

Abstract
The diagnosis of pulmonary arterial hypertension (PAH) is frequently delayed. We hypothesized that circulating angiogenic modulatory protein levels might correspond with vascular remodeling activity and serve as sensitive biomarkers of PAH. Levels of soluble endoglin (sEng), soluble vascular endothelial growth factor receptor-1 (sVEGFR1), N-terminal brain natriuretic peptide (NT-proBNP), C-reactive protein (CRP), and other biomarkers were measured in peripheral blood from 97 PAH patients, 16 first-degree relatives of idiopathic or heritable pulmonary arterial hypertension (HPAH) patients, and 56 controls, and correlated with disease, functional class, hemodynamic parameters, exercise capacity, and transplant-free survival. Endoglin expression was analyzed in lung tissues of six individuals with idiopathic or HPAH and four individuals without PAH. Levels of sEng, sVEGFR1, CRP, and NT-proBNP were elevated in Group I PAH of diverse etiologies, with sEng performing better than NT-proBNP in detecting PAH (receiver operator characteristic-area-under-the curve [ROC-AUC] of 0.82 ± 0.03 vs. 0.71 ± 0.05, P = 0.016). While sEng, sVEGFR1, and NT-proBNP correlated with New York Heart Association (NYHA) class, sEng levels were more sensitive than NT-proBNP in detecting NYHA Class I-II disease (ROC-AUC of 0.88 ± 0.05 vs. 0.67 ± 0.08, P = 0.028). sEng, sVEGFR1, CRP, and NT-proBNP predicted transplant-free survival by univariate Cox regression. After adjusting for NT-proBNP levels, each of the other three markers predicted transplant-free survival. In multivariate analysis, sEng and CRP were independent predictors of survival. Endoglin expression was markedly enhanced in the microvascular endothelium and endovascular lesions of PAH versus control lung tissues. Circulating angiogenic proteins sEng and sVEGFR1 are sensitive markers of prognosis and function in Group I PAH, including mildly symptomatic disease, and may provide unique noninvasive data reflecting underlying remodeling activity.
Keywords: angiogenesis, biomarkers, endoglin, pulmonary arterial hypertension, VEGF receptor-1
Pulmonary arterial hypertension (PAH) is defined by an elevated mean pulmonary artery pressure (mPAP) of ≥25 mmHg at rest with a left ventricular end diastolic pressure ≤15 mmHg, and a pulmonary vascular resistance (PVR) of >3 Wood units (WU).[1] PAH affects approximately 20 individuals per million adults with a 9-15% mortality rate in the first year despite current therapy.[2,3,4,5,6] In the World Health Organization (WHO) classification, Group I PAH includes idiopathic pulmonary arterial hypertension (IPAH) occurring in the absence of cardiopulmonary disease, a subset of which is heritable pulmonary arterial hypertension (HPAH), or associated pulmonary arterial hypertension (APAH) with connective tissue disease (CTD), stimulant drug use, congenital heart disease (CHD), portal hypertension, or human immunodeficiency virus (HIV).[1,7]
Dysregulated endothelial cell and myofibroblast proliferation, smooth muscle hypertrophy, intimal fibrosis, and enhanced thrombosis—features evident in obstructive and obliterative arteriolar and multi-channeled “plexiform” lesions of PAH—have been interpreted by some to reflect an underlying process of disordered angiogenesis.[8,9] Endovascular PAH lesions are often accompanied by perivascular myelomonocytic infiltrates, suggesting a role of inflammation.[10,11,12] While these features are found in end-stage PAH, it is unclear if dysregulated angiogenesis or vascular inflammation are present in early PAH or contribute to its progression.
Recent observations have provided further evidence for dysregulated angiogenic activity in PAH. Administration of imatinib, an inhibitor of platelet-derived growth factor receptor beta (PDGF-Rβ), which regulates smooth muscle proliferation and pericyte recruitment, attenuates PH in rats, and possibly in humans.[13,14,15] In contrast, administering SU-5416, an antiangiogenic inhibitor of vascular endothelial growth factor receptor (VEGF-R) and PDGF-Rβ signaling, triggers severe pulmonary vascular remodeling and pulmonary hypertension (PH) in rodents.[16] The potent PDGF-Rβ and SRC inhibitor dasatinib attenuates PH in rats,[17] whereas chronic dasatinib administration has been associated with the development of PAH in man.[18,19] Angiogenic signaling cascades have important, albeit complex, roles in the development and progression of PAH. We hypothesize that surrogates of angiogenic activity could provide insight into disease activity and progression as biomarkers of PAH.
As noninvasive adjuncts to diagnostic right heart catheterization, circulating biomarkers have been evaluated in relationship to clinical severity, response to therapy, and survival in PAH,[1,20] ideally providing additional risk stratification for clinical interventions. The most extensively validated PAH biomarkers, B-type natriuretic peptide (BNP) and its pro-peptide N-terminal brain natriuretic peptide (NT-proBNP), predict survival and function, but, as surrogates of right ventricular myocyte stretch, are insensitive for mildly symptomatic disease.[21,22,23] Numerous other candidate biomarkers have been explored in PAH; however, their utility above and beyond BNP in prognosis and management have not been confirmed.[24] In a cohort of 97 individuals with Group I PAH and 56 age- and gender-matched unaffected controls, we evaluated several circulating markers of systemic and vascular inflammation, C-reactive protein (CRP), osteoprotegerin (OPG), and interleukin (IL)-15, and two markers of endothelial activation and angiogenic activity, soluble endoglin (sEng) and soluble VEGF receptor-1 (sVEGFR1). We evaluated these biomarkers in relationship to functional status and survival, hypothesizing that surrogates of angiogenesis and inflammation might yield novel functional or prognostic data, and detect minimally symptomatic disease not captured by current biomarkers of PAH.
MATERIALS AND METHODS
Study population
Plasma samples were obtained from 97 patients with confirmed diagnoses of PAH by WHO criteria (mPAP ≥25 mmHg at rest with a pulmonary capillary wedge pressure ≤15 mmHg and a PVR >3 WUs).[1] This cohort included patients with idiopathic or HPAH, as well as PAH associated with CTD, CHD, portal hypertension, drugs and toxins, HIV, and Gaucher's disease. Samples were also collected from 16 first-degree relatives of IPAH or HPAH patients. Control samples were obtained from 56 healthy individuals. Patients, relatives, and healthy controls were recruited at the Pulmonary Hypertension Association Scientific Sessions, the California PH Forum Conference, and Stanford University Medical Center, with informed consent and Institutional Review Board (IRB) approval from Partners Health-care, Inc. (Boston, Mass., USA), University of Utah, LDS Hospital and Intermountain Medical Center (Salt Lake City, Utah, USA), and Stanford University Medical Center (Stanford, Calif., USA).
Serum samples and biomarker measurements
Serum samples were collected from peripheral blood and stored at –80°C until analyzed. Serum analytes were measured by sandwich enzyme-linked immunosorbent assay (ELISA) according to manufacturers’ protocols, including OPG (Duoset, R&D Systems, Minneapolis, Minn., USA), high-sensitivity CRP, sEng, sVEGFR1 (Quantikine, R&D Systems), and IL-15 (Quantiglo, R&D Systems), or by competition ELISA in the case of NT-proBNP (Alpco Diagnostics, Salem, N.H.). All analytes were measured at least in duplicate, and quantified in comparison to dilutions of recombinant protein standards and standardized serum samples.
Clinical and hemodynamic parameters
New York Heart Association (NYHA) functional class, 6-Min Walk Distance (6MWD), pulmonary function tests, echocardiographic measurements of left and right ventricular function and Doppler-based estimates of right ventricular systolic pressure, medical therapies, and invasive hemodynamic data assessed contemporaneously with blood sampling (within three months before or after sample collection) were obtained by chart review with informed consent. PVR was calculated as (mean pulmonary arterial pressure – pulmonary capillary wedge pressure)/(cardiac output) and expressed in WUs. Survival data was obtained retrospectively both from hospital records and the social security death index.
Immunohistochemistry
Paraffin sections were obtained from explanted lung tissue of IPAH patients (n = 3), HPAH patients (n = 3), or controls (explanted lungs lacking pulmonary vascular disease, n = 2; untransplanted normal donor lungs, n = 1; and normal lung tissues, n = 1). After deparaffinization, antigen unmasking, and quenching of endogenous peroxidase activity (3% H2O2 in methanol), slides were blocked with 5% normal goat serum in Tris-buffered saline with 0.1% Tween-20, incubated with biotinylated goat anti-human endoglin (R&D Systems, 1:50) overnight at 4°C, and incubated with streptavidin-horse radish peroxidase (LSAB2, Dako, Carpinteria, Calif., USA) at room temperature for 30 minutes followed by diaminobenzidine reagent (immPACT DAB, Vector Laboratories). Sections were counterstained in hematoxylin, mounted, and imaged using an Olympus BX63 microscope equipped with a DP-25 color camera and cellSens Dimension 1.6 software (Olympus, Tokyo, Japan).
Statistical analyses
Statistical analyses were performed using STATA 8.0 (StataCorp, College Station, Tex., USA) or GraphPad Prism 5.0 (San Diego, Calif., USA). Pairwise comparisons of plasma biomarker levels were made in the control population, PAH patients, and first-degree relatives using the Mann-Whitney statistic. Normality of biomarker levels and clinical variables were assessed by the Shapiro-Wilk test. Continuous variables were analyzed with either the Student's t-test or Mann-Whitney statistic as appropriate, and Fisher's exact test for categorical variables. Receiver operator characteristic (ROC) curves were derived and area-under-the curve (AUC) analysis performed to compare performance of biomarkers in predicting presence or absence of disease. Associations between biomarker levels were examined using Spearman's correlation. One-way analysis of variance (ANOVA) was used to determine associations between normally distributed (NT-proBNP, sEng, sVEGFR1, and OPG) biomarker levels and NYHA functional class, while Kruskal-Wallis testing was used for non-normally distributed biomarker levels (CRP and IL-15). Survival characteristics were compared using the Kaplan–Meier method with Log Rank testing. Cox proportional hazards models were employed to identify univariate and multivariate predictors of transplant-free survival. Values are reported as mean ± SEM unless noted, with P values <0.05 considered significant.
RESULTS
Study population
Table 1 illustrates the baseline demographics of the control, PAH, and first-degree relative groups. The gender of patients in the PAH group reflected the known female predominance of PAH. Control individuals were selected to reflect a similar female predominance. The ages of the PAH and control populations were 48.9 ± 13.1 and 47.4 ± 8.0, respectively (P = 0.47). Among the disease group, 51.6% of cases were idiopathic (IPAH, n = 50), 21.6% associated with CTD (APAH-CTD, n = 21), 12.4% associated with stimulant drugs and toxins (APAH-Stim, n = 12), 6.2% associated with CHD (APAH-CHD, n = 6), and 8.2% associated with portal hypertension or other conditions (APAH-Other, n = 8). While the patients presented across the spectrum of functional status, the majority exhibited NYHA Class II or III symptoms at the time of evaluation (Table 2). Hemodynamic parameters were obtained from right heart catheterization, with an average mPAP of 53 ± 13 mmHg, cardiac index of 2.7 ± 1.0 L/min/m2, and PVR of 11.5 ± 6.0 WUs in the 97 PAH patients. These patients received currently approved PAH-directed therapies including prostacyclin analogues, endothelin receptor antagonists (ETRA), and phosphodiesterase-type 5 (PDE5) inhibitors. More than 50% received combination therapy and a similar proportion received chronic anticoagulation.
Table 1.
Population demographics and pulmonary arterial hypertension etiology
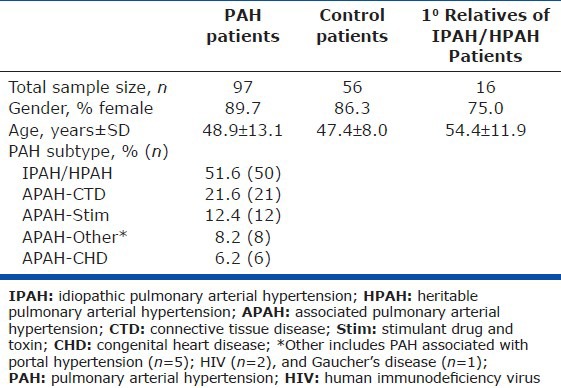
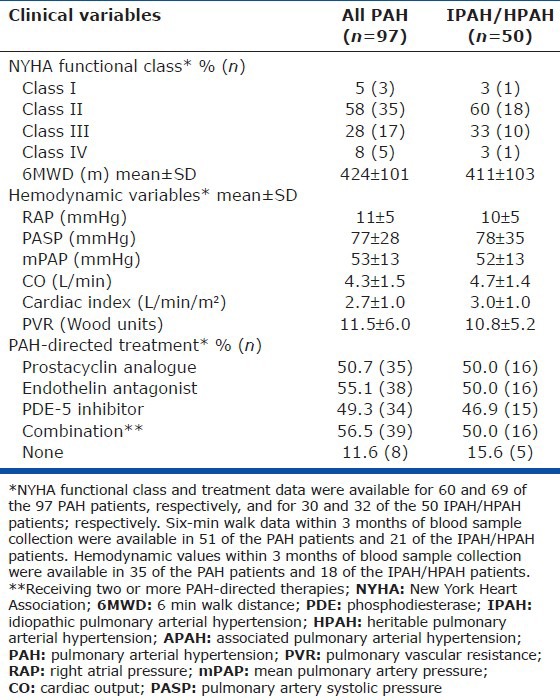
Table 2.
Clinical, hemodynamic, and treatment characteristics of pulmonary arterial hypertension patients and the idiopathic pulmonary arterial hypertension subgroup
Levels of CRP, NT-proBNP, sEng, and sVEGFR1, but not OPG or IL-15 were elevated in PAH
Baseline levels of biomarkers in our control population fell within the range of normal values previously reported.[21,25,26,27,28] Serum levels of CRP (P < 0.0001), NT-proBNP (P = 0.0008), sEng (P < 0.0001), and sVEGFR1 (P < 0.0001) were elevated in individuals with PAH compared to control individuals of similar age and gender (Fig. 1 and Table 3). However, no differences in the levels of OPG and IL-15 were observed between the PAH cohort and controls. None of the PAH-directed therapies taken in isolation or in combination (i.e., prostacyclins, ETRA, or PDE5 inhibitors) were associated with biomarker levels, except that patients receiving prostacyclin analogues exhibited reduced sVEGFR1 levels (121 ± 10 vs. 172 ± 14 pg/mL, P = 0.005).
Figure 1.
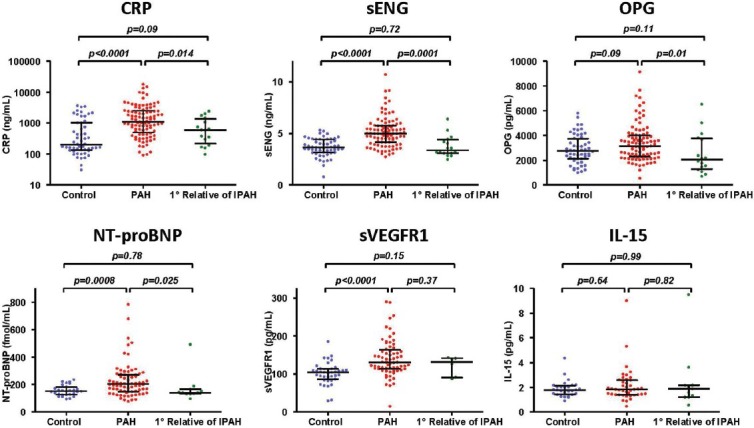
C-reactive protein (CRP), N-terminal brain natriuretic peptide (NT-proBNP), soluble endoglin (sEng), and soluble VEGF receptor-1 (sVEGFR1) are elevated in pulmonary arterial hypertension (PAH). Levels of the biomarker analyzed are depicted among control individuals, PAH patients, and the first-degree relatives of either idiopathic pulmonary arterial hypertension or heritable pulmonary arterial hypertension patients, with median, top and bottom quartile values indicated as shown.
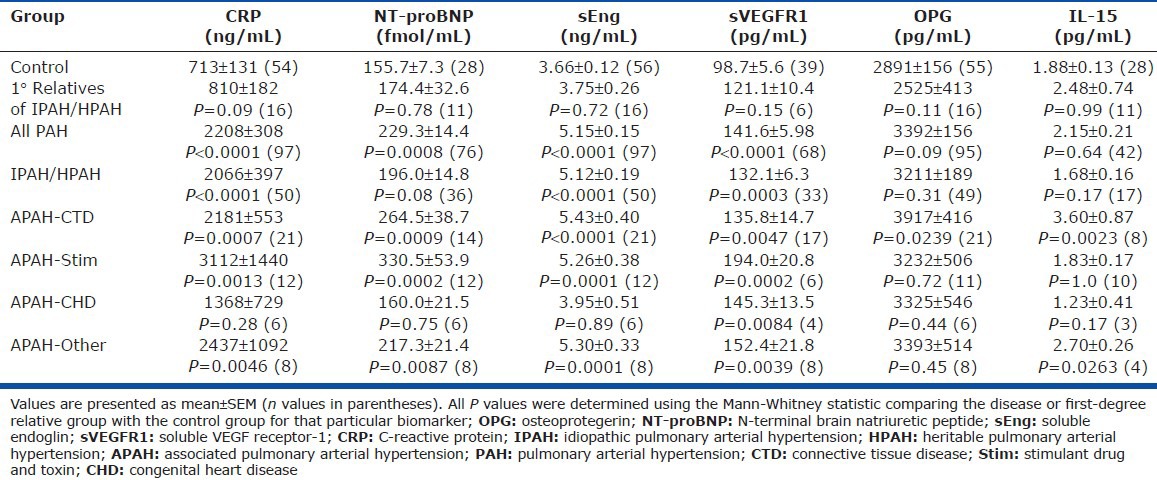
Table 3.
Serum levels of inflammatory markers, angiogenic modulators, and natriuretic peptides in controls, pulmonary arterial hypertension patients, and 1° relatives of idiopathic pulmonary arterial hypertension
Levels of sEng were more sensitive and specific for PAH than NT-proBNP
Consistent with prior reports,[21,22,23] elevated NT-proBNP predicted PAH with an ROC-AUC of 0.71 ± 0.05 (Fig. 2). Only sEng was superior to NT-proBNP in predicting the presence of PAH (AUC 0.82 ± 0.03, P = 0.016), while sVEGFR1 (0.79 ± 0.05) and CRP (0.77 ± 0.04) were similar to NT-proBNP. An equally weighted combination of sEng and NT-proBNP (calculated as the linear sum of each biomarker value's Z score, i.e., [value − mean]/(standard deviation) yielded an AUC of 0.87 ± 0.04, which was not significantly better than sEng alone.
Figure 2.
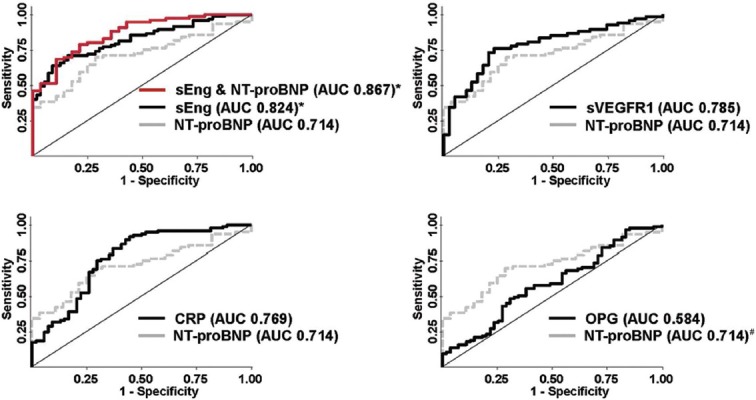
Soluble endoglin (sEng) is a better predictor of the presence of pulmonary arterial hypertension than N-terminal brain natriuretic peptide (NT-proBNP). Receiver operator characteristic curves for sEng, soluble VEGF receptor-1 (sVEGFR1), C-reactive protein (CRP), and osteoprotegerin (OPG) are depicted in relationship to NT-proBNP as a reference biomarker. Areas under the curve (AUC ± SEM) were determined for sEng (0.824 ± 0.033), sVEGFR1 (0.785 ± 0.045), CRP (0.769 ± 0.042), NT-proBNP (0.714 ± 0.051), and OPG (0.584 ± 0.049), *P<0.05 versus NT-proBNP; #P<0.05 versus OPG.
Relationship of biomarker levels and underlying etiology of PAH
This cohort included patients with distinct etiologies of WHO Group I PAH disease (Table 1). The mean and distribution of each biomarker studied in relationship to underlying disease etiology is shown numerically (Table 3) and graphically (Fig. 3). Circulating levels of sVEGFR1 were elevated in all categories of Group I PAH, whereas levels of sEng and CRP were increased in all groups studied (IPAH, APAH-CTD, APAH-toxin, and APAH-other) with the exception of CHD. As observed among PAH patients as a whole, among IPAH and HPAH patients sEng performed better than NT-proBNP in predicting disease (AUC of 0.86 ± 0.04 vs. 0.63 ± 0.07, P = 0.008).
Figure 3.
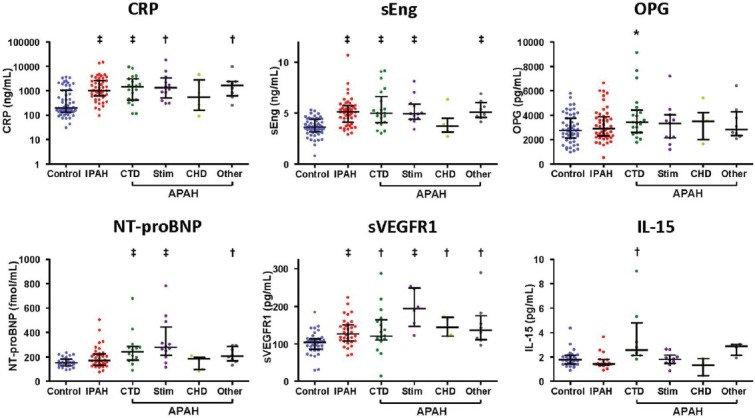
Angiogenic biomarkers are elevated in diverse etiologies of group I pulmonary arterial hypertension (PAH). Levels of inflammatory markers (C-reactive protein [CRP], IL-15, and osteoprotegerin [OPG]), angiogenic markers (soluble endoglin [sEng] and soluble VEGF receptor-1 [sVEGFR1]) and N-terminal brain natriuretic peptide (NT-proBNP) levels are depicted among the control population, and patients with PAH of various etiologies: idiopathic or heritable PAH (IPAH), patients with PAH associated with connective tissue disease (CTD), stimulant drug or toxin (Stim), congenital heart disease (CHD), or other forms (portopulmonary, human immunodeficiency virus, and Gaucher's disease) of PAH (*P<0.05, †P<0.01, ‡P<0.001 as compared to control group), with median, top and bottom quartile values indicated as shown.
Levels of OPG and IL-15 were elevated in APAH-CTD
Elevated OPG levels were not associated with PAH; however, among APAH-CTD patients, OPG levels were increased (3917 ± 416 pg/mL vs. 2891 ± 156 pg/mL in controls, P = 0.02). Similarly, elevated IL-15 levels were not associated with PAH, but were increased in APAH-CTD patients (3.60 ± 0.87 vs. 1.88 ± 0.13 pg/mL, P = 0.002), underscoring the known association between IL-15 and rheumatologic disorders (Fig. 1 and Table 3).[27,28] By ROC analysis, OPG and IL-15 levels were sensitive for the presence of PAH among individuals with APAH-CTD (AUCs of 0.67 ± 0.07 and 0.86 ± 0.07, respectively).
Biomarkers were not elevated in first-degree relatives of patients with IPAH or HPAH
The levels of each of the biomarkers tested were not different between the first-degree relatives of IPAH and HPAH patients and controls (Table 3).
Association of biomarker levels with functional class
Elevated NT-proBNP (P = 0.03), sEng (P = 0.02), and sVEGFR1 (P = 0.004) levels were associated with increasing NYHA Class, while OPG (Fig. 4), CRP, and IL-15 levels were not (data not shown). Levels of NT-proBNP, sEng, and sVEGFR1 were elevated among patients of NYHA Class I or II status; however, sEng was a significantly better predictor of the presence of disease among NYHA Class I-II patients than NT-proBNP by ROC analysis (AUC 0.88 ± 0.05 vs. 0.67 ± 0.08 respectively, P = 0.028). Among these markers, NT-proBNP levels also demonstrated an association with 6MWD (r = −0.37, P = 0.018).
Figure 4.
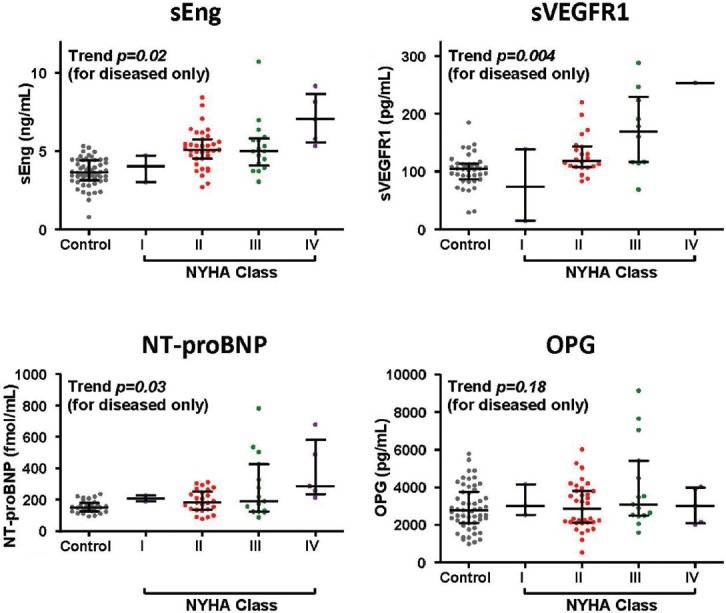
Soluble endoglin (sENG), soluble VEGF receptor-1 (sVEGFR1), and N-terminal brain natriuretic peptide (NT-proBNP) levels in pulmonary arterial hypertension (PAH) patients are associated with New York Heart Association (NYHA) Class. Levels of biomarkers are depicted for control individuals and PAH patients grouped by NYHA Class, with median, top and bottom quartile values indicated as shown. Trend analysis was performed with the one-way analysis of variance for the diseased population.
sEng correlates highly with sVEGFR1, but not NT-proBNP and CRP
Among individuals with PAH, the strongest correlation between the biomarkers existed between levels of sEng and sVEGFR1 (r = 0.39, P = 0.001, Table 4, Fig. 5). In contrast, sEng did not correlate with CRP or NT-proBNP.
Table 4.
Correlations among pulmonary arterial hypertension biomarkers
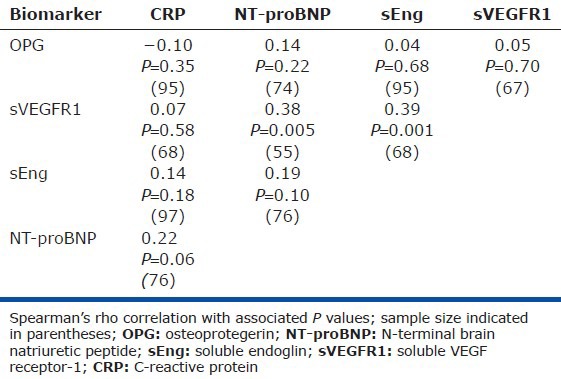
Figure 5.
Soluble endoglin (sEng) levels correlate with levels of soluble VEGF receptor-1 (sVEGFR1). (A-C) Circulating levels of sEng correlated with levels of sVEGFR1 (r=0.39, P=0.001), but did not correlate with levels of C-reactive protein (CRP) or N-terminal brain natriuretic peptide (NT-proBNP).
Predictors of survival in PAH
Median follow-up for the PAH patients was 3.2 years (interquartile range, IQR 3.0-4.2), while 5-year transplant-free survival was 73%. Of clinical and hemodynamic variables, univariate predictors of transplant-free survival included NYHA, 6MWD, right atrial pressure, cardiac index, and PVR (Table 5). Age was not a predictor of transplant-free survival in our PAH population (Cox HR 1.03 per decade, P = 0.88). Analyzed as continuous variables, biomarkers that predicted transplant-free survival included NT-proBNP, sEng, sVEGFR1, and CRP with Cox hazard ratios (HR) of 1.43 (for every 100 fmol/mL increase, 95% confidence interval [CI] 1.11-1.85, P = 0.005), 1.42 (for every 1.0 ng/mL increase, 1.08-1.87, P = 0.013), 1.14 (for every 10 pg/mL increase, 1.02-1.27, P = 0.025), and 1.11 (for every 500 ng/mL increase, 1.05-1.17, P < 0.001), respectively. Each of these biomarkers remained as significant predictors of transplant-free survival after adjusting for age and gender.
Table 5.
Univariate predictors of transplant-free survival in pulmonary arterial hypertension
Kaplan-Meier survival analysis was performed on biomarkers dichotomized about the median value (Fig. 6). Median values for NT-proBNP, sEng, sVEGFR1, and CRP were 200 fmol/mL, 4.97 ng/mL, 131 pg/mL, and 1100 ng/mL, respectively. PAH patients with an NT-proBNP ≤200 fmol/mL exhibited a 74% reduced hazard rate for death or transplant (Cox HR 0.26, 95% CI 0.08-0.82, P = 0.02) compared to those with NT-proBNP >200 fmol/mL. Similarly, PAH patients with either a sEng level ≤4.97 ng/mL, sVEGFR1 ≤131 pg/mL, or CRP level ≤1100 ng/mL exhibited improved transplant-free survival with a Cox HR of 0.33 (95% CI 0.11-1.0, P = 0.05), 0.13 (95% CI 0.02-1.0, P = 0.05), and 0.22 (95% CI 0.06-0.78, P = 0.02), respectively.
Figure 6.
Soluble endoglin (sEng), soluble VEGF receptor-1 (sVEGFR1), N-terminal brain natriuretic peptide (NT-proBNP), and C-reactive protein (CRP) predict transplant-free survival in pulmonary arterial hypertension. Kaplan–Meier survival curves are depicted for sEng, sVEGFR1, NT-proBNP, and CRP, all of which were dichotomized about their respective median value in our cohort. Median values for sEng, sVEGFR1, NT-proBNP, and CRP in this pulmonary arterial hypertension population were 4.97 ng/mL, 131 pg/mL, 200 fmol/mL, and 1100 ng/mL, respectively. Transplant-free survival curves were compared using the Log Rank test.
In multivariate Cox regression analysis, sEng and CRP were independent predictors of transplant free survival with a Cox HR of 1.40 (for every 1.0 ng/mL increase, 1.04-1.90, P = 0.028) and 1.10 (for every 500 ng/mL increase, 1.04-1.16, P < 0.001), respectively. Right atrial pressure (RAP) correlated with NT-proBNP (r = 0.42, P = 0.02) but did not correlate with any of the other biomarkers suggesting that sEng, sVEGFR1, and CRP predict prognosis in PAH independent of hemodynamic right ventricular compromise. Furthermore, after adjusting for NT-proBNP levels, sEng, sVEGFR1, and CRP each predicted transplant-free survival with Cox regression analysis.
Endoglin expression is enhanced in human endovascular lesions of PAH
Given that sEng may derive from the cleavage of membrane endoglin in endothelial cells,[29] we examined expression of endoglin in lung tissues from patients with IPAH or HPAH, and individuals without pulmonary vascular disease. Endoglin was expressed prominently in the endothelium of large, medium, and small arteries, arterioles, and venules, and to a much lesser extent in the adventitia, but not the media of the pulmonary vessels (Fig. 7A). No difference in endoglin staining between patients and controls was observed in large and medium-sized pulmonary arteries, whereas small pulmonary arteries (diameter < 40 μm) and peri-bronchiolar arteries exhibited increased endoglin expression in IPAH and HPAH (Fig. 7B and C). Most impressively, intraluminal channels of plexiform lesions exhibited intense expression of endoglin in both IPAH and HPAH lung tissues.
Figure 7.
Endoglin expression is increased in the pulmonary vascular endothelium of pulmonary arterial hypertension (PAH) patients. (A) Immunofluorescent staining of normal human lung reveals endoglin expression (green) in the endothelium of muscularized arterioles (ma) and non-MA and venules (nm), which does not overlap with expression of vascular or bronchial (br) smooth muscle α-actin depicted in red (scale bar=50 μm). (B) Endoglin (stained in brown using 3,3’-diaminobenzidine, DAB, with hematoxylin counterstain in blue) was expressed in the endothelium of large, medium and small vessels of control lungs, expressed to an equal extent in the large and medium vessels of idiopathic pulmonary arterial hypertension (IPAH) and heritable pulmonary arterial hypertension (HPAH) lungs, enhanced in the small (diameter <40 μm) vessels of IPAH and HPAH lungs, and markedly expressed in the luminal endothelium of plexiform lesions (scale bar=40 μm). (C) Endoglin expression, quantified by DAB staining intensity, was substantially increased in vessels with a diameter of <40 μm in lungs of individuals with IPAH or HPAH, as compared to vessels of the same size analyzed in lung tissues obtained from individuals with airway disease without PAH (values shown as mean±SEM, P<0.001 vs. controls).
DISCUSSION
PAH is a multifactorial disease involving abnormal vascular tone, endothelial dysfunction, inflammation, dysregulated angiogenesis, and enhanced thrombosis.[9,10] In this study, we postulated that circulating markers of angiogenic activity or inflammation might serve as sensitive markers of PAH disease severity and prognosis, and due to their ability to reflect vascular remodeling activity, perform better than conventional biomarkers. Widely used markers such as BNP and NT-proBNP correlate closely with functional status, therapeutic responses, and outcomes in PAH,[22,23] but are limited by the fact that right ventricular distention reflected by these markers occurs in relatively advanced disease. In fact, the potent anti-angiogenic molecule sEng had the best overall performance based upon ROC analysis among mildly symptomatic (NYHA Class I-II) patients, suggesting sensitivity even with modest disease burden. sEng, sVEGFR1, and NT-proBNP predicted functional class and disease severity and, along with CRP, predicted transplant-free survival in PAH by univariate analysis. After adjusting for NT-proBNP levels, sEng, sVEGFR1, and CRP each still predicted survival in PAH, indicating that these biomarkers predict prognosis in PAH above and beyond right ventricular compromise. Only sEng and CRP independently predicted survival based on multivariate analysis, however. Importantly, endoglin expression was enhanced in the microvascular endothelium of IPAH and HPAH lung tissues as compared to non-PAH control lungs, and was intensely expressed in plexiform lesions. Endothelial-derived anti-angiogenic proteins such as sEng and sVEGFR1, known to be released in the contexts of endothelial activation and vascular remodeling, appear to be more sensitive and specific in Group I PAH than previously described markers, and may represent surrogates of angiogenic remodeling activity in PAH. The elevation of anti-angiogenic markers in Group I PAH of diverse etiologies suggests dysregulated angiogenesis may be a common feature of these conditions.
Endoglin and VEGFR1 are expressed in the vascular endothelium where they transduce prosurvival and proproliferative signals required for vascular homeostasis and angiogenesis.[30,31,32] In contrast, the soluble extracellular domains of endoglin and VEGFR1 appear to exert potent antiangiogenic effects. Endoglin, induced with endothelial activation, serves as a coreceptor for bone morphogenetic protein (BMP) ligands such as BMP9 and BMP10 which promote endothelial survival and quiescence.[33,34] sEng, which blocks signaling of these ligands, potently inhibits tumor xenograft vascularization in vivo.[35] Similarly, sVEGFR1 is a truncated splice variant of VEGFR1 that inhibits angiogenesis and induces endothelial dysfunction by sequestering VEGF and placental growth factor.[32,36] Both sEng and sVEGFR1 are markedly elevated in patients with pre-ecclampsia, and in addition to being sensitive biomarkers of this disease, appear to contribute to the underlying pathophysiology.[25,37] The elevation of sEng and sVEGFR1 seen in PAH may also reflect endothelial activation and proliferation common to these two conditions. While sVEGFR1 has been found to be elevated in patients with PH associated with sickle cell disease,[38] and sEng was found to be elevated among individuals with scleroderma and PAH, we found increased levels of sEng and sVEGFR1 in diverse Group I PAH patients, including those with minimal symptoms. The observation of markedly enhanced endoglin expression in plexiform lesions, thought to be sites of dysregulated angiogenesis in PAH,[9,10] would support the interpretation that circulating sEng reflects endothelial activation, proliferation, and active pulmonary vascular remodeling. sEng may be released from sites of active remodeling by the activity of matrix metalloproteinase-14 (MMP-14), reported to regulate endoglin cleavage in carcinoma tissues.[29] We did not find elevated vascular expression of MMP-14 in normal or PAH lung tissues (data not shown), suggesting that other mechanisms might contribute. Endoglin is expressed in the failing left ventricle, and its soluble isoform is detected in the circulation after subjecting the left ventricle to pressure overload.[39] Circulating endoglin can also be detected in the context of endothelial microparticles, as others have observed in PAH.[40] Our current approach, however, did not distinguish between the expression of sEng as a secreted isoform versus proteolytic cleavage product, or as a ventricular- or endothelium-derived fragment.
In our study, levels of circulating sEng and sVEGFR1 in PAH patients were 2- to 3-fold increased as compared to controls, lower than levels reported in pre-ecclampsia.[25] If these antiangiogenic proteins derive from a pulmonary vascular bed that is obliterated due to extensive remodeling in PAH, the observed increases in these proteins may reflect proportionally greater expression by a diminished arteriolar tree. Since hypoxia itself can induce endoglin cleavage, elevated levels of endoglin associated with PAH could simply reflect hypoxemia. We did not observe increased expression of endoglin in the vessels of lungs obtained from individuals with known hypoxemic airway disease without PAH, as compared to lung tissues without airway disease or PAH, decreasing the chance of such a nonspecific finding (Fig. 7).
The performance of sEng as a biomarker might suggest a pathophysiologic role in the progression of PAH, but further work in appropriate models will be needed to ascertain whether antiangiogenic factors such as sEng are causal, protective, or secondary to pulmonary vascular remodeling. Future studies are also needed to determine whether risk stratification based on the novel antiangiogenic biomarkers should impact the therapeutic approach taken for individual patients.
Elevated OPG levels have previously been reported among individuals with IPAH,[41,42] whereas OPG was only elevated among APAH-CTD patients in our cohort. OPG levels predict mortality associated with systemic cardiovascular disease, atherosclerosis, or diabetes mellitus.[26,43,44,45,46] OPG, being a key modulator of tumor necrosis factor-α signaling and osteoclast function, may reflect states of vascular inflammation and bone remodeling, as well as vascular calcification, itself a marker of cardiovascular risk.[47,48] IL-15, an inflammatory cytokine observed to be elevated in systemic sclerosis with thoracic involvement,[27] is also thought to contribute to the pathophysiology of atherosclerosis and systemic vascular disease.[49,50,51] Since the levels of these markers were not measured in CTD patients lacking PAH, it is possible that the elevation of OPG and IL-15 among APAH-CTD patients reflects systemic inflammation associated with autoimmunity.
In contrast, CRP, an acute phase reactant and established biomarker of cardiovascular risk used in stratification of patients for coronary disease primary prevention,[52] was a sensitive marker of Group I PAH disease, consistent with previous reports.[53] While some groups have not found CRP to predict survival in PAH,[54] our study validates CRP as an important predictor of long-term prognosis in PAH. CRP and sEng were independent predictors of transplant-free survival, suggesting that these markers may reflect distinct aspects of PAH progression or severity, a notion supported by the lack of correlation between these two markers (Table 4).
None of the biomarkers assayed were significantly elevated in the relatively small cohort of asymptomatic first-degree relatives of IPAH and HPAH patients, suggesting these biomarkers may be surrogates of disease activity rather than disease potential. However, several of the biomarkers exhibited variability among controls and first-degree relatives, with CRP trending toward being elevated in first-degree relatives. Monitoring long-term outcomes in a larger number of first-degree relatives, combined with screening for genetic risk-factors such as bone morphogenetic protein type II receptor (BMPR2) mutations, could determine if these markers correlate with subsequent disease in susceptible populations.
Several factors limit the interpretation of these results. While our study contained a relatively large cohort of Group I PAH patients, conclusions about the relevance of these markers to specific etiologies of PAH or to first-degree relatives of IPAH patients are limited by the relatively small sample size for each category. Our study exhibited a higher female predominance than other PAH cohorts such as the REVEAL registry (90% vs. 79%). This relative enrichment of female patients may have been a function of the self-referral mechanism for enrollment, or due to chance. It should be noted that our PAH cohort is similar to other published populations of Group I PAH with regard to age, etiology of disease, functional status, and hemodynamic variables. Moreover, the control cohort was chosen to match the female predominance of the disease cohort (Table 1). In a small number of patients, we were able to measure most, but not all, of the biomarkers (NT-proBNP, sVEGFR1, and IL-15) due to insufficient quantities of blood. Therefore, correlations were obtained by comparing only patients for which both values were available ( ). Since the majority of patients in this study were enrolled in a nonclinical setting (Pulmonary Hypertension Association Scientific Sessions), clinical imaging, functional class, and hemodynamic assessments were obtained contemporaneously (±3 months by our definition) rather than simultaneously with blood sampling, and carried a significant missing data rate (Table 2). Given that CRP and OPG are elevated in other forms of vascular disease, further studies would be needed to control for the presence or absence of diabetes, cerebrovascular disease, and coronary artery disease as confounding variables. Our finding that sEng or sVEGFR1 are useful diagnostic and prognostic tools for PAH will need to be confirmed in validation cohorts.
In summary, this study has identified the potential role of two angiogenic modulatory proteins, sEng and sVEGFR1, as biomarkers of Group I PAH, and validated the role of CRP in the assessment and prognosis of PAH. Our findings on sEng and sVEGFR1 provide further evidence of an important role for dysregulated angiogenesis in the pathophysiology of PAH. The ability of these novel biomarkers to detect PAH even in minimally symptomatic patients and to predict long-term survival suggests that they could reflect pulmonary vascular remodeling activity at earlier stages, and perhaps be used to expedite treatment during critical periods when the outcome of the disease may be most amenable to modification.
ACKNOWLEDGMENTS
The authors thank Drs. A. Waxman and A. Wang for helpful discussions, M.L. Everett for technical assistance, and Dr. E.T.H. Yeh for generously providing facilities for portions of this work. RM, SPF, RTZ, CGE, and PBY contributed to conception, data acquisition, interpretation, and drafting of manuscript. PZ, JWS, DYD, MS, RS, CSL, WP contributed to data acquisition and revision. RM, SPF, RTZ, RNC, NWM, CGE, and PBY provided critical review and approval of final manuscript.
Footnotes
Source of Support: This work was supported by American Heart Association 11FTF7290032 (Rajeev Malhotra), the NIH (HL007208, Rajeev Malhotra; HL079943 and AR057374, Paul B. Yu), the Pulmonary Hypertension Association (Paul B. Yu), the Howard Hughes Medical Institute (Paul B. Yu), the Leducq Foundation (Paul B. Yu, Nicholas W. Morrell), Cambridge National Institute of Health Research Biomedical Research Centre (Nicholas W. Morrell), Brigham and Women’s Hospital Department of Medicine and Cardiovascular Division (Paul B. Yu) and the Vera Moulton Wall Center for Pulmonary Vascular Disease (Roham T. Zamanian)
Conflict of Interest: None declared.
REFERENCES
- 1.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Elliott CG, Barst RJ, Seeger W, Porres-Aguilar M, Brown LM, Zamanian RT, et al. Worldwide physician education and training in pulmonary hypertension: Pulmonary vascular disease: The global perspective. Chest. 2010;137:85S–94. doi: 10.1378/chest.09-2816. [DOI] [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: Results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 5.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9. doi: 10.1183/09031936.00092306. [DOI] [PubMed] [Google Scholar]
- 6.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103–10. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 7.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1998;114:225S–30. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 9.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–74. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 10.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–85. [PMC free article] [PubMed] [Google Scholar]
- 11.Heath D. Pulmonary hypertension in pulmonary parenchymal disease. Cardiovasc Clin. 1972;4:79–96. [PubMed] [Google Scholar]
- 12.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 13.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–3. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 15.ten Freyhaus H, Dumitrescu D, Berghausen E, Vantler M, Caglayan E, Rosenkranz S. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs. 2012;21:119–34. doi: 10.1517/13543784.2012.632408. [DOI] [PubMed] [Google Scholar]
- 16.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–54. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 17.Pullamsetti SS, Berghausen EM, Dabral S, Tretyn A, Butrous E, Savai R, et al. Role of Src tyrosine kinases in experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2012;32:1354–65. doi: 10.1161/ATVBAHA.112.248500. [DOI] [PubMed] [Google Scholar]
- 18.Hennigs JK, Keller G, Baumann HJ, Honecker F, Kluge S, Bokemeyer C, et al. Multi tyrosine kinase inhibitor dasatinib as novel cause of severe pre-capillary pulmonary hypertension? BMC Pulm Med. 2011;11:30. doi: 10.1186/1471-2466-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125:2128–37. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 20.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 21.Elstein D, Nir A, Klutstein M, Rudensky B, Zimran A. C-reactive protein and NT-proBNP as surrogate markers for pulmonary hypertension in Gaucher disease. Blood Cells Mol Dis. 2005;34:201–5. doi: 10.1016/j.bcmd.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Blyth KG, Groenning BA, Mark PB, Martin TN, Foster JE, Steedman T, et al. NT-pro BNP can be used to detect right ventricular systolic dysfunction in pulmonary hypertension. Eur Respir J. 2007;29:737–44. doi: 10.1183/09031936.00095606. [DOI] [PubMed] [Google Scholar]
- 23.Fijalkowska A, Kurzyna M, Torbicki A, Szewczyk G, Florczyk M, Pruszczyk P, et al. Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest. 2006;129:1313–21. doi: 10.1378/chest.129.5.1313. [DOI] [PubMed] [Google Scholar]
- 24.Heresi GA. Clinical perspective: Biomarkers in pulmonary arterial hypertension. Int J Clin Pract Suppl. 2011;169:5–7. doi: 10.1111/j.1742-1241.2010.02598.x. [DOI] [PubMed] [Google Scholar]
- 25.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–9. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 26.Rasmussen LM, Tarnow L, Hansen TK, Parving HH, Flyvbjerg A. Plasma osteoprotegerin levels are associated with glycaemic status, systolic blood pressure, kidney function and cardiovascular morbidity in type 1 diabetic patients. Eur J Endocrinol. 2006;154:75–81. doi: 10.1530/eje.1.02049. [DOI] [PubMed] [Google Scholar]
- 27.Wuttge DM, Wildt M, Geborek P, Wollheim FA, Scheja A, Akesson A. Serum IL-15 in patients with early systemic sclerosis: A potential novel marker of lung disease. Arthritis Res Ther. 2007;9:R85. doi: 10.1186/ar2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki J, Morimoto S, Amano H, Tokano Y, Takasaki Y, Hashimoto H. Serum levels of interleukin 15 in patients with rheumatic diseases. J Rheumatol. 2001;28:2389–91. [PubMed] [Google Scholar]
- 29.Hawinkels LJ, Kuiper P, Wiercinska E, Verspaget HW, Liu Z, Pardali E, et al. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010;70:4141–50. doi: 10.1158/0008-5472.CAN-09-4466. [DOI] [PubMed] [Google Scholar]
- 30.Cheifetz S, Bellón T, Calés C, Vera S, Bernabeu C, Massagué J, et al. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–30. [PubMed] [Google Scholar]
- 31.Gougos A, St Jacques S, Greaves A, O’Connell PJ, d’Apice AJ, Bühring HJ, et al. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int Immunol. 1992;4:83–92. doi: 10.1093/intimm/4.1.83. [DOI] [PubMed] [Google Scholar]
- 32.Wu FT, Stefanini MO, Mac Gabhann F, Kontos CD, Annex BH, Popel AS. A systems biology perspective on sVEGFR1: Its biological function, pathogenic role and therapeutic use. J Cell Mol Med. 2010;14:528–52. doi: 10.1111/j.1582-4934.2009.00941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López-Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2010;299:H959–74. doi: 10.1152/ajpheart.01251.2009. [DOI] [PubMed] [Google Scholar]
- 34.Toporsian M, Gros R, Kabir MG, Vera S, Govindaraju K, Eidelman DH, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res. 2005;96:684–92. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- 35.Castonguay R, Werner ED, Matthews RG, Presman E, Mulivor AW, Solban N, et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J Biol Chem. 2011;286:30034–46. doi: 10.1074/jbc.M111.260133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–83. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 37.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–58. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ataga KI, Brittain JE, Jones SK, May R, Delaney J, Strayhorn D, et al. Association of soluble fms-like tyrosine kinase-1 with pulmonary hypertension and haemolysis in sickle cell disease. Br J Haematol. 2011;152:485–91. doi: 10.1111/j.1365-2141.2010.08410.x. [DOI] [PubMed] [Google Scholar]
- 39.Kapur NK, Wilson S, Yunis AA, Qiao X, Mackey E, Paruchuri V, et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation. 2012;125:2728–38. doi: 10.1161/CIRCULATIONAHA.111.080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakouboula B, Morel O, Faure A, Zobairi F, Jesel L, Trinh A, et al. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:536–43. doi: 10.1164/rccm.200706-840OC. [DOI] [PubMed] [Google Scholar]
- 41.Lawrie A, Waterman E, Southwood M, Evans D, Suntharalingam J, Francis S, et al. Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am J Pathol. 2008;172:256–64. doi: 10.2353/ajpath.2008.070395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Condliffe R, Pickworth JA, Hopkinson K, Walker SJ, Hameed AG, Suntharaligam J, et al. Serum osteoprotegerin is increased and predicts survival in idiopathic pulmonary arterial hypertension. Pulm Circ. 2012;2:21–7. doi: 10.4103/2045-8932.94819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guldiken B, Guldiken S, Turgut B, Turgut N, Demir M, Celik Y, et al. Serum osteoprotegerin levels in patients with acute atherothrombotic stroke and lacunar infarct. Thromb Res. 2007;120:511–6. doi: 10.1016/j.thromres.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 44.Jono S, Ikari Y, Shioi A, Mori K, Miki T, Hara K, et al. Serum osteoprotegerin levels are associated with the presence and severity of coronary artery disease. Circulation. 2002;106:1192–4. doi: 10.1161/01.cir.0000031524.49139.29. [DOI] [PubMed] [Google Scholar]
- 45.Ueland T, Jemtland R, Godang K, Kjekshus J, Hognestad A, Omland T, et al. Prognostic value of osteoprotegerin in heart failure after acute myocardial infarction. J Am Coll Cardiol. 2004;44:1970–6. doi: 10.1016/j.jacc.2004.06.076. [DOI] [PubMed] [Google Scholar]
- 46.Dhore CR, Cleutjens JP, Lutgens E, Cleutjens KB, Geusens PP, Kitslaar PJ, et al. Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2001;21:1998–2003. doi: 10.1161/hq1201.100229. [DOI] [PubMed] [Google Scholar]
- 47.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 48.Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15:457–75. doi: 10.1016/j.cytogfr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Angiolillo AL, Kanegane H, Sgadari C, Reaman GH, Tosato G. Interleukin-15 promotes angiogenesis in vivo. Biochem Biophys Res Commun. 1997;233:231–7. doi: 10.1006/bbrc.1997.6435. [DOI] [PubMed] [Google Scholar]
- 50.Kaibe M, Ohishi M, Ito N, Yuan M, Takagi T, Terai M, et al. Serum interleukin-15 concentration in patients with essential hypertension. Am J Hypertens. 2005;18:1019–25. doi: 10.1016/j.amjhyper.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 51.Wuttge DM, Eriksson P, Sirsjö A, Hansson GK, Stemme S. Expression of interleukin-15 in mouse and human atherosclerotic lesions. Am J Pathol. 2001;159:417–23. doi: 10.1016/S0002-9440(10)61712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lloyd-Jones DM, Liu K, Tian L, Greenland P. Narrative review: Assessment of C-reactive protein in risk prediction for cardiovascular disease. Ann Intern Med. 2006;145:35–42. doi: 10.7326/0003-4819-145-1-200607040-00129. [DOI] [PubMed] [Google Scholar]
- 53.Quarck R, Nawrot T, Meyns B, Delcroix M. C-reactive protein: A new predictor of adverse outcome in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;53:1211–8. doi: 10.1016/j.jacc.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 54.Heresi GA, Tang WH, Aytekin M, Hammel J, Hazen SL, Dweik RA. Sensitive cardiac troponin I predicts poor outcomes in pulmonary arterial hypertension. Eur Respir J. 2012;39:939–44. doi: 10.1183/09031936.00067011. [DOI] [PubMed] [Google Scholar]