

Abstract
The MET tyrosine kinase has been identified as a susceptibility gene in patients with autism spectrum disorders. MET is expressed in the forebrain during prenatal and postnatal development. After birth, MET participates in dendritic outgrowth and circuit formation. Alterations in neuronal development, particularly in the cerebral cortex, may contribute to the pathology of developmental disorders, including autism. Patients with autism can exhibit abnormal cortical volumes and head circumferences. We tested the hypothesis that impaired Met signaling during development alters forebrain structure. We have utilized a conditional mutant mouse line which expresses a kinase-dead Met restricted to the cerebral cortex and hippocampal structures. In these mice, we have used magnetic resonance imaging (MRI) to analyze the structure of the cerebral cortex and related structures across postnatal development. We found that the rostral cortex, caudal hippocampus, dorsal striatum, thalamus, and corpus callosum were all larger in adult, but not juvenile, mutant mice relative to control mice. The specificity of the changes suggests that aberrant expansion of the forebrain is consistent with continued axonal and dendritic growth, potentially leading to improper circuit formation and maintenance.
Abbreviations: ANOVA, analysis of variance; bp, base pairs; CC, corpus callosum; ctx, cerebral cortex; HGF, hepatocyte growth factor; H,hipp, hippocampus; K–S, Kolmogorov–Smirnov; mm, millimeter; MRI, magnetic resonance imaging; P, postnatal day; PCR, polymerase chain reaction; RARE, rapid acquisition relaxation enhancement; SEM, standard error of the mean; str, striatum; th,thal, thalamus
Keywords: Met tyrosine kinase, MRI, Autism, Frontal cortex, Mouse, Corpus callosum
Introduction
Autism spectrum disorder (ASD) is characterized by communication and social interaction impairments, as well as repetitive behaviors and narrow interests (APA, 2000). Autisms are highly heritable (Folstein and Rosen-Sheidley, 2001; Hallmayer et al., 2011). Many single gene mutations, such as those underlying Fragile X (FMR1), Rett (MECP2) and tuberous sclerosis (TSC1 and TSC2) syndromes, lead to autistic behaviors in a significant fraction of patients (Bailey et al., 2001; Gutknecht, 2001; Lewis et al., in press; Percy, 2011; Smalley et al., 1992). However, no single gene has been identified for idiopathic autism, but multiple susceptibility factors are being identified (Anney et al., in press; Griswold et al., 2012). ASDs show considerable variability in both symptoms and pathology, and it is possible that different genetic susceptibilities in combination with environmental insults may underlie the observed variations (Abrahams and Geschwind, 2010; Hallmayer et al., 2011; Stoltenberg et al., 2010; Tassone et al., 2011; Williams and Casanova, 2011).
Studies have shown alterations in brain size in ASD (Amaral et al., 2008; Redcay and Courchesne, 2005; Stigler et al., 2011). Regions such as the frontal and temporal cortex (Carper et al., 2002; Hazlett et al., 2006), hippocampus (Schumann et al., 2004), and caudate (Hollander et al., 2005; Sears et al., 1999) have been shown to be expanded in autism, while the thalamus is reduced (Tsatsanis et al., 2003). Several studies have also found increases in white matter volume in autistic patients, particularly in radiate white matter (Freitag et al., 2009; Herbert et al., 2003; Herbert et al., 2004). Notably, the alterations in brain size in ASD appear to be age-dependent, with increases in brain size being reported more consistently in very young patients, leveling off during childhood and adolescence, and possible increased volume in some brain structures into adulthood (Amaral et al., 2008; Aylward et al., 2002; Carper et al., 2002; Courchesne et al., 2011; Duerden et al., 2012; Hazlett et al., 2011; Piven et al., 1995; Redcay and Courchesne, 2005).
Met (also known as c-Met) is a receptor tyrosine kinase which binds the high-affinity ligand hepatocyte growth factor (HGF) (Naldini et al., 1991). Recently, decreased Met function has been found to increase the risk of developing autism spectrum disorders (Campbell et al., 2006; Sousa et al., 2009; Thanseem et al., 2010). Met is crucial during normal development (Birchmeier and Gherardi, 1998), and mediates cell survival, migration, proliferation, and the formation of neuronal processes (Gutierrez et al., 2004; Maina et al., 1997; Powell et al., 2001; Powell et al., 2003b). Importantly, both Met and its ligand, HGF, are expressed in the brain during development (Achim et al., 1997; Bae et al., 2010; Honda et al., 1995; Judson et al., 2009; Martins et al., 2007; Martins and Powell, 2011; Powell et al., 2001). Met transcript expression is prominent in the cerebral cortex, hippocampus, and amygdala, while Met protein can also be detected in white matter tracts such as the corpus callosum (Judson et al., 2009). Changes in Met signaling during development could therefore affect neuronal number as well as the complexity of the neuropil (Bae et al., 2010; Martins et al., 2007; Martins and Powell, 2011; Powell et al., 2003b). Indeed, inactivation of Met in the cerebral cortex has been shown to alter the strength of local excitatory connections between layers 2/3 and 5 (Qiu et al., 2011). Changes in Met function during development may therefore alter the structure or connectivity of cortical and subcortical structures, and ultimately impact their function.
Despite progress in the understanding of the neurobiology of autism, and the role Met plays in neurodevelopment, it remains unclear how disruption of HGF–Met signaling could contribute to defects like those seen in ASD. Anatomical studies in humans likely reflect the effects of multiple different changes at the genetic level, as reflected in the considerable heterogeneity observed between studies. Furthermore, human studies have generally not attempted to correlate changes in brain structure to specific genetic variations. To this end, we have utilized a mouse line in which Met is inactivated specifically in the cerebral cortex and hippocampus using a Cre-loxP recombination strategy. This allows us to examine the effects of loss of Met function on a constant genetic background. Unlike global Met null mutants (Bladt et al., 1995), Met–Emx1 mice live to adulthood (Judson et al., 2009), allowing us to examine brain structure during the postnatal and adult periods. Here we report long-term specific structural changes after altering Met signaling in the embryonic mouse dorsal telencephalon.
Material and methods
Animals
Emx1-Cre (K. Jones, University of Colorado) and Met-fx (S. Thorgeirsson, National Cancer Institute, NIH) mice were generous gifts from collaborators and backcrossed onto the C57BL/6J strain obtained from the Jackson Laboratory (Bar Harbor, ME). Mice used in these experiments were littermates resulting from matings between non-sibling heterozygotes (fx/+:Cre). All mice were genotyped via PCR using the following primer sets: Met-fx primers 5′-TTA GGC AAT GAG GTG TCC CAC-3′ and 5′-CCA GGT GGC TTC AAA TTC TAA GG- 3′ (380 bp for the floxed allele and 300 bp for wildtype); Emx1-Cre primers 5′-CAC CCT GTT ACG TAT AGC CG-3′ and 5′-GAG TCA TCC TTA GCG CCG TA-3′ (320 bp). For this study control mice were wildtype (+/+) and mutant mice were homozygous for the Met mutation, Met(fx/fx)/Emx1(Cre), written as Met–Emx1.
MRI imaging
At postnatal day 30 (P30) or after postnatal day 90 (adult), mice were transcardially perfused with 0.9% saline followed by 4% buffered paraformaldehyde. The fixed brains were subsequently scanned via MRI. All experiments were performed on a Bruker Biospec 7.0 T 30 cm horizontal bore scanner equipped with a 12 cm inner diameter gradient and a gradient strength of 400 mT/m using Paravision 5.0 software. A Bruker 1H surface coil array was used as the receiver and a Bruker 72 mm linear-volume coil as the transmitter. Three-dimensional (3D) T2-weighted images were acquired using a rapid acquisition with relaxation enhancement (RARE) sequence (TEeff/TR = 60/1500 ms, rare factor 16 , number of averages 3) with a field of view of 25.6 × 25.6 × 18 mm3 for a 100 μm3 spatial resolution. The total acquisition time for each sample was approximately 3 h and 36 min.
Area and volume estimation
Post-imaging analysis for volume and area was performed using NIH ImageJ software (version 1.43u). Fig. 1 shows the boundaries of the regions of interest that were measured in the coronal plane (Fig. 1A–C), as well as in the mid-sagittal plane (Fig. 1D). Measurements of the whole brain included all structures from the rostral extent of the cerebral cortex (bregma ~ 4.0 mm, (Paxinos and Franklin, 2001)) to the caudal brainstem-spinal cord juncture (bregma ~ 8.5 mm). The area of the cerebral cortex was measured from pia to white matter, with boundaries defined rostrally by the emergence of the olfactory bulb, caudally by the plane in which the deep white matter (dorsal hippocampal commissure) was no longer visible, and ventrally by the rhinal sulcus, end of the CC, or level of the anterior commissure (from caudal to rostral). Measurements of the hippocampus include both the upper and lower leaves. The hippocampus is bordered dorsally and laterally by the corpus callosum (CC), lateral ventricles, or fimbria, and ventrally and medially by the thalamus, third ventricle, alveus hippocampus, brachium superior colliculus, or optic tract.
Fig. 1.
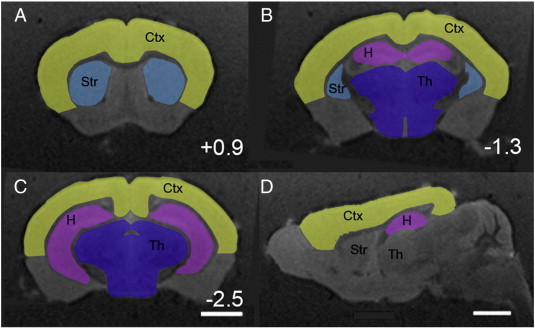
Schematics representing the areas included in measurements of the cerebral cortex, hippocampus, striatum, thalamus and corpus callosum in coronal sections at bregma = + 0.94 mm (A) bregma = − 1.36 mm (B) and bregma = − 2.56 mm (C) and in a parasagittal section (D). Bar = 2 mm (A–C) and 2 mm (D).
Measurements of the striatum include the caudate and putamen. The boundaries of the striatum were defined medially by the lateral ventricles and internal capsule, laterally and dorsally by the external capsule, and ventrally by the anterior commissure. Measurements of the thalamus include both thalamic and hypothalamic nuclei. The boundaries of this region were defined laterally by the internal capsule and cerebral peduncle, dorsally by the hippocampus, and ventrally by the ventral surface of the brain. Measurements of the thalamus did not include the third ventricle or posterior commissure.
Data analysis
Four brains were imaged for each of the P30 control, P30 Met–Emx1, adult control, and Met–Emx1 groups. The images analyzed represent 100 μm thick sections, with a voxel size of 0.1 × 0.1 × 0.1 mm. With the exception of measurements of the whole brain, area measurements were taken separately for each hemisphere. The distributions of cross-sectional areas across different rostrocaudal levels were compared between control and Met–Emx1 mice at each age with Kolmogorov–Smirnov (K–S). For volume comparisons, measurements were made for the left and right hemispheres for each structure. The overall volume of each structure was compared using a three-way ANOVA in SigmaPlot12 (Systat Software, San Jose, CA) followed by a Bonferroni t-test as appropriate. Cortical thickness and surface area were compared between adult control and Met–Emx1 mice using a Student's t test.
Results
Alterations in gray-matter structures in adult, but not juvenile, Met–Emx1 mice
Met was inactivated in the cerebral cortex and hippocampus by the selective removal of the intracellular signaling domain (encoded by exon 16) (Huh et al., 2004), using a Cre-loxP recombination strategy with the Emx1-Cre mouse driver (Gorski et al., 2002). The Met–Emx1 mice were viable in agreement with the previous reports (Judson et al., 2009; Judson et al., 2010; Qiu et al., 2011). Initial histological examination suggested altered cortical thickness and prompted an MRI structural study of uncut fixed brains.
Whole brain — total brain weight was measured for all groups (P30 control = 0.444 ± 0.01 g, P30 Met–Emx1 = 0.421 ± 0.02 g, adult control = 0.411 ± 0.07, adult Met–Emx1 = 0.514 ± 0.06 g).
Two-way ANOVA found no overall effect of genotype (F(1,15) = 2.548, p = 0.136) or age (F(1,15) = 1.395, p = 0.260). A genotype × age interaction was found (F(1,15) = 6.082, p = 0.030), with significant differences between P30 and adult Met–Emx1 brains (p = 0.024) and between adult control and adult Met–Emx1 brains (p = 0.014). These data suggest that while neither the Met allele nor age individually alters total brain weight, the combination contributes to an overall increased brain size.
The cross-sectional area of the entire brain was estimated. Cross sectional area of the whole brain was similar for control and Met–Emx1 mice at P30 (Fig. 2A, D = 0.0527, p = 0.514, K–S test). The cross-sectional area was increased in adult Met–Emx1 animals compared to control (Fig. 2B, D = 0.2345, p < 0.001, K–S test). No significant effects of either genotype (F(1,15) = 3.031, p = 0.107) or age (F(1,15) = 2.395, p = 0.148, two-way ANOVA) were found on total brain volume. The interaction of genotype × age was significant at the p < 0.1 level (F(1,15) = 4.418, p = 0.057). In control mice, whole brain volume did not change from P30 to adult, from 406.1 ± 13.4 mm3 to 391.5 ± 19.3 mm3 (p = 0.702, Bonferroni t test). By contrast, a significant increase in volume was observed in the Met–Emx1 mice, from 396.6 ± 15.3 mm3 at P30 to 492.7 ± 44.6 mm3 in adults (p = 0.024, Bonferroni t test). Similar to the total brain weight, the combination of the Met allele and age lead to increased brain volume in the adult Met–Emx1 mice.
Fig. 2.
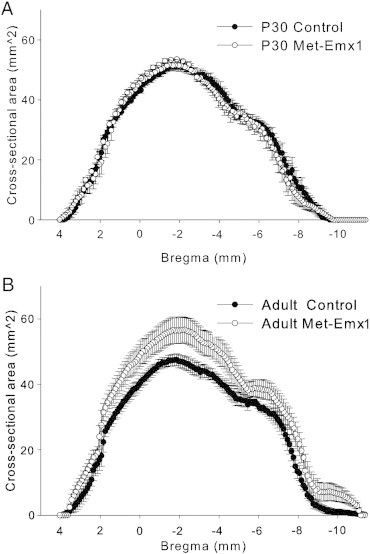
Loss of Met function alters whole-brain cross-sectional area. The average cross sectional area of the brain is plotted versus estimated bregma level for P30 (A) and adult (B) animals. The cross sectional area of the whole brain is similar between P30 control and Met–Emx1 mice (D = 0.0527, p = 0.514, K–S test). However, the brains of adult Met–Emx1 mice are larger than control brains (D = 0.2345, p < 0.001, K–S test). Data represent n = 4 brains per genotype and age, points represent the mean areas with the error bars signifying the SEM.
Cerebral cortex
We initially focused on the cerebral cortex and hippocampus, the areas which were targeted by Emx1-Cre expression (Gorski et al., 2003). The brains from control juvenile (P30) mice looked similar to Met–Emx1 mice, whereas the cerebral cortex in the adult Met–Emx1 mouse appeared slightly larger overall (Fig. 3A–H). The distributions of areas were not significantly different for the juvenile control and Met–Emx1 mice (Fig. 3I, D = 0.1076, p = 0.067, K–S test). Although in frontal regions (bregma levels > 2.5 mm), the areas in the Met–Emx1 mice were larger on average than in control mice. In the adult mice, the distributions of cerebral cortical areas were significantly different between control and Met–Emx1 mice (Fig. 3J, D = 0.2200, p < 0.001, K–S).
Fig. 3.
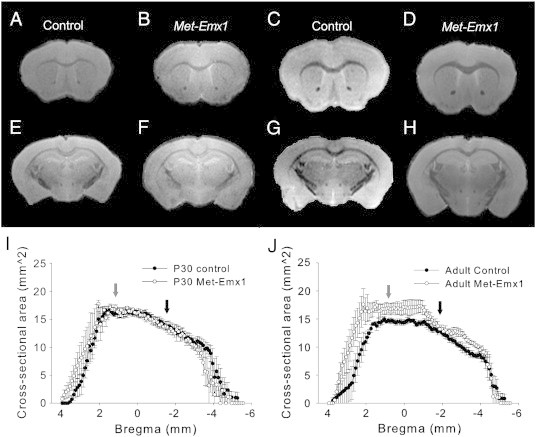
Structural differences in the cerebral cortex of Met–Emx1 mice are age specific. (A–H) Representative MRI images at rostral (A–D, bregma = + 1.04 mm) and caudal (E–H, bregma = − 1.66 mm) levels. Panels A, B, E, and F were obtained from P30 mice, whereas C, D, G, and H were from adult mice. (I and J) The cross-sectional area of the cerebral cortex is plotted versus estimated bregma level for P30 (I) and adult (J) animals. The overall distributions of area are the same for control and Met–Emx1 mice at P30 (D = 0.1076, p = 0.067, K–S test). Yet, the distributions are significantly different in control and Met–Emx1 mice at adulthood (D = 0.2200, p < 0.001, K–S test). Arrows denote the levels for which the rostral (gray arrows) or caudal (black arrows) images are shown. Data represent n = 4 brains per genotype and age, points represent the mean areas with the error bars signifying the SEM.
Total cortical volume was estimated for control (96.98 ± 2.59 mm3) and Met–Emx1 mice (97.30 ± 3.48 mm3) at P30. In the adult mice, the total cortical volumes were 89.69 ± 4.01 mm3 (control) and 98.06 ± 3.47 mm3 (Met–Emx1). There was a significant main effect of genotype (F(1,31) = 9.290, p = 0.006), but not of age (F(1,31) = 0.720, p = 0.404), or hemisphere (F(1,31) = 0.133, p = 0.719, three-way ANOVA) on cortical volume. There was also a significant genotype × age interaction (F(1,31) = 9.671, p = 0.005). The cortical volume data were in agreement with the cross-sectional area distributions.
The comparison of the cross-sectional areas showed an in increase in the Met–Emx1 mice in the frontal and parietal cortical regions. The frontal cortical areas are associated with executive functions and emotional processing, which are altered in ASD. Therefore, we divided the dataset into rostral and caudal regions (at bregma = − 1 mm), to specifically examine frontal areas. At P30, rostral and caudal volumes were similar between genotypes (control, rostral: 57.89 ± 2.42 mm3and caudal: 39.10 ± 4.40 mm3; Met–Emx1, rostral: 62.88 ± 4.77 mm3 and caudal: 34.43 ± 4.33 mm3). In adult Met–Emx1 mice the rostral half of cerebral cortex appeared larger than in control mice (control: 50.41 ± 4.03 mm3, Met–Emx1: 63.06 ± 4.77 mm3), while the caudal half remains similar (control: 39.28 ± 2.44 mm3, Met–Emx1: 34.99 ± 4.06 mm3). At rostral levels, there was no main effect of genotype (F(1,31) = 1.219, p = 0.281) or age (F(1,31) = 1.365, p = 0.254), but there was a significant genotype × age interaction effect (F(1,31) = 5.691, p = 0.025), again implying a combination of genotype and age leads to increased size. At caudal levels, we found no main effects of genotype (F(1,31) = 1.476, p = 0.236), age (F(1,31) = 0.310, p = 0.580) or genotype × age interaction (F(1,31) = 0.01, p = 0.972).
We next sought to determine which dimensions were expanded in the adult Met–Emx1 cortex to account for the increase in volume. We first measured the thickness of the cortex from pia to white matter at the level of the barrel fields (bregma 0.34 mm to − 1.96 mm) (Paxinos and Franklin, 2001). We found no significant difference in the thickness of the cortex at these levels in either the right (control: 1.15 ± 0.03 mm, Met–Emx1: 1.26 ± 0.06 mm, p = 0.17, Student's t test) or left hemispheres (control: 1.17 ± 0.03 mm, Met–Emx1: 1.24 ± 0.05 mm, p = 0.28, Student's t test). Next we measured the tangential length of the cortical surface in each image, to determine the surface area of the cortex. The total surface area of the cortex is larger in Met–Emx1 mice (115.63 ± 5.52 mm2) than in controls (97.18 ± 3.47 mm2, p = 0.049, Student's t test).
Lastly, we compared the rostrocaudal distance spanned by the cerebral cortex. This appeared to be slightly longer in adult Met–Emx1 mice (8.12 ± 0.13 mm) compared to adult control (7.70 ± 0.15 mm), and this difference was significant at the p < 0.1 level (p = 0.09, Student's t test). No difference in rostrocaudal length was observed in P30 mice (control: 7.80 ± 0.10 mm; Met–Emx1: 7.53 ± 0.21 mm; p = 0.32, Student's t test). In summary, the cerebral cortex was larger in the adult Met–Emx1 mouse. This difference appears to be due to a tangential, but not radial, expansion of the cortex. There may also be a rostrocaudal expansion of the cortex in the Met–Emx1 mouse.
Hippocampus
We measured the hippocampus, which also expresses Emx1-cre and may be affected by loss of Met signaling (Gorski et al., 2002; Guo et al., 2000). Lamination and subfields were grossly similar in all mice (Fig. 4A–H). Estimates of cross-sectional area showed no difference at P30 (Fig. 4I, D = 0.1113, p = 0.426, K–S test), but a significant difference was found in the distribution of areas between adult control and Met–Emx1 animals (Fig. 4J, D = 0.2560, p = 0.001, K–S test). This difference in the adult also translated to a difference in the total volume of the hippocampus (control: 13.67 ± 0.44 mm3, Met–Emx1: 20.02 ± 2.94 mm3, p = 0.013, Bonferroni t test). We found no main effect of either genotype (F(1,31) = 2.950, p = 0.099), or age (F(1,31) = 0.898, p = 0.353), but there was a significant age × genotype interaction effect (F(1,31) = 5.335, p = 0.030, ANOVA). Like the total brain and cerebral cortex analyses, the overall increase in hippocampal volume is due to the combination of small effects of genotype and age.
Fig. 4.
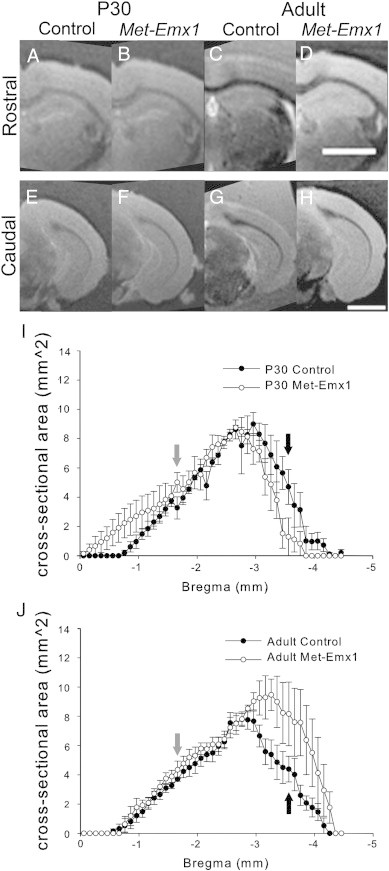
Loss of Met function alters hippocampal area in adult mice. Met–Emx1 and control mice have similar hippocampal cross-sectional areas at P30 (D = 0.1113, p = 0.426, K–S test). The distributions of areas are significantly different between adult Met–Emx1 and control mice (D = 0.2560, p = 0.001, K–S test). Representative images at rostral (A–D, approximate bregma = − 1.66) and caudal (E–H, approximate bregma = − 3.56) levels. The average cross-sectional area of the hippocampus (upper and lower leaves) is plotted versus estimated bregma level for P30 (I) and adult (J) animals. Arrows denote the levels for which the rostral (gray arrows) or caudal (black arrows) images are shown. Data represent n = 4 brains per genotype and age, points represent the mean areas with the error bars signifying the SEM. Scale bar = 2 mm.
Frontal cortical regions project to the caudal portion of the hippocampus. When only the caudal portion (caudal to bregma < − 2.50 mm) is considered, there was no main effect of genotype (F(1,31) = 0.561, p = 0.461) or age (F(1,31) = 4.208, p = 0.056), but there was a significant interaction between age and genotype (F(1,31) = 12.880, p = 0.001). In control mice, the caudal portion of the hippocampus had similar volumes at both P30 and in adulthood (p = 0.38, Bonferroni t test). But in the Met–Emx1 mice the caudal portion of the hippocampus was significantly larger in adult, as compared to P30 mice (p = 0.029, Bonferroni t-test). For the rostral portion (bregma > − 2.50) of the hippocampus, no significant effect of genotype (F(1,31) = 2.321, p = 0.141), age (F(1,31) = 0.758, p = 0.393), or any genotype × age interaction (F(1,31) = 0.523, p = 0.476) was found. The increase in hippocampal volume therefore appears to be restricted to the caudal hippocampus.
Striatum
The cross-sectional area of striatum of Met–Emx1 animals was found to be similar to control at P30 (Fig. 5A, D = 0.0572, p = 0.984, K–S test). However, the distributions between genotypes were different in the adult (Fig. 5B, D = 0.2157, p = 0.002, K–S test). The total striatal volume was calculated, and at P30 control mice had a volume of 16.60 ± 0.87 mm3 and Met–Emx1 mice had a volume of 16.44 ± 0.95 mm3. For adult mice, the volumes were 16.96 ± 0.57 mm3 for control mice and 21.95 ± 2.02 mm3 for Met–Emx1 mice. We found main effects of genotype (F(1,31) = 7.559, p = 0.011) and age (F(1,31) = 11.172, p = 0.003), and a significant genotype × age interaction (F(1,31) = 8.570, p = 0.011). Striatal volume was increased in adult Met–Emx1 mice relative to adult control mice (p = 0.002, Bonferroni t test) and P30 Met–Emx1 mice (p = 0.003, Bonferroni t test).
Fig. 5.
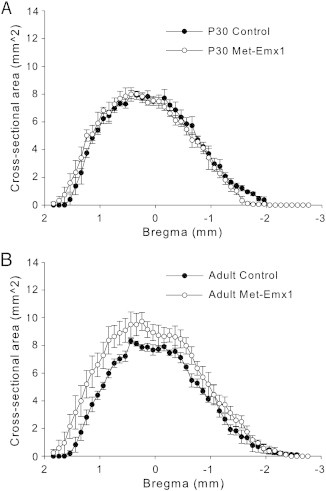
The striatum of adult Met–Emx1 mice is larger than the striatum of control mice. Average cross-sectional area of the striatum is plotted versus estimated bregma level for P30 (A) and adult (B) animals. Overall the distribution of area is significantly different in adult control and Met–Emx1 mice (D = 0.2157, p = 0.002, K–S test) but not between P30 control and Met–Emx1 mice (D = 0.0572, p = 0.984, K–S test). Data represent n = 4 brains per genotype and age, points represent the mean areas with the error bars signifying the SEM.
Thalamus
We next measured the area of the thalamus, which followed a similar pattern to the other structures. No difference was observed at P30 between control and Met–Emx1 mice (Fig. 6A, D = 0.2002, p = 0.056, K–S test), but a significant increase was found in the thalamus of adult Met–Emx1 compared to adult control mice (Fig. 6B, D = 0.3960, p < 0.001, K–S test). We also calculated the volume of the thalamus. At P30, thalamic volume is 26.04 ± 1.79 mm3 in control mice and 24.86 ± 1.48 mm3 in Met–Emx1 mice. In the adult, the volume of the thalamus is 20.87 ± 3.34 mm3 in control mice and 29.41 ± 2.09 mm3 in Met–Emx1 mice. We found no main effect of genotype (F(1,31) = 3.256, p = 0.084) or age (F(1,31) = 0.025, p = 0.875). Yet, the genotype × age interaction (F(1,31) = 9.504, p = 0.005) indicated a difference among the dataset. Post hoc analysis revealed that the thalamic volume of the adult Met–Emx1 mice was significantly different from adult control mice (p = 0.04, Bonferroni t test).
Fig. 6.
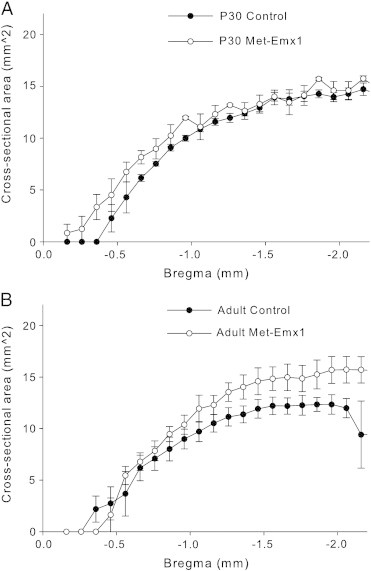
Loss of Met function alters thalamic area in adult mice. Average cross-sectional area of the thalamus is plotted versus estimated bregma level for P30 (A) and adult (B) animals. Overall the distribution of area is significantly different in control and Met–Emx1 mice in adulthood (D = 0.3960, p < 0.001, K–S test), but not at P30 (D = 0.2002, p = 0.056, K–S test). Data represent n = 4 brains per genotype and age, points represent the mean areas with the error bars signifying the SEM.
Increased thickness of the corpus callosum in adult, but not juvenile, Met–Emx1 mice
Met has previously been implicated in neurite extension, as well as axonal outgrowth and survival (Hamanoue et al., 1996; Maina et al., 1997). Therefore, we measured the thickness of the CC at the midline across the rostrocaudal extent, (bregma = + 1.14 mm to − 2.96 mm, Fig. 7A–D). We found no difference between Met–Emx1 and control mice at P30 (Fig. 7E, D = 0.0723, p = 0.893, K–S test). However, adult Met–Emx1 mice have a significantly thicker CC than control mice (Fig. 7F, D = 0.2239, p = 0.001, K–S test). The difference appears greatest towards the rostral end (bregma levels + 0.1 to 1.0 mm) of the CC (Fig. 7B). For example, at bregma + 0.74 mm, the thickness of the CC is 0.32 ± 0.03 mm in control mice and 0.54 mm ± 0.08 mm in Met–Emx1 mice, and, at bregma − 1.06, the CC was 0.20 ± 0.02 mm in control mice and 0.24 ± 0.04 mm in Met–Emx1 mice.
Fig. 7.
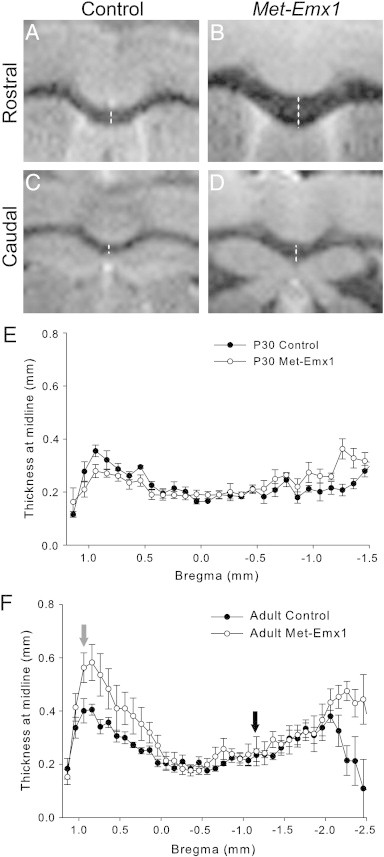
The corpus callosum is expanded at rostral levels in adult Met–Emx1 mice. (A–D) Representative MRI images at approximate bregma = + 0.94 mm (A, B) and − 1.16 mm (C, D) levels. Dotted line demonstrates the location of the thickness measurement. The average thickness of the corpus callosum at the midline is plotted versus estimated bregma level for P30 (E) animals and adult (F) mice. Arrows denote the level at which images are shown for rostral (black) and caudal (gray) levels. The distribution of thickness is significantly different in control and Met–Emx1 mice in adulthood (D = 0.2239, p = 0.001, K–S test), but not at P30 (D = 0.0723, p = 0.893, K–S test). Data represent n = 4 brains per genotype, points represent the mean areas with the error bars signifying the SEM.
Discussion
Our results demonstrate widespread alterations in forebrain structure in adult mice with impaired Met signaling. In the adult Met–Emx1 mice, we observed an expansion in the rostral pole, in frontal cortical areas, and an enlargement in the caudal hippocampal areas that connect to the frontal regions. The striatum and thalamus are expanded as well, but more evenly across the rostrocaudal extent. None of the differences between adult Met–Emx1 and control mice were detected in juvenile (P30) mice. The differences observed in the adult mice support the idea of a progressive defect.
The late postnatal nature of the enlargement of these brain areas could be due to continued growth in the Met–Emx1 mice, or a lack of normal pruning. The cerebral cortex and hippocampus are directly targeted by the Emx1-Cre driver (Gorski et al., 2002), so alterations in those structures could be due to either increased cell numbers or size of neurons or glia (Geisert et al., 2002; Groszer et al., 2001), or increases in the size or number of axonal or dendritic processes. The expression pattern of the Emx1-Cre driver is largely uniform with regards to the rostrocaudal extent of the cerebral cortex (Gorski et al., 2003), excluding the possibility that the differential effects are due to driver expression. However, Met expression has been reported to be in a low rostral — high caudal pattern during the first postnatal month (Judson et al., 2009). In another mutant mouse, the urokinase plasminogen activator receptor null, Met levels were reported to be selectively decreased in embryonic frontal and parietal cortical areas leading to seizures and impaired cognition (Bae et al., 2010; Bissonette et al., 2010; Powell et al., 2003a), providing support that loss of Met has the greatest consequences in frontal regions. Frontal and limbic structures continue to develop through adolescence. The enlarged frontal structures in the Met–Emx1 mice may represent the compensatory changes due to abnormal ontogeny of the frontal pole.
Changes in the subcortical structures, the striatum and thalamus, are perhaps best explained by an increase in white matter volume, as reflected in the increased thickness of the CC. The CC is composed primarily of contralaterally projecting axons from cortical pyramidal neurons, both cortico-cortical and sub-cortical projections. An increase in the thickness of the CC could be caused by an increase in the number, size, or branching of axonal processes originating from affected cells, by increased myelination, or some combination of these factors. Both the striatum and thalamus exchange reciprocal connections with the cortex, including the frontal cortical areas which we found to be expanded in Met–Emx1 mice. Notably, the ventral hippocampus also has reciprocal connections with the frontal cortex (Fanselow and Dong, 2010). The ventral division of the hippocampus is found in more caudal sections where the hippocampus appears to be expanded in adult Met–Emx1 mice, raising the possibility that the change in hippocampal size may be due to altered cortical afferents rather than direct effects of loss of Met signaling in the hippocampus.
Changes in both white and gray matter volume have been detected in autistic humans. The brain is slightly smaller in individuals with ASD at birth and undergoes a period of early over-growth, leading to increased brain volume and head circumference within the first few years of life (Courchesne et al., 2011; Redcay and Courchesne, 2005). A study of autistic patients and controls from ages 18 months to 5 years found increased cortical volume, gray matter volume, and white matter volume in autistic patients, with no change in cortical thickness (Hazlett et al., 2011). The authors therefore attributed the increase in cortical volume to the increased surface area (volume ÷ thickness). After the initial period of overgrowth, the discrepancy in growth rate diminishes, and consequently brain volume and head circumference values approach those of typically developing children (Redcay and Courchesne, 2005). For example, an MRI study using individuals with a mean age near 10 years found no difference in total brain volume, nor in gray or white matter volume in autistic patients, although they did find an increase in cortical thickness at this age (Hardan et al., 2006). While most studies focus on childhood, some studies that included older children and adults report overall increases in brain size (Aylward et al., 1999; Piven et al., 1995), raising the possibility that such increases could persist into adulthood in some cases. The earliest time we examined in the mice was P30 which represents the juvenile, closer to the 10 year old children, and this presents a limitation to our study that we do not have an earlier structural data.
Studies focusing on white matter structures using diffusion tensor imaging found different changes in autistic patients depending on age. While one study on children between 1.8 and 3.3 years found increased fractional anisotropy in the genu and splenium of the CC, as well as in portions of the internal and external capsules and the forceps minor (Ben Bashat et al., 2007), others in a slightly older population have found reduced fractional anisotropy values in the CC and frontal white matter tracts (Alexander et al., 2007; Barnea-Goraly et al., 2004). While some reports have shown decreases in CC size (Freitag et al., 2009; Piven et al., 1997), or no change in autistic subjects (Herbert et al., 2004; Piven et al., 1997), others have found increased white matter volume in autistic individuals (Freitag et al., 2009; Herbert et al., 2003; Herbert et al., 2004). None of the studies were correlated with genetic information, and likely represent spectrum of diverse genetic and environmental factors leading to the ASD diagnosis.
The structural changes we observed in the brains of adult Met–Emx1 mice may resemble some of those reported in autistic patients (Amaral et al., 2008). While the cause in each case remains uncertain, alterations in white matter structures seem to make a contribution in both cases. Despite this similarity, the time course of the alterations is divergent between the two species. In addition to simple species differences, a few factors may contribute to this divergence. Most notably, the Met–Emx1 mice undergo what we believe to be a complete loss of the kinase activity of Met in the cells affected by the Emx1-Cre driver, namely neurons and glia in the cerebral cortex and hippocampus. Changes in the striatum and thalamus are likely due to the cortical and hippocampal afferents to these regions. By contrast, the Met polymorphism associated with autism in humans leads to decreased transcription of MET (Campbell et al., 2006), presumably affecting all brain areas which normally express MET. Since Met function is not completely ablated in the brains of humans with the polymorphism, there is the possibility that changes in the regulation of the pathway could eventually compensate for reduced transcription, as is found with MET in the cancer environment (Straussman et al., 2012). Similarly, the complete loss of Met in our mouse model may lead to compensatory changes in HGF levels, or other related pathways, that do not occur after the partial reduction in Met signaling seen in humans. Finally, our dataset relies on separate animals for each time point, and it was not a longitudinal study, and so conclusions regarding developmental trajectory are limited by group data. By pairing the imaging study with a single mutation and a defined phenotype, we were able to view the contribution of a single gene. The human situation is far more complex, yet multiple allelic variants may contribute to different stages of the ASD phenotype over the lifespan of the individual. Future studies will focus on the cellular and molecular underpinnings of the anatomical changes seen in these mice.
Acknowledgments
We would like to acknowledge the contributions of Drs. Su Xu, Da Shi, and Rao Gullapalli for assistance with the collection of MRI images. Funded by NIH R01 DA018826.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- Abrahams B.S., Geschwind D.H. Connecting genes to brain in the autism spectrum disorders. Archives of Neurology. 2010;67:395–399. doi: 10.1001/archneurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achim C.L., Katyal S., Wiley C.A., Shiratori M., Wang G., Oshika E., Petersen B.E., Li J.M., Michalopoulos G.K. Expression of HGF and cMet in the developing and adult brain. Brain Research. Developmental Brain Research. 1997;102:299–303. doi: 10.1016/s0165-3806(97)00108-9. [DOI] [PubMed] [Google Scholar]
- Alexander A.L., Lee J.E., Lazar M., Boudos R., DuBray M.B., Oakes T.R., Miller J.N., Lu J., Jeong E.K., McMahon W.M., Bigler E.D., Lainhart J.E. Diffusion tensor imaging of the corpus callosum in autism. NeuroImage. 2007;34:61–73. doi: 10.1016/j.neuroimage.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Amaral D.G., Schumann C.M., Nordahl C.W. Neuroanatomy of autism. Trends in Neurosciences. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Anney, R., Klei, L., Pinto, D., Almeida, J., Bacchelli, E., Baird, G., Bolshakova, N., Bolte, S., Bolton, P.F., Bourgeron, T., Brennan, S., Brian, J., Casey, J., Conroy, J., Correia, C., Corsello, C., Crawford, E.L., de Jonge, M., Delorme, R., Duketis, E., Duque, F., Estes, A., Farrar, P., Fernandez, B.A., Folstein, S.E., Fombonne, E., Gilbert, J., Gillberg, C., Glessner, J.T., Green, A., Green, J., Guter, S.J., Heron, E.A., Holt, R., Howe, J.L., Hughes, G., Hus, V., Igliozzi, R., Jacob, S., Kenny, G.P., Kim, C., Kolevzon, A., Kustanovich, V., Lajonchere, C.M., Lamb, J.A., Law-Smith, M., Leboyer, M., Le Couteur, A., Leventhal, B.L., Liu, X.Q., Lombard, F., Lord, C., Lotspeich, L., Lund, S.C., Magalhaes, T.R., Mantoulan, C., McDougle, C.J., Melhem, N.M., Merikangas, A., Minshew, N.J., Mirza, G.K., Munson, J., Noakes, C., Papanikolaou, K., Pagnamenta, A.T., Parrini, B., Paton, T., Pickles, A., Posey, D.J., Poustka, F., Ragoussis, J., Regan, R., Renshaw, K., Roberts, W., Roeder, K., Roge, B., Rutter, M.L., Schlitt, S., Shah, N., Sheffield, V.C., Soorya, L., Sousa, I., Stoppioni, V., Sykes, N., Tancredi, R., Thompson, A.P., Thomson, S., Tryfon, A., Tsiantis, J., Van Engeland, H., Vincent, J.B., Volkmar, F., Vorstman, J., Wallace, S., Wing, K., Wittemeyer, K., Wood, S., Zurawiecki, D., Zwaigenbaum, L., Bailey, A.J., Battaglia, A., Cantor, R.M., Coon, H., Cuccaro, M.L., Dawson, G., Ennis, S., Freitag, C.M., Geschwind, D.H., Haines, J.L., Klauck, S.M., McMahon, W.M., Maestrini, E., Miller, J., Monaco, A.P., Nelson, S.F., Nurnberger Jr., J.I., Oliveira, G., Parr, J.R., Pericak-Vance, M.A., Piven, J., Schellenberg, G.D., Scherer, S.W., Vicente, A.M., Wassink, T.H., Wijsman, E.M., Betancur, C., Buxbaum, J.D., Cook, E.H., Gallagher, L., Gill, M., Hallmayer, J., Paterson, A.D., Sutcliffe, J.S., Szatmari, P., Vieland, V.J., Hakonarson, H., Devlin, B., in press. Individual common variants exert weak effects on risk for autism spectrum disorders. Human Molecular Genetics. (Electronic publication ahead of print). http://dx.doi.org/10.1093/hmg/dds301.
- APA . American Pyschiatric Association; Washington, DC: 2000. Diagnostic and Statistical Manual of Mental Disorders DSM-IV-TR. [Google Scholar]
- Aylward E.H., Minshew N.J., Field K., Sparks B.F., Singh N. Effects of age on brain volume and head circumference in autism. Neurology. 2002;59:175–183. doi: 10.1212/wnl.59.2.175. [DOI] [PubMed] [Google Scholar]
- Aylward E.H., Minshew N.J., Goldstein G., Honeycutt N.A., Augustine A.M., Yates K.O., Barta P.E., Pearlson G.D. MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology. 1999;53:2145–2150. doi: 10.1212/wnl.53.9.2145. [DOI] [PubMed] [Google Scholar]
- Bae M.H., Bissonette G.B., Mars W.M., Michalopoulos G.K., Achim C.L., Depireux D.A., Powell E.M. Hepatocyte growth factor (HGF) modulates GABAergic inhibition and seizure susceptibility. Experimental Neurology. 2010;221:129–135. doi: 10.1016/j.expneurol.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D.B., Jr., Hatton D.D., Skinner M., Mesibov G. Autistic behavior, FMR1 protein, and developmental trajectories in young males with fragile X syndrome. Journal of Autism and Developmental Disorders. 2001;31:165–174. doi: 10.1023/a:1010747131386. [DOI] [PubMed] [Google Scholar]
- Barnea-Goraly N., Kwon H., Menon V., Eliez S., Lotspeich L., Reiss A.L. White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biological Psychiatry. 2004;55:323–326. doi: 10.1016/j.biopsych.2003.10.022. [DOI] [PubMed] [Google Scholar]
- Ben Bashat D., Kronfeld-Duenias V., Zachor D.A., Ekstein P.M., Hendler T., Tarrasch R., Even A., Levy Y., Ben Sira L. Accelerated maturation of white matter in young children with autism: a high b value DWI study. NeuroImage. 2007;37:40–47. doi: 10.1016/j.neuroimage.2007.04.060. [DOI] [PubMed] [Google Scholar]
- Birchmeier C., Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends in Cell Biology. 1998;8:404–410. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- Bissonette G.B., Bae M.H., Suresh T., Jaffe D.E., Powell E.M. Astrocyte-mediated hepatocyte growth factor/scatter factor supplementation restores GABAergic interneurons and corrects reversal learning deficits in mice. Journal of Neuroscience. 2010;30:2918–2923. doi: 10.1523/JNEUROSCI.5268-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladt F., Riethmacher D., Isenmann S., Aguzzi A., Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- Campbell D.B., Sutcliffe J.S., Ebert P.J., Militerni R., Bravaccio C., Trillo S., Elia M., Schneider C., Melmed R., Sacco R., Persico A.M., Levitt P. A genetic variant that disrupts MET transcription is associated with autism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carper R.A., Moses P., Tigue Z.D., Courchesne E. Cerebral lobes in autism: early hyperplasia and abnormal age effects. NeuroImage. 2002;16:1038–1051. doi: 10.1006/nimg.2002.1099. [DOI] [PubMed] [Google Scholar]
- Courchesne E., Campbell K., Solso S. Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Research. 2011;1380:138–145. doi: 10.1016/j.brainres.2010.09.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerden E.G., Mak-Fan K.M., Taylor M.J., Roberts S.W. Regional differences in grey and white matter in children and adults with autism spectrum disorders: an activation likelihood estimate (ALE) meta-analysis. Autism Research. 2012;5:49–66. doi: 10.1002/aur.235. [DOI] [PubMed] [Google Scholar]
- Fanselow M.S., Dong H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein S.E., Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nature Reviews Genetics. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- Freitag C.M., Luders E., Hulst H.E., Narr K.L., Thompson P.M., Toga A.W., Krick C., Konrad C. Total brain volume and corpus callosum size in medication-naive adolescents and young adults with autism spectrum disorder. Biological Psychiatry. 2009;66:316–319. doi: 10.1016/j.biopsych.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisert E.E., Jr., Williams R.W., Geisert G.R., Fan L., Asbury A.M., Maecker H.T., Deng J., Levy S. Increased brain size and glial cell number in CD81-null mice. The Journal of Comparative Neurology. 2002;453:22–32. doi: 10.1002/cne.10364. [DOI] [PubMed] [Google Scholar]
- Gorski J.A., Balogh S.A., Wehner J.M., Jones K.R. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- Gorski J.A., Talley T., Qiu M., Puelles L., Rubenstein J.L., Jones K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. Journal of Neuroscience. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold A.J., Ma D., Cukier H.N., Nations L.D., Schmidt M.A., Chung R.H., Jaworski J.M., Salyakina D., Konidari I., Whitehead P.L., Wright H.H., Abramson R.K., Williams S.M., Menon R., Martin E.R., Haines J.L., Gilbert J.R., Cuccaro M.L., Pericak-Vance M.A. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Human Molecular Genetics. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M., Erickson R., Scripture-Adams D.D., Lesche R., Trumpp A., Zack J.A., Kornblum H.I., Liu X., Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Guo H., Hong S., Jin X.L., Chen R.S., Avasthi P.P., Tu Y.T., Ivanco T.L., Li Y. Specificity and efficiency of Cre-mediated recombination in Emx1-Cre knock-in mice. Biochemical and Biophysical Research Communications. 2000;273:661–665. doi: 10.1006/bbrc.2000.2870. [DOI] [PubMed] [Google Scholar]
- Gutierrez H., Dolcet X., Tolcos M., Davies A. HGF regulates the development of cortical pyramidal dendrites. Development. 2004;131:3717–3726. doi: 10.1242/dev.01209. [DOI] [PubMed] [Google Scholar]
- Gutknecht L. Full-genome scans with autistic disorder: a review. Behavior Genetics. 2001;31:113–123. doi: 10.1023/a:1010218227600. [DOI] [PubMed] [Google Scholar]
- Hallmayer J., Cleveland S., Torres A., Phillips J., Cohen B., Torigoe T., Miller J., Fedele A., Collins J., Smith K., Lotspeich L., Croen L.A., Ozonoff S., Lajonchere C., Grether J.K., Risch N. Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanoue M., Takemoto N., Matsumoto K., Nakamura T., Nakajima K., Kohsaka S. Neurotrophic effect of hepatocyte growth factor on central nervous system neurons in vitro. Journal of Neuroscience Research. 1996;43:554–564. doi: 10.1002/(SICI)1097-4547(19960301)43:5<554::AID-JNR5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hardan A.Y., Muddasani S., Vemulapalli M., Keshavan M.S., Minshew N.J. An MRI study of increased cortical thickness in autism. The American Journal of Psychiatry. 2006;163:1290–1292. doi: 10.1176/appi.ajp.163.7.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett H.C., Poe M.D., Gerig G., Smith R.G., Piven J. Cortical gray and white brain tissue volume in adolescents and adults with autism. Biological Psychiatry. 2006;59:1–6. doi: 10.1016/j.biopsych.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Hazlett H.C., Poe M.D., Gerig G., Styner M., Chappell C., Smith R.G., Vachet C., Piven J. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Archives of General Psychiatry. 2011;68:467–476. doi: 10.1001/archgenpsychiatry.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert M.R., Ziegler D.A., Deutsch C.K., O'Brien L.M., Lange N., Bakardjiev A., Hodgson J., Adrien K.T., Steele S., Makris N., Kennedy D., Harris G.J., Caviness V.S., Jr. Dissociations of cerebral cortex, subcortical and cerebral white matter volumes in autistic boys. Brain. 2003;126:1182–1192. doi: 10.1093/brain/awg110. [DOI] [PubMed] [Google Scholar]
- Herbert M.R., Ziegler D.A., Makris N., Filipek P.A., Kemper T.L., Normandin J.J., Sanders H.A., Kennedy D.N., Caviness V.S., Jr. Localization of white matter volume increase in autism and developmental language disorder. Annals of Neurology. 2004;55:530–540. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- Hollander E., Anagnostou E., Chaplin W., Esposito K., Haznedar M.M., Licalzi E., Wasserman S., Soorya L., Buchsbaum M. Striatal volume on magnetic resonance imaging and repetitive behaviors in autism. Biological Psychiatry. 2005;58:226–232. doi: 10.1016/j.biopsych.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Honda S., Kagoshima M., Wanaka A., Tohyama M., Matsumoto K., Nakamura T. Localization and functional coupling of HGF and c-Met/HGF receptor in rat brain: implication as neurotrophic factor. Brain Research. Molecular Brain Research. 1995;32:197–210. doi: 10.1016/0169-328x(95)00075-4. [DOI] [PubMed] [Google Scholar]
- Huh C.G., Factor V.M., Sanchez A., Uchida K., Conner E.A., Thorgeirsson S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson M.C., Bergman M.Y., Campbell D.B., Eagleson K.L., Levitt P. Dynamic gene and protein expression patterns of the autism-associated met receptor tyrosine kinase in the developing mouse forebrain. The Journal of Comparative Neurology. 2009;513:511–531. doi: 10.1002/cne.21969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson M.C., Eagleson K.L., Wang L., Levitt P. Evidence of cell-nonautonomous changes in dendrite and dendritic spine morphology in the met-signaling-deficient mouse forebrain. The Journal of Comparative Neurology. 2010;518:4463–4478. doi: 10.1002/cne.22467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, W.W., Sahin, M., Scherrer, B., Peters, J.M., Suarez, R.O., Vogel-Farley, V.K., Jeste, S.S., Gregas, M.C., Prabhu, S.P., Nelson III, C.A., Warfield, S.K., in press. Impaired lan-guage pathways in tuberous sclerosis complex patients with autism spectrum dis-orders. Cerebral Cortex. (Electronic publication ahead of print). http://dx.doi.org/10.1093/cercor/bhs135. [DOI] [PMC free article] [PubMed]
- Maina F., Hilton M.C., Ponzetto C., Davies A.M., Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes & Development. 1997;11:3341–3350. doi: 10.1101/gad.11.24.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins G.J., Plachez C., Powell E.M. Loss of embryonic MET signaling alters profiles of hippocampal interneurons. Developmental Neuroscience. 2007;29:143–158. doi: 10.1159/000096219. [DOI] [PubMed] [Google Scholar]
- Martins G.J., Powell E.M. Genetic disruption of Met signaling impairs GABAergic striatal development and cognition. Neuroscience. 2011;176:199–209. doi: 10.1016/j.neuroscience.2010.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L., Weidner K.M., Vigna E., Gaudino G., Bardelli A., Ponzetto C., Narsimhan R.P., Hartmann G., Zarnegar R., Michalopoulos G.K. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO Journal. 1991;10:2867–2878. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G., Franklin K.B.J. 2nd ed. Academic Press; San Diego: 2001. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
- Percy A.K. Rett syndrome: exploring the autism link. Archives of Neurology. 2011;68:985–989. doi: 10.1001/archneurol.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piven J., Arndt S., Bailey J., Havercamp S., Andreasen N.C., Palmer P. An MRI study of brain size in autism. The American Journal of Psychiatry. 1995;152:1145–1149. doi: 10.1176/ajp.152.8.1145. [DOI] [PubMed] [Google Scholar]
- Piven J., Bailey J., Ranson B.J., Arndt S. An MRI study of the corpus callosum in autism. The American Journal of Psychiatry. 1997;154:1051–1056. doi: 10.1176/ajp.154.8.1051. [DOI] [PubMed] [Google Scholar]
- Powell E.M., Campbell D.B., Stanwood G.D., Davis C., Noebels J.L., Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. Journal of Neuroscience. 2003;23:622–631. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell E.M., Mars W.M., Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Powell E.M., Muhlfriedel S., Bolz J., Levitt P. Differential regulation of thalamic and cortical axonal growth by hepatocyte growth factor/scatter factor. Developmental Neuroscience. 2003;25:197–206. doi: 10.1159/000072268. [DOI] [PubMed] [Google Scholar]
- Qiu S., Anderson C.T., Levitt P., Shepherd G.M. Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated Met receptor tyrosine kinase. The Journal of Neuroscience: The official journal of the Society for Neuroscience. 2011;31:5855–5864. doi: 10.1523/JNEUROSCI.6569-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redcay E., Courchesne E. When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biological Psychology. 2005;58:1–9. doi: 10.1016/j.biopsych.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Schumann C.M., Hamstra J., Goodlin-Jones B.L., Lotspeich L.J., Kwon H., Buonocore M.H., Lammers C.R., Reiss A.L., Amaral D.G. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. Journal of Neuroscience. 2004;24:6392–6401. doi: 10.1523/JNEUROSCI.1297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears L.L., Vest C., Mohamed S., Bailey J., Ranson B.J., Piven J. An MRI study of the basal ganglia in autism. Progress in Neuropsychopharmacology & Biological Psychiatry. 1999;23:613–624. doi: 10.1016/s0278-5846(99)00020-2. [DOI] [PubMed] [Google Scholar]
- Smalley S.L., Tanguay P.E., Smith M., Gutierrez G. Autism and tuberous sclerosis. Journal of Autism and Developmental Disorders. 1992;22:339–355. doi: 10.1007/BF01048239. [DOI] [PubMed] [Google Scholar]
- Sousa I., Clark T.G., Toma C., Kobayashi K., Choma M., Holt R., Sykes N.H., Lamb J.A., Bailey A.J., Battaglia A., Maestrini E., Monaco A.P. MET and autism susceptibility: family and case–control studies. European Journal of Human Genetics. 2009;17:749–758. doi: 10.1038/ejhg.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stigler K.A., McDonald B.C., Anand A., Saykin A.J., McDougle C.J. Structural and functional magnetic resonance imaging of autism spectrum disorders. Brain Research. 2011;1380:146–161. doi: 10.1016/j.brainres.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltenberg C., Schjolberg S., Bresnahan M., Hornig M., Hirtz D., Dahl C., Lie K.K., Reichborn-Kjennerud T., Schreuder P., Alsaker E., Oyen A.S., Magnus P., Suren P., Susser E., Lipkin W.I. The autism birth cohort: a paradigm for gene-environment-timing research. Molecular Psychiatry. 2010;15:676–680. doi: 10.1038/mp.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straussman R., Morikawa T., Shee K., Barzily-Rokni M., Qian Z.R., Du J., Davis A., Mongare M.M., Gould J., Frederick D.T., Cooper Z.A., Chapman P.B., Solit D.B., Ribas A., Lo R.S., Flaherty K.T., Ogino S., Wargo J.A., Golub T.R. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F., Qi L., Zhang W., Hansen R.L., Pessah I.N., Hertz-Picciotto I. MAOA, DBH, and SLC6A4 variants in CHARGE: a case–control study of autism spectrum disorders. Autism Research. 2011;4:250–261. doi: 10.1002/aur.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanseem I., Nakamura K., Miyachi T., Toyota T., Yamada S., Tsujii M., Tsuchiya K.J., Anitha A., Iwayama Y., Yamada K., Hattori E., Matsuzaki H., Matsumoto K., Iwata Y., Suzuki K., Suda S., Kawai M., Sugihara G., Takebayashi K., Takei N., Ichikawa H., Sugiyama T., Yoshikawa T., Mori N. Further evidence for the role of MET in autism susceptibility. Neurosciences Research. 2010;68:137–141. doi: 10.1016/j.neures.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Tsatsanis K.D., Rourke B.P., Klin A., Volkmar F.R., Cicchetti D., Schultz R.T. Reduced thalamic volume in high-functioning individuals with autism. Biological Psychiatry. 2003;53:121–129. doi: 10.1016/s0006-3223(02)01530-5. [DOI] [PubMed] [Google Scholar]
- Williams E.L., Casanova M.F. Above genetics: lessons from cerebral development in autism. Translational Neuroscience. 2011;2:106–120. doi: 10.2478/s13380-011-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]