

Abstract
Most subtypes of pulmonary arterial hypertension (PAH) are characterized by a greater susceptibility to disease among females, although females with PAH appear to live longer after diagnosis. While this “estrogen paradoxȍ of enhanced female survival despite increased female susceptibility remains a mystery, recent progress has begun to shed light upon the interplay of sex hormones, the pathogenesis of pulmonary hypertension, and the right ventricular response to stress. For example, emerging data in humans and experimental models suggest that estrogens or differential sex hormone metabolism may modify disease risk among susceptible subjects, and that estrogens may interact with additional local factors such as serotonin to enhance the potentially damaging chronic effects of estrogens on the pulmonary vasculature. Regardless, it remains unclear why not all estrogenic compounds behave equally, nor why estrogens appear to be protective in certain settings but detrimental in others. The contribution of androgens and other compounds, such as dehydroepiandrosterone, to pathogenesis and possibly treatment must be considered as well. In this review, we will discuss the recent understandings on how estrogens, estrogen metabolism, dehydroepiandrosterone, and additional susceptibility factors may all contribute to the pathogenesis or potentially to the treatment of pulmonary hypertension, by evaluating current human, cell-based, and experimental model data.
Keywords: pulmonary hypertension, estrogen, dehydroepiandrosterone, serotonin, bone morphogenetic protein receptor type II
Pulmonary hypertension (PH) is the inappropriate elevation of pulmonary artery pressure which can ultimately result in right ventricular (RV) dysfunction and failure. PH may occur due to a wide variety of clinical conditions, the majority of which are manifested histopathologically by medial hypertrophy of the pulmonary arterioles with intimal proliferation.[1,2] In the current classification scheme for PH, PAH is considered Category 1, and is divided into multiple disease subgroups including idiopathic PAH (IPAH), heritable PAH (HPAH), and PAH-associated (APAH) with a variety of other systemic diseases or drug/toxin exposures.[3] Nearly all subtypes of PAH share a common feature: A higher female than male prevalence among adult patients.[4,5,6] However, the precise mechanisms of this gender disparity remain elusive, as does the impact of gender upon RV response to PH and, ultimately, survival.
Long recognized,[7] the skewed gender ratio was confirmed in 1987 by a large National Institutes of Health (NIH) registry, which reported a female to male ratio of 1.7:1.[8] However, the precise prevalence and incidence rates of PAH substratified according to gender, as well as clinical outcomes, remain incompletely understood in the current era. Recent epidemiologic studies, such as the Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL), have begun to shed light on the gender inequity and suggest differences between disease initiation and progression. REVEAL, an observational cohort study of PAH cases in North America,[9] recently reported that 79.5% of prevalent adult PAH patients were females, with a female predominance across virtually all subtypes of PAH.[4,10] Investigating incident cases, UK and Ireland investigators recently reported that 69.9% of incident PAH patients from 2001-2009 were females.[11] However, while data from France support the high prevalence of female cases (65.3%) for most subtypes of PAH, among incident cases, the percentage of females was only 57.0%.[6]
The potential difference in prevalence and incidence according to gender, at least in the French cohort, suggests that females live longer than males after diagnosis of PAH. Initial prospectively collected data from France support this conclusion, with improved survival at three years for females compared to males.[12,13] Similarly, REVEAL investigators retrospectively found that men over 60 years of age had poorer survival compared with younger males and compared to all females.[10] While comprehensive prospective data will emerge over the coming years to address specifics with regard to the gender epidemiology of PH, the suggestion that females get disease more frequently but survive longer underscores the complexity of gender in the pathogenesis and course of PH; and, it highlights the need to more precisely understand the pathologic (and potentially protective) consequences of gender. The prospect that gender-associated factors, which promote disease onset may differentially impact disease outcome is particularly interesting given the prevailing opinion that while PAH is a pulmonary vascular disease, survival is dictated by RV adaptation to load stress. This incongruous finding has been termed the “estrogen paradox in PH.”[14]
The epidemiology of PH with regard to gender is but one component of the evolving story around gender in this devastating disease. The issue has attracted renewed focus in the past few years, providing fertile ground for the intersection of in vitro, animal model, and human studies to ultimately improve our understanding of not only the factors which promote disease, but also those which modify survival. While chromosomal differences (XX versus XY) certainly exist between the genders, an obvious source of variation is the production, metabolism, and response to sex hormones, which will be the particular focus of this review.
SEX HORMONES AND HUMAN PAH
The impact of sex hormones on the pulmonary vasculature and right heart is complex and incompletely understood. For example, while acutely vasodilatory, estrogens may have deleterious consequences upon the pulmonary vasculature with chronic excess exposure. Thus, differential exposure to sex hormones is an active area of investigation, particularly in PAH research.
Estrogens and androgens are vasoactive compounds in the pulmonary vasculature with variable effects on cell proliferation and apoptosis. Several previous reports described an association between pharmacologically (and natural increase via pregnancy) enhanced estrogen exposure and PAH.[15,16,17,18] More recently, Sweeney and colleagues determined that 81% of women with PAH reported prior use of any female hormone therapy, including 70% reporting exogenous hormone use for greater than 10 years.[19] Meanwhile, in a select cohort of portal hypertension patients, Roberts and colleagues found that both elevated plasma 17β-estradiol (estradiol, E2) levels and genetic variations in estrogen signaling are associated with portopulmonary PAH (PPHTN).[20] However, comprehensive measures of parent compound estrogen and androgen levels in incident and prevalent PH cases are lacking.
While both female and male sex hormones may contribute to pathogenesis (or protection), the metabolic products of sex hormone metabolism are biologically active and may also contribute. As with parent compound sex hormones, the complete effect of the various sex hormone metabolites on the pulmonary vasculature and right ventricle has yet to be determined, but may be highly relevant. For example, variability in metabolism may account for the apparent contradictory influences of estrogens discussed below, perhaps via differential sex hormone receptor activation and signaling.
The majority of estrogen and androgen metabolism occurs via the Cytochrome P450 (CYP) system, although there may be organ-specific differences.[21] CYPs constitute a gene superfamily that plays an essential role in the metabolism of exogenous chemicals present in the diet and environment, as well as endogenous substances such as parent compound estrogens and androgens.[22,23] For the metabolism of estrogens, the initial step is typically an oxidative process, via CYP1B1 and other CYP enzymes (Fig. 1). CYP1B1 and several other CYPs are highly expressed in the lung. CYPs oxidize estrogens to 2-hydroxy (2-OHE1/2) and 4-hydroxy (4-OHE1/2) estrogens in the initial metabolic step[24] to produce “2-estrogens” and “4-estrogens,” or, oxidation of estrogens by hydroxylation can occur at the C-16 position, predominantly resulting in 16α-hydroxyestrone (16α-OHE1) for the ultimate generation of “16-estrogens.”[25,26] While full details on the molecular consequences of exposure to the various estrogen metabolites on the pulmonary vasculature are needed, it appears that “2-estrogens" are typically anti-mitogenic, while “16-estrogens" stimulate cellular proliferation by constitutively activating the estrogen receptors. In addition to being more mitogenic, “16-estrogens" may also be more detrimental due to a genotoxic effect via the formation of unstable DNA adducts.[27,28] As a result, one might ultimately hypothesize that individuals who produce more “16-estrogens” are at increased risk of diseases that result from both the mitogenic and genotoxic effects of estrogens.[29,30,31,32,33,34]
Figure 1.
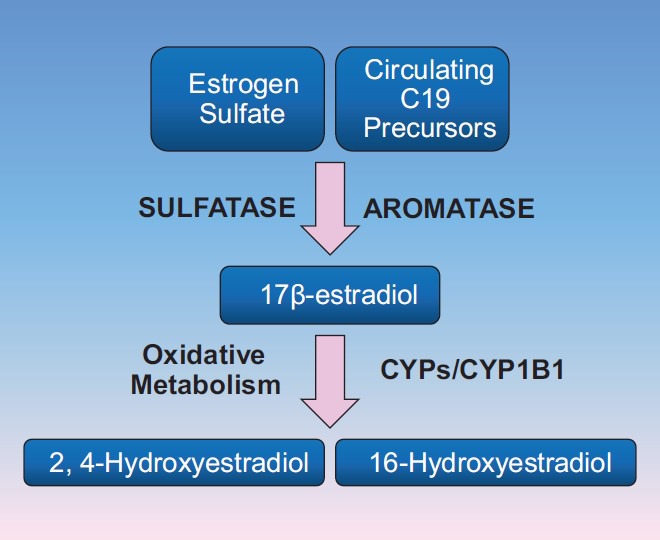
Simplified schematic of estrogen metabolism. Parent compound estrogens are metabolized via oxidative metabolism to hydroxestradiol forms in the initial step of metabolism. Hydroxylation may occur at position C2, C4, or C16. This step of oxidative metabolism determines the nature of the biologic effects of the metabolites of 17β-estradiol (E2). These products are most commonly cleared from circulation via methylation, as demonstrated in (Figure 5.
The detection of a germline mutation in the gene bone morphogenetic protein receptor type 2 (BMPR2) gene confers the greatest risk of PAH, although the factors modifying this risk are unclear. Penetrance of PAH among BMPR2 mutation carriers is incomplete and differs according to gender: Female penetrance is ~42%, while male penetrance is ~14%.[35] In an effort to determine genetic modifiers of BMPR2-associated PAH, West et al. studied expression array data, verified by quantitative PCR, in BMPR2 mutation carriers’ EBV-immortalized lymphocytes, and found that CYP1B1 gene expression was reduced 10-fold among female BMPR2 mutation carriers with PAH compared to healthy females.[36] This finding prompted the hypothesis that among female BMPR2 mutation carriers, those with PAH were more likely to possess genetic variants associated with altered CYP1B1 activity, as well as measurable alterations in estrogen metabolites, compared to those without PAH.
To test this hypothesis, targeted functional variants in CYP1B1 were evaluated in a cohort of 140 BMPR2 mutation carriers (86 females, 54 males). Genetic polymorphisms were chosen based upon previous association with alterations in protein function, including CYP1B1 Asn453Ser (N453S).[37] As hypothesized, among females there was a four-fold higher penetrance of PAH (P = 0.005) among subjects homozygous for the wild-type genotype (N/N) of CYP1B1 Asn453Ser (N453S); no difference was detected among males. This genotype had previously been associated with alterations in in vitro CYP1B1 activity and with differences in breast cancer risk in humans.[38] A nested study of a small number of those BMPR2 mutation carriers supported this finding. Specifically, female BMPR2 mutation carriers with PAH had a significantly lower ratio of 2-hydroxyestrogens (2-OHE1/2): 16α-hydroxyestrone (16α-OHE1) compared to unaffected BMPR2 mutation carriers (P = 0.006) carriers.[39] In an effort to explore the biology of this association, it was subsequently demonstrated that parent compound estrogens, and “16-estrogens,” directly reduce BMPR2 gene expression; additionally, this effect occurred at least in part via direct estrogen receptor alpha (ERα) binding to the BMPR2 gene promoter.[40] Taken together, these results support the hypothesis that variations in estrogen metabolism may contribute to the development of PAH among humans at heightened genetic risk (BMPR2 gene mutation), although the precise molecular mechanisms and relevance to variations in estrogen receptor signaling need to be clarified.
ESTROGEN RECEPTOR SUBTYPES, SIGNALING, AND REGULATION
Two classical estrogen receptors (ERs) exist: ERα and ERβ.[41,42,43,44,45] In addition, an orphan G-protein coupled receptor (GPR30) can also bind estrogen, but seems to be primarily responsible for mediating acute (nongenomic) estrogen effects.[46] ERα and ERβ are steroid hormone receptors that are coded by different genes on different chromosomes. ERα is a 66 kDa protein, while ERβ is 59 kDa. Both ERs exhibit a Deoxyribonucleic acid (DNA)-binding domain, a ligand-binding domain, and an amino terminal transcriptional control domain (AF-1), through which they interact with regulatory binding proteins. The DNA-binding domain and the ligand-binding domain are 97% and 60%, respectively, homologous between both ERs. However, significant differences exist between ERα and ERβ in the AF-1 domain. In addition, the two ERs differ in their tissue distribution, and both ERα and -β have been found to have specific and nonredundant functions in the reproductive, cardiovascular, respiratory, central nervous, immune, and skeletal systems. At least two splice variants for ERα (ERα-36 and ERα-46) and four splice variants for ERβ (ERβ2, -4, -5, Δexon5) have been described, thereby, further contributing to the complexity of ER effects.[44]
While initially described to be located in the cytoplasm and nucleus, more recent evidence suggests that both ERα and ERβ can also be located in the cell membrane, where they mediate nongenomic estrogen signaling.[47] In addition, ERβ has also been found in the mitochondria, where it is purported to modulate mitochondrial DNA transcription.[48,49] These diverse cellular locations reflect the different existing mechanisms and how ERs mediate their various effects.[41,42,43,44,45] Briefly, four major signaling pathways exist (Fig. 2). The classical (genomic or direct) pathway of ER signaling follows estrogen binding to cytoplasmatic ERα or ERβ, resulting in dissociation of chaperone proteins away from the ER with subsequent ER conformational change and dimerization of the estrogen-ER complex with another estrogen-ER complex. This is followed by translocation of the dimerized estrogen-ER unit to the nucleus, where it then binds to estrogen responsive elements (EREs) and acts as a classical transcription factor, with its function being modified by co-activators and co-repressors. In the tethered pathway, the estrogen-ER-complex dimer—instead of directly binding to EREs—indirectly affects gene regulation by binding to other transcription factors. Thirdly, ER activation, dimerization, and nuclear translocation can also occur in an estrogen-independent fashion through direct phosphorylation by nonestrogen ligands (usually a growth factor, e.g., epidermal growth factor[1,50]. Lastly, recent evidence suggests that ERα or ERβ can also be located on the cell membrane, where they, upon estrogen binding, directly activate kinases or other second messengers (e.g. phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK)), thereby, leading to rapid cellular affects (e.g. endothelial nitric oxide synthase (eNOS) or ion channel activation) without affecting gene expression (referred to as the non-genomic pathway).[47] In contrast to genomic effects, which occur over several hours, non-genomic effects occur within seconds to minutes. Non-genomic ER signaling appears to be particularly important in the cardiovascular system, and in the cellular response to acute injury.[51,52,53,54,55,56,57,58]
Figure 2.
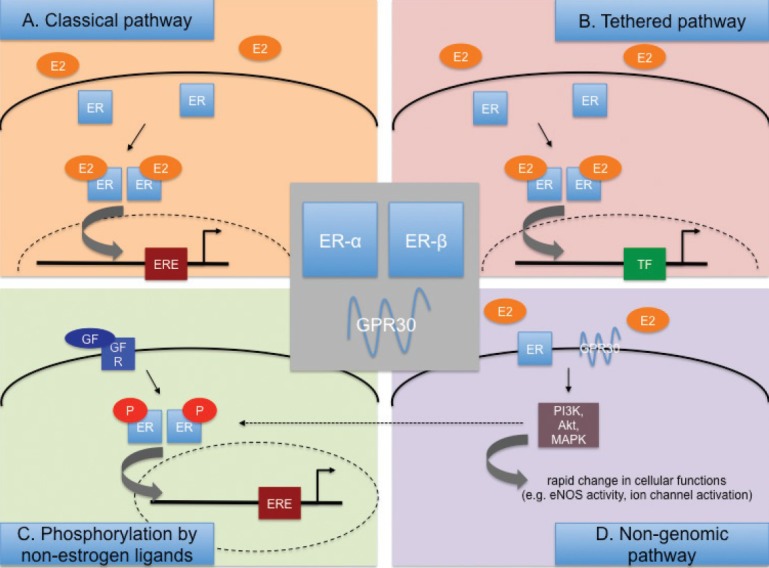
Major estrogen receptor (ER)-mediated signaling pathways engaged by 17β-estradiol (E2). (A) and (B) E2 diffuses through the cell membrane and interacts with ER-α or –β, followed by formation of E2-ER homo- or heterodimers. The E2-ER complexes then translocate into the nucleus to interact with estrogen response elements (ERE; A) or with other transcription factors (TF; B). (C) ERs are directly phosphorylated by growth factors (GF). (D) E2 can interact with membrane-bound classical ERs or GPR30, leading to rapid changes in protein function without immediately altering genomic pathways.
Multiple factors regulate ER expression and activity.[41] For example, estrogen increases ER transcription, while progesterone, vitamin D and the ERα variant ERα-36 have been reported to decrease expression of ER. However, estrogen binding to the ER may also stimulate its ubiquitination and proteosomal degradation. Furthermore, methylation of the ER promoter reduces ER mRNA levels, and transcription factors or enzymes such as myocyte enhancer factor 2 or class II histone deacetylase may downregulate ERα expression. In addition, post-translational ER modification occurs, with S-nitrolysation inhibiting, and acteylation enhancing ER transcriptional activity. Consequently, ER expression and activity are significantly affected by sex and variations in endogenous sex hormone levels (e.g., menstrual cycle and menopause), as well as other factors, such as age and disease. Furthermore, ERα or ERβ splice variants are responsible for significant variations in ER effects, and ER splice variants can silence signaling of other ERs. For example, ERβ2 dimerizes with preferentially ERα, thereby, facilitating degradation of the latter.[59]
The role of estrogen receptors in the pulmonary vasculature (from animal to human, and from health to disease)
Both ERα and ERβ are present in endothelial cells and smooth muscle cells of the systemic vasculature, where they have been implicated in mediating protective estrogen effects in the setting of acute and chronic vascular injury. Nevertheless, ERα seems to mediate most of the protective effects of estrogen on injured blood vessels.[60,61] Estrogen is also critical for maintaining the homeostasis of the normal endothelium, with effects being mediated by nongenomic activation of both classical ERs as well as G protein-coupled receptor 30 (GPR30).[62,63,64] However, genomic mechanisms are also involved. Along those lines, several studies have associated single nucleotide polymorphisms (SNPs) in the Estrogen receptor alpha (ESR1) gene (the gene encoding ERα) with myocardial infarction, hypertension, and stroke,[65,66,67] while, in hypertensive women, SNPs in Estrogen receptor beta (ESR2) (encoding ERβ) have been linked to increases in left ventricular mass and hypertrophy.[68]
In the lung, ERα and -β are expressed in pulmonary artery endothelial cells (PAECs),[69,70,71] smooth muscle cells,[70,72] airway epithelium,[70,73] type 1 and 2 alveolar cells,[74] and alveolar macrophages.[71,75] Several studies suggest more abundant expression of ERβ over ERα in the lung,[74,75,76,77] while others suggest a more predominant role for ERα.[71] It is, therefore, conceivable that the predominance of one ER over the other may depend on the clinical context. Factors such as sex, age, hormonal status, and the presence of disease likely also play a significant role. Lastly, ER expression and function may be species-specific. It should also be noted that divergent results between studies may be due to the fact that some studies look at ER transcript, while others investigate ER protein. In general, the role and expression of ERα and ERβ in the pulmonary vascular system is not as well described as in the systemic vasculature, and most of the currently available information comes from studies using animal tissues. These studies (reviewed below) demonstrate that both ERα and ERβ are present and physiologically active in lung blood vessels.
In vitro and ex vivo studies of lung vascular ER signaling. In studies of simian virus, 40-transformed rat lung vascular endothelial cells (TRLECs), Hisamoto et al. demonstrated that E2-induced eNOS activation through a nongenomic, ERα-mediated, and Akt-dependent mechanism.[51] Another group reported that E2 increases eNOS activity in fetal lamb PAECs through activation of ERβ in caveolae.[52] Also, in fetal PAECs, Sherman et al. demonstrated that E2 enhances prostacyclin synthesis in fetal PAEC through an ERβ- and calcium-dependent, but tyrosine kinase/MAPK-independent, mechanism.[53] Lahm et al. have previously shown that, in isolated rat pulmonary artery rings, administration of the selective ERα-agonist propyl pyrazole triol (PPT) attenuates phenylephrine-induced vasocontraction, while administration of the selective ERβ-agonist diarylpropionitrile (DPN) attenuated hypoxia-induced vasocontraction.[78] Of note, these effects were abolished after administration of the nitric oxide synthase (NOS) inhibitor L-NG-Nitroarginine Methyl Ester (L-NAME), suggesting that the effects of the ER-subtype selective agonists are endothelium- and NO-dependent. Since activation of either ER subtype resulted in rapid (within 20-30 minutes of administration) vasocontraction, the authors concluded that these effects were mediated through the nongenomic pathway. This observation of NO-dependent, nongenomic vasorelaxation is consistent with reports of nongenomic, ER-mediated NO release in the systemic vasculature.[54]
In vivo studies of lung vascular ER signaling. Immunohistochemical investigations have demonstrated both ERα and -β in PAECs of normal rats and mice, respectively.[70,71] With regards to PAH, a recent study by Umar et al.[79] demonstrated that administration of E2 to male rats in a monocrotaline model attenuated established severe PAH. Interestingly, E2 protection was lost after coadministration of the selective ERβ-antagonist 4-[2-Phenyl-5,7-bis(trifluoromethyl) pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP), while treatment with an ERβ-agonist (DPN) replicated E2's effects on the pulmonary vasculature and RV. The authors, therefore, concluded that E2 protection in their model was mediated by an ERβ-mediated mechanism. ER-mediated protection was also demonstrated in a study by Lahm et al. in which E2 was administered to male rats with hypoxia-induced PH (HPH).[80] In this study, E2 attenuated hemodynamic alterations, hypoxia-induced erythrocytosis, and pulmonary vascular and RV remodeling in an ER-dependent fashion, as evidenced by loss of E2 protection after coadministration of the nonselective ER-antagonist fulvestrant. E2 protection was associated with ER-mediated decreases in lung and RV ERK1/2 activation. Similar ER-mediated effects were confirmed in isolated rat PAECs. Importantly, E2 exerted differential effects in normoxic and hypoxic cells. In particular, E2 decreased Extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) activation and increased expression of the cell cycle inhibitor p27Kip1 and the autophagy marker Microtubule-associated protein 1A/1B-light chain 3-phosphatidylethanolamine conjugate (LC3-II) in hypoxic (1% O2 for 48 hours), but not normoxic, rat PAECs. These effects were accompanied by a decrease in PAEC secretion of vascular endothelial growth factor (VEGF) and inhibitory E2 effects on cell proliferation during hypoxia only. Differential, context-specific effects of E2 on proliferative processes may, therefore, explain at least in part the protective effects of estrogens in animal models of PH. Of note, E2 effects on hemodynamics and RV mass in the Lahm study were not attenuated with ERβ-antagonist cotreatment, while coadministration of ERα-antagonist 1-methyl-4-phenyl pyridinium (MPP) lead to a significant decrease in E2 protection. Interestingly, animals cotreated with E2 and ERα-antagonist exhibited a significant decrease in cardiac output, suggesting a predominantly RV-directed mechanism of E2 and ERα action. On the other hand, E2 effects on certain remodeling and signaling parameters (PA muscularization, PAEC ERK1/2 activation) were attenuated after either selective ERα- or selective ERβ-blockade, suggesting that both ERs need to be functional for E2 to exert protective effects on these endpoints. However, neither ERα nor ERβ-blockade attenuated E2 effects on RV capillarization or RV ERK signaling. This suggests a potential redundant role of the two ERs with regards to RV remodeling/capillarization in HPH. The authors of this study also evaluated lung vascular ER expression in normal and HPH rats. While ERα expression was not affected by two weeks of hypobaric hypoxia (Patm= 362 mmHg), there was evidence of hypoxia-induced pulmonary vascular upregulation of ERβ (demonstrated by immunohistochemistry). ERβ expression appeared most pronounced in PAECs.
Studies of ER signaling in humans. With regards to human pulmonary vascular disease, a recent study of genome-wide Ribonucleic acid (RNA) expression profiling in lungs of patients with PAH or PH secondary to idiopathic pulmonary fibrosis (IPF-PH)[81] demonstrated upregulation of ESR1 in PAH subjects relative to normal controls and IPF-PH. Segregation of PAH and normal control samples into females and males showed that both female and male PAH had a higher mean expression of ESR1 than female or male controls. However, the experimental groups in this study were relatively small (n = 4-11). ESR1 has also been involved in the development of PPHTN. In the previously mentioned study of patients with advanced liver disease by Roberts et al.,[20] SNPs in the ESR1 gene were (amongst SNPs in other genes) associated with an increased risk of PPHTN development. While these studies suggest an association between ESR1 and PAH/PPHTN, it is not known if this represents a pathogenic mechanism of PAH/PPHTN development versus a consequence of the disease (e.g., a compensatory mechanism for impaired ERα or ERβ signaling, or for other abnormalities in E2 signaling pathways).
Taken together, the role of the ERs in the development of PAH at this point remains unclear. While in vitro, ex vivo, and in vivo studies in monocrotaline-PH and HPH suggest potential protective effects of ERα and/or ERβ, the two currently available human studies suggest an association between ESR1 and PAH/PPHTN development. However, cause and effect in human PAH remain unclear. Further mechanistic animal studies in angioproliferative PH models, as well as further epidemiologic studies in PAH patients, will hopefully help shed further light on this issue.
The role of estrogen receptors in the right ventricle
Very little is known about the role of the ER in the right ventricle. Multiple studies in different types of acute or chronic left ventricular injury have demonstrated ERα- or ERβ-mediated protective effects of E2 on functional parameters and markers of myocardial metabolism, inflammation, and apoptosis.[82,83,84,85] These effects likely occur through E2 action on cardiomyocytes and endothelial cells, as well as progenitor cells and inflammatory cells.[41] Given that RV systolic function in both healthy and PAH-affected women is superior relative to men, and correlates with E2 levels,[86,87] it is conceivable that E2 also has direct, ER-mediated RV cardioprotective effects. Such a notion is supported by the studies by Umar et al.[79] and Lahm et al.,[80] in which E2 exerted beneficial effects on RV function. The first study (using a monocrotaline model of PH) suggested an ERβ-mediated mechanism of action, while involvement of ERs in the second study (performed in an HPH model) was more complex. While the authors of the latter study demonstrated E2-mediated increases in CO that were attenuated after nonselective ER-blockade as well as selective ERα-blockade, E2 effects on RV capillarization as well as RV ERK1/2 activation were only attenuated after nonselective ER-blockade, but not after ERα- or ERβ-selective blockade. This may suggest potential redundant functions of each ER subtype in mediating E2 effects on these endpoints. To date, no study has investigated the role of the ER in the RV in human PAH. Since RV function in healthy individuals correlates with E2 levels,[87] and given the currently existing animal studies of ER-mediated E2 action in both the LV and RV, it is likely that E2 affects function in the human RV at least in part via ER.
In conclusion, ER signaling is very complex, and not only regulated on a transcriptional and translational level, but also affected by the presence or absence of coactivators, corepressors, and splice variants, as well as by epigenetic phenomena, tissue distribution, and factors like age, sex, hormonal status, and disease. The role of the ER in PAH development, though likely important, remains poorly defined. Additional investigations of ER signaling in advanced animal models of PH and in PAH patients may help further elucidate the molecular mechanisms of sex differences in PAH, and may facilitate the development of novel nonhormonal, targeted therapies for PAH patients of either sex.
EXPERIMENTAL PH: BMP SIGNALING AND ESTROGENS
BMPR2-estrogen interaction
Enhanced estrogenic activity appears to be a risk factor for human PAH, and a BMPR2 gene mutation is a roughly ×100,000 risk factor for PAH; thus, an important question is whether these two factors are independent or interacting. Recent data suggests that estrogen and Bone morphogenetic protein (BMP) signaling interact in complex ways. Broadly, estrogens suppress BMP signaling through transcriptional and nontranscriptional means, while BMP signaling modulates the mode of estrogen signaling.
There is an increasing body of literature from the osteoporosis and breast cancer fields examining interaction between BMP signaling and estrogen. Estrogen suppression of the BMP pathway appears to be through nongenomic estrogen receptor signaling: E2 suppresses BMP2-induced differentiation of osteoblasts; this was true even in mice with an ERα mutated to prevent DNA binding.[88] Estrogen can also suppress BMP4[89,90] and BMP7[91] expression in different tissue types. This consensus of estrogen inhibition of BMP signaling is not perfect; however, E2 may increase BMP6 expression in some breast cancer cell lines.[92]
There is also evidence from the literature that BMP signaling modulates estrogens. In breast cancer cells, BMP2 induces expression of an estrogen splice variant that lacks transcriptional activation domains, and is restricted to noncanonical signaling.[93] In lactotrophs, BMP directly inhibits ER transcription,[94] and induces prolactin expression[95] through direct SMAD binding of the promoters. In oocytes, BMP6 enhances E2 synthesis but not progesterone.[96]
In summary, in the non-PAH literature, there is substantial evidence that the typical role of estrogens is to suppress the BMP pathway, particularly through nongenomic signaling, while the BMP pathway has extensive and complex regulation of estrogens and estrogen receptors.
Effect of BMPR2 mutation on estrogen receptor trafficking
BMPR2 normally regulates steroid hormone receptor trafficking,[97] and BMPR2 mutation results in defects in glucocorticoid receptor (GR) trafficking.[98] GR trafficking is regulated by cofilin phosphorylation,[99] and BMPR2 directly binds and regulates the primary protein which phosphorylates cofilin, LIM kinase 1 (LIMK1).[100,101] The most likely mechanism for BMPR2 regulation of steroid hormone receptor trafficking is thus through direct interaction with LIMK1 and consequent regulation of cofilin. However, this mechanism has not been explicitly tested, and BMPR2 may also regulate receptor shuttling through its direct regulation of Src[102] or Dynein light chain Tctex-type 1 (DYNLT1).[103]
BMPR2 mutant pulmonary microvascular endothelial cells (PMVECs) show dysregulation of ERα trafficking; by estrogen response element luciferase reporter assay, BMPR2 mutant PMVECs have dramatically reduced canonical (genomic) response to either E2 or 16α-OHE1 signal (Pulmonary Circulation 2013, in press). By immunohistochemistry, instead of the movement of ERα to the nucleus caused by E2 in control PMVEC, in BMPR2 mutants ERα appears to move to the cell surface (Fig. 3). While this has not been explicitly tested, this implies that in the context of BMPR2 mutation, estrogens preferentially signal through nongenomic means.
Figure 3.
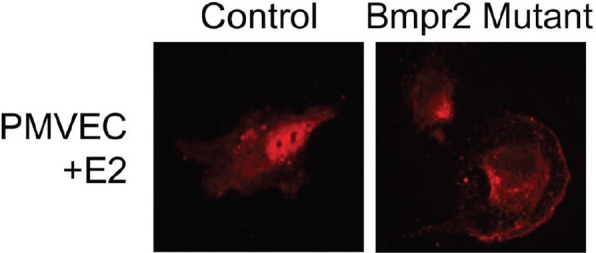
BMPR2 mutant PMVEC show dysregulation of ERα trafficking. Immunohistochemistry studies demonstrate abnormal movement of ERα in BMPR2 mutant cells. While in control PMVEC (left) the ERα moves to the nucleus with E2 stimulation, ERα appears to move to the cell surface in BMPR2 mutant PMVEC cells (right) stimulated by E2 exposure.
16α-OHE1 worsens penetrance in BMPR2 mutant mice and suppresses BMPR2 protein levels in control mice
Increased 16α-OHE1 is correlated with increased penetrance in human heritable PAH patients, both female[39] and male (Fessel et al., Pulmonary Circulation 2013, submitted; title: “Interaction of BMPR2 and Estrogenic Compounds in PAH”). To determine whether this correlation implied causation, Fessel et al. treated control mice or mice with two different inducible BMPR2 mutations with 16α-OHE1 in osmotic pumps. They reported that 16α-OHE1 doubled penetrance of significantly increased pulmonary vascular resistance (defined as PVR increase of at least 50% from controls). Physiologically, this appeared to be caused by a decrease in patent vessels, based on perfusion by contrast agent.
In whole lung from control mice, 16α-OHE1 suppressed BMPR2 protein levels three-fold and SMAD 1/5/8 phosphorylation two-fold, without a change in BMPR2 expression. These changes were also seen to a lesser extent in kidney. Estrogens regulate the lysosome through a nontransciptional mechanism[104] and BMPR2 protein levels are primarily regulated through lysosomal degradation.[105] While this hasn’t been explicitly shown, the simplest hypothesis for estrogen regulation of BMPR2 protein levels is thus through lysosomal activation.
However, this estrogen-mediated suppression of SMAD phosphorylation and BMPR2 protein levels was not the explanation for worsened penetrance in mice overexpressing BMPR2 dominant negatives; presumably because the dominant negative acts as a decoy for lysosomal degradation, 16α-OHE1 treatment did not change already suppressed SMAD phosphorylation in these mice. There must be an additional mechanism by which 16α-OHE1 worsens BMPR2-mediated PAH.
Gene expression arrays suggest 16α-OHE1 causes vascular injury response
To try to uncover this additional mechanism, Fessel et al. performed gene expression arrays on lungs from mice with and without BMPR2R899X mutation, and with and without 16α-OHE1 treatment. Only mice with normal right ventricular systolic pressure (RVSP) were used to avoid expression signature caused by high pressure. It was found that, as expected based on the suppression of BMPR2 by 16α-OHE1 in control mice, that gene expression in control mice treated with 16α-OHE1 was shifted somewhat toward the pattern in untreated BMPR2 mutants. Fessel et al. also found that 16α-OHE1 treatment had the positive effects that are normally associated with estrogen treatment, including suppression of traditional cytokine signaling and inflammatory cell recruitment (Fessel et al., Pulmonary Circulation 2013, submitted; title: “Interaction of BMPR2 and Estrogenic Compounds in PAH”). However, it had additional effects, most of which had previously been reported in the literature for other systems but which had not previously been considered in the context of PAH. Chronic 16α-OHE1 treatment resulted in increased angiogenesis-related genes, alterations in metabolic genes (including a 3× increase in the insulin resistance gene Resistin), increased platelet and thrombosis related genes, and alterations in the Wnt pathway consistent with induction of angiogenesis.[106] In sum, it appears that in addition to suppressing BMPR2 protein levels directly, 16α-OHE1 is inducing vascular injury and activation of angiogenesis pathways, potentially through increasing insulin resistance.
2-Methoxyestradiol (2-ME) treatment does not prevent BMPR2-related PAH
2-ME ameliorates both hypoxic and monocrotaline-related PAH through vasodilation and inhibition of cytokines.[14] Estrogens have the same molecular effect in BMPR2-related PAH: Based on array data, elevated cytokine expression is suppressed, and based on tail-cuff data, systemic pressures have statistically significant decrease in the presence of chronic 2-ME. However, despite this, 2-ME was not capable of preventing BMPR2-related PAH.
BMPR2R899X mutant or control mice were implanted with pumps containing vehicle, 2-ME alone, 16α-OHE1 alone, or a mixture of 2-ME and 16α-OHE1 for four weeks, after a prior two weeks of induction of mutation. BMPR2R899X mutant treated with 2-ME alone had a nonsignificant trend toward worse PVR than mutant mice treated with vehicle; those treated with 2-ME in addition to 16α-OHE1 had a nonsignificant trend toward improvement compared with 16α-OHE1 alone (although still worse than mutant mice with vehicle).
In sum, these results suggest that 2-ME effect in the context of BMPR2 mutation is essentially a partial agonist: 2-ME has higher affinity for the estrogen receptors, but 16α-OHE1, when it binds, does so covalently.[107,108] Thus, 2-ME may have a slight beneficial effect in the context of 16α-OHE1 by lowering the rate of covalent binding through competition for receptors. However, on its own, it has the same negative effects as 16α-OHE1 on insulin resistance, even though it continues to have the beneficial effects commonly reported (vasodilation and suppression of inflammation).
In conclusion, the BMP pathway and estrogen have multiple avenues of mutual regulation (Fig. 4). Classical estrogen signaling leads to transcriptional suppression of BMP ligands and receptors.[40,89,90,91] BMPR2 is an important determinant of steroid hormone sensitivity,[97] and properly functioning BMPR2 promotes classical estrogen signaling through assisting translocation of the complexes to the nucleus. Mutant BMPR2 prevents classical estrogen signaling, and promotes cell surface ER, associated with nongenomic signaling (Fig. 4). Nongenomic ER signaling suppresses BMP signaling,[88] most likely through induction of lysosomes[104] which regulate BMPR2 protein level[105] (estrogen reduces BMPR2 protein levels [in press]. Functionally, then increased estrogen signaling leads to decreased BMP signaling through multiple mechanisms. In addition, while estrogen signaling is vasodilatory and anti-inflammatory, it also promotes PAH in the context of BMPR2 mutation through promoting insulin resistance,[109] angiogenesis,[110] and thrombosis.[111,112]
Figure 4.
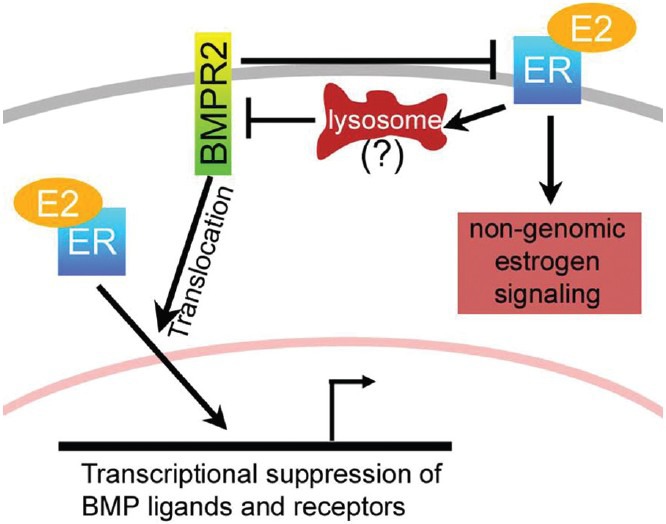
The BMP pathway and estrogen have multiple avenues of mutual regulation.
EXPERIMENTAL PH: EFFECTS OF GENDER, ESTRADIOL METABOLITES, AND PROGESTINS
Numerous studies indicate that E2 and female gender have protective effects in experimental PH, mainly hypoxia- and monocrotaline-induced PH (MCT-PH).[2] Yet, as previously noted, an “Estrogen Paradoxȍ exists in PAH as it occurs more frequently in women although women with PAH have better right ventricular function and survival compared to men with PAH.[10,12,14,113] The apparent contradictions posed by the effects of female gender and estrogens in experimental PH versus human PAH may be explained by the complexity of E2 metabolism and limitations of the experimental models used.[14,114]
Estrogen metabolites and the vasculature
The oxidative metabolism of E2 determines the nature of the biologic effects of this endogenous estrogen. C16 hydroxylation leads to the production of estriol (16α-hydroxyestradiol) and 16α-OHE1, very active estrogens.[115] The C4 hydroxylation leads to formation of catechol estrogen 4-hydroxyestradiol with significant estrogenic activity, an unstable but very reactive metabolite which induces oxidative stress and formation of DNA adducts. Importantly, the metabolites of the 16-hydroxylation and 4-hydroxylation pathways have significant proinflammatory,[116] mitogenic,[117] and angiogenic properties.[118] In contrast to the 4- and 16-hydroxylation pathways, C2 hydroxylation leads to the formation of 2-hydroxyestradiol (2-OHE), a metabolite with very low affinity for estrogen receptors, which is quickly cleared (t/2 = 90 minutes) from the plasma by prompt O-methylation which is catalyzed by catechol-O-methyl transferase.[119] The product of this reaction is 2-ME, a metabolite with no reported estrogenic activity.[120] The known biological effects of 2-ME are mediated by estrogen receptor-independent mechanisms[121] and 2-ME exhibits strong antimitogenic, antiangiogenic and anti-inflammatory effects (Fig. 5).[120,121,122]
Figure 5.
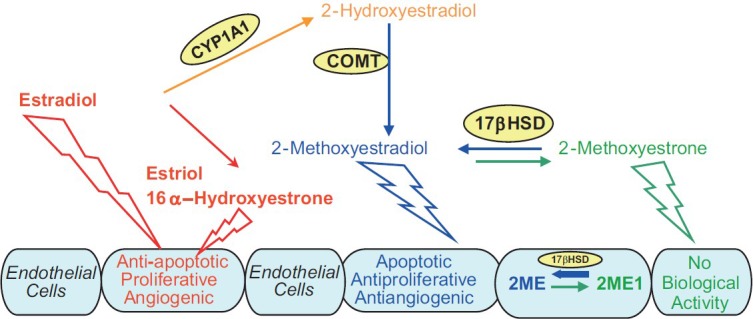
Crossroads of estradiol metabolism in PAH: Activities of 2-hydroxylation, 16alpha-hydroxylation, and 17beta-HSD pathways may determine the overall biological effects of estradiol in PAH.
Several lines of evidence strongly suggested that some estradiol metabolites may have vascular protective effects and that the protective effects of E2 are mediated, at least in part, by its downstream metabolites, products of the 2-hydroxylation pathway.[121] In support of this hypothesis, in vivo studies demonstrated that (1) 2-ME, its metabolic precursor 2-OHE, and its synthetic analogue 2-ethoxyestradiol (2-EE), attenuate vascular and renal dysfunction, and 2-ME has antiangiogenic, anti-inflammatory, and antiproliferative effects in obese, diabetic ZSF1 rats;[123,124] (2) 2-OHE attenuates renal disease in puromycin aminonucleoside nephropathy;[125] and (3) both 2-OHE and 2-ME reduce vascular and cardiac remodeling induced by chronic nitric oxide synthase inhibition.[126] Furthermore, all three, 2-ME, 2-OHE, and 2-EE inhibit cardiac fibrosis and remodeling in an isoproterenol-induced cardiac hypertrophy rat model,[127] and 2-ME inhibits injury-induced neointimal proliferation and smooth muscle cell growth in rats.[128]
Estrogen metabolites and experimental PH
The first evidence that estradiol metabolites may influence the development of PH comes from studies by Tofovic and colleagues[129] who demonstrated that, in male rats with MCT-PH, 2-OHE and 2-ME attenuate the development and 2-ME retards the progression of disease, and that both 2-OHE and 2-ME attenuate lungs inflammatory response and right ventricular and pulmonary vascular remodeling. Notably, ovariectomy (OVX) exacerbates MCT-PH or bleomycin-induced PH and pulmonary fibrosis, and treatment of OVX pulmonary hypertensive animals with 2-ME attenuates pulmonary vascular remodeling, RV hypertrophy, and pulmonary inflammation and fibrosis and eliminates the mortality due to ovariectomy.[130,131] Furthermore, 2-ME attenuates the development and retards the progression of hypoxia-induced PH in male rats[132] and has synergistic effects with sildenafil and bosentan in attenuating pulmonary vascular remodeling and pulmonary inflammation and in reducing mortality in MCT-PH male rats.[133] Similar effects in male rats with MCT-PH are seen with synthetic analog 2-EE.[127] The beneficial effects of E2 in MCT-PH are associated with increased lung NO and prostacyclin levels and reduced endothelin levels, and the inhibitory effects on of E2 on vascular remodeling are linked to down-regulated expression of phosphorylated Akt and up-regulated expression of cyclooxygenase-2.[134] 2-ME is decisively more potent than E2 in increasing prostacyclin synthesis and release, in inhibiting endothelin synthesis in endothelial cells (ECs), and in stimulating NO release from endothelium.[14,121] Likewise, in systemic and pulmonary vascular smooth muscle cells, 2-ME exerts stronger antimitotic effects than E2 itself,[14,121] and inhibition of vascular remodeling by 2-ME is also associated with down-regulated expression of phosphorylated Akt and up-regulated expression of cyclooxygenase-2.[128] Thus, at least in part, the beneficial effects of E2 in experimental PH may be mediated by its major non-estrogenic metabolite 2-ME.
However, MCT- and hypoxia-induced PH may not be the best models for studying the complexities of estrogen pharmacology in human PAH.[14,114] The hallmark vascular change in hypoxia- and MCT-induced PH is medial thickening and neomuscularization, with only mild endothelial damage. This is in contrast to the marked endothelial involvement in severe PAH in humans characterized by occlusive proliferation of endothelial cells and formation of plexifom lesions. Estradiol and 2-ME have opposing effects on ECs:E2 is promitogenic, proangiogenic, and antiapoptotic, whereas 2-ME is antimitogenic, antiangiogenic, and proapoptotic. This may have significant ramifications for the overall effects of estrogens in experimental PH, and the overall effects of gender and E2 in experimental PH may depend on the model system used.[14,114] Therefore, it was suggested[129] that the effects of estrogens and their metabolites should be studied in a model that more closely mimics vascular (endothelial) alterations seen in human PAH, i.e., rat model of angioproliferative PH induced by administration of VEGFR2 antagonist SU5416 and exposure to hypoxia (Sugen [Su5416] plus hypoxia [SuHx rats].[135]
Opposite to the gender effects in hypoxia- and MCT-induced PAH, in this model of experimental PH, female gender does not protect against PH.[136] Female rats rather develop more severe disease and, when compared to males, have increased lung weight and collagen content, as well as more severe nitrosative and oxidative lung injury.[137,138] Plexiform lesions are detected in female animals as early as three weeks after administration of SU5416 and exposure to hypoxia.[138] Male rats develop predominantly occlusive vasculopathy with sporadic presence of plexiform lesions, whereas female rats develop numerous plexiform lesions with sporadic presence of necrotizing arteritis (Grade 6 lesions, Heath-Edwards classification).[137,138] Also, in opposite to the effects in MCT- and hypoxia-induced PH, in the female model of angioproliferative PH, OVX does not exacerbate but rather tends to attenuate the disease; furthermore, in OVX SuHx rats, rescue treatment with E2 exacerbates vascular lesions and PH. Notably, similar to human PAH, in the angioproliferative PH model (SuHx rats), estrogens induce RV-pulmonary vascular uncoupling; despite more severe PH, females had significantly reduced RV hypertrophy compared to males, and OVX exacerbates the increase in RV mass, effects that are reversed by exogenous E2.[137,138]
2-ME is extensively metabolized by type-2 17β-hydroxysteroid dehydrogenase (17β-HSD-2) to 2-methoxyestrone (2-ME1), a metabolite largely lacking antimitogenic activity in cardiovascular cells. In this regard, in human pulmonary artery smooth muscle cells (hPASMCs) and lung fibroblasts (hLFs), at high pharmacological concentrations (10μM), 2-ME1 had only modest (–10 to –20%) antimitogenic effects.[139] Notably, overexpression and increased activity of 17β-HSD-2 abolishes the antimitogenic effects of 2-ME in tumor cells,[140] suggesting that intracellular conversion to its much less active metabolite may significantly influence the biological effects of 2-ME. Importantly, 2-ME1 can be converted back to 2-ME by 17β-HSD type-1 and thereby exhibit cardiovascular effects. All-trans retinoic acid (atRA) increases 17β-HSD type-1 expression and activity[141,142] and would, thereby, be expected to increase the conversion of 2-ME1 to 2-ME. Indeed, in the presence of 1mM of atRA (a concentration that does not affect cell growth) 2-ME1 strongly and in a concentration-dependent manner (10 ng-10 μM) inhibits growth of hPASMCs and hLFs (–20 to –80%).[139] Initial studies have confirmed the importance of 17β-HSD in experimental PH. In male MCT-PH rats, 2-ME1 attenuates the development of PH, RV hypertrophy, and pulmonary vascular remodeling, suggesting that in vivo significant conversion of 2-ME1 back to 2-ME takes place in PAH.[139] Furthermore, in male rats with MCT-PH, 2-ME and atRA have synergistic effects in ameliorating PH and in inhibiting vascular remodeling.[143] These studies strongly suggest that 2-ME1 to 2-ME conversion takes place in vivo and that the 17β-hydroxysteroid dehydrogenase metabolic pathway influences the development of experimental PH.
Progestins and experimental PH
The effects of progesterone in experimental PH have not been fully studied. This is somewhat surprising because progesterone receptors are present both in intact human ECs and in modified ECs within plexiform lesions from patients with PAH.[144,145] Furthermore, natural progesterone inhibits proliferation of ECs, and this effect involves a reduction in cyclin-dependent kinase activity, and the altered expression of cyclin E and A in accordance with G1 arrest.[146] Finally, progesterone exerts vasodilatory properties in different vascular beds including the pulmonary circulation of rats.[147] This would suggest that progesterone may be protective in PH. Indeed, in OVX rats with MCT-PH, progesterone attenuates PH and RV hypertrophy, reduces pulmonary inflammation, inhibits pulmonary vascular remodeling, and reduces mortality.[148] Similar effects are seen with medroxyprogesterone, whereas tibolone, a combined progestin/estrogen compound, prevents the development of disease and eliminates MCT-induced late mortality in OVX rats.[149] Although initial studies in estrogen deficient (OVX) rats suggest that progesterone may have beneficial effects in experimental PH, further evaluation of the role of progesterone in experimental PH, and more importantly its interaction with estrogens in angioproliferative PH, are warranted.
EXPERIMENTAL PH: SEROTONERGIC ACTIVITY AND ESTROGENS
Serotonin is a potent inducer of PASMC proliferation and vasoconstriction and has been implicated in the development of both experimental and clinical PAH. This important contributor to the development and pathogenesis of PAH was first recognized by the associated increase in the incidence of PAH in patients prescribed the appetite suppressant drug, dexfenfluramine.[150] Dexfenfluramine acts as an indirect serotonergic agonist as it is a substrate for the serotonin transporter (SERT) through which it enters the cell and causes release of intracellular serotonin (via the SERT).[151] Dexfenfluramine also reduces serotonin reuptake into cells.[152] The development of dexfenfluramine-induced PAH is dependent on serotonin synthesis as PAH is attenuated in mice devoid of tryptophan hydroxylase 1 (TPH1), the rate limiting enzyme in the synthesis of peripheral serotonin.[153] Recently, a similar drug methamphetamine was associated with increased remodeling of the pulmonary vasculature with increased expression of the 5-HT1B receptor and SERT.[154] This indicates the importance of understanding the underlying mechanisms of this family of drugs to prevent future drugs that act by a similar mechanism entering the drug market.
There is substantial evidence suggesting that TPH1 activity contributes to experimental PAH.[155,156,157,158,159] Pulmonary arterial endothelial cell expression of TPH1 is increased in both experimental and clinical PAH, increasing local serotonin production which can act in a paracrine fashion on underlying PASMCs to facilitate proliferation via SERT, the 5-HT1B and 5-HT2A receptors.[158,160] Multiple studies have also implicated the SERT in the pathobiology of PAH. For example, overexpression of the human SERT gene in mice promotes the spontaneous development of a PAH phenotype which is exaggerated by hypoxia.[161] In contrast, mice deficient in SERT develop less severe PAH phenotypes compared to mice that overexpress SERT.[162] Importantly, SERT expression is increased in PASMCs in patients with PAH.[163] Serotonin can mediate PASMC proliferation by transactivation of the platelet-derived growth factor receptor (PDGFRβ) through SERT.[164] Although inhibition of SERT may offer some therapeutic value, extracellular concentrations of serotonin available to bind with the 5-HT1B receptor may increase, which mediates vasoconstriction in human pulmonary arteries and proliferation of human PASMCs.[165,166] Furthermore, there is evidence for cross-talk between SERT and the 5-HT1B receptor. For example, nuclear transport of phosphorylated extracellular regulated kinase (ERK) by the 5-HT1B receptor is mediated by SERT and is necessary for the upregulation of the S100 calcium binding protein, S100A4/mts1, involved in proliferation and migration of PASMCs.[166] Therefore, taken together, a greater therapeutic effect may be offered by a combined inhibition of SERT and the 5-HT1B receptor, as demonstrated in hypoxia-induced PAH and in SERT+ mice.[167]
Both idiopathic pulmonary arterial hypertension (IPAH) and heritable pulmonary arterial hypertension (HPAH) occur more frequently in females compared to males, as discussed previously. For example, in recent epidemiological studies in the UK/Ireland and the USA, ~70% and 80% of the patients studied were female, respectively.[11,168] The reason for this disparity is unclear and is under-investigated. An understanding of this gender difference would certainly lead to novel therapeutic strategies. Paradoxically, mortality is higher in male patients.[12,169] Research into why PAH occur more frequently in females has been hampered by a lack of relevant animal models, as male rodents typically develop a more severe disease phenotype. For example, male rats develop more severe hypoxia-induced PAH compared to their female counterparts.[170] Additionally, there is evidence that E2 attenuates the development of a PAH phenotype in both the hypoxic and monocrotaline models of the disease.[79,80,171]
Recently, MacLean et al. demonstrated that only female SERT+ mice and female mice overexpressing the S100A4/mts1 gene develop a spontaneous PAH phenotype and that dexfenfluramine-induced PAH is also female selective.[172,173,174] The development of PAH in all these models is dependent on elevated serotonin activity and in all these models, estrogen is also a major contributing factor. This suggests the hypothesis that serotonin is facilitating the damaging effects of estrogens.
Consistent with this, E2 induces proliferation of human PASMCs, increases the expression of TPH1, SERT, and the 5-HT1B receptor, and in line with this, inhibitors of serotonergic signaling have been found to attenuate E2-induced proliferation.[17] This evidence supports an estrogen-serotonin axis mediating the development of PAH. Microarray studies have identified a gene specific to estrogen metabolism that is upregulated in pulmonary arteries of female SERT+ mice, CYP1B1, a major extra-hepatic estrogen metabolizing enzyme.[175] It has subsequently been shown that CYP1B1 expression is upregulated in pulmonary arteries of both IPAH and HPAH, and in two independent animal models, the SUGEN-hypoxic and hypoxic models of PAH, regardless of gender.[176] In contrast, CYP1B1 expression is ten-fold lower in Epstein-Barr virus immortalized B lymphocytes obtained from PAH affected patients harboring a mutation in the BMPR2 gene.[36] The underlying cause of this discrepancy is intriguing and yet to be fully understood but may be attributable to alterations in gene expression induced by the Epstein Barr Virus[177] or attributable to tissue specific differences. Furthermore, a CYP1B1 genetic polymorphism Asn453ΰSer (N453S), associated with increased degradation of CYP1B1,[178] is more prevalent in unaffected female BMPR2 mutation carriers, compared with affected mutation carriers where the wildtype genotype is more common. However, in males carrying a BMPR2 mutation the wildtype genotype predominates in both unaffected and affected carriers.[39] This may therefore contribute to the decreased penetrance in patients with this mutation. Genetic polymorphisms of CYP1B1 are associated with altered catalytic properties, which may affect its metabolic profile and biological activity.[179] Individual status may, therefore, defer or infer susceptibility.
Aromatase, CYP1B1, and Catechol-O-methyl transferase (COMT) are key enzymes in the synthesis and metabolism of estrogen. The formation of E2 from C19 steroids is catalyzed by aromatase, a cytochrome P450 enzyme encoded by the CYP19A1 gene. A genetic polymorphism of aromatase leading to increased plasma estrogen levels has been associated with an increased risk of PPHTN.[20] As aromatase inhibitors are already widely used as cancer treatment, this makes it a particularly attractive target. Recent unpublished results from MacLean et al. show that the aromatase inhibitor anastrozole can reverse both hypoxia and SUGEN/hypoxic PAH in female mice. A recent, large-scale epidemiological study reported a greater association of PAH among postmenopausal females and in obesity, and at this age comorbidities are increasingly prevalent.[11] In both men and women, extragonadal sites such as the adipose tissue have the capacity to synthesize estrogen via aromatase from circulating C19 precursors.[180] In postmenopausal women, once the ovaries have stopped producing estrogens, the adipose tissue increases its production of estrogen.[181] Aromatase inhibitors could therefore be effective in a wide range of patients regardless of age or gender.
Local increased aromatase activity provides estrogen as a substrate for metabolizing enzymes. Estrogens are metabolized to both pro- and antiproliferative metabolites by various CYP enzymes. As discussed above, CYP1B1 primarily metabolizes estrogens to the 4-OHE1/2 by hydroxylation at the C4 position and to a lesser extent to 2- and the 16-OHE1/2.[182,183] The catechol estrogens (the 2- and 4-OHE1/2) can then be further metabolized to the methoxyestrogens by the ubiquitously expressed Catechol-O-methyltransferase (COMT). Importantly, hypoxia, serotonin, and estrogen all increase the expression of CYP1B1 and are all factors that have been implicated in PAH. E2 and estrone are present in the lung, and it is, therefore, conceivable that increased estrogen metabolism to proproliferative metabolites by upregulation of CYP1B1 in response to hypoxia, serotonin, and/or estrogen could occur (Fig. 6).[175,176,184] Additionally, CYP1B1 expression and activity is upregulated in response to shear stress, which may implicate a prolonged contribution of CYP1B1 to the pathogenesis of PAH, by persistence of increased CYP1B1 in the face of increased pulmonary arterial pressures, increased circulating estrogens, and locally produced estrogens.[185]
Figure 6.
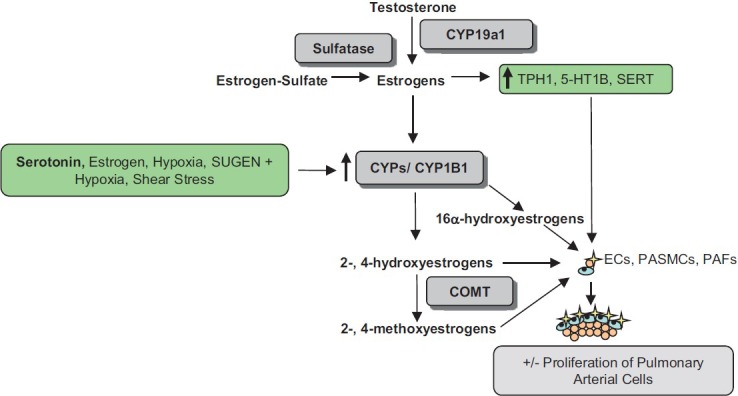
The influence of serotonin and estrogen in the development of PAH. Estrogens can increase the expression of TPH1, the 5-HT1B receptor and SERT. Serotonin, shear stress, hypoxia, SUGEN + hypoxia and estrogen all increase the expression of CYP1B1 (and perhaps other CYP enzymes) which can metabolize estrogen to both pro- and anti-proliferative metabolites, some via COMT. Changes in this system that favour accumulation of pro-proliferative metabolites in the face of decreased protective metabolites would favour proliferation of PASMCs and contribute to pulmonary vascular remodeling.
CYP1B1 in PAH may be an attractive therapeutic target and, recently, White et al. established a direct pathological role for CYP1B1 metabolism on the development of PAH.[175] Mice devoid of functional CYP1B1 (CYP1B1-/- mice) and chronic inhibition by a highly selective CYP1B1 inhibitor 2,3’,4,5’-tetramethoxystilbene (TMS) are protected against the development of hypoxia-induced PAH, independent of sex. This effect was also convincingly observed in the SUGEN-hypoxic model and, importantly, TMS also inhibited the formation of occluded pulmonary arteries. To further elucidate potential mechanisms by which CYP1B1 metabolism was causative on disease development, White et al. assessed proliferation to the 2-, 4-, and 2-OHE1/2 in human PASMCs. 16α-OHE1 stimulated the most profound proliferation of PASMCs and this effect was 100-fold more pronounced in PAH-PASMCs. To establish a direct relationship of 16α-OHE1 in vivo, mice were chronically dosed with 16α-OHE1 which induced a PAH phenotype.[175] Although 16α-OHE1 is a minor CYP1B1 metabolite, increased CYP1B1 expression would increase the production of all its metabolites, including 16α-OHE1. There is also the possibility that simultaneous changes in aromatase, COMT, and CYP1B1 activity could drive estrogen metabolism toward an accumulation of damaging metabolites. Additionally, 16α-OHE1 levels are increased in affected BMPR2 mutation carriers compared to unaffected carriers.[39] The estrogen metabolism pathway is complex, tissue specific, and dependent on the presence and absence of different enzymes, and more work teasing out its precise function in the pulmonary vasculature is essential.
Estrogen metabolism may be integral to a wide span of diseases and may offer targets for therapeutic intervention in the management of PAH. The exact mechanisms by which aromatase, CYP1B1, and COMT influence disease development and progression merits further investigation. Gender, locally produced estrogens, and serotonin together with increased shear stress may interact to facilitate the development of PAH via CYP1B1 activity and are suggestive as “second hit factors” that may influence the development of PAH in genetically-susceptible individuals and may also be causative in idiopathic cases of PAH.
EXPERIMENTAL PH: DEHYDROEPIANDROSTERONE (DHEA)
DHEA is a naturally occurring C-19 steroid that is synthesized mainly by the adrenal cortex only in humans and close primate relatives (other species, including rodents, have very low levels of DHEA produced by other organs, such as testis, ovary, and brain).[186,187] DHEA and its sulfated ester, DHEA sulfate (DHEA-S), are produced in larger quantities than any other circulating steroid hormone. Most circulating DHEA is in the form of DHEA-S, which functions as a less active reservoir for DHEA (DHEA-S can be converted to DHEA by a ubiquitous enzyme, steroid sulfatase, in peripheral tissues; Fig. 7). These two steroids show a distinct life history secretion pattern; blood levels of DHEA-S are very high at birth and fall precipitously to near zero by about six months of age, they then rise to peak values in men around age 25, and finally undergo a progressive steep decline over age. Levels in females are lower but follow the same pattern.[188,189,190] Although DHEA serves as a precursor to both androgens and estrogens (Fig. 7),[191] there is no obvious role for DHEA/DHEA-S in the blood levels of sex steroid hormones in normal men and premenopausal women.[192]
Figure 7.
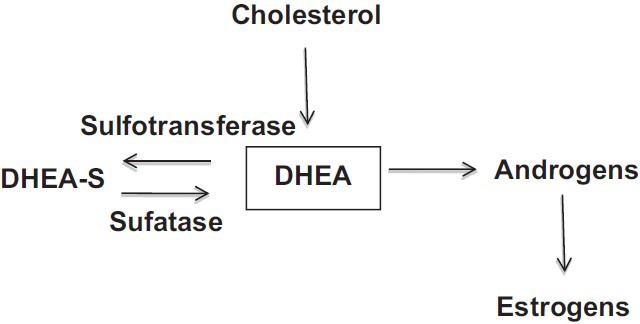
Synthesis and metabolism of dehydroepiandrosterone (DHEA).
Epidemiological observations and animal experiments indicate that DHEA has a wide variety of beneficial biological and physiological effects, including prevention of obesity, diabetes, cancer, and cardiovascular diseases.[193,194] However, the mechanisms of the cardiovascular protective effects of DHEA, such as antiproliferative, proapoptotic, anti-inflammatory, and antioxidant effects, have not been fully elucidated. There are several known biochemical actions by which some of its beneficial properties might be explained (Table 1). The best-described action of DHEA is inhibition of glucose-6-phosphatase dehydrogenase (G-6-PDH),[195,196] which decreases NADPH production by the pentose phosphate pathway, and thereby modulates tissue redox state. This mechanism can partly explain its antiproliferative effect via reduced RNA/DNA productions[194,197] and its acute vasodilatory effect via opening of K+ channels.[199] A decrease in NADPH levels plays a major role in DHEA's antioxidant effect by reducing O2- production via NADPH oxidase.[196] Another well-described action is activation of endothelial nitric oxide synthase (eNOS),[200] which exerts vasodilatory and antiproliferative effects. DHEA has been reported to bind to a specific endothelial cell membrane receptor coupled to Gai2,3 to activate eNOS.[201] It has also recently been reported that DHEA stimulates Akt-eNOS signaling pathway through activation/upregulation of sigma-1 receptor.[202] Other known actions of DHEA include activation of peroxisome proliferator-activated receptors [PPARs]; which is involved in fat metabolism and anti-inflammation/-proliferation)[203,204] and inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase[205] (which is involved in cholesterol metabolism and cell cycle arrest and growth inhibition probably through decreased RhoA/Rho kinase signaling activity). Finally, it has been reported that DHEA is a poor agonist for estrogen receptors, and several studies have demonstrated that the antiproliferative effects of DHEA are independent of either the estrogen or androgen receptors.[206] However, since a specific receptor for DHEA has not yet been isolated and fully characterized, it is still not clear whether part of DHEA's effects are mediated through the androgen/estrogen receptors or through conversion of DHEA to these hormones. Taken together, all these actions of DHEA are closely related to the pathogenesis of PH and could act against its development.
Table 1.
Major mechanisms of action for dehydroepiandrosterone

DHEA and pulmonary circulation
Little is known about the effects of DHEA on the pulmonary circulation. Farrukh et al.[206] and Gupte et al.[198] have shown in vitro that DHEA is a pulmonary vasodilator and inhibits acute hypoxic pulmonary vasoconstriction due at least partly to opening of potassium channels. In the latter study, it is shown that vasodilatory effects of pentose phosphate pathway inhibitors, including DHEA, are mediated through opening of voltage-gated K+ channels due to reduction of tissue NADPH levels and a change in redox status.[198] The levels of DHEA/DHEA-S in PH patients over time have not been determined, but the recent MESA-RV Study found that higher DHEA levels were associated with increased RV mass and stroke volume in women.[87]
Effects of chronic DHEA treatment on experimental PH
Prompted by the epidemiological and experimental findings in the systemic and pulmonary circulation, studies have been performed to determine whether DHEA is effective for the treatment of PH in various experimental models. There are numerous animal models of PH available for various research purposes, but they are now divided roughly into two groups based on the presence or absence of occlusive neointimal lesions, one of the hallmarks of PAH. One group is conventional, which includes the most frequently used models that essentially lack occlusive lesions, such as chronic hypoxia-induced and monocrotaline-injected models. The other group encompasses relatively newer models that develop occlusive neointimal lesions, including monocrotaline-injected pneumonectomized rats[207] and Sugen5416 injection and hypoxia-exposed rats.[135]
Chronically hypoxic PH. Hampl et al.[208] and Bonnet et al.[209] in the same year reported that chronic treatment with DHEA-S or DHEA inhibited and reversed chronic HPH (increase in pulmonary arterial pressure and RV hypertrophy) in rats. Bonnet et al. showed that the beneficial effects of DHEA treatment were associated with increased expression and function of pulmonary artery Ca2+ activated K+ channels. Oka et al. subsequently reported that dietary DHEA (0.3 and 1% DHEA containing food) dose-dependently, completely prevented and effectively reversed the HPH.[210] In agreement with previous reports in the systemic circulation,[211,212] the chronic DHEA treatment improved endothelium-dependent (acetylcholine-induced) relaxation in isolated hypertensive pulmonary arteries. This improvement was accompanied by activation and upregulation of soluble guanylate cyclase but not eNOS, which is not consistent with the findings in the systemic circulation. It is not clear whether this difference simply reflects the difference between the systemic and pulmonary circulations, but the results suggest that DHEA can modulate activity/expression of soluble guanylate cyclase. It should be noted in this study that while DHEA attenuated chronic HPH with increased plasma DHEA levels in a dose-dependent fashion, plasma estradiol and testosterone levels were elevated only in the higher dose group (1% DHEA). These findings suggest that protective effects of DHEA against HPH are at least partly independent of its conversion to estrogen/testosterone.
In addition, another study reported that chronic DHEA-S treatment inhibited the development of HPH in older mice, and prevented them from age-related frailty induced by HPH.[213] The authors emphasized the similarity in this and a human study, where a strong inverse correlation between natural DHEA/DHEA-S blood levels and the ten-year mortality in old male smokers and ex-smokers, and suggested clinical trials with DHEA in patients with PH and chronic obstructive pulmonary disease. The same group subsequently showed that DHEA decreased hypoxia inducible factor-1a accumulation under hypoxic conditions in vitro, and suggested this might explain why DHEA was effective in treating chronic HPH.[214] The beneficial effects of DHEA/DHEA-S appear to involve multiple actions including K+ channel activation, upregulation of soluble guanylate cyclase, and downregulation of hypoxia inducible factor-1a. Collectively, there is an increasing body of experimental evidence that supports that DHEA/DHEA-S is an effective agent in preventing and reversing CH-PH. In fact, a recent preliminary report of an ongoing clinical trial in chronic obstructive lung disease-associated PH patients revealed that chronic DHEA treatment (200 mg/day for 12 weeks) improved their exercise tolerance and hemodynamic parameters.[215]
Other models. The drugs currently used to treat PH patients were first tested and found effective in conventional PH models, i.e., chronically hypoxic and/or monocrotaline-injected pulmonary hypertensive rats, and many new agents continue to be evaluated in these models. While the information acquired from studies of these models advances our understanding of the molecular mechanisms of pulmonary vasoconstriction and vascular remodeling, a major limitation is that these animals do not develop the occlusive neointimal and plexiform pulmonary arterial lesions that are the hallmarks of severe human pulmonary arterial hypertension. There are newer models of PH that develop a closer phenotype to human pulmonary arterial hypertension than the conventional models. These models are usually induced by multiple insults/hits. In contrast to the conventional models, these models develop occlusive neointimal lesions in distal pulmonary arteries. Oka et al. tested effects of chronic DHEA treatment on two of these models, i.e., monocrotaline-injected pneumonectmized (MCT-PN) and Sugen 5416 (SU, a vascular endothelial growth factor receptor blocker)-injected followed by exposure to hypoxia and subsequent re-exposure to normoxia (SuHx) rats:
(a) MCT-PN: This model was first introduced by Okada et al. in 1997 based on a two-hit theory, i.e., endothelial injury by MCT combined with increased blood flow by unilateral pneumonectomy.[207] In contrast to its effects in normal rats, MCT injection causes severe PH accompanied not only by medial wall thickening but also by neointimal lesions in small pulmonary arteries in PN rats. Homma et al. reported that a high dose of three-week dietary DHEA treatment (1% DHEA containing food) completely prevented the development of MCT-induced severe PH, RV hypertrophy, and pulmonary vascular remodeling (medial wall thickening and occlusive neointimal formation) in left PN rats with no adverse systemic hemodynamic effects.[216] Protein analyses of lungs from PN rats revealed that MCT injection caused the following: (1) Increased RhoA activity accompanied by markedly increased 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and slightly decreased soluble guanylyl cyclase (sGC) expression; (2) constitutive activation (cleavage) of Rho-associated coiled-coil protein kinase 1 (ROCK I) associated with caspase-3 activation; (3) increased ROCK II expression; and (4) increased ROCK activity. Chronic DHEA treatment nearly normalized the increased RhoA/ROCK signaling pathway activity, which was associated with inhibition of caspase-3 activation, HMG-CoA reductase overexpression, and preservation of sGC expression. DHEA, when started after the PH was fully developed, also completely inhibited the progression of MCT-induced severe PH, and more importantly, strikingly improved survival (30 vs. 100% for DHEA-untreated vs. -treated). These results showed that chronic DHEA treatment nearly completely prevented the development of MCT-induced severe PH, and that inhibition of RhoA/ROCK signaling, apparently by multiple mechanisms including downregulation of HMG-CoA reductase and inhibition of ROCK I cleavage, was involved in the impressive protective effects of DHEA. In addition, a recent study suggests that downregulation of Src/STAT3 signaling may be involved in the inhibitory effect of DHEA against RhoA/ROCK signaling[217]
(b) SuHx: In 2001, Taraseviciene-Stewart et al. first described a rat model of severe PH induced by a combination of vascular endothelial growth factor (VEGF) receptor blockade (by Sugen 5416) and chronic hypoxic exposure.[135] A single injection of Sugen 5416 followed by a three-week exposure to hypoxia (10% O2) resulted in the development of severe PH that was accompanied by occlusive neointimal lesions in precapillary small pulmonary arteries. Importantly, the PH is progressive even when the animals are re-exposed to normoxia. Recent work with this model shows that after three weeks of exposure to hypoxia and an additional two to 10 weeks in normoxia (5–13 weeks after Sugen 5416 injection), the severe pulmonary hypertensive animals develop over time a variety of pulmonary arterial lesions including medial wall thickening (Heath-Edwards Grade 1) and neointimal (Grades 2 and 3) and complex plexiform lesions (Grade 4) that are histologically indistinguishable from those found in human PH.[218] In addition, hemodynamic data show that RVSP appears to reach its maximum (~100 mmHg) around three to five weeks after Sugen 5416 injection and stays at about the same high level thereafter. At the five-week time point, cardiac output decreases to approximately 50% of that of the normal, and is further reduced at the 13-week time point. Reflecting this reduction in cardiac input, calculated total pulmonary vascular resistance increases further at this very late time point, even though the RVSP does not increase, suggesting that the vascular disorder is still progressing. This hemodynamic time-course pattern is very similar to that observed in patients with PAH, and notably, the PH in these animals is also resistant to treatment with drugs conventionally used to treat the human disease.
Alzoubi et al. have tested effects of DHEA in this severe PAH model. The authors found that a five-week dietary treatment with a high dose DHEA (1%), starting from the three-week time point when RVSP is near the maximum level (>80 mmHg), slowed the progression but did not reverse the severe PH (RVSP = 70 ± 7 and 97 ± 5 mmHg, at eight-week time point with and without DHEA treatment, respectively). Likewise, DHEA treatment significantly reduced the severity of RV hypertrophy and the neointimal lesion formation in small pulmonary arteries, but it did not normalize them. It is, however, notable that DHEA treatment impressively improved the impaired cardiac index to near normal levels, and the calculated total pulmonary vascular resistance was dramatically reduced. The authors hypothesized that this impressive improvement of cardiac function might be due not only to decreases in RVSP and RV pressure overload, but also to direct effects of DHEA on cardiomyocytes. Because it has been reported that increased reactive oxygen species (ROS) are involved in the impaired RV function in this model,[219] and because DHEA is a potent antioxidant, the authors looked at ROS signals in the heart. They found that ROS signals were markedly increased in RV but not LV from PH rats (at the eight-week time point), and DHEA treatment effectively reduced the signal. These results suggest that DHEA is effective in treating this severe PH model, not only by improving abnormalities in the hypertensive pulmonary arteries but also by directly protecting cardiomyocytes as an antioxidant.[220]
In conclusion, high doses of DHEA are effective against various experimental models of PH. Multiple mechanisms, including upregulation of Nitric oxide cyclic guanosine monophosphate (NO-cGMP) and inhibition of ROCK signaling pathways, are involved in its beneficial effects, and DHEA may have a direct protective effect on impaired RV function. Taken together, DHEA appears to be a promising agent for the treatment of PH as a monotherapy. DHEA is an over-the-counter food supplement and generally has no major side effects, but its safety at very high doses is not known. Because DHEA can be metabolized to androgens and estrogens, sex hormone-dependent cancer development may be a concern for its high dose chronic treatment. Although, effective doses in humans might be much lower because of very high basal levels of DHEA/DHEA-S compared to those of rodents, and in fact a recent preliminary clinical trial shows that a relatively low dose (200 mg/day) improves 6-Minute Walk Distance as well as hemodynamic parameters in patients with PH owing to chronic obstructive lung disease.[215] One may need to evaluate synthetic derivatives of DHEA such as HE3286 (Triolex) that is not metabolized to sex steroids and retains potent anti-inflammatory, and possibly other properties, of DHEA.[221]
CONCLUSIONS AND FUTURE DIRECTION
Despite improvements in our understanding of sex hormone physiology and estrogen metabolism in PH, it remains unclear why estrogens appear to be protective in certain settings (HPH, MCT-PH) but detrimental in others (SERT+, S100A4/mts1+). Effects of E2, its receptors, and its metabolites may be context- and/or compartment-specific, and vary depending on oxygen tension, ER distribution, estrogen metabolism, duration of exposure, and biochemical milieu. The emerging paradigm of “good” and “bad” estrogens and altered estrogen metabolism suggests that PAH may be a condition with an imbalance between beneficial and detrimental estrogens. Such alterations may be provoked or aggravated in the setting of genetic disposition (BMPR2 mutation) or other “first hitsȍ (PPHTN, altered serotonin homeostasis); this could explain why protective E2 effects in classical PH models are not recapitulated in others. Further research will need to decipher the molecular basis of these disparate estrogen effects. Detailed investigations of estrogen metabolites in human PAH using mass spectroscopy would be helpful, but are currently limited by lack of standardized techniques. The “estrogen paradoxȍ of increased female susceptibility, yet better survival, still remains a mystery. It appears that women-though being more prone to developing the disease-tolerate PAH better than men, and recent research suggests that female PAH patients may have better RV function,[86] more favorable baseline hemodynamics,[169] and a better response to certain therapeutic interventions.[222] Further studies of the mechanisms underlying these phenomena are needed. DHEA appears uniformly protective in PAH models, while the effects of male sex hormones are only poorly characterized. The investigations of sex hormone effects reviewed here, as well as results of future studies in PAH models and patients, will hopefully allow for the development of targeted therapies that ultimately will benefit PAH patients of either sex.
Footnotes
Source of Support: Career Development Award Support NIH 5K23HL098743 (EDA)
Conflict of Interest: None declared.
REFERENCES
- 1.Rich S, Rabinovitch M. Diagnosis and treatment of secondary (non-category 1) pulmonary hypertension. Circulation. 2008;118:2190–9. doi: 10.1161/CIRCULATIONAHA.107.723007. [DOI] [PubMed] [Google Scholar]
- 2.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: Baseline characteristics from the reveal registry. Chest. 2010;137:376–87. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 5.Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:1527–38. doi: 10.1016/j.jacc.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 6.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in france: Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 7.Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med. 1951;11:686–705. doi: 10.1016/0002-9343(51)90020-4. [DOI] [PubMed] [Google Scholar]
- 8.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 9.McGoon MD, Krichman A, Farber HW, Barst RJ, Raskob GE, Liou TG, et al. Design of the reveal registry for us patients with pulmonary arterial hypertension. Mayo Clin Proc. 2008;83:923–31. doi: 10.4065/83.8.923. [DOI] [PubMed] [Google Scholar]
- 10.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (reveal) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 11.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the united kingdom and ireland. Am J Respir Crit Care Med. 2012;186:790–6. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- 12.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 13.Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–55. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 14.Tofovic SP. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. 2010;56:696–708. doi: 10.1097/FJC.0b013e3181f9ea8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleiger RE, Boxer M, Ingham RE, Harrison DC. Pulmonary hypertension in patients using oral contraceptives. A report of six cases. Chest. 1976;69:143–7. doi: 10.1378/chest.69.2.143. [DOI] [PubMed] [Google Scholar]
- 16.Morse JH, Horn EM, Barst RJ. Hormone replacement therapy: A possible risk factor in carriers of familial primary pulmonary hypertension. Chest. 1999;116:847. doi: 10.1378/chest.116.3.847. [DOI] [PubMed] [Google Scholar]
- 17.Irey NS, Manion WC, Taylor HB. Vascular lesions in women taking oral contraceptives. Arch Pathol. 1970;89:1–8. [PubMed] [Google Scholar]
- 18.Irey NS, Norris HJ. Intimal vascular lesions associated with female reproductive steroids. Arch Pathol. 1973;96:227–34. [PubMed] [Google Scholar]
- 19.Sweeney L, Voelkel NF. Estrogen exposure, obesity and thyroid disease in women with severe pulmonary hypertension. Eur J Med Res. 2009;14:433–42. doi: 10.1186/2047-783X-14-10-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835–42. doi: 10.1164/rccm.200809-1472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parl FF, Dawling S, Roodi N, Crooke PS. Estrogen metabolism and breast cancer: A risk model. Ann N Y Acad Sci. 2009;1155:68–75. doi: 10.1111/j.1749-6632.2008.03676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez FJ. The molecular biology of cytochrome p450s. Pharmacol Rev. 1988;40:243–88. [PubMed] [Google Scholar]
- 23.Nebert DW, Russell DW. Clinical importance of the cytochromes p450. Lancet. 2002;360:1155–62. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- 24.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome p450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–24. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 26.Nebert DW. Elevated estrogen 16 alpha-hydroxylase activity: Is this a genotoxic or nongenotoxic biomarker in human breast cancer risk? J Natl Cancer Inst. 1993;85:1888–91. doi: 10.1093/jnci/85.23.1888. [DOI] [PubMed] [Google Scholar]
- 27.Roy D, Cai Q, Felty Q, Narayan S. Estrogen-induced generation of reactive oxygen and nitrogen species, gene damage, and estrogen-dependent cancers. J Toxicol Environ Health B Crit Rev. 2007;10:235–57. doi: 10.1080/15287390600974924. [DOI] [PubMed] [Google Scholar]
- 28.Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muti P, Bradlow HL, Micheli A, Krogh V, Freudenheim JL, Schunemann HJ, et al. Estrogen metabolism and risk of breast cancer: A prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology. 2000;11:635–40. doi: 10.1097/00001648-200011000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Belous AR, Hachey DL, Dawling S, Roodi N, Parl FF. Cytochrome p450 1b1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res. 2007;67:812–7. doi: 10.1158/0008-5472.CAN-06-2133. [DOI] [PubMed] [Google Scholar]
- 31.Eliassen AH, Missmer SA, Tworoger SS, Hankinson SE. Circulating 2-hydroxy- and 16alpha-hydroxy estrone levels and risk of breast cancer among postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2008;17:2029–35. doi: 10.1158/1055-9965.EPI-08-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaaks R, Rinaldi S, Key TJ, Berrino F, Peeters PH, Biessy C, et al. Postmenopausal serum androgens, oestrogens and breast cancer risk: The european prospective investigation into cancer and nutrition. Endocr Relat Cancer. 2005;12:1071–82. doi: 10.1677/erc.1.01038. [DOI] [PubMed] [Google Scholar]
- 33.Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among postmenopausal women. J Natl Cancer Inst. 2004;96:1856–65. doi: 10.1093/jnci/djh336. [DOI] [PubMed] [Google Scholar]
- 34.Muti P, Westerlind K, Wu T, Grimaldi T, De Berry J, 3rd, Schunemann H, et al. Urinary estrogen metabolites and prostate cancer: A case-control study in the united states. Cancer Causes Control. 2002;13:947–55. doi: 10.1023/a:1021986811425. [DOI] [PubMed] [Google Scholar]
- 35.Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, Robbins IM, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:892–6. doi: 10.1164/rccm.201205-0886OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.West J, Cogan J, Geraci M, Robinson L, Newman J, Phillips JA, et al. Gene expression in bmpr2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics. 2008;1:45. doi: 10.1186/1755-8794-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agundez JA. Cytochrome p450 gene polymorphism and cancer. Curr Drug Metab. 2004;5:211–24. doi: 10.2174/1389200043335621. [DOI] [PubMed] [Google Scholar]
- 38.Greenlee H, Chen Y, Kabat GC, Wang Q, Kibriya MG, Gurvich I, et al. Variants in estrogen metabolism and biosynthesis genes and urinary estrogen metabolites in women with a family history of breast cancer. Breast Cancer Res Treat. 2007;102:111–7. doi: 10.1007/s10549-006-9308-7. [DOI] [PubMed] [Google Scholar]
- 39.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, et al. Alterations in oestrogen metabolism: Implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009;34:1093–9. doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ. 2012;3:6. doi: 10.1186/2042-6410-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy E. Estrogen signaling and cardiovascular disease. Circ Res. 2011;109:687–96. doi: 10.1161/CIRCRESAHA.110.236687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science. 2005;308:1583–7. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 43.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–11. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 44.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: How do they signal and what are their targets. Physiol Rev. 2007;87:905–31. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 45.Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Genazzani AR. Genomic and non-genomic effects of estrogens on endothelial cells. Steroids. 2004;69:537–42. doi: 10.1016/j.steroids.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh YC, Yu HP, Frink M, Suzuki T, Choudhry MA, Schwacha MG, et al. G protein-coupled receptor 30-dependent protein kinase a pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma-hemorrhage. Am J Pathol. 2007;170:1210–8. doi: 10.2353/ajpath.2007.060883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-oh kinase. Nature. 2000;407:538–41. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedram A, Razandi M, Wallace DC, Levin ER. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell. 2006;17:2125–37. doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen actions on mitochondria--physiological and pathological implications. Mol Cell Endocrinol. 2008;290:51–9. doi: 10.1016/j.mce.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF, et al. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci U S A. 1996;93:12626–30. doi: 10.1073/pnas.93.22.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y, et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2001;276:3459–67. doi: 10.1074/jbc.M005036200. [DOI] [PubMed] [Google Scholar]
- 52.Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. Erbeta has nongenomic action in caveolae. Mol Endocrinol. 2002;16:938–46. doi: 10.1210/mend.16.5.0827. [DOI] [PubMed] [Google Scholar]
- 53.Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME, Pfister SL, et al. Estrogen acutely activates prostacyclin synthesis in ovine fetal pulmonary artery endothelium. Am J Respir Cell Mol Biol. 2002;26:610–6. doi: 10.1165/ajrcmb.26.5.4528. [DOI] [PubMed] [Google Scholar]
- 54.Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–6. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaul PW. Rapid activation of endothelial nitric oxide synthase by estrogen. Steroids. 1999;64:28–34. doi: 10.1016/s0039-128x(98)00105-6. [DOI] [PubMed] [Google Scholar]
- 56.Yu HP, Hsieh YC, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, et al. Mechanism of the nongenomic effects of estrogen on intestinal myeloperoxidase activity following trauma-hemorrhage: Up-regulation of the pi-3k/Akt pathway. J Leukoc Biol. 2007;82:774–80. doi: 10.1189/jlb.0307182. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki T, Yu HP, Hsieh YC, Choudhry MA, Bland KI, Chaudry IH. Estrogen-mediated activation of non-genomic pathway improves macrophages cytokine production following trauma-hemorrhage. J Cell Physiol. 2008;214:662–72. doi: 10.1002/jcp.21255. [DOI] [PubMed] [Google Scholar]
- 58.Ba ZF, Lu A, Shimizu T, Szalay L, Schwacha MG, Rue LW, 3rd, et al. 17beta-estradiol modulates vasoconstriction induced by endothelin-1 following trauma-hemorrhage. Am J Physiol Heart Circ Physiol. 2007;292:H245–50. doi: 10.1152/ajpheart.00809.2006. [DOI] [PubMed] [Google Scholar]
- 59.Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, et al. Molecular cloning and characterization of human estrogen receptor betacx: A potential inhibitor ofestrogen action in human. Nucleic Acids Res. 1998;26:3505–12. doi: 10.1093/nar/26.15.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, et al. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res. 2002;90:1087–92. doi: 10.1161/01.res.0000021114.92282.fa. [DOI] [PubMed] [Google Scholar]
- 61.Toutain CE, Filipe C, Billon A, Fontaine C, Brouchet L, Guery JC, et al. Estrogen receptor alpha expression in both endothelium and hematopoietic cells is required for the accelerative effect of estradiol on reendothelialization. Arterioscler Thromb Vasc Biol. 2009;29:1543–50. doi: 10.1161/ATVBAHA.109.192849. [DOI] [PubMed] [Google Scholar]
- 62.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–30. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–43. doi: 10.1074/jbc.R110.191791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, et al. Regulatory role of g protein-coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–91. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peter I, Shearman AM, Zucker DR, Schmid CH, Demissie S, Cupples LA, et al. Variation in estrogen-related genes and cross-sectional and longitudinal blood pressure in the framingham heart study. J Hypertens. 2005;23:2193–200. doi: 10.1097/01.hjh.0000188728.66183.92. [DOI] [PubMed] [Google Scholar]
- 66.Shearman AM, Cooper JA, Kotwinski PJ, Humphries SE, Mendelsohn ME, Housman DE, et al. Estrogen receptor alpha gene variation and the risk of stroke. Stroke. 2005;36:2281–2. doi: 10.1161/01.STR.0000181088.76518.ec. [DOI] [PubMed] [Google Scholar]
- 67.Shearman AM, Cooper JA, Kotwinski PJ, Miller GJ, Humphries SE, Ardlie KG, et al. Estrogen receptor alpha gene variation is associated with risk of myocardial infarction in more than seven thousand men from five cohorts. Circ Res. 2006;98:590–2. doi: 10.1161/01.RES.0000210578.62102.a6. [DOI] [PubMed] [Google Scholar]
- 68.Peter I, Shearman AM, Vasan RS, Zucker DR, Schmid CH, Demissie S, et al. Association of estrogen receptor beta gene polymorphisms with left ventricular mass and wall thickness in women. Am J Hypertens. 2005;18:1388–95. doi: 10.1016/j.amjhyper.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 69.Venkov CD, Rankin AB, Vaughan DE. Identification of authentic estrogen receptor in cultured endothelial cells. A potential mechanism for steroid hormone regulation of endothelial function. Circulation. 1996;94:727–33. doi: 10.1161/01.cir.94.4.727. [DOI] [PubMed] [Google Scholar]
- 70.Hamidi SA, Dickman KG, Berisha H, Said SI. 17beta-estradiol protects the lung against acute injury: Possible mediation by vasoactive intestinal polypeptide. Endocrinology. 2011;152:4729–37. doi: 10.1210/en.2011-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vegeto E, Cuzzocrea S, Crisafulli C, Mazzon E, Sala A, Krust A, et al. Estrogen receptor-alpha as a drug target candidate for preventing lung inflammation. Endocrinology. 2010;151:174–84. doi: 10.1210/en.2009-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karas RH, Patterson BL, Mendelsohn ME. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation. 1994;89:1943–50. doi: 10.1161/01.cir.89.5.1943. [DOI] [PubMed] [Google Scholar]
- 73.Jia S, Zhang X, He DZ, Segal M, Berro A, Gerson T, et al. Expression and function of a novel variant of estrogen receptor-alpha36 in murine airways. Am J Respir Cell Mol Biol. 2011;45:1084–9. doi: 10.1165/rcmb.2010-0268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patrone C, Cassel TN, Pettersson K, Piao YS, Cheng G, Ciana P, et al. Regulation of postnatal lung development and homeostasis by estrogen receptor beta. Mol Cell Biol. 2003;23:8542–52. doi: 10.1128/MCB.23.23.8542-8552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Suzuki T, Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Schwacha MG, et al. Tissue compartment-specific role of estrogen receptor subtypes in immune cell cytokine production following trauma-hemorrhage. J Appl Physiol. 2007;102:163–8. doi: 10.1152/japplphysiol.00964.2006. [DOI] [PubMed] [Google Scholar]
- 76.Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-alpha (eralpha) and estrogen receptor-beta (erbeta) messenger ribonucleic acid in the wild-type and eralpha-knockout mouse. Endocrinology. 1997;138:4613–21. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- 77.Mollerup S, Jorgensen K, Berge G, Haugen A. Expression of estrogen receptors alpha and beta in human lung tissue and cell lines. Lung Cancer. 2002;37:153–9. doi: 10.1016/s0169-5002(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 78.Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Tan J, et al. Selective estrogen receptor-{alpha} and estrogen receptor-{beta} agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1486–93. doi: 10.1152/ajpregu.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184:715–23. doi: 10.1164/rccm.201101-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, et al. 17beta-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–980. doi: 10.1164/rccm.201107-1293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, et al. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H1235–48. doi: 10.1152/ajpheart.00254.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, et al. Estrogen receptor beta mediates increased activation of pi3k/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R972–8. doi: 10.1152/ajpregu.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. 2006;290:H2204–9. doi: 10.1152/ajpheart.01219.2005. [DOI] [PubMed] [Google Scholar]
- 84.Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1597–606. doi: 10.1152/ajpregu.00825.2009. [DOI] [PubMed] [Google Scholar]
- 85.Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in s-nitrosylation of proteins involved in cardioprotection. Circulation. 2009;120:245–54. doi: 10.1161/CIRCULATIONAHA.109.868729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kawut SM, Al-Naamani N, Agerstrand C, Rosenzweig EB, Rowan C, Barst RJ, et al. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest. 2009;135:752–9. doi: 10.1378/chest.08-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, et al. Sex hormones are associated with right ventricular structure and function: The mesa-right ventricle study. Am J Respir Crit Care Med. 2011;183:659–67. doi: 10.1164/rccm.201007-1027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Almeida M, Martin-Millan M, Ambrogini E, Bradsher R, 3rd, Han L, Chen XD, et al. Estrogens attenuate oxidative stress and the differentiation and apoptosis of osteoblasts by DNA-binding-independent actions of the eralpha. J Bone Miner Res. 2010;25:769–81. doi: 10.1359/jbmr.091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Min HJ, Lee MJ, Kim JY, Cho SW, Park HD, Lee SI, et al. Alteration of bmp-4 and runx2 expression patterns in mouse temporomandibular joint after ovariectomy. Oral Dis. 2007;13:220–7. doi: 10.1111/j.1601-0825.2006.01270.x. [DOI] [PubMed] [Google Scholar]
- 90.van den Wijngaard A, Mulder WR, Dijkema R, Boersma CJ, Mosselman S, van Zoelen EJ, et al. Antiestrogens specifically up-regulate bone morphogenetic protein-4 promoter activity in human osteoblastic cells. Mol Endocrinol. 2000;14:623–33. doi: 10.1210/mend.14.5.0463. [DOI] [PubMed] [Google Scholar]
- 91.Monroe DG, Jin DF, Sanders MM. Estrogen opposes the apoptotic effects of bone morphogenetic protein 7 on tissue remodeling. Mol Cell Biol. 2000;20:4626–34. doi: 10.1128/mcb.20.13.4626-4634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang M, Yan JD, Zhang L, Wang Q, Lu SJ, Zhang J, et al. Activation of bone morphogenetic protein-6 gene transcription in mcf-7 cells by estrogen. Chin Med J (Engl) 2005;118:1629–36. [PubMed] [Google Scholar]
- 93.Wang D, Huang P, Zhu B, Sun L, Huang Q, Wang J. Induction of estrogen receptor alpha-36 expression by bone morphogenetic protein 2 in breast cancer cell lines. Mol Med Report. 2012;6:591–6. doi: 10.3892/mmr.2012.945. [DOI] [PubMed] [Google Scholar]
- 94.Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, et al. Smad4 as a transcription corepressor for estrogen receptor alpha. J Biol Chem. 2003;278:15192–200. doi: 10.1074/jbc.M212332200. [DOI] [PubMed] [Google Scholar]
- 95.Giacomini D, Paez-Pereda M, Stalla J, Stalla GK, Arzt E. Molecular interaction of bmp-4, tgf-beta, and estrogens in lactotrophs: Impact on the prl promoter. Mol Endocrinol. 2009;23:1102–14. doi: 10.1210/me.2008-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ebeling S, Topfer D, Weitzel JM, Meinecke B. Bone morphogenetic protein-6 (bmp-6): mRNA expression and effect on steroidogenesis during in vitro maturation of porcine cumulus oocyte complexes. Reprod Fertil Dev. 2011;23:1034–42. doi: 10.1071/RD11027. [DOI] [PubMed] [Google Scholar]
- 97.Donn R, Berry A, Stevens A, Farrow S, Betts J, Stevens R, et al. Use of gene expression profiling to identify a novel glucocorticoid sensitivity determining gene, bmprii. FASEB J. 2007;21:402–14. doi: 10.1096/fj.06-7236com. [DOI] [PubMed] [Google Scholar]
- 98.West J, Niswender KD, Johnson JA, Pugh ME, Gleaves L, Fessel JP, et al. A potential role for insulin resistance in experimental pulmonary hypertension. Eur Respir J. 2012 doi: 10.1183/09031936.00030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruegg J, Holsboer F, Turck C, Rein T. Cofilin 1 is revealed as an inhibitor of glucocorticoid receptor by analysis of hormone-resistant cells. Mol Cell Biol. 2004;24:9371–82. doi: 10.1128/MCB.24.21.9371-9382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, et al. Direct signaling by the bmp type ii receptor via the cytoskeletal regulator limk1. J Cell Biol. 2003;162:1089–98. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L474–84. doi: 10.1152/ajplung.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wong WK, Knowles JA, Morse JH. Bone morphogenetic protein receptor type ii c-terminus interacts with c-src: Implication for a role in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2005;33:438–46. doi: 10.1165/rcmb.2005-0103OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Machado RD, Rudarakanchana N, Atkinson C, Flanagan JA, Harrison R, Morrell NW, et al. Functional interaction between bmpr-ii and tctex-1, a light chain of dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2003;12:3277–86. doi: 10.1093/hmg/ddg365. [DOI] [PubMed] [Google Scholar]
- 104.Burlando B, Marchi B, Panfoli I, Viarengo A. Essential role of Ca2+-dependent phospholipase a2 in estradiol-induced lysosome activation. Am J Physiol Cell Physiol. 2002;283:C1461–8. doi: 10.1152/ajpcell.00429.2001. [DOI] [PubMed] [Google Scholar]
- 105.Durrington HJ, Upton PD, Hoer S, Boname J, Dunmore BJ, Yang J, et al. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type ii. J Biol Chem. 2010;285:37641–9. doi: 10.1074/jbc.M110.132415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, et al. Bone morphogenetic protein 2 induces pulmonary angiogenesis via wnt-beta-catenin and wnt-rhoa-rac1 pathways. J Cell Biol. 2009;184:83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Swaneck GE, Fishman J. Covalent binding of the endogenous estrogen 16 alpha-hydroxyestrone to estradiol receptor in human breast cancer cells: Characterization and intranuclear localization. Proc Natl Acad Sci USA. 1988;85:7831–5. doi: 10.1073/pnas.85.21.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miyairi S, Maeda K, Oe T, Kato T, Naganuma A. Effect of metal ions on the stable adduct formation of 16alpha-hydroxyestrone with a primary amine via the heyns rearrangement. Steroids. 1999;64:252–8. doi: 10.1016/s0039-128x(98)00118-4. [DOI] [PubMed] [Google Scholar]
- 109.Caja S, Puerta M. Control by reproduction-related hormones of resistin expression and plasma concentration. Horm Metab Res. 2007;39:501–6. doi: 10.1055/s-2007-982515. [DOI] [PubMed] [Google Scholar]
- 110.Helmestam M, Andersson H, Stavreus-Evers A, Brittebo E, Olovsson M. Tamoxifen modulates cell migration and expression of angiogenesis-related genes in human endometrial endothelial cells. Am J Pathol. 2012;180:2527–35. doi: 10.1016/j.ajpath.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 111.Tchaikovski SN, Rosing J. Mechanisms of estrogen-induced venous thromboembolism. Thromb Res. 2010;126:5–11. doi: 10.1016/j.thromres.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 112.Anderson JA, Weitz JI. Hypercoagulable states. Clin Chest Med. 2010;31:659–73. doi: 10.1016/j.ccm.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 113.Escribano-Subias P, Blanco I, Lopez-Meseguer M, Lopez-Guarch CJ, Roman A, Morales P, et al. Survival in pulmonary hypertension in Spain: Insights from the spanish registry. Eur Respir J. 2012;40:596–603. doi: 10.1183/09031936.00101211. [DOI] [PubMed] [Google Scholar]
- 114.Tofovic SP, Jackson EK. Complexities of oestradiol pharmacology in pulmonary arterial hypertension. Eur Respir J. 2012 doi: 10.1183/09031936.00164912. [DOI] [PubMed] [Google Scholar]
- 115.Fishman J, Martucci C. Biological properties of 16 alpha-hydroxyestrone: Implications in estrogen physiology and pathophysiology. J Clin Endocrinol Metab. 1980;51:611–5. doi: 10.1210/jcem-51-3-611. [DOI] [PubMed] [Google Scholar]
- 116.Zuckerman SH, Ahmari SE, Bryan-Poole N, Evans GF, Short L, Glasebrook AL. Estriol: A potent regulator of tnf and IL-6 expression in a murine model of endotoxemia. Inflammation. 1996;20:581–97. doi: 10.1007/BF01488797. [DOI] [PubMed] [Google Scholar]
- 117.Lippert C, Seeger H, Mueck AO. The effect of endogenous estradiol metabolites on the proliferation of human breast cancer cells. Life Sci. 2003;72:877–83. doi: 10.1016/s0024-3205(02)02305-6. [DOI] [PubMed] [Google Scholar]
- 118.Cullinan-Bove K, Koos RD. Vascular endothelial growth factor/vascular permeability factor expression in the rat uterus: Rapid stimulation by estrogen correlates with estrogen-induced increases in uterine capillary permeability and growth. Endocrinology. 1993;133:829–37. doi: 10.1210/endo.133.2.8344219. [DOI] [PubMed] [Google Scholar]
- 119.Ball P, Emons G, Kayser H, Teichmann J. Metabolic clearance rates of catechol estrogens in rats. Endocrinology. 1983;113:1781–3. doi: 10.1210/endo-113-5-1781. [DOI] [PubMed] [Google Scholar]
- 120.Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-methoxyestradiol, a promising anticancer agent. Pharmacotherapy. 2003;23:165–72. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]
- 121.Dubey RK, Tofovic SP, Jackson EK. Cardiovascular pharmacology of estradiol metabolites. J Pharmacol Exp Ther. 2004;308:403–9. doi: 10.1124/jpet.103.058057. [DOI] [PubMed] [Google Scholar]
- 122.Sutherland TE, Anderson RL, Hughes RA, Altmann E, Schuliga M, Ziogas J, et al. 2-methoxyestradiol--a unique blend of activities generating a new class of anti-tumour/anti-inflammatory agents. Drug Discov Today. 2007;12:577–84. doi: 10.1016/j.drudis.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 123.Tofovic SP, Dubey RK, Jackson EK. 2-hydroxyestradiol attenuates the development of obesity, the metabolic syndrome, and vascular and renal dysfunction in obese zsf1 rats. J Pharmacol Exp Ther. 2001;299:973–7. [PubMed] [Google Scholar]
- 124.Zhang X, Jia Y, Jackson EK, Tofovic SP. 2-methoxyestradiol and 2-ethoxyestradiol retard the progression of renal disease in aged, obese, diabetic zsf1 rats. J Cardiovasc Pharmacol. 2007;49:56–63. doi: 10.1097/FJC.0b013e31802cb88e. [DOI] [PubMed] [Google Scholar]
- 125.Tofovic SP, Dubey R, Salah EM, Jackson EK. 2-hydroxyestradiol attenuates renal disease in chronic puromycin aminonucleoside nephropathy. J Am Soc Nephrol. 2002;13:2737–47. doi: 10.1097/01.asn.0000031804.77546.f5. [DOI] [PubMed] [Google Scholar]
- 126.Tofovic SP, Salah EM, Dubey RK, Melhem MF, Jackson EK. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic nitric oxide synthase inhibition. J Cardiovasc Pharmacol. 2005;46:25–35. doi: 10.1097/01.fjc.0000162765.89437.ae. [DOI] [PubMed] [Google Scholar]
- 127.Tofovic SP, Zhang X, Zhu H, Jackson EK, Rafikova O, Petrusevska G. 2-ethoxyestradiol is antimitogenic and attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling. Vascul Pharmacol. 2008;48:174–83. doi: 10.1016/j.vph.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 128.Barchiesi F, Jackson EK, Fingerle J, Gillespie DG, Odermatt B, Dubey RK. 2-methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circ Res. 2006;99:266–74. doi: 10.1161/01.RES.0000233318.85181.2e. [DOI] [PubMed] [Google Scholar]
- 129.Tofovic SP, Salah EM, Mady HH, Jackson EK, Melhem MF. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol. 2005;46:430–7. doi: 10.1097/01.fjc.0000175878.32920.17. [DOI] [PubMed] [Google Scholar]
- 130.Tofovic SP, Zhang X, Jackson EK, Dacic S, Petrusevska G. 2-methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul Pharmacol. 2006;45:358–67. doi: 10.1016/j.vph.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 131.Tofovic SP, Zhang X, Jackson EK, Zhu H, Petrusevska G. 2-methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul Pharmacol. 2009;51:190–7. doi: 10.1016/j.vph.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tofovic SP, Zhang X, Jones T, Jackson EK, Petrusevska G. 2-methoxyestradiol attenuates the development and retards the progression of chronic hypoxia-induced pulmonary hypertension in rats. Circulation. 2005;112:II98–9. 2005. [Google Scholar]
- 133.Tofovic SP, Jones TJ, Bilan VP, Jackson EK, Petrusevska G. Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J Cardiovasc Pharmacol. 2010;56:475–83. doi: 10.1097/FJC.0b013e3181f215e7. [DOI] [PubMed] [Google Scholar]
- 134.Yuan PW, Wu WH, Gao L, Zheng ZQ, Liu D, Mei HY, et al. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, PGI2 AND ET-1 pathways. Eur Respir J. 2012 [Google Scholar]
- 135.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–38. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 136.Tofovic SP, Rafikova O. Preventive and therapeutic effects of 2-methoxyestradiol, but not estradiol, in severe occlusive pulmonary arterial hypertension in female rats. Am J Respir Crit Care Med. 2009:179. 1_MeetingAbstracts.A1802. [Google Scholar]
- 137.Tofovic SP, Rafikova O, Champion H. Estrogens exacerbates development of occlusive pulmonary arterial hypertension and formation of plexiform lesions. Am J Crit Care Med. 2012:185. 1_MeetingAbstracts.A6803. [Google Scholar]
- 138.Rafikova O, Rafikov R, Kumar S, Sharma S, Aggarwal S, Schneider F, et al. Bosentan inhibits oxidative and nitrosative stress and rescues occlusive pulmonaryhypertension. Free Radic Biol Med. 2013;56:28–43. doi: 10.1016/j.freeradbiomed.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tofovic SP, Zhu H, Jackson EK, Rafikova O. 2-methoxyestrone inhibits vascular remodeling and attenuates monocrotaline-induced pulmonary hypertension. Eur Respir J. 2008;32:S164. doi: 10.1016/j.vph.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 140.Newman SP, Ireson CR, Tutill HJ, Day JM, Parsons MF, Leese MP, et al. The role of 17beta-hydroxysteroid dehydrogenases in modulating the activity of 2-methoxyestradiol in breast cancer cells. Cancer Res. 2006;66:324–30. doi: 10.1158/0008-5472.CAN-05-2391. [DOI] [PubMed] [Google Scholar]
- 141.Bayard F, Clamens S, Meggetto F, Blaes N, Delsol G, Faye JC. Estrogen synthesis, estrogen metabolism, and functional estrogen receptors in rat arterial smooth muscle cells in culture. Endocrinology. 1995;136:1523–9. doi: 10.1210/endo.136.4.7895662. [DOI] [PubMed] [Google Scholar]
- 142.Piao YS, Peltoketo H, Jouppila A, Vihko R. Retinoic acids increase 17 beta-hydroxysteroid dehydrogenase type 1 expression in JEG-3 and T47D cells, but the stimulation is potentiated by epidermal growth factor, 12-o-tetradecanoylphorbol-13-acetate, and cyclic adenosine 3’,5’-monophosphate only in jeg-3 cells. Endocrinology. 1997;138:898–904. doi: 10.1210/endo.138.3.5008. [DOI] [PubMed] [Google Scholar]
- 143.Tofovic SP, Jackson EK, Rafikova O. Synergistic effects of 2-methoxyestradiol and retinoic acid on amelioration of monocrotaline-induced pulmonary hypertension. Am J Res Crit Care Med. 2008;177:A592. [Google Scholar]
- 144.Welter BH, Hansen EL, Saner KJ, Wei Y, Price TM. Membrane-bound progesterone receptor expression in human aortic endothelial cells. J Histochem Cytochem. 2003;51:1049–55. doi: 10.1177/002215540305100808. [DOI] [PubMed] [Google Scholar]
- 145.Barberis MC, Veronese S, Bauer D, De Juli E, Harari S. Immunocytochemical detection of progesterone receptors. A study in a patient with primary pulmonary hypertension. Chest. 1995;107:869–72. doi: 10.1378/chest.107.3.869. [DOI] [PubMed] [Google Scholar]
- 146.Vazquez F, Rodriguez-Manzaneque JC, Lydon JP, Edwards DP, O’Malley BW, Iruela-Arispe ML. Progesterone regulates proliferation of endothelial cells. J Biol Chem. 1999;274:2185–92. doi: 10.1074/jbc.274.4.2185. [DOI] [PubMed] [Google Scholar]
- 147.English KM, Jones RD, Jones TH, Morice AH, Channer KS. Gender differences in the vasomotor effects of different steroid hormones in rat pulmonary and coronary arteries. Horm Metab Res. 2001;33:645–52. doi: 10.1055/s-2001-18689. [DOI] [PubMed] [Google Scholar]
- 148.Tofovic PS, Zhang X, Petrusevska G. Progesterone inhibits vascular remodeling and attenuates monocrotaline-induced pulmonary hypertension in estrogen-deficient rats. Prilozi. 2009;30:25–44. [PubMed] [Google Scholar]
- 149.Jones T, Zhang X, Jackson EK, Tofovic SP. Medroxyprogesterone acetate attenuates and tibolone prevents the development of monocrotaline-induced pulmonary hypertension. FASEB J. 2006;20:A402. [Google Scholar]
- 150.Rochefort GY, Lemaire MC, Eder V, Hanton G, Hyvelin JM, Bonnet P, et al. Dexfenfluramine does not worsen but moderates progression of chronic hypoxia-induced pulmonary hypertension. Eur J Pharmacol. 2006;550:149–54. doi: 10.1016/j.ejphar.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 151.Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates. Implications for primary pulmonary hypertension. Circulation. 1999;100:869–75. doi: 10.1161/01.cir.100.8.869. [DOI] [PubMed] [Google Scholar]
- 152.Eddahibi S, Adnot S, Frisdal E, Levame M, Hamon M, Raffestin B. Dexfenfluramine-associated changes in 5-hydroxytryptamine transporter expression and development of hypoxic pulmonary hypertension in rats. J Pharmacol Exp Ther. 2001;297:148–54. [PubMed] [Google Scholar]
- 153.Dempsie Y, Morecroft I, Welsh DJ, MacRitchie NA, Herold N, Loughlin L, et al. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation. 2008;117:2928–37. doi: 10.1161/CIRCULATIONAHA.108.767558. [DOI] [PubMed] [Google Scholar]
- 154.Liu M, Wang Y, Wang HM, Bai Y, Zhang XH, Sun YX, et al. Fluoxetine attenuates chronic methamphetamine-induced pulmonary arterial remodeling: Possible involvement of serotonin transporter and serotonin 1B receptor. Basic Clin Pharmacol Toxicol. 2013;112:77–82. doi: 10.1111/j.1742-7843.2012.00933.x. [DOI] [PubMed] [Google Scholar]
- 155.Abid S, Houssaini A, Chevarin C, Marcos E, Tissot CM, Gary-Bobo G, et al. Inhibition of gut- and lung-derived serotonin attenuates pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol. 2012;303:L500–8. doi: 10.1152/ajplung.00049.2012. [DOI] [PubMed] [Google Scholar]
- 156.Izikki M, Hanoun N, Marcos E, Savale L, Barlier-Mur AM, Saurini F, et al. Tryptophan hydroxylase 1 knockout and tryptophan hydroxylase 2 polymorphism: Effects on hypoxic pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1045–52. doi: 10.1152/ajplung.00082.2007. [DOI] [PubMed] [Google Scholar]
- 157.Morecroft I, Dempsie Y, Bader M, Walther DJ, Kotnik K, Loughlin L, et al. Effect of tryptophan hydroxylase 1 deficiency on the development of hypoxia-induced pulmonary hypertension. Hypertension. 2007;49:232–6. doi: 10.1161/01.HYP.0000252210.58849.78. [DOI] [PubMed] [Google Scholar]
- 158.Morecroft I, White K, Caruso P, Nilsen M, Loughlin L, Alba R, et al. Gene therapy by targeted adenovirus-mediated knockdown of pulmonary endothelial TPH1 attenuates hypoxia-induced pulmonary hypertension. Mol Ther. 2012;20:1516–28. doi: 10.1038/mt.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ciuclan L, Hussey MJ, Burton V, Good R, Duggan N, Beach S, J, et al. Imatinib attenuates hypoxia-induced pulmonary arterial hypertension pathology via reduction in 5-hydroxytryptamine through inhibition of tryptophan hydroxylase 1 expression. Am J Respir Crit Care Med. 2013;187:78–89. doi: 10.1164/rccm.201206-1028OC. [DOI] [PubMed] [Google Scholar]
- 160.Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: Critical role for serotonin-induced smooth muscle hyperplasia. Circulation. 2006;113:1857–64. doi: 10.1161/CIRCULATIONAHA.105.591321. [DOI] [PubMed] [Google Scholar]
- 161.MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, et al. Overexpression of the 5-hydroxytryptamine transporter gene: Effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation. 2004;109:2150–5. doi: 10.1161/01.CIR.0000127375.56172.92. [DOI] [PubMed] [Google Scholar]
- 162.Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, et al. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest. 2000;105:1555–62. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marcos E, Fadel E, Sanchez O, Humbert M, Dartevelle P, Simonneau G, et al. Serotonin-induced smooth muscle hyperplasia in various forms of human pulmonary hypertension. Circ Res. 2004;94:1263–70. doi: 10.1161/01.RES.0000126847.27660.69. [DOI] [PubMed] [Google Scholar]
- 164.Liu Y, Li M, Warburton RR, Hill NS, Fanburg BL. The 5-ht transporter transactivates the pdgfbeta receptor in pulmonary artery smooth muscle cells. FASEB J. 2007;21:2725–34. doi: 10.1096/fj.06-8058com. [DOI] [PubMed] [Google Scholar]
- 165.Morecroft I, Heeley RP, Prentice HM, Kirk A, MacLean MR. 5-hydroxytryptamine receptors mediating contraction in human small muscular pulmonary arteries: Importance of the 5-ht1b receptor. Br J Pharmacol. 1999;128:730–4. doi: 10.1038/sj.bjp.0702841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lawrie A, Spiekerkoetter E, Martinez EC, Ambartsumian N, Sheward WJ, MacLean MR, et al. Interdependent serotonin transporter and receptor pathways regulate s100a4/mts1, a gene associated with pulmonary vascular disease. Circ Res. 2005;97:227–35. doi: 10.1161/01.RES.0000176025.57706.1e. [DOI] [PubMed] [Google Scholar]
- 167.Morecroft I, Pang L, Baranowska M, Nilsen M, Loughlin L, Dempsie Y, et al. In vivo effects of a combined 5-HT1B receptor/sert antagonist in experimental pulmonary hypertension. Cardiovasc Res. 2010;85:593–603. doi: 10.1093/cvr/cvp306. [DOI] [PubMed] [Google Scholar]
- 168.McGoon MD, Miller DP. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur Respir Rev. 2012;21:8–18. doi: 10.1183/09059180.00008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the post-bmpr2 era. Pulm Circ. 2011;1:305–19. doi: 10.4103/2045-8932.87293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240:H62–72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- 171.Xu DQ, Luo Y, Liu Y, Wang J, Zhang B, Xu M, et al. Beta-estradiol attenuates hypoxic pulmonary hypertension by stabilizing the expression of p27kip1 in rats. Respir Res. 2010;11:182. doi: 10.1186/1465-9921-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res. 2011;90:373–82. doi: 10.1093/cvr/cvq408. [DOI] [PubMed] [Google Scholar]
- 173.Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, et al. Development of pulmonary arterial hypertension in mice over-expressing s100a4/mts1 is specific to females. Respir Res. 2011;12:159. doi: 10.1186/1465-9921-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dempsie Y, Morecroft I, Nilsen M, Loughlin L, MacLean MR. The effects of gender on the development of dexfenfluramine-induced pulmonary arterial hypertension in mice. Am J Respir Crit Care Med. 2009;179:A1808. [Google Scholar]
- 175.White K, Loughlin L, Maqbool Z, Nilsen M, McClure J, Dempsie Y, et al. Serotonin transporter, sex, and hypoxia: Microarray analysis in the pulmonary arteries of mice identifies genes with relevance to human PAH. Physiol Genomics. 2011;43:417–37. doi: 10.1152/physiolgenomics.00249.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, et al. Activity of the estrogen-metabolizing enzyme cytochrome p450 1b1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–98. doi: 10.1161/CIRCULATIONAHA.111.062927. [DOI] [PubMed] [Google Scholar]
- 177.Carter KL, Cahir-McFarland E, Kieff E. Epstein-barr virus-induced changes in b-lymphocyte gene expression. J Virol. 2002;76:10427–36. doi: 10.1128/JVI.76.20.10427-10436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bandiera S, Weidlich S, Harth V, Broede P, Ko Y, Friedberg T. Proteasomal degradation of human CYP1B1: Effect of the asn453ser polymorphism on the post-translational regulation of cyp1b1 expression. Mol Pharmacol. 2005;67:435–43. doi: 10.1124/mol.104.006056. [DOI] [PubMed] [Google Scholar]
- 179.Hanna IH, Dawling S, Roodi N, Guengerich FP, Parl FF. Cytochrome p450 1b1 (CYP1B1) pharmacogenetics: Association of polymorphisms with functional differences in estrogen hydroxylation activity. Cancer Res. 2000;60:3440–4. [PubMed] [Google Scholar]
- 180.Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. 2003;86:225–30. doi: 10.1016/s0960-0760(03)00360-1. [DOI] [PubMed] [Google Scholar]
- 181.Misso ML, Jang C, Adams J, Tran J, Murata Y, Bell R, et al. Adipose aromatase gene expression is greater in older women and is unaffected by postmenopausal estrogen therapy. Menopause. 2005;12:210–5. doi: 10.1097/00042192-200512020-00016. [DOI] [PubMed] [Google Scholar]
- 182.Badawi AF, Cavalieri EL, Rogan EG. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16alpha-hydroxylation of 17beta-estradiol. Metabolism. 2001;50:1001–3. doi: 10.1053/meta.2001.25592. [DOI] [PubMed] [Google Scholar]
- 183.Lee AJ, Cai MX, Thomas PE, Conney AH, Zhu BT. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology. 2003;144:3382–98. doi: 10.1210/en.2003-0192. [DOI] [PubMed] [Google Scholar]
- 184.Meireles SI, Esteves GH, Hirata R, Peri S, Devarajan K, Slifker M, et al. Early changes in gene expression induced by tobacco smoke: Evidence for the importance of estrogen within lung tissue. Cancer Prev Res (Phila) 2010;3:707–17. doi: 10.1158/1940-6207.CAPR-09-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Conway DE, Sakurai Y, Weiss D, Vega JD, Taylor WR, Jo H, et al. Expression of CYP1A1 and CYP1B1 in human endothelial cells: Regulation by fluid shear stress. Cardiovasc Res. 2009;81:669–77. doi: 10.1093/cvr/cvn360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tariq SH, Kamel H, Morley JE. Dehydroepiandrosterone and pregnenolone. In: Meikle AW, editor. Endocrine Replacement Therapy in Clinical Practice. Totowa, NJ: Humana Press; 2003. pp. 307–368. [Google Scholar]
- 187.Johnson MD, Bebb RA, Sirrs SM. Uses of dhea in aging and other disease states. Ageing Res Rev. 2002;1:29–41. doi: 10.1016/s0047-6374(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 188.Orentreich N, Brind JL, Rizer RL, Vogelman JH. Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J Clin Endocrinol Metab. 1984;59:551–5. doi: 10.1210/jcem-59-3-551. [DOI] [PubMed] [Google Scholar]
- 189.Orentreich N, Brind JL, Vogelman JH, Andres R, Baldwin H. Long-term longitudinal measurements of plasma dehydroepiandrosterone sulfate in normal men. J Clin Endocrinol Metab. 1992;75:1002–4. doi: 10.1210/jcem.75.4.1400863. [DOI] [PubMed] [Google Scholar]
- 190.Baulieu EE. Dehydroepiandrosterone (DHEA): A fountain of youth? J Clin Endocrinol Metab. 1996;81:3147–51. doi: 10.1210/jcem.81.9.8784058. [DOI] [PubMed] [Google Scholar]
- 191.Labrie F, Luu-The V, Labrie C, Simard J. DHEA and its transformation into androgens and estrogens in peripheral target tissues: Intracrinology. Front Neuroendocrinol. 2001;22:185–212. doi: 10.1006/frne.2001.0216. [DOI] [PubMed] [Google Scholar]
- 192.MacDonald PC, Edman CD, Kerber IJ, Siiteri PK. Plasma precursors of estrogen. III. Conversion of plasma dehydroisoandrosterone to estrogen in young nonpregnant women. Gynecol Invest. 1976;7:165–75. [PubMed] [Google Scholar]
- 193.Barrett-Connor E, Goodman-Gruen D. The epidemiology of dheas and cardiovascular disease. Ann N Y Acad Sci. 1995;774:259–70. doi: 10.1111/j.1749-6632.1995.tb17386.x-i1. [DOI] [PubMed] [Google Scholar]
- 194.Williams JR. The effects of dehydroepiandrosterone on carcinogenesis, obesity, the immune system, and aging. Lipids. 2000;35:325–31. doi: 10.1007/s11745-000-0529-7. [DOI] [PubMed] [Google Scholar]
- 195.Levy HR. Glucose-6-phosphate dehydrogenases. Adv Enzymol Relat Areas Mol Biol. 1979;48:97–192. doi: 10.1002/9780470122938.ch3. [DOI] [PubMed] [Google Scholar]
- 196.Schwartz AG, Pashko LL. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res Rev. 2004;3:171–87. doi: 10.1016/j.arr.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 197.Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998;273:10609–17. doi: 10.1074/jbc.273.17.10609. [DOI] [PubMed] [Google Scholar]
- 198.Gupte SA, Li KX, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: Involvement of voltage-gated potassium channels. J Pharmacol Exp Ther. 2002;301:299–305. doi: 10.1124/jpet.301.1.299. [DOI] [PubMed] [Google Scholar]
- 199.Simoncini T, Mannella P, Fornari L, Varone G, Caruso A, Genazzani AR. Dehydroepiandrosterone modulates endothelial nitric oxide synthesis via direct genomic and nongenomic mechanisms. Endocrinology. 2003;144:3449–55. doi: 10.1210/en.2003-0044. [DOI] [PubMed] [Google Scholar]
- 200.Liu D, Dillon JS. Dehydroepiandrosterone activates endothelial cell nitric-oxide synthase by a specific plasma membrane receptor coupled to G alpha(i2,3) J Biol Chem. 2002;277:21379–88. doi: 10.1074/jbc.M200491200. [DOI] [PubMed] [Google Scholar]
- 201.Bhuiyan MS, Tagashira H, Fukunaga K. Dehydroepiandrosterone-mediated stimulation of sigma-1 receptor activates akt-enos signaling in the thoracic aorta of ovariectomized rats with abdominal aortic banding. Cardiovasc Ther. 2011;29:219–30. doi: 10.1111/j.1755-5922.2010.00196.x. [DOI] [PubMed] [Google Scholar]
- 202.Karbowska J, Kochan Z. Effect of dhea on endocrine functions of adipose tissue, the involvement of ppar gamma. Biochem Pharmacol. 2005;70:249–57. doi: 10.1016/j.bcp.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 203.Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappab signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–41. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- 204.Schulz S, Nyce JW. Inhibition of protein isoprenylation and p21ras membrane association by dehydroepiandrosterone in human colonic adenocarcinoma cells in vitro. Cancer Res. 1991;51:6563–7. [PubMed] [Google Scholar]
- 205.Williams MR, Ling S, Dawood T, Hashimura K, Dai A, Li H, et al. Dehydroepiandrosterone inhibits human vascular smooth muscle cell proliferation independent of ars and ers. J Clin Endocrinol Metab. 2002;87:176–81. doi: 10.1210/jcem.87.1.8161. [DOI] [PubMed] [Google Scholar]
- 206.Farrukh IS, Peng W, Orlinska U, Hoidal JR. Effect of dehydroepiandrosterone on hypoxic pulmonary vasoconstriction: A Ca(2+)-activated K(+)-channel opener. Am J Physiol. 1998;274:L186–95. doi: 10.1152/ajplung.1998.274.2.L186. [DOI] [PubMed] [Google Scholar]
- 207.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Pathol. 1997;151:1019–25. [PMC free article] [PubMed] [Google Scholar]
- 208.Hampl V, Bibova J, Povysilova V, Herget J. Dehydroepiandrosterone sulphate reduces chronic hypoxic pulmonary hypertension in rats. Eur Respir J. 2003;21:862–5. doi: 10.1183/09031936.03.00084503. [DOI] [PubMed] [Google Scholar]
- 209.Bonnet S, Dumas-de-La-Roque E, Begueret H, Marthan R, Fayon M, Dos Santos P, et al. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA. 2003;100:9488–93. doi: 10.1073/pnas.1633724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, et al. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res. 2007;74:377–87. doi: 10.1016/j.cardiores.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hayashi T, Esaki T, Muto E, Kano H, Asai Y, Thakur NK, et al. Dehydroepiandrosterone retards atherosclerosis formation through its conversion to estrogen: The possible role of nitric oxide. Arterioscler Thromb Vasc Biol. 2000;20:782–92. doi: 10.1161/01.atv.20.3.782. [DOI] [PubMed] [Google Scholar]
- 212.Kawano H, Yasue H, Kitagawa A, Hirai N, Yoshida T, Soejima H, et al. Dehydroepiandrosterone supplementation improves endothelial function and insulin sensitivity in men. J Clin Endocrinol Metab. 2003;88:3190–5. doi: 10.1210/jc.2002-021603. [DOI] [PubMed] [Google Scholar]
- 213.Debonneuil EH, Quillard J, Baulieu EE. Hypoxia and dehydroepiandrosterone in old age: A mouse survival study. Respir Res. 2006;7:144. doi: 10.1186/1465-9921-7-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Dessouroux A, Akwa Y, Baulieu EE. DHEA decreases HIF-1alpha accumulation under hypoxia in human pulmonary artery cells: Potential role in the treatment of pulmonary arterial hypertension. J Steroid Biochem Mol Biol. 2008;109:81–9. doi: 10.1016/j.jsbmb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 215.Dumas de La Roque E, Savineau JP, Metivier AC, Billes MA, Kraemer JP, Doutreleau S, et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): A pilot study. Ann Endocrinol (Paris) 2012;73:20–5. doi: 10.1016/j.ando.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 216.Homma N, Nagaoka T, Karoor V, Imamura M, Taraseviciene-Stewart L, Walker LA, et al. Involvement of RHOA/RHO kinase signaling in protection against monocrotaline-induced pulmonary hypertension in pneumonectomized rats by dehydroepiandrosterone. Am J Physiol Lung Cell Mol Physiol. 2008;295:L71–8. doi: 10.1152/ajplung.90251.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the src/stat3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H1798–809. doi: 10.1152/ajpheart.00654.2011. [DOI] [PubMed] [Google Scholar]
- 218.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–54. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 219.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–60. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 220.Alzoubi A, Toba M, Abe K, O’Neill KD, Rocic P, Fagan KA, et al. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol Apr. 2013 Apr 12; doi: 10.1152/ajpheart.00746.2012. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 221.Ahlem C, Auci D, Mangano K, Reading C, Frincke J, Stickney D, et al. HE3286: A novel synthetic steroid as an oral treatment for autoimmune disease. Ann N Y Acad Sci. 2009;1173:781–90. doi: 10.1111/j.1749-6632.2009.04798.x. [DOI] [PubMed] [Google Scholar]
- 222.Gabler NB, French B, Strom BL, Liu Z, Palevsky HI, Taichman DB, et al. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest. 2012;141:20–6. doi: 10.1378/chest.11-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]