.Devidas S., Guggino W.B. The cystic fibrosis transmembrane conductance regulator and ATP. Current Opinion in Cell Biology. 1997;9:547–552. doi: 10.1016/s0955-0674(97)80032-4. [DOI] [PubMed] [Google Scholar]
- 50.Pasyk E.A., Foskett J.K. Cystic fibrosis transmembrane conductance regulator-associated ATP and adenosine 3′-phosphate 5′-phosphosulfate channels in endoplasmic reticulum and plasma membranes. Journal of Biological Chemistry. 1997;272:7746–7751. doi: 10.1074/jbc.272.12.7746. [DOI] [PubMed] [Google Scholar]
- 51.Cantiello H.F., Jackson G.R., Jr., Grosman C.F., Prat A.G., Borkan S.C., Wang Y., Reisin I.L., O'Riordan C.R., Ausiello D.A., Electrodiffusional ATP. movement through the cystic fibrosis transmembrane conductance regulator. American journal of physiology. 1998;274:C799–809. doi: 10.1152/ajpcell.1998.274.3.C799. [DOI] [PubMed] [Google Scholar]
- 52.Abraham E.H., Sterling K.M., Kim R.J., Salikhova A.Y., Huffman H.B., Crockett M.A., Johnston N., Parker H.W., Boyle W.E., Jr., Hartov A., Demidenko E., Efird J., Kahn J., Grubman S.A., Jefferson D.M., Robson S.C., Thakar J.H., Lorico A., Rappa G., Sartorelli A.C., Okunieff P. Erythrocyte membrane ATP binding cassette (ABC) proteins: MRP1 and CFTR as well as CD39 (ecto-apyrase) involved in RBC ATP transport and elevated blood plasma ATP of cystic fibrosis. Blood cells, Molecules and Diseases. 2001;27:165–180. doi: 10.1006/bcmd.2000.0357. [DOI] [PubMed] [Google Scholar]
- 53.Cantiello H.F., Electrodiffusional ATP. movement through CFTR and other ABC transporters. Pflugers Archiv. 2001;443:S22–27. doi: 10.1007/s004240100639. [DOI] [PubMed] [Google Scholar]
- 54.Reigada D., Mitchell C.H. Release of ATP from retinal pigment epithelial cells involves both CFTR and vesicular transport. American Journal of Physiology Cell Physiology. 2005;288:C132–140. doi: 10.1152/ajpcell.00201.2004. [DOI] [PubMed] [Google Scholar]
- 55.Schwiebert E.M., Benos D.J., Egan M.E., Stutts M.J., Guggino W.B. CFTR is a conductance regulator as well as a chloride channel. Physiological Reviews. 1999;79:S145–166. doi: 10.1152/physrev.1999.79.1.S145. [DOI] [PubMed] [Google Scholar]
- 56.Alzamora R., King J.D., Jr., Hallows K.R. CFTR regulation by phosphorylation. Methods in Molecular Biology. 2011;741:471–488. doi: 10.1007/978-1-61779-117-8_29. [DOI] [PubMed] [Google Scholar]
- 57.Fischer H., Machen T.E. The tyrosine kinase p60c-src regulates the fast gate of the cystic fibrosis transmembrane conductance regulator chloride channel. Biophysical Journal. 1996;71:3073–3082. doi: 10.1016/S0006-3495(96)79501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allan B.J., Izutsu K.T., Ramsey B.W., Schubert M.M., Ensign W.Y., Truelove E.L. Cyclic nucleotide responses in control and cystic fibrosis labial glands. American Journal of Physiology. 1990;258:R1320–1326. doi: 10.1152/ajpregu.1990.258.6.R1320. [DOI] [PubMed] [Google Scholar]
- 59.Faria D., Schreiber R., Kunzelmann K. CFTR is activated through stimulation of purinergic P2Y2 receptors. Pflugers Archiv. 2009;457:1373–1380. doi: 10.1007/s00424-008-0606-2. [DOI] [PubMed] [Google Scholar]
- 60.Li C., Naren A.P. Analysis of CFTR interactome in the macromolecular complexes. Methods in Molecular Biology. 2011;741:255–270. doi: 10.1007/978-1-61779-117-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naren A.P., Cobb B., Li C., Roy K., Nelson D., Heda G.D., Liao J., Kirk K.L., Sorscher E.J., Hanrahan J., Clancy J.P. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W., Penmatsa H., Ren A., Punchihewa C., Lemoff A., Yan B., Fujii N., Naren A.P. Functional regulation of cystic fibrosis transmembrane conductance regulator-containing macromolecular complexes: a small-molecule inhibitor approach. Biochemical Journal. 2011;435:451–462. doi: 10.1042/BJ20101725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li C., Naren A.P. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacology and Therapeutics. 2005;108:208–223. doi: 10.1016/j.pharmthera.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 64.Buchwald M., Sood R., Auerbach W. Regulation of expression of CFTR in human intestinal epithelial cells. Advances in Experimental Medicine and Biology. 1991;290:241–250. doi: 10.1007/978-1-4684-5934-0_24. (discussion 242–250) [DOI] [PubMed] [Google Scholar]
- 65.Breuer W., Kartner N., Riordan J.R., Cabantchik Z.I. Induction of expression of the cystic fibrosis transmembrane conductance regulator. Journal of Biological Chemistry. 1992;267:10465–10469. [PubMed] [Google Scholar]
- 66.McDonald R.A., Matthews R.P., Idzerda R.L., McKnight G.S. Basal expression of the cystic fibrosis transmembrane conductance regulator gene is dependent on protein kinase A activity. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7560–7564. doi: 10.1073/pnas.92.16.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Besancon F., Przewlocki G., Baro I., Hongre A.S., Escande D., Edelman A. Interferon-gamma downregulates CFTR gene expression in epithelial cells. American Journal of Physiology. 1994;267:C1398–1404. doi: 10.1152/ajpcell.1994.267.5.C1398. [DOI] [PubMed] [Google Scholar]
- 68.Mularoni A., Beck L., Sadir R., Adessi G.L., Nicollier M. Down-regulation by progesterone of CFTR expression in endometrial epithelial cells: a study by competitive RT-PCR. Biochemical and Biophysical Research Communications. 1995;217:1105–1111. doi: 10.1006/bbrc.1995.2883. [DOI] [PubMed] [Google Scholar]
- 69.Cafferata E.G., González-Guerrico A.M., Giordano L., Pivetta O.H., Santa-Coloma T.A. Interleukin-1beta regulates CFTR expression in human intestinal T84 cells. Biochimica et Biophysica Acta. 2000;1500:241–248. doi: 10.1016/s0925-4439(99)00105-2. [DOI] [PubMed] [Google Scholar]
- 70.Cafferata E.G., González-Guerrico A.M., Pivetta O.H., Santa-Coloma T.A. NF-kappaB activation is involved in regulation of cystic fibrosis transmembrane conductance regulator (CFTR) by interleukin-1beta. Journal of Biological Chemistry. 2001;276:15441–15444. doi: 10.1074/jbc.M010061200. [DOI] [PubMed] [Google Scholar]
- 71.Cafferata E.G.A. Regulación del gen CFTR (afectado en fibrosis quística) en células de carcinoma de colon humano T84. Thesis dissertation School of Pharmacy and Biochemistry, University of Buenos Aires, 2002:1–106. [Google Scholar]
- 72.Osika E., Cavaillon J.M., Chadelat K., Boule M., Fitting C., Tournier G., Clement A. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. European Respiratory Journal. 1999;14:339–346. doi: 10.1034/j.1399-3003.1999.14b17.x. [DOI] [PubMed] [Google Scholar]
- 73.Rochwerger L., Buchwald M. Stimulation of the cystic fibrosis transmembrane regulator expression by estrogen in vivo. Endocrinology. 1993;133:921–930. doi: 10.1210/endo.133.2.7688293. [DOI] [PubMed] [Google Scholar]
- 74.Rowlands D.K., Tsang L.L., Cui Y.G., Chung Y.W., Chan L.N., Liu C.Q., James T., Chan H.C. Upregulation of cystic fibrosis transmembrane conductance regulator expression by oestrogen and Bak Foong Pill in mouse uteri. Cell Biology International. 2001;25:1033–1035. doi: 10.1006/cbir.2001.0746. [DOI] [PubMed] [Google Scholar]
- 75.Longo M., Brama M., Marino M., Bernardini S., Korach K.S., Wetsel W.C., Scandurra R., Faraggiana T., Spera G., Baron R., Teti A., Migliaccio S. Interaction of estrogen receptor alpha with protein kinase C alpha and c-Src in osteoblasts during differentiation. Bone. 2004;34:100–111. doi: 10.1016/j.bone.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 76.Chotirmall S.H., Greene C.M., Oglesby I.K., Thomas W., O'Neill S.J., Harvey B.J., McElvaney N.G. 17Beta-estradiol inhibits IL-8 in cystic fibrosis by up-regulating secretory leucoprotease inhibitor. American Journal of Respiratory and Critical Care Medicine. 2010;182:62–72. doi: 10.1164/rccm.201001-0053OC. [DOI] [PubMed] [Google Scholar]
- 77.Dong Y.J., Chao A.C., Kouyama K., Hsu Y.P., Bocian R.C., Moss R.B., Gardner P. Activation of CFTR chloride current by nitric oxide in human T lymphocytes. EMBO Journal. 1995;14:2700–2707. doi: 10.1002/j.1460-2075.1995.tb07270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jilling T., Haddad I.Y., Cheng S.H., Matalon S. Nitric oxide inhibits heterologous CFTR expression in polarized epithelial cells. American Journal of Physiology. 1999;277:L89–96. doi: 10.1152/ajplung.1999.277.1.L89. [DOI] [PubMed] [Google Scholar]
- 79.Texereau J., Marullo S., Hubert D., Coste J., Dusser D.J., Dall'Ava-Santucci J., Dinh-Xuan A.T. Nitric oxide synthase 1 as a potential modifier gene of decline in lung function in patients with cystic fibrosis. Thorax. 2004;59:156–158. doi: 10.1136/thorax.2003.006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Skinn A.C., MacNaughton W.K. Nitric oxide inhibits cAMP-dependent CFTR trafficking in intestinal epithelial cells. American Journal of Physiology Gastrointestinal and Liver Physiology. 2005;289:G739–744. doi: 10.1152/ajpgi.00425.2004. [DOI] [PubMed] [Google Scholar]
- 81.Baudouin-Legros M., Brouillard F., Cougnon M., Tondelier D., Leclerc T., Edelman A. Modulation of CFTR gene expression in HT-29 cells by extracellular hyperosmolarity. American Journal of Physiology—Cell Physiology. 2000;278:C49–56. doi: 10.1152/ajpcell.2000.278.1.C49. [DOI] [PubMed] [Google Scholar]
- 82.Qu F., Liu H.J., Xiang Y., Tan Y.R., Liu C., Zhu X.L., Qin X.Q. Activation of CFTR trafficking and gating by vasoactive intestinal peptide in human bronchial epithelial cells. Journal of Cellular Biochemistry. 2011;112:902–908. doi: 10.1002/jcb.22999. [DOI] [PubMed] [Google Scholar]
- 83.Alcolado N., Conrad D.J., Rafferty S., Chappe F.G., Chappe V.M. VIP-dependent increase in F508del-CFTR membrane localization is mediated by PKCepsilon. American Journal of Physiology—Cell Physiology. 2011;301:C53–65. doi: 10.1152/ajpcell.00568.2009. [DOI] [PubMed] [Google Scholar]
- 84.Mendes A.I., Matos P., Moniz S., Luz S., Amaral M.D., Farinha C.M., Jordan P. Antagonistic regulation of cystic fibrosis transmembrane conductance regulator cell surface expression by protein kinases WNK4 and spleen tyrosine kinase. Molecular and Cellular Biology. 2011;31:4076–4086. doi: 10.1128/MCB.05152-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Luz S., Kongsuphol P., Mendes A.I., Romeiras F., Sousa M., Schreiber R., Matos P., Jordan P., Mehta A., Amaral M.D., Kunzelmann K., Farinha C.M. Contribution of casein kinase 2 and spleen tyrosine kinase to CFTR trafficking and protein kinase A-induced activity. Molecular and Cellular Biology. 2011;31:4392–4404. doi: 10.1128/MCB.05517-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez-Guerrico A.M., Cafferata E.G., Radrizzani M., Marcucci F., Gruenert D., Pivetta O.H., Favaloro R.R., Laguens R., Perrone S.V., Gallo G.C., Santa-Coloma T.A. Tyrosine kinase c-Src constitutes a bridge between cystic fibrosis transmembrane regulator channel failure and MUC1 overexpression in cystic fibrosis. Journal of Biological Chemistry. 2002;277:17239–17247. doi: 10.1074/jbc.M112456200. [DOI] [PubMed] [Google Scholar]
- 87.A.G., Valdivieso, Modulación de la expresión del gen MTND4 mitocondrial mediada por la actividad del CFTR, Thesis dissertation, School of Natural and Exact Sciences, University of Buenos Aires, 2009, p. 1–118
- 88.Taminelli G.L., Sotomayor V., Valdivieso A.G., Teiber M.L., Marín M.C., Santa-Coloma T.A. CISD1 codifies a mitochondrial protein upregulated by the CFTR channel. Biochemical and Biophysical Research Communications. 2008;365:856–862. doi: 10.1016/j.bbrc.2007.11.076. [DOI] [PubMed] [Google Scholar]
- 89.Estell K., Braunstein G., Tucker T., Varga K., Collawn J.F., Schwiebert L.M. Plasma membrane CFTR regulates RANTES expression via its C-terminal PDZ-interacting motif. Molecular and Cellular Biology. 2003;23:594–606. doi: 10.1128/MCB.23.2.594-606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Srivastava M., Eidelman O., Pollard H.B. Pharmacogenomics of the cystic fibrosis transmembrane conductance regulator (CFTR) and the cystic fibrosis drug CPX using genome microarray analysis. Molecular Medicine. 1999;5:753–767. [PMC free article] [PubMed] [Google Scholar]
- 91.Srivastava M., Eidelman O., Pollard H.B. cDNA microarrays for pharmacogenomic analysis of cystic fibrosis. Methods in Molecular Medicine. 2002;70:21–29. doi: 10.1385/1-59259-187-6:21. [DOI] [PubMed] [Google Scholar]
- 92.Galvin P., Clarke L.A., Harvey S., Amaral M.D. Microarray analysis in cystic fibrosis. Journal of Cystic Fibrosis. 2004;3(Suppl 2):29–33. doi: 10.1016/j.jcf.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 93.Srivastava M., Eidelman O., Jozwik C., Paweletz C., Huang W., Zeitlin P.L., Pollard H.B. Serum proteomic signature for cystic fibrosis using an antibody microarray platform. Molecular Genetics and Metabolism. 2006;87:303–310. doi: 10.1016/j.ymgme.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 94.Valdivieso A.G., Clauzure M., Marín M.C., Taminelli G.L., Massip Copiz M.M., Sánchez F., Schulman G., Teiber M.L., Santa-Coloma T.A. The mitochondrial complex I activity is reduced in cells with impaired Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) function. PLoS ONE. 2012;7(11):e48059. doi: 10.1371/journal.pone.0048059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clauzure M., Sanchez F., Valdivieso A.G., Massip Copiz M.M., Taminelli G.L., Schulman G., Teiber M.L., Santa-Coloma T.A. Abstract 596. Regulación de la actividad mitocondrial en Fibrosis Quística. Abstracts of the 56th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina 16–19 November 2011. Medicina (Buenos Aires) 2012;71(Suppl. III):236. [Google Scholar]
- 96.Teiber M.L., Valdivieso A.G., Massip Copiz M.M., Clauzure M., Sánchez F., Schulman G., Taminelli G.L., Pagano E.S., Santa-Coloma T.A. Abstract 188. La inhibición de la actividad o expresión del canal CFTR disminuye el potencial de membrana mitocondrial e induce apoptosis. Abstracts of the 55th annual meeting of the Argentine society for clinical research. Mar del Plata, Buenos Aires, Argentina; 17–20 November 2010. Medicina (Buenos Aires) 2010;70(Suppl. II):109. [Google Scholar]
- 97.Valdivieso A.G., Taminelli G.L., Marín M.C., Pagano E.S., Teiber M.L., Santa-Coloma T.A. Abstract 529. La inhibición del transporte de Cl- a través del canal CFTR modula la actividad del complejo I mitocondrial. Abstracts of the 53th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 19–22 November 2008. Medicina (Buenos Aires) 2008;68(Suppl. II):209. [Google Scholar]
- 98.Taminelli G.L., Valdivieso A.G., Marín M.C., Teiber M.L., Pagano E.S., Santa-Coloma T.A. Abstract 537. Caracterización parcial de una proteína nuclear de localización mitocondrial denominada CISD1 cuya expresión es modulada por el canal de Cl- CFTR. Abstracts of the 53th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 19–22 November 2008. Medicina (Buenos Aires) 2008;68(Suppl. II):211. [Google Scholar]
- 99.Valdivieso A.G., Taminelli G.L., Santa-Coloma T.A. Abstract 621. La Falla del CFTR en fibrosis quística reduce la actividad del complejo I mitocondrial. Abstracts of the 52th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 21–24 November 2007. Medicina (Buenos Aires) 2007;67(Suppl. III):240. [Google Scholar]
- 100.Valdivieso A.G., Marcucci F., Taminelli G.L., González-Guerrico A.M., Alvarez S., Teiber M.L., Dankert M.A., Santa-Coloma T.A. The expression of the mitochondrial gene MT-ND4 is downregulated in cystic fibrosis. Biochemical and Biophysical Research Communications. 2007;356:805–809. doi: 10.1016/j.bbrc.2007.03.057. [DOI] [PubMed] [Google Scholar]
- 101.Taminelli G.L., Valdivieso A.G., Santa-Coloma T.A. Abstract 625. Expresión diferencial en fibrosis quística de una nueva proteína nuclear de localización mitocondrial denominada CISD1. Abstracts of the 52th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 21–24 November 2007. Medicina (Buenos Aires) 2007;67(Suppl. III):241. [Google Scholar]
- 102.Valdivieso A.G., Taminelli G.L., Teiber M.L., Dankert M.A., Santa-Coloma T.A. Abstract CB-P82. Down-regulation of the mitochondrial gene ND4 in Cystic Fibrosis. Abstracts of the 42th annual meeting of the Argentine society for biochemistry and molecular biology research. Rosario, Argentina; 12–15 November 2006. Abstract Book. 2006;81 [Google Scholar]
- 103.Taminelli G.L., Valdivieso A.G., Teiber M.L., Dankert M.A., Santa-Coloma T.A. Abstract CB-P81. ZCD1 is a new mitochondrial-located protein down-regulated in cystic fibrosis. Abstracts of the 42th annual meeting of the Argentine society for biochemistry and molecular biology research. Rosario, Argentina; 12–15 November 2006. Abstracts Book. 2006;81 [Google Scholar]
- 104.Valdivieso A.G., Taminelli G.L., Tironi Farinati A.C.F., Reyes G.B., Calabró V., Dankert M.A., Santa-Coloma T.A. Abstract 451. Regulación del gen ND4 mitocondrial mediada por el canal de cloruro CFTR. Abstracts of the 50th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 29 November – 2 December 2005. Medicina (Buenos Aires) 2005;65(Suppl. II):167. [Google Scholar]
- 105.Taminelli G.L., Valdivieso A.G., Marín M.C., Tironi Farinati A.C.F., Calabró V., Dankert M.A., Santa-Coloma T.A. Abstract 449. Caracterización de ZCD1, un gen nuclear de localización mitocondrial disminuido en Fibrosis Quística. Abstracts of the 50th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 29 November – 2 December 2005. Medicina (Buenos Aires) 2005;65(Suppl. II):166. [Google Scholar]
- 106.Valdivieso A.G., Marcucci F., Taminelli G.L., Reyes G.B., Dankert M.A., Santa-Coloma T.A. Abstract 308. El canal de cloruro CFTR, afectado en Fibrosis Quística, regula la proteína ND4,una subunidad del Complejo I mitocondrial que es clave para el transporte de electrones. Abstracts of the 49th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina;16–20 November 2004. Medicina (Buenos Aires) 2004;64(Suppl. II):163. [Google Scholar]
- 107.Taminelli G.L., Sotomayor V., Valdivieso A.G., Reyes G.B., Dankert M.A., Santa-Coloma T.A. Abstract 309. KLPX es una proteína de localización mitocondrial, con un dominio “Zn finger” similar al de algunas kinesinas y cuya expresión estí disminuida en fibrosis quística. Abstracts of the 49th annual meeting of the Argentine society for clinical investigation. Mar delPlata, Buenos Aires, Argentina; 16–20 November2004. Medicina (Buenos Aires) 2004;64(Suppl. II):163. [Google Scholar]
- 108.Shapiro B.L. Evidence for a mitochondrial lesion in cystic fibrosis. Life Sciences. 1989;44:1327–1334. doi: 10.1016/0024-3205(89)90389-5. [DOI] [PubMed] [Google Scholar]
- 109.Shapiro B.L. Mitochondrial dysfunction, energy expenditure, and cystic fibrosis. Lancet. 1988;2:289. doi: 10.1016/s0140-6736(88)92591-3. [DOI] [PubMed] [Google Scholar]
- 110.Shapiro B.L., Lam L.F., Feigal R.J. Mitochondrial NADH dehydrogenase in cystic fibrosis: enzyme kinetics in cultured fibroblasts. American Journal of Human Genetics. 1982;34:846–852. [PMC free article] [PubMed] [Google Scholar]
- 111.Shapiro B.L., Feigal R.J., Lam L.F. Mitrochondrial NADH dehydrogenase in cystic fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:2979–2983. doi: 10.1073/pnas.76.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feigal R.J., Shapiro B.L. Altered intracellular calcium in fibroblasts from patients with cystic fibrosis and heterozygotes. Pediatric Research. 1979;13:764–768. doi: 10.1203/00006450-197906000-00009. [DOI] [PubMed] [Google Scholar]
- 113.Feigal R.J., Shapiro B.L. Mitochondrial calcium uptake and oxygen consumption in cystic fibrosis. Nature. 1979;278:276–277. doi: 10.1038/278276a0. [DOI] [PubMed] [Google Scholar]
- 114.Shapiro B.L., Lam L.F. Intracellular calcium in cystic fibrosis heterozygotes. Life Sciences. 1987;40:2361–2366. doi: 10.1016/0024-3205(87)90510-8. [DOI] [PubMed] [Google Scholar]
- 115.Feigal R.J., Shapiro B.L. Cystic fibrosis—a lethal exocrinopathy with altered mitochondrial calcium metabolism. Annals of the New York Academy of Sciences. 1986;488:82–98. [PubMed] [Google Scholar]
- 116.Shapiro B.L., Lam L.F. Calcium and age in fibroblasts from control subjects and patients with cystic fibrosis. Science. 1982;216:417–419. doi: 10.1126/science.7071590. [DOI] [PubMed] [Google Scholar]
- 117.Feigal R.J., Tomczyk M.S., Shapiro B.L. The calcium abnormality in cystic fibrosis mitochondria: relative role of respiration and ATP hydrolysis. Life Sciences. 1982;30:93–98. doi: 10.1016/0024-3205(82)90640-3. [DOI] [PubMed] [Google Scholar]
- 118.Shapiro B.L., Feigal R.J., Laible N.J., Biros M.H., Warwick W.J. Doubling time alpha-aminoisobutyrate transport and calcium exchange in cultured fibroblasts from cystic fibrosis and control subjects. Clinica Chimica Acta. 1978;82:125–131. doi: 10.1016/0009-8981(78)90035-9. [DOI] [PubMed] [Google Scholar]
- 119.Shapiro B.L., Smith Q.T., Warwick W.J. Letter: serum glutathione reductase and cystic fibrosis. Pediatric Research. 1976;10:250. doi: 10.1203/00006450-197604000-00011. [DOI] [PubMed] [Google Scholar]
- 120.Shapiro B.L., Smith Q.T., Warick W.J. Serum glutathione reductase and cystic fibrosis. Pediatric Research. 1975;9:885–888. doi: 10.1203/00006450-197512000-00003. [DOI] [PubMed] [Google Scholar]
- 121.Shapiro B.L., Smith Q.T., Martinez A. Letter: white-cell glutathione reductase in cystic fibrosis. Lancet. 1974;2:1020–1021. doi: 10.1016/s0140-6736(74)92124-2. [DOI] [PubMed] [Google Scholar]
- 122.Shapiro B.L., Smith Q.T., Warwick W.J. Red cell glutathione and glutathione reductase in cystic fibrosis. Proceedings of the Society for Experimental Biology and Medicine. 1973;144:181–183. doi: 10.3181/00379727-144-37552. [DOI] [PubMed] [Google Scholar]
- 123.Antonowicz I., Sippell W.G., Shwachman H. Cystic fibrosis: lysosomal and mitochondrial enzyme activities of lymphoid cell lines. Pediatric Research. 1972;6:803–812. doi: 10.1203/00006450-197211000-00001. [DOI] [PubMed] [Google Scholar]
- 124.Congdon P.J., Littlewood J.M., Aggarwal R.K., Shapiro H. Glucose 6-phosphate dehydrogenase deficiency and cystic fibrosis. Postgraduate Medical Journal. 1981;57:453–454. doi: 10.1136/pgmj.57.669.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shapiro B.L., Lam L.F., Fast L.H. Premature senescence in cultured skin fibroblasts from subjects with cystic fibrosis. Science. 1979;203:1251–1253. doi: 10.1126/science.424752. [DOI] [PubMed] [Google Scholar]
- 126.Wotman S., Mandel I.D., Mercadante J., Denning C.R. Parotid and submaxillary calcium in human cystic fibrosis. Archives of Oral Biology. 1971;16:663–665. doi: 10.1016/0003-9969(71)90070-7. [DOI] [PubMed] [Google Scholar]
- 127.Lobeck C.C. Cystic fibrosis. In: Stanbury J.B., Wyngaarden J.B., Frederickson D.S., editors. McGraw-Hill; New York: 1972. p. 1605. (The metabolic basis of inherited disease). Chapter 68. [Google Scholar]
- 128.Botelho S.Y., Goldstein A.M., Rosenlund M.L. Tear sodium, potassium, chloride, and calcium at various flow rates: children with cystic fibrosis and unaffected siblings with and without corneal staining. Journal of Pediatrics. 1973;83:601–606. doi: 10.1016/s0022-3476(73)80221-5. [DOI] [PubMed] [Google Scholar]
- 129.Allars H.M., Blomfield J., Rush A.R., Brown J.M. Colloid and crystal formation in parotid saliva of cystic fibrosis patients and non-cystic fibrosis subjects. I. Physicochemistry. Pediatric Research. 1976;10:578–584. doi: 10.1203/00006450-197606000-00005. [DOI] [PubMed] [Google Scholar]
- 130.Blomfield J., Rush A.R., Allars H.M., Brown J.M. Parotid gland function in children with cystic fibrosis and child control subjects. Pediatric Research. 1976;10:574–578. doi: 10.1203/00006450-197606000-00004. [DOI] [PubMed] [Google Scholar]
- 131.von Ruecker A.A., Bertele R., Harms H.K. Calcium metabolism and cystic fibrosis: mitochondrial abnormalities suggest a modification of the mitochondrial membrane. Pediatric Research. 1984;18:594–599. doi: 10.1203/00006450-198407000-00005. [DOI] [PubMed] [Google Scholar]
- 132.Waller R.L., Brattin W.J., Dearborn D.G. Cytosolic free calcium concentration and intracellular calcium distribution in lymphocytes from cystic fibrosis patients. Life Sciences. 1984;35:775–781. doi: 10.1016/0024-3205(84)90347-3. [DOI] [PubMed] [Google Scholar]
- 133.Dechecchi M.C., Girella E., Cabrini G., Berton G. The Km of NADH dehydrogenase is decreased in mitochondria of cystic fibrosis cells. Enzyme. 1988;40:45–50. doi: 10.1159/000469141. [DOI] [PubMed] [Google Scholar]
- 134.Rommens J.M., Zengerling S., Burns J., Melmer G., Kerem B.S., Plavsic N., Zsiga M., Kennedy D., Markiewicz D., Rozmahel R., Riordan J.R., Buchwald M., Tsui L.C. Identification and regional localization of DNA markers on chromosome 7 for the cloning of the cystic fibrosis gene. American Journal of Human Genetics. 1988;43:645–663. [PMC free article] [PubMed] [Google Scholar]
- 135.Picci L., Brentagni L., Mastella G., Scarso E., Pizzochero P., Mattiazzo P., Chiandetti L., Anglani F., Zacchello F. 2D-electrophoresis of mitochondrial proteins from cystic fibrosis patients. Advances in Experimental medicine and Biology. 1991;290:379–381. doi: 10.1007/978-1-4684-5934-0_44. [DOI] [PubMed] [Google Scholar]
- 136.de Meer K., Jeneson J.A., Gulmans V.A., van der Laag J., Berger R. Efficiency of oxidative work performance of skeletal muscle in patients with cystic fibrosis. Thorax. 1995;50:980–983. doi: 10.1136/thx.50.9.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Day B.J., van Heeckeren A.M., Min E., Velsor L.W. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infection and Immunity. 2004;72:2045–2051. doi: 10.1128/IAI.72.4.2045-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liang P., Pardee A.B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 139.Cafferata E.G., González-Guerrico A.M., Pivetta O.H., Santa-Coloma T.A. Identification by differential display of a mRNA specifically induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) in T84 human colon carcinoma cells. Cell and Molecular Biology (Noisy-le-grand) 1996;42:797–804. [PubMed] [Google Scholar]
- 140.Cafferata E.G., González-Guerrico A., Pivetta O.H., Santa-Coloma T.A. Abstract M99. Identificación mediante differential display de genes especóficamente regulados por diferentes factores que afectan la expresión del CFTR(canal de cloruro afectado en Fibrosis Quística). Abstracts of the 31st annual meeting of the Argentine society for biochemistry and molecular biology research. Villa Giardino, Córdoba, Argentina; 13-18 November 1995. Abstracts Book. 1995 [Google Scholar]
- 141.A.M. González-Guerrico, Expresión de genes asociados a fibrosis quística. Thesis dissertation, School of Exact and Natural Sciences, University of Buenos Aires; 2001, p. 1–120
- 142.Eidelman O., Zhang J., Srivastava M., Pollard H.B. Cystic fibrosis and the use of pharmacogenomics to determine surrogate endpoints for drug discovery. American Journal of Pharmacogenomics. 2001;1:223–238. doi: 10.2165/00129785-200101030-00006. [DOI] [PubMed] [Google Scholar]
- 143.Pollard H.B., Eidelman O., Jacobson K.A., Srivastava M. Pharmacogenomics of cystic fibrosis. Molecular Interventions. 2001;1:54–63. [PMC free article] [PubMed] [Google Scholar]
- 144.Ichikawa J.K., Norris A., Bangera M.G., Geiss G.K., van't Wout A.B., Bumgarner R.E., Lory S. Interaction of Pseudomonas aeruginosa with epithelial cells: identification of differentially regulated genes by expression microarray analysis of human cDNAs. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9659–9664. doi: 10.1073/pnas.160140297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xu Y., Clark J.C., Aronow B.J., Dey C.R., Liu C., Wooldridge J.L., Whitsett J.A. Transcriptional adaptation to CFTR deficiency. Journal of Biological Chemistry. 2002;12:12. doi: 10.1074/jbc.M210277200. [DOI] [PubMed] [Google Scholar]
- 146.Xu Y., Clark J.C., Aronow B.J., Dey C.R., Liu C., Wooldridge J.L., Whitsett J.A. Transcriptional adaptation to cystic fibrosis transmembrane conductance regulator deficiency. Journal of Biological Chemistry. 2003;278:7674–7682. doi: 10.1074/jbc.M210277200. [DOI] [PubMed] [Google Scholar]
- 147.McCarthy V.A., Harris A. The CFTR gene and regulation of its expression. Pediatric Pulmonology. 2005;40:1–8. doi: 10.1002/ppul.20199. [DOI] [PubMed] [Google Scholar]
- 148.Ogilvie V., Passmore M., Hyndman L., Jones L., Stevenson B., Wilson A., Davidson H., Kitchen R.R., Gray R.D., Shah P., Alton E.W., Davies J.C., Porteous D.J., Boyd A.C. Differential global gene expression in cystic fibrosis nasal and bronchial epithelium. Genomics. 2011;98:327–336. doi: 10.1016/j.ygeno.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 149.Jozwik C.E., Pollard H.B., Srivastava M., Eidelman O., Fan Q., Darling T.N., Zeitlin P.L. Antibody microarrays: analysis of cystic fibrosis. Methods in Molecular Biology. 2012;823:179–200. doi: 10.1007/978-1-60327-216-2_12. [DOI] [PubMed] [Google Scholar]
- 150.Sotomayor V., Marcucci F., Santa-Coloma T.A. Abstract 281. CFTR-RG2, un nuevo miembro de la familia de kinesinas regulado por CFTR. Abstracts of the 47th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 20–23 November 2002. Medicina (Buenos Aires) 2002;62:457. [Google Scholar]
- 151.Sotomayor V., Marcucci F., Santa-Coloma T.A. Abstract 445. Identificación de genes regulados por la actividad del CFTR, canal de cloruro afectado en la fibrosis quística (FQ). Abstracts of the 48th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 19–22 November 2003. Medicina (Buenos Aires) 2003;63:631–635. [Google Scholar]
- 152.V. Sotomayor, Identificación de genes regulados por la actividad del CFTR, canal de cloruro afectado en la fibrosis quística. Thesis (Lic. in Biological Sciences). Department of Biology, School of Exact and Natural Sciences, University of Buenos Aires; 2003, p. 1–56
- 153.Marcucci F., González-Guerrico A.M., Cafferata E., Radrizzani M., Alvarez S., Santa-Coloma T.A. Abstract P031. Identification by differential display of mitochondrial ND4 as a CFTR-dependent gene. Abstracts of the 37th annual meeting of the Argentine society for biochemistry and molecular biology research. Villa Carlos Paz, Córdoba, Argentina, 2001. Biocell. 2001;25(Supl. II):49. [Google Scholar]
- 154.Marcucci F., Sotomayor V., Reyes B., Santa-Coloma T.A. Abstract 282. Identificación mediante display diferencial de ND4 mitocondrial como un gen dependiente de CFTR, canal afectado en fibrosis quística. Abstracts of the 47th annual meeting of the Argentine society for clinical investigation. Mar del Plata, Buenos Aires, Argentina; 20–23 November 2002. Medicina (Buenos Aires) 2002;62(5):457. [Google Scholar]
- 155.F. Marcucci, Identificación de genes regulados por CFTR, canal de cloruro afectado en fibrosis quística. Thesis (Lic. in Biology). Department of Biology, School of Exact and Natural Sciences, University of Buenos Aires; 2003, p. 1–82
- 156.Marchler-Bauer A., Panchenko A.R., Shoemaker B.A., Thiessen P.A., Geer L.Y., Bryant S.H. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Research. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schultz J., Milpetz F., Bork P., Ponting C.P. SMART, a simple modular architecture research tool: identification of signaling domains. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Strausberg R.L., Feingold E.A., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F., Zeeberg B., Buetow K.H., Schaefer C.F., Bhat N.K., Hopkins R.F., Jordan H., Moore T., Max S.I., Wang J., Hsieh F., Diatchenko L., Marusina K., Farmer A.A., Rubin G.M., Hong L., Stapleton M., Soares M.B., Bonaldo M.F., Casavant T.L., Scheetz T.E., Brownstein M.J., Usdin T.B., Toshiyuki S., Carninci P., Prange C., Raha S.S., Loquellano N.A., Peters G.J., Abramson R.D., Mullahy S.J., Bosak S.A., McEwan P.J., McKernan K.J., Malek J.A., Gunaratne P.H., Richards S., Worley K.C., Hale S., Garcia A.M., Gay L.J., Hulyk S.W., Villalon D.K., Muzny D.M., Sodergren E.J., Lu X., Gibbs R.A., Fahey J., Helton E., Ketteman M., Madan A., Rodrigues S., Sanchez A., Whiting M., Young A.C., Shevchenko Y., Bouffard G.G., Blakesley R.W., Touchman J.W., Green E.D., Dickson M.C., Rodriguez A.C., Grimwood J., Schmutz J., Myers R.M., Butterfield Y.S., Krzywinski M.I., Skalska U., Smailus D.E., Schnerch A., Schein J.E., Jones S.J., Marra M.A. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J.P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann N., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J.C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R.H., Wilson R.K., Hillier L.W., McPherson J.D., Marra M.A., Mardis E.R., Fulton L.A., Chinwalla A.T., Pepin K.H., Gish W.R., Chissoe S.L., Wendl M.C., Delehaunty K.D., Miner T.L., Delehaunty A., Kramer J.B., Cook L.L., Fulton R.S., Johnson D.L., Minx P.J., Clifton S.W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J.F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R.A., Muzny D.M., Scherer S.E., Bouck J.B., Sodergren E.J., Worley K.C., Rives C.M., Gorrell J.H., Metzker M.L., Naylor S.L., Kucherlapati R.S., Nelson D.L., Weinstock G.M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D.R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H.M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R.W., Federspiel N.A., Abola A.P., Proctor M.J., Myers R.M., Schmutz J., Dickson M., Grimwood J., Cox D.R., Olson M.V., Kaul R., Shimizu N., Kawasaki K., Minoshima S., Evans G.A., Athanasiou M., Schultz R., Roe B.A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W.R., de la Bastide M., Dedhia N., Blocker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J.A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D.G., Burge C.B., Cerutti L., Chen H.C., Church D., Clamp M., Copley R.R., Doerks T., Eddy S.R., Eichler E.E., Furey T.S., Galagan J., Gilbert J.G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L.S., Jones T.A., Kasif S., Kaspryzk A., Kennedy S., Kent W.J., Kitts P., Koonin E.V., Korf I., Kulp D., Lancet D., Lowe T.M., McLysaght A., Mikkelsen T., Moran J.V., Mulder N., Pollara V.J., Ponting C.P., Schuler G., Schultz J., Slater G., Smit A.F., Stupka E., Szustakowski J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y.I., Wolfe K.H., Yang S.P., Yeh R.F., Collins F., Guyer M.S., Peterson J., Felsenfeld A., Wetterstrand K.A., Patrinos A., Morgan M.J., de Jong P., Catanese J.J., Osoegawa K., Shizuya H., Choi S., Chen Y.J. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 160.Derosa G., Maffioli P. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonists on glycemic control, lipid profile and cardiovascular risk. Current Molecular Pharmacology. 2011;5(2):272–281. doi: 10.2174/1874467211205020272. [DOI] [PubMed] [Google Scholar]
- 161.Colca J.R., McDonald W.G., Waldon D.J., Leone J.W., Lull J.M., Bannow C.A., Lund E.T., Mathews W.R. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. American Journal of Physiology—Endocrinology and Metabolism. 2004;286:E252–260. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 162.Hou X., Liu R., Ross S., Smart E.J., Zhu H., Gong W. Crystallographic studies of human MitoNEET. Journal of Biological Chemistry. 2007;282(46):33242–33246. doi: 10.1074/jbc.C700172200. [DOI] [PubMed] [Google Scholar]
- 163.Lin J., Zhou T., Ye K., Wang J. Crystal structure of human mitoNEET reveals distinct groups of iron sulfur proteins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14640–14645. doi: 10.1073/pnas.0702426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Paddock M.L., Wiley S.E., Axelrod H.L., Cohen A.E., Roy M., Abresch E.C., Capraro D., Murphy A.N., Nechushtai R., Dixon J.E., Jennings P.A. MitoNEET is a uniquely folded 2Fe-2S outer mitochondrial membrane protein stabilized by pioglitazone. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14342–14347. doi: 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wiley S.E., Paddock M.L., Abresch E.C., Gross L., van der Geer P., Nechushtai R., Murphy A.N., Jennings P.A., Dixon J.E. The outer mitochondrial membrane protein mitoNEET contains a novel redox-active 2Fe-2S cluster. Journal of Biological Chemistry. 2007;282:23745–23749. doi: 10.1074/jbc.C700107200. [DOI] [PubMed] [Google Scholar]
- 166.Wiley S.E., Murphy A.N., Ross S.A., van der Geer P., Dixon J.E. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5318–5323. doi: 10.1073/pnas.0701078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tan G., Landry A.P., Dai R., Wang L., Lu J., Ding H. Competition of zinc ion for the [2Fe-2S] cluster binding site in the diabetes drug target protein mitoNEET. Biometals. 2012;25(6):1177–1184. doi: 10.1007/s10534-012-9580-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bieganski R.M., Yarmush M.L. Novel ligands that target the mitochondrial membrane protein mitoNEET. Journal of Molecular Graphics and Modelling. 2011;29:965–973. doi: 10.1016/j.jmgm.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zuris J.A., Harir Y., Conlan A.R., Shvartsman M., Michaeli D., Tamir S., Paddock M.L., Onuchic J.N., Mittler R., Cabantchik Z.I., Jennings P.A., Nechushtai R. Facile transfer of [2Fe–2S] clusters from the diabetes drug target mitoNEET to an apo-acceptor protein. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(32):13047–13052. doi: 10.1073/pnas.1109986108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhou T., Lin J., Feng Y., Wang J. Binding of reduced nicotinamide adenine dinucleotide phosphate destabilizes the iron–sulfur clusters of human mitoNEET. Biochemistry. 2010;49:9604–9612. doi: 10.1021/bi101168c. [DOI] [PubMed] [Google Scholar]
- 171.Zuris J.A., Ali S.S., Yeh H., Nguyen T.A., Nechushtai R., Paddock M.L., Jennings P.A. NADPH inhibits [2Fe-2S] cluster protein transfer from diabetes drug target MitoNEET to an apo-acceptor protein. Journal of Biological Chemistry. 2012;287:11649–11655. doi: 10.1074/jbc.M111.319731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chomyn A. Mitochondrial genetic control of assembly and function of complex I in mammalian cells. Journal of Bioenergetics and Biomembranes. 2001;33:251–257. doi: 10.1023/a:1010791204961. [DOI] [PubMed] [Google Scholar]
- 173.Carroll J., Fearnley I.M., Skehel J.M., Shannon R.J., Hirst J., Walker J.E. Bovine complex I is a complex of 45 different subunits. Journal of Biological Chemistry. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 174.Hofhaus G., Attardi G. Lack of assembly of mitochondrial DNA-encoded subunits of respiratory NADH dehydrogenase and loss of enzyme activity in a human cell mutant lacking the mitochondrial ND4 gene product. EMBO Journal. 1993;12:3043–3048. doi: 10.1002/j.1460-2075.1993.tb05973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hofhaus G., Attardi G. Efficient selection and characterization of mutants of a human cell line which are defective in mitochondrial DNA-encoded subunits of respiratory NADH dehydrogenase. Molecular and Cellular Biology. 1995;15:964–974. doi: 10.1128/mcb.15.2.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bai Y., Hajek P., Chomyn A., Chan E., Seo B.B., Matsuno-Yagi A., Yagi T., Attardi G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. Journal of Biological Chemistry. 2001;276:38808–38813. doi: 10.1074/jbc.M106363200. [DOI] [PubMed] [Google Scholar]
- 177.Cardol P., Matagne R.F., Remacle C. Impact of mutations affecting ND mitochondria-encoded subunits on the activity and assembly of complex I in Chlamydomonas. Implication for the structural organization of the enzyme. Journal of Molecular Biology. 2002;319:1211–1221. doi: 10.1016/S0022-2836(02)00407-2. [DOI] [PubMed] [Google Scholar]
- 178.Bourges I., Ramus C., Mousson de Camaret B., Beugnot R., Remacle C., Cardol P., Hofhaus G., Issartel J.P. Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochemical Journal. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Qi X., Sun L., Lewin A.S., Hauswirth W.W., Guy J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Investigative Ophthalmology and Visual Science. 2007;48:1–10. doi: 10.1167/iovs.06-0789. [DOI] [PubMed] [Google Scholar]
- 180.Baracca A., Solaini G., Sgarbi G., Lenaz G., Baruzzi A., Schapira A.H., Martinuzzi A., Carelli V. Severe impairment of complex I-driven adenosine triphosphate synthesis in leber hereditary optic neuropathy cybrids. Archives of Neurology. 2005;62:730–736. doi: 10.1001/archneur.62.5.730. [DOI] [PubMed] [Google Scholar]
- 181.Majander A., Huoponen K., Savontaus M.L., Nikoskelainen E., Wikstrom M. Electron transfer properties of NADH:ubiquinone reductase in the ND1/3460 and the ND4/11778 mutations of the Leber hereditary optic neuroretinopathy (LHON) FEBS Letters. 1991;292:289–292. doi: 10.1016/0014-5793(91)80886-8. [DOI] [PubMed] [Google Scholar]
- 182.Kelly-Aubert M., Trudel S., Fritsch J., Nguyen-Khoa T., Baudouin-Legros M., Moriceau S., Jeanson L., Djouadi F., Matar C., Conti M., Ollero M., Brouillard F., Edelman A. GSH monoethyl ester rescues mitochondrial defects in cystic fibrosis models. Human Molecular Genetics. 2011;20:2745–2759. doi: 10.1093/hmg/ddr173. [DOI] [PubMed] [Google Scholar]
- 183.Antigny F., Norez C., Becq F., Vandebrouck C. CFTR and Ca signaling in cystic fibrosis. Frontiers in Pharmacology. 2011;2:67. doi: 10.3389/fphar.2011.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Donnell G.N., Cleland R.S. Intestinal atresia or stenosis in the newborn associated with fibrocystic disease of the pancreas. California Medicine. 1961;94:165–170. [PMC free article] [PubMed] [Google Scholar]
- 185.Marmar J., Barbero G.J., Sibinga M.S. The pattern of parotid gland secretion in cystic fibrosis of the pancreas. Gastroenterology. 1966;50:551–556. [PubMed] [Google Scholar]
- 186.Blomfield J., Dascalu J., van Lennep E.W., Brown J.M. Hypersecretion of zymogen granules in the pathogenesis of cystic fibrosis. Gut. 1973;14:558–565. doi: 10.1136/gut.14.7.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Antigny F., Girardin N., Raveau D., Frieden M., Becq F., Vandebrouck C. Dysfunction of mitochondria Ca2+ uptake in cystic fibrosis airway epithelial cells. Mitochondrion. 2009;9:232–241. doi: 10.1016/j.mito.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 188.Ribeiro C.M., Paradiso A.M., Carew M.A., Shears S.B., Boucher R.C. Cystic fibrosis airway epithelial Ca2+ i signaling: the mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. Journal of Biological Chemistry. 2005;280:10202–10209. doi: 10.1074/jbc.M410617200. [DOI] [PubMed] [Google Scholar]
- 189.Tabary O., Boncoeur E., de Martin R., Pepperkok R., Clement A., Schultz C., Jacquot J. Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cellular Signalling. 2006;18:652–660. doi: 10.1016/j.cellsig.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 190.Galli F., Battistoni A., Gambari R., Pompella A., Bragonzi A., Pilolli F., Iuliano L., Piroddi M., Dechecchi M.C., Cabrini G. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochimica et Biophysica Acta. 2012;1822:690–713. doi: 10.1016/j.bbadis.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 191.l'Hoste S., Chargui A., Belfodil R., Corcelle E., Duranton C., Rubera I., Poujeol C., Mograbi B., Tauc M., Poujeol P. CFTR mediates apoptotic volume decrease and cell death by controlling glutathione efflux and ROS production in cultured mice proximal tubules. American Journal of Physiology—Renal Physiology. 2010;298:F435–453. doi: 10.1152/ajprenal.00286.2009. [DOI] [PubMed] [Google Scholar]
- 192.Rottner M., Tual-Chalot S., Mostefai H.A., Andriantsitohaina R., Freyssinet J.M., Martinez M.C. Increased oxidative stress induces apoptosis in human cystic fibrosis cells. PLoS One. 2011;6:e24880. doi: 10.1371/journal.pone.0024880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Velsor L.W., Kariya C., Kachadourian R., Day B.J. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. American Journal of Respiratory Cell and Molecular Biology. 2006;35:579–586. doi: 10.1165/rcmb.2005-0473OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang Y., Li X., Grassme H., Doring G., Gulbins E. Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. Journal of Immunology. 2010;184:5104–5111. doi: 10.4049/jimmunol.0902851. [DOI] [PubMed] [Google Scholar]
- 195.Luciani A., Villella V.R., Esposito S., Brunetti-Pierri N., Medina D., Settembre C., Gavina M., Pulze L., Giardino I., Pettoello-Mantovani M., D'Apolito M., Guido S., Masliah E., Spencer B., Quaratino S., Raia V., Ballabio A., Maiuri L. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature Cell Biology. 2010;12:863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 196.Thevenod F. Multifaceted CFTR: novel role in ROS signaling and apoptotic cell death—a commentary on “CFTR mediates cadmium-induced apoptosis through modulation of ROS levels in mouse proximal tubule cells”. Free Radical Biology and Medicine. 2009;46:1014–1016. doi: 10.1016/j.freeradbiomed.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 197.L'Hoste S., Chargui A., Belfodil R., Duranton C., Rubera I., Mograbi B., Poujeol C., Tauc M., Poujeol P. CFTR mediates cadmium-induced apoptosis through modulation of ROS level in mouse proximal tubule cells. Free Radical Biology and Medicine. 2009;46:1017–1031. doi: 10.1016/j.freeradbiomed.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 198.Ahmad S., Nichols D.P., Strand M., Rancourt R.C., Randell S.H., White C.W., Ahmad A. SERCA2 regulates non-CF and CF airway epithelial cell response to ozone. PLoS One. 2011;6:e27451. doi: 10.1371/journal.pone.0027451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kamdar O., Le W., Zhang J., Ghio A.J., Rosen G.D., Upadhyay D. Air pollution induces enhanced mitochondrial oxidative stress in cystic fibrosis airway epithelium. FEBS Letters. 2008;582:3601–3606. doi: 10.1016/j.febslet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jendrossek V., Grassme H., Mueller I., Lang F., Gulbins E. Pseudomonas aeruginosa-induced apoptosis involves mitochondria and stress-activated protein kinases. Infection and Immunity. 2001;69:2675–2683. doi: 10.1128/IAI.69.4.2675-2683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cleeter M.W., Cooper J.M., Schapira A.H. Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: evidence for free radical involvement. Journal of Neurochemistry. 1992;58:786–789. doi: 10.1111/j.1471-4159.1992.tb09789.x. [DOI] [PubMed] [Google Scholar]
- 202.Esposito L.A., Melov S., Panov A., Cottrell B.A., Wallace D.C. Mitochondrial disease in mouse results in increased oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liang F.Q., Godley B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Experimental Eye Research. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- 204.Van Houten B., Woshner V., Santos J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 205.Escames G., Lopez L.C., Garcia J.A., Garcia-Corzo L., Ortiz F., Acuna-Castroviejo D. Mitochondrial DNA and inflammatory diseases. Human Genetics. 2012;131:161–173. doi: 10.1007/s00439-011-1057-y. [DOI] [PubMed] [Google Scholar]
- 206.Patel A.S., Morse D., Choi A.M. Regulation and functional significance of autophagy in respiratory cell biology and disease. American Journal of Respiratory Cell and Molecular Biology. 2012;48(1):1–9. doi: 10.1165/rcmb.2012-0282TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Markossian K.A., Kurganov B.I. Protein folding, misfolding, and aggregation. Formation of inclusion bodies and aggresomes. Biochemistry (Mosc) 2004;69:971–984. doi: 10.1023/b:biry.0000043539.07961.4c. [DOI] [PubMed] [Google Scholar]
- 208.Luciani A., Villella V.R., Esposito S., Brunetti-Pierri N., Medina D.L., Settembre C., Gavina M., Raia V., Ballabio A., Maiuri L. Cystic fibrosis: a disorder with defective autophagy. Autophagy. 2011;7:104–106. doi: 10.4161/auto.7.1.13987. [DOI] [PubMed] [Google Scholar]
- 209.Abdulrahman B.A., Khweek A.A., Akhter A., Caution K., Kotrange S., Abdelaziz D.H., Newland C., Rosales-Reyes R., Kopp B., McCoy K., Montione R., Schlesinger L.S., Gavrilin M.A., Wewers M.D., Valvano M.A., Amer A.O. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7:1359–1370. doi: 10.4161/auto.7.11.17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Luciani A., Villella V.R., Esposito S., Gavina M., Russo I., Silano M., Guido S., Pettoello-Mantovani M., Carnuccio R., Scholte B., De Matteis A., Maiuri M.C., Raia V., Luini A., Kroemer G., Maiuri L. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DeltaF508 cystic fibrosis transmembrane conductance regulator. Autophagy. 2012;8:1657–1672. doi: 10.4161/auto.21483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gao L., Kim K.J., Yankaskas J.R., Forman H.J. Abnormal glutathione transport in cystic fibrosis airway epithelia. American Journal of Physiology. 1999;277:L113–118. doi: 10.1152/ajplung.1999.277.1.L113. [DOI] [PubMed] [Google Scholar]
- 212.Linsdell P., Hanrahan J.W. Glutathione permeability of CFTR. American Journal of Physiology. 1998;275:C323–326. doi: 10.1152/ajpcell.1998.275.1.C323. [DOI] [PubMed] [Google Scholar]
- 213.Velsor L.W., van Heeckeren A., Day B.J. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. American Journal of Physiology Lung Cellular and Molecular Physiology. 2001;281:L31–38. doi: 10.1152/ajplung.2001.281.1.L31. [DOI] [PubMed] [Google Scholar]
- 214.Fernandez-Checa J.C., Garcia-Ruiz C., Ookhtens M., Kaplowitz N. Impaired uptake of glutathione by hepatic mitochondria from chronic ethanol-fed rats. Tracer kinetic studies in vitro and in vivo and susceptibility to oxidant stress. Journal of Clinical Investigation. 1991;87:397–405. doi: 10.1172/JCI115010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Martensson J., Jain A., Frayer W., Meister A. Glutathione metabolism in the lung: inhibition of its synthesis leads to lamellar body and mitochondrial defects. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:5296–5300. doi: 10.1073/pnas.86.14.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Martensson J., Meister A. Mitochondrial damage in muscle occurs after marked depletion of glutathione and is prevented by giving glutathione monoester. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:471–475. doi: 10.1073/pnas.86.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Trudel S., Kelly M., Fritsch J., Nguyen-Khoa T., Therond P., Couturier M., Dadlez M., Debski J., Touqui L., Vallee B., Ollero M., Edelman A., Brouillard F. Peroxiredoxin 6 fails to limit phospholipid peroxidation in lung from Cftr-knockout mice subjected to oxidative challenge. PLoS One. 2009;4:e6075. doi: 10.1371/journal.pone.0006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang Y., Manevich Y., Feinstein S.I., Fisher A.B. Adenovirus-mediated transfer of the 1-cys peroxiredoxin gene to mouse lung protects against hyperoxic injury. American Journal of Physiology Lung Cellular and Molecular Physiology. 2004;286:L1188–1193. doi: 10.1152/ajplung.00288.2003. [DOI] [PubMed] [Google Scholar]
- 219.Wang Y., Phelan S.A., Manevich Y., Feinstein S.I., Fisher A.B. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. American Journal of Respiratory Cell and Molecular Biology. 2006;34:481–486. doi: 10.1165/rcmb.2005-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fisher A.B., Dodia C., Manevich Y., Chen J.W., Feinstein S.I. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. Journal of Biological Chemistry. 1999;274:21326–21334. doi: 10.1074/jbc.274.30.21326. [DOI] [PubMed] [Google Scholar]
- 221.Madarasi A., Lugassi A., Greiner E., Holics K., Biro L., Mozsary E. Antioxidant status in patients with cystic fibrosis. Annals of Nutrition and Metabolism. 2000;44:207–211. doi: 10.1159/000046685. [DOI] [PubMed] [Google Scholar]
- 222.Percival S.S., Bowser E., Wagner M. Reduced copper enzyme activities in blood cells of children with cystic fibrosis. American Journal of Clinical Nutrition. 1995;62:633–638. doi: 10.1093/ajcn/62.3.633. [DOI] [PubMed] [Google Scholar]
- 223.Percival S.S., Kauwell G.P., Bowser E., Wagner M. Altered copper status in adult men with cystic fibrosis. Journal of the American College of Nutrition. 1999;18:614–619. doi: 10.1080/07315724.1999.10718896. [DOI] [PubMed] [Google Scholar]
- 224.Gallagher A.M., Gottlieb R.A. Proliferation, not apoptosis, alters epithelial cell migration in small intestine of CFTR null mice. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2001;281:G681–687. doi: 10.1152/ajpgi.2001.281.3.G681. [DOI] [PubMed] [Google Scholar]
- 225.Jendrossek V., Fillon S., Belka C., Muller I., Puttkammer B., Lang F. Apoptotic response of Chang cells to infection with Pseudomonas aeruginosa strains PAK and PAO-I: molecular ordering of the apoptosis signaling cascade and role of type IV pili. Infection and Immunity. 2003;71:2665–2673. doi: 10.1128/IAI.71.5.2665-2673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kirschnek S., Gulbins E. Phospholipase A2 functions in Pseudomonas aeruginosa-induced apoptosis. Infection and Immunity. 2006;74:850–860. doi: 10.1128/IAI.74.2.850-860.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Rottner M., Kunzelmann C., Mergey M., Freyssinet J.M., Martinez M.C. Exaggerated apoptosis and NF-kappaB activation in pancreatic and tracheal cystic fibrosis cells. FASEB Journal. 2007;21:2939–2948. doi: 10.1096/fj.06-7614com. [DOI] [PubMed] [Google Scholar]
- 228.Schwarzer C., Fu Z., Patanwala M., Hum L., Lopez-Guzman M., Illek B., Kong W., Lynch S.V., Machen T.E. Pseudomonas aeruginosa biofilm-associated homoserine lactone C12 rapidly activates apoptosis in airway epithelia. Cellular Microbiology. 2012;14:698–709. doi: 10.1111/j.1462-5822.2012.01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Barriere H., Poujeol C., Tauc M., Blasi J.M., Counillon L., Poujeol P. CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. American Journal of Physiology Cell Physiology. 2001;281:C810–824. doi: 10.1152/ajpcell.2001.281.3.C810. [DOI] [PubMed] [Google Scholar]
- 230.Bals R., Weiner D.J., Wilson J.M. The innate immune system in cystic fibrosis lung disease. Journal of Clinical Investigation. 1999;103:303–307. doi: 10.1172/JCI6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Conese M. Cystic fibrosis and the innate immune system: therapeutic implications. Endocrine, Metabolic and Immune Disorders Drug Targets. 2011;11:8–22. doi: 10.2174/187153011794982022. [DOI] [PubMed] [Google Scholar]
- 232.Kronborg G., Hansen M.B., Svenson M., Fomsgaard A., Hoiby N., Bendtzen K. Cytokines in sputum and serum from patients with cystic fibrosis and chronic Pseudomonas aeruginosa infection as markers of destructive inflammation in the lungs. Pediatric Pulmonology. 1993;15:292–297. doi: 10.1002/ppul.1950150506. [DOI] [PubMed] [Google Scholar]
- 233.Kronborg G. Lipopolysaccharide (LPS), LPS-immune complexes and cytokines as inducers of pulmonary inflammation in patients with cystic fibrosis and chronic Pseudomonas aeruginosa lung infection. APMIS Supplementum. 1995;50:1–30. [PubMed] [Google Scholar]
- 234.Dai Y., Dean T.P., Church M.K., Warner J.O., Shute J.K. Desensitisation of neutrophil responses by systemic interleukin 8 in cystic fibrosis. Thorax. 1994;49:867–871. doi: 10.1136/thx.49.9.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Schuster A., Haarmann A., Wahn V. Cytokines in neutrophil-dominated airway inflammation in patients with cystic fibrosis. European Archives of Oto-Rhino-Laryngology Supplement. 1995;1:S59–60. doi: 10.1007/BF02484436. [DOI] [PubMed] [Google Scholar]
- 236.Salva P.S., Doyle N.A., Graham L., Eigen H., Doerschuk C.M. TNF-alpha, IL-8, soluble ICAM-1, and neutrophils in sputum of cystic fibrosis patients. Pediatric Pulmonology. 1996;21:11–19. doi: 10.1002/(SICI)1099-0496(199601)21:1<11::AID-PPUL2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 237.Koller D.Y., Nething I., Otto J., Urbanek R., Eichler I. Cytokine concentrations in sputum from patients with cystic fibrosis and their relation to eosinophil activity. American Journal of Respiratory and Critical Care Medicine. 1997;155:1050–1054. doi: 10.1164/ajrccm.155.3.9116985. [DOI] [PubMed] [Google Scholar]
- 238.Palfreyman R.W., Watson M.L., Eden C., Smith A.W. Induction of biologically active interleukin-8 from lung epithelial cells by Burkholderia (Pseudomonas) cepacia products. Infection and Immunity. 1997;65:617–622. doi: 10.1128/iai.65.2.617-622.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Nixon L.S., Yung B., Bell S.C., Elborn J.S., Shale D.J. Circulating immunoreactive interleukin-6 in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1998;157:1764–1769. doi: 10.1164/ajrccm.157.6.9704086. [DOI] [PubMed] [Google Scholar]
- 240.Karpati F., Hjelte F.L., Wretlind B. TNF-alpha and IL-8 in consecutive sputum samples from cystic fibrosis patients during antibiotic treatment. Scandinavian Journal of Infectious Diseases. 2000;32:75–79. doi: 10.1080/00365540050164263. [DOI] [PubMed] [Google Scholar]
- 241.McGarvey L.P., Dunbar K., Martin S.L., Brown V., Macmahon J., Ennis M., Elborn J.S. Cytokine concentrations and neutrophil elastase activity in bronchoalveolar lavage and induced sputum from patients with cystic fibrosis, mild asthma and healthy volunteers. Journal of Cystic Fibrosis. 2002;1:269–275. doi: 10.1016/s1569-1993(02)00098-x. [DOI] [PubMed] [Google Scholar]
- 242.Colombo C., Costantini D., Rocchi A., Cariani L., Garlaschi M.L., Tirelli S., Calori G., Copreni E., Conese M. Cytokine levels in sputum of cystic fibrosis patients before and after antibiotic therapy. Pediatric Pulmonology. 2005;40:15–21. doi: 10.1002/ppul.20237. [DOI] [PubMed] [Google Scholar]
- 243.Sagel S.D., Kapsner R.K., Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatric Pulmonology. 2005;39:224–232. doi: 10.1002/ppul.20165. [DOI] [PubMed] [Google Scholar]
- 244.Mackerness K.J., Jenkins G.R., Bush A., Jose P.J. Characterisation of the range of neutrophil stimulating mediators in cystic fibrosis sputum. Thorax. 2008;63:614–620. doi: 10.1136/thx.2007.089359. [DOI] [PubMed] [Google Scholar]
- 245.Nichols D., Chmiel J., Berger M. Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clinical Reviews in Allergy and Immunology. 2008;34:146–162. doi: 10.1007/s12016-007-8039-9. [DOI] [PubMed] [Google Scholar]
- 246.Martin S.L., Moffitt K.L., McDowell A., Greenan C., Bright-Thomas R.J., Jones A.M., Webb A.K., Elborn J.S. Association of airway cathepsin B and S with inflammation in cystic fibrosis. Pediatric Pulmonology. 2010;45:860–868. doi: 10.1002/ppul.21274. [DOI] [PubMed] [Google Scholar]
- 247.Cohen T.S., Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nature Medicine. 2012;18:509–519. doi: 10.1038/nm.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Becker K.A., Henry B., Ziobro R., Tummler B., Gulbins E., Grassme H. Role of CD95 in pulmonary inflammation and infection in cystic fibrosis. Journal of Molecular Medicine (Berlin) 2012;90(9):1011–1023. doi: 10.1007/s00109-012-0867-2. [DOI] [PubMed] [Google Scholar]
- 249.Su X., Looney M.R., Su H.E., Lee J.W., Song Y., Matthay M.A. Role of CFTR expressed by neutrophils in modulating acute lung inflammation and injury in mice. Inflammation Research. 2011;60:619–632. doi: 10.1007/s00011-011-0313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Regamey N., Jeffery P.K., Alton E.W., Bush A., Davies J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax. 2011;66:624–629. doi: 10.1136/thx.2009.134106. [DOI] [PubMed] [Google Scholar]
- 251.Carroll T.P., Greene C.M., Taggart C.C., McElvaney N.G., ONeill S.J. Interleukin-1, neutrophil elastase, and lipopolysaccharide: key pro-inflammatory stimuli regulating inflammation in cystic fibrosis. Current Respiratory Medicine Reviews. 2005;1:43–67. [Google Scholar]
- 252.Meissner A., Yang J., Kroetsch J.T., Sauve M., Dax H., Momen A., Noyan-Ashraf M.H., Heximer S., Husain M., Lidington D., Bolz S.S. Tumor necrosis factor-α-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation. 2012;125(22):2739–2750. doi: 10.1161/CIRCULATIONAHA.111.047316. [DOI] [PubMed] [Google Scholar]
- 253.Nakamura H., Yoshimura K., Bajocchi G., Trapnell B.C., Pavirani A., Crystal R.G. Tumor necrosis factor modulation of expression of the cystic fibrosis transmembrane conductance regulator gene. FEBS Letters. 1992;314:366–370. doi: 10.1016/0014-5793(92)81507-i. [DOI] [PubMed] [Google Scholar]
- 254.Elborn J.S., Norman D., Delamere F.M., Shale D.J. In vitro tumor necrosis factor-alpha secretion by monocytes from patients with cystic fibrosis. American Journal of Respiratory Cell and Molecular Biology. 1992;6:207–211. doi: 10.1165/ajrcmb/6.2.207. [DOI] [PubMed] [Google Scholar]
- 255.Brown M.A., Morgan W.J., Finley P.R., Scuderi P. Circulating levels of tumor necrosis factor and interleukin-1 in cystic fibrosis. Pediatric Pulmonology. 1991;10:86–91. doi: 10.1002/ppul.1950100209. [DOI] [PubMed] [Google Scholar]
- 256.Vij N., Mazur S., Zeitlin P.L. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS One. 2009;4:e4664. doi: 10.1371/journal.pone.0004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lopez-Armada M.J., Carames B., Martin M.A., Cillero-Pastor B., Lires-Dean M., Fuentes-Boquete I., Arenas J., Blanco F.J. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthritis and Cartilage. 2006;14:1011–1022. doi: 10.1016/j.joca.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 258.Clauzure M., Valdivieso A.G., Sánchez F., Taminelli G.L., Pagano E.S., Massip Copiz M.M., Schulman G., Teiber M.L., Santa-Coloma T.A. Abstract 182. Efecto de IL-1β en la regulación de la actividad mitocondrial en Fibrosis Quística. Abstracts of the 55th annual meeting of the Argentine society for clinical research. Mar del Plata, Buenos Aires, Argentina; 17–20 November 2010. Medicina (Buenos Aires) 2010;70(Suppl. II):107. [Google Scholar]
- 259.Bartling T.R., Drumm M.L. Oxidative stress causes IL8 promoter hyperacetylation in cystic fibrosis airway cell models. American Journal of Respiratory Cell and Molecular Biology. 2009;40:58–65. doi: 10.1165/rcmb.2007-0464OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Montemurro P., Mariggio M.A., Barbuti G., Cassano A., Vincenti A., Serio G., Guerra L., Diana A., Santostasi T., Polizzi A., Fumarulo R., Casavola V., Manca A., Conese M. Increase in interleukin-8 production from circulating neutrophils upon antibiotic therapy in cystic fibrosis patients. Journal of Cystic Fibrosis. 2012;11(6):518–524. doi: 10.1016/j.jcf.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 261.Saadane A., Eastman J., Berger M., Bonfield T.L. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. Journal of Inflammation (London) 2011;8:26. doi: 10.1186/1476-9255-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kaza S.K., McClean S., Callaghan M. IL-8 released from human lung epithelial cells induced by cystic fibrosis pathogens Burkholderia cepacia complex affects the growth and intracellular survival of bacteria. International Journal of Medical Microbiology. 2011;301:26–33. doi: 10.1016/j.ijmm.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 263.Brandolini L., Sergi R., Caselli G., Boraschi D., Locati M., Sozzani S., Bertini R. Interleukin-1 beta primes interleukin-8-stimulated chemotaxis and elastase release in human neutrophils via its type I receptor. European Cytokine Network. 1997;8:173–178. [PubMed] [Google Scholar]
- 264.Bonfield T.L., Panuska J.R., Konstan M.W., Hilliard K.A., Hilliard J.B., Ghnaim H., Berger M. Inflammatory cytokines in cystic fibrosis lungs. American Journal of Respiratory and Critical Care Medicine. 1995;152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 265.Dean T.P., Dai Y., Shute J.K., Church M.K., Warner J.O. Interleukin-8 concentrations are elevated in bronchoalveolar lavage, sputum, and sera of children with cystic fibrosis. Pediatric Research. 1993;34:159–161. doi: 10.1203/00006450-199308000-00010. [DOI] [PubMed] [Google Scholar]
- 266.Mizunoe S., Shuto T., Suzuki S., Matsumoto C., Watanabe K., Ueno-Shuto K., Suico M.A., Onuki K., Gruenert D.C., Kai H. Synergism Between Interleukin (IL)-17 and Toll-like Receptor 2 and 4 Signals to Induce IL-8 Expression in Cystic Fibrosis Airway Epithelial Cells. Journal of Pharmacological Sciences. 2012;118:512–520. doi: 10.1254/jphs.11240fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Derycke L., Zhang N., Holtappels G., Dutre T., Bachert C. IL-17A as a regulator of neutrophil survival in nasal polyp disease of patients with and without cystic fibrosis. Journal of Cystic Fibrosis. 2012;11:193–200. doi: 10.1016/j.jcf.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 268.Tan H.L., Regamey N., Brown S., Bush A., Lloyd C.M., Davies J.C. The Th17 pathway in cystic fibrosis lung disease. American Journal of Respiratory and Critical Care Medicine. 2011;184:252–258. doi: 10.1164/rccm.201102-0236OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Brodlie M., McKean M.C., Johnson G.E., Anderson A.E., Hilkens C.M., Fisher A.J., Corris P.A., Lordan J.L., Ward C. Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. European Respiratory Journal. 2011;37:1378–1385. doi: 10.1183/09031936.00067110. [DOI] [PubMed] [Google Scholar]
- 270.Chen Y., Thai P., Zhao Y.H., Ho Y.S., DeSouza M.M., Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. Journal of Biological Chemistry. 2003;278:17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 271.Brennan P., O'Neill L.A. Effects of oxidants and antioxidants on nuclear factor kappa B activation in three different cell lines: evidence against a universal hypothesis involving oxygen radicals. Biochimica et Biophysica Acta. 1995;1260:167–175. doi: 10.1016/0167-4781(94)00186-7. [DOI] [PubMed] [Google Scholar]
- 272.Brennan S. Innate immune activation and cystic fibrosis. Paediatric Respiratory Reviews. 2008;9:271–279. doi: 10.1016/j.prrv.2008.05.008. (quiz 279-280) [DOI] [PubMed] [Google Scholar]
- 273.Filosto M., Tonin P., Vattemi G., Savio C., Rizzuto N., Tomelleri G. Transcription factors c-Jun/activator protein-1 and nuclear factor-kappa B in oxidative stress response in mitochondrial diseases. Neuropathology and Applied Neurobiology. 2003;29:52–59. doi: 10.1046/j.1365-2990.2003.00411.x. [DOI] [PubMed] [Google Scholar]
- 274.Konstan M.W., Berger M. Current understanding of the inflammatory process in cystic fibrosis: onset and etiology. Pediatric Pulmonology. 1997;24:137–142. doi: 10.1002/(sici)1099-0496(199708)24:2<137::aid-ppul13>3.0.co;2-3. (discussion 159-161) [DOI] [PubMed] [Google Scholar]
- 275.Yang J., Eiserich J.P., Cross C.E., Morrissey B.M., Hammock B.D. Metabolomic profiling of regulatory lipid mediators in sputum from adult cystic fibrosis patients. Free Radical Biology and Medicine. 2012;53(1):160–171. doi: 10.1016/j.freeradbiomed.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.van de Weert-van Leeuwen P.B., Slieker M.G., Hulzebos H.J., Kruitwagen C.L., van der Ent C.K., Arets H.G. Chronic infection and inflammation affect exercise capacity in cystic fibrosis. European Respiratory Journal. 2012;39:893–898. doi: 10.1183/09031936.00086211. [DOI] [PubMed] [Google Scholar]
- 277.Regamey N., Tsartsali L., Hilliard T.N., Fuchs O., Tan H.L., Zhu J., Qiu Y.S., Alton E.W., Jeffery P.K., Bush A., Davies J.C. Distinct patterns of inflammation in the airway lumen and bronchial mucosa of children with cystic fibrosis. Thorax. 2012;67:164–170. doi: 10.1136/thoraxjnl-2011-200585. [DOI] [PubMed] [Google Scholar]
- 278.Rebeyrol C., Saint-Criq V., Guillot L., Riffault L., Corvol H., Chadelat K., Ray D.W., Clement A., Tabary O., Le Rouzic P. Glucocorticoids reduce inflammation in cystic fibrosis bronchial epithelial cells. Cellular signalling. 2012;24:1093–1099. doi: 10.1016/j.cellsig.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 279.Everett H., McFadden G. Apoptosis: an innate immune response to virus infection. Trends in Microbiology. 1999;7:160–165. doi: 10.1016/s0966-842x(99)01487-0. [DOI] [PubMed] [Google Scholar]
- 280.McWhirter S.M., Tenoever B.R., Maniatis T. Connecting mitochondria and innate immunity. Cell. 2005;122:645–647. doi: 10.1016/j.cell.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 281.Seth R.B., Sun L., Chen Z.J. Antiviral innate immunity pathways. Cell Research. 2006;16:141–147. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- 282.Scott I. Mitochondrial factors in the regulation of innate immunity. Microbes and Infection. 2009;11:729–736. doi: 10.1016/j.micinf.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Temkin V., Karin M. From death receptor to reactive oxygen species and c-Jun N-terminal protein kinase: the receptor-interacting protein 1 odyssey. Immunological Reviews. 2007;220:8–21. doi: 10.1111/j.1600-065X.2007.00560.x. [DOI] [PubMed] [Google Scholar]
- 284.Rudiger A., Stotz M., Singer M. Cellular processes in sepsis. Swiss Medical Weekly. 2008;138:629–634. doi: 10.4414/smw.2008.12319. [DOI] [PubMed] [Google Scholar]
- 285.Yeretssian G., Labbe K., Saleh M. Molecular regulation of inflammation and cell death. Cytokine. 2008;43:380–390. doi: 10.1016/j.cyto.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 286.Ting J.P., Duncan J.A., Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–290. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Arnoult D., Carneiro L., Tattoli I., Girardin S.E. The role of mitochondria in cellular defense against microbial infection. Seminars in Immunology. 2009;21:223–232. doi: 10.1016/j.smim.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 288.Delgado M., Singh S., De Haro S., Master S., Ponpuak M., Dinkins C., Ornatowski W., Vergne I., Deretic V. Autophagy and pattern recognition receptors in innate immunity. Immunological Reviews. 2009;227:189–202. doi: 10.1111/j.1600-065X.2008.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Tal M.C., Iwasaki A. Autophagic control of RLR signaling. Autophagy. 2009;5:749–750. doi: 10.4161/auto.5.5.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Deretic V. Autophagy of intracellular microbes and mitochondria: two sides of the same coin? F1000 Biology Reports. 2010;2:45. doi: 10.3410/B2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kohchi C., Inagawa H., Nishizawa T., Soma G. ROS and innate immunity. Anticancer Research. 2009;29:817–821. [PubMed] [Google Scholar]
- 292.Finkel T. Signal transduction by mitochondrial oxidants. Journal of Biological Chemistry. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Manfredi A.A., Rovere-Querini P. The mitochondrion—a Trojan horse that kicks off inflammation? New England Journal of Medicine. 2010;362:2132–2134. doi: 10.1056/NEJMcibr1003521. [DOI] [PubMed] [Google Scholar]
- 294.Arnoult D., Soares F., Tattoli I., Girardin S.E. Mitochondria in innate immunity. EMBO Reports. 2011;12:901–910. doi: 10.1038/embor.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.West A.P., Shadel G.S., Ghosh S. Mitochondria in innate immune responses. Nature Reviews Immunology. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Bird L. Innate immunity: linking mitochondria and microbes to inflammasomes. Nature Reviews Immunology. 2012;12:229. doi: 10.1038/nri3195. [DOI] [PubMed] [Google Scholar]
- 297.Cloonan S.M., Choi A.M. Mitochondria: commanders of innate immunity and disease? Current Opinion in Immunology. 2012;24:32–40. doi: 10.1016/j.coi.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 298.Kepp O., Galluzzi L., Kroemer G. Mitochondrial control of the NLRP3 inflammasome. Nature Immunology. 2011;12:199–200. doi: 10.1038/ni0311-199. [DOI] [PubMed] [Google Scholar]
- 299.Tschopp J. Mitochondria: sovereign of inflammation? European Journal of immunology. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 300.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 301.Gelman M.S., Kannegaard E.S., Kopito R.R. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. Journal of Biological Chemistry. 2002;277:11709–11714. doi: 10.1074/jbc.M111958200. [DOI] [PubMed] [Google Scholar]
- 302.Devenish R.J. Autophagy and the evasion of host defense: a new variation on the theme for Burkholderia cepacia? Autophagy. 2011;7:1269–1270. doi: 10.4161/auto.7.11.17941. [DOI] [PubMed] [Google Scholar]
- 303.Choi A.J., Ryter S.W. Autophagy in inflammatory diseases. International Journal of Cell Biology. 2011;2011:732798. doi: 10.1155/2011/732798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Haspel J.A., Choi A.M. Autophagy: a core cellular process with emerging links to pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2011;184:1237–1246. doi: 10.1164/rccm.201106-0966CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Ryter S.W., Nakahira K., Haspel J.A., Choi A.M. Autophagy in pulmonary diseases. Annual Review of Physiology. 2012;74:377–401. doi: 10.1146/annurev-physiol-020911-153348. [DOI] [PubMed] [Google Scholar]
- 306.Gavrilin M.A., Abdelaziz D.H., Mostafa M., Abdulrahman B.A., Grandhi J., Akhter A., Abu Khweek A., Aubert D.F., Valvano M.A., Wewers M.D., Amer A.O. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. Journal of Immunology. 2012;188:3469–3477. doi: 10.4049/jimmunol.1102272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Tang A., Sharma A., Jen R., Hirschfeld A.F., Chilvers M.A., Lavoie P.M., Turvey S.E. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS One. 2012;7:e37689. doi: 10.1371/journal.pone.0037689. [DOI] [PMC free article] [PubMed] [Google Scholar]