

Abstract
Pulmonary endothelium is a major metabolic organ affecting pulmonary and systemic vascular homeostasis. Brain death (BD)-induced physiologic and metabolic derangements in donors’ lungs, in the absence of overt lung pathology, may cause pulmonary dysfunction and compromise post-transplant graft function. To explore the impact of BD on pulmonary endothelium, we estimated pulmonary capillary endothelium-bound (PCEB)-angiotensin converting enzyme (ACE) activity, a direct and quantifiable index of pulmonary endothelial function, in eight brain-dead patients and ten brain-injured mechanically ventilated controls. No subject suffered from acute lung injury or any other overt lung pathology. Applying indicator-dilution type techniques, we measured single-pass transpulmonary percent metabolism (%M) and hydrolysis (v) of the synthetic, biologically inactive, and highly specific for ACE substrate 3H-benzoyl-Phe-Ala-Pro, under first order reaction conditions, and calculated lung functional capillary surface area (FCSA). Substrate %M (35 ± 6.8%) and v (0.49 ± 0.13) in BD patients were decreased as compared to controls (55.9 ± 4.9, P = 0.033 and 0.9 ± 0.15, P = 0.033, respectively), denoting decreased pulmonary endothelial enzyme activity at the capillary level; FCSA, a reflection of endothelial enzyme activity per vascular bed, was also decreased (BD patients: 1,563 ± 562 mL/min vs 4,235 ± 559 in controls; P = 0.003). We conclude that BD is associated with subtle pulmonary endothelial injury, expressed by decreased PCEB-ACE activity. The applied indicator-dilution type technique provides direct and quantifiable indices of pulmonary endothelial function at the bedside that may reveal the existence of preclinical lung pathology in potential lung donors.
Keywords: angiotensin converting enzyme, brain death, pulmonary endothelium
Lung transplantation is often the only available treatment option for patients with end-stage vascular and other lung disease. However, despite the advances in surgical techniques and pharmacologic management, a significant proportion of patients do not benefit from transplantation, due to severe early allograft dysfunction; this may account for the death of 20% of recipients in the first few weeks after transplantation.[1] Although several factors may contribute to the adverse prognosis of lung transplant recipients, there is strong evidence that preclinical lung injury is already present in donor lungs before their retrieval.[2] Potential lung donors are generally patients admitted in the Intensive Care Unit (ICU), who progress to brain death (BD) following irreversible cessation of brainstem function;[3,4] such patients are considered at high risk for development of lung injury due to trauma, mechanical ventilation, aspiration, or infection. Additionally, the process of BD itself can damage the lung directly and jeopardize its function post-transplantation.[5]
BD may cause pulmonary dysfunction secondary to α-adrenergic stimulation and hemodynamic derangements of the pulmonary capillaries.[4,6] Evidence also suggests that BD results in a systemic inflammatory response by the release of potent proinflammatory mediators into the systemic circulation[7] that could induce preclinical lung injury and undermine graft survival.[2] However, human studies have thus far focused on the alveolar epithelium and capillary barrier function; direct in vivo evidence on the contribution of pulmonary endothelium in such a BD-induced subtle lung injury is still missing.[3,6]
Pulmonary endothelium (PE) is a major metabolic organ that warrants the maintenance of systemic and pulmonary circulation homeostasis.[8,9] PE may be affected by either the BD-induced inflammatory response and/or the above mentioned hemodynamic perturbations and shear stress; the latter have been shown to upregulate various endothelial inflammatory pathways,[6] including reactive oxygen species generation, nuclear factor-κB (NF-κB) activation, and upregulation of adhesion molecules and pro- or anti-inflammatory cytokines.[10,11,12,13]
To investigate the role of BD as a factor causing preclinical lung injury, we estimated pulmonary endothelial function in BD subjects. We hypothesized that BD may induce pulmonary endothelial dysfunction, denoted by pulmonary endothelial angiotensin converting enzyme (ACE) activity reduction, as a result of the BD-triggered inflammatory response. To this end, we compared pulmonary capillary endothelium-bound-ACE (PCEB-ACE) activity and plasma inflammatory mediator levels in BD patients and brain-injured mechanically ventilated controls. ACE is expressed as an ectoenzyme on the PE surface, and PCEB-ACE activity may be measured by means of indicator dilution techniques that allow quantifiable assessments of (1) the enzyme activity at the capillary endothelial level and (2) the functional capillary surface area (FCSA) which is available for reaction.[14,15,16,17] Early PCEB-ACE activity reduction has been documented in various animal models of acute lung injury (ALI) as well as in patients with ALI and acute respiratory distress syndrome (ARDS).[9,16,18,19] In this study, we found that PCEB-ACE activity in BD patients with no evidence of ALI or other overt lung pathology was reduced compared to mechanically ventilated brain-injured patients with functioning brainstem.
MATERIALS AND METHODS
Study population
The study was conducted in compliance with the Declaration of Helsinki and its protocol was reviewed and approved by our Institutional Ethics Committee. Informed written consent was obtained from subjects’ next of kin. Eighteen patients were enrolled in the study; they were all hospitalized in a mixed (i.e., medical and surgical) ICU of a general hospital. All patients had catheters placed in either the subclavian or the internal jugular vein and in the radial artery, as part of their routine treatment. Eight patients had developed BD (BD group), and ten patients who suffered from brain trauma or injury but never developed BD served as controls. Patients’ traumatic or medical injuries were diagnosed by neurologists and/or neurosurgeons based on computerized tomographies of the brain. No subject had thoracic or lung trauma, ALI, or any other overt lung pathology. Descriptive data consisting of demographics, diagnosis, clinical and laboratory data, and lung injury score (LIS)[20] were recorded. Chest X-ray (CXR) score, a LIS component, was independently measured. CXR score ranges from 0 to 4, depending on the absence (0) or presence of alveolar consolidations confined to one (1) up to all four lung quadrants (4).[20] CXR scoring was performed by two “blind” nonstudy-related intensivists. Most BD subjects exhibited mild elevations of aspartate aminotransferase (AST), and two exhibited mild elevations of alanine aminotransferase (ALT) in serum; no BD patient exhibited elevated circulating bilirubin or creatine levels. Thus no BD subject suffered from overt liver or renal failure.
BD diagnosis had been confirmed when an irreversible catastrophic structural brain lesion resulted in unresponsiveness to noxious pain stimuli and to abolition of brainstem reflexes (papillary light responses, corneal reflexes, vestibulo-ocular tests, tracheobronchial stimulation) in the absence of hypothermia, metabolic or electrolyte disturbances, and depressant drugs. Testing for apnea was performed twice, with 24 hours in between, using previously described guidelines after all other prespecified brain-death criteria had been fulfilled.[21] Patients were announced brain dead by a medical team that included a neurologist or a neurosurgeon, an anesthetist, and the treating attending intensivist, in compliance with Greek regulations.
Laboratory measurements
Immediately prior to PCEB-ACE activity measurements, venous blood was obtained by venous puncture into EDTA-containing tubes and was immediately placed on ice before centrifugation at 1000 × g at 4°C for 20 minutes. Plasma samples were then aliquoted and stored at -80°C until processed. Tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and -8 concentrations were measured in duplicate by flow cytometry using cytometric bead array (CBA) technology after staining with monoclonal antibodies and passage through a FACSCalibur flow cytometric device (Becton Dickinson, Cockeysville, Md., USA). The lowest limits of detection were as follows: 1.1 pg/mL for TNF-α; 1.7 pg/mL for IL-6; and 2.5 pg/mL for IL-8.[22] C-reactive protein (CRP) was measured using an immunoturbometric assay (Tina-quart C-reactive protein, Roche Diagnostics GmbH, Mannheim, Germany). S-100b protein was measured by an immunoluminometric technique with a commercially available kit (LIA-mat Sangtec 100, AB Sangtec Medical, Bromma, Sweden).[23] Procalcitonin (PCT) levels were determined by means of a specific and ultrasensitive immunoluminometric assay (Liaison Brahms procalcitonin, Diagnostica, Berlin, Germany).
Determination of PCEB-ACE activity
Determination of PCEB-ACE activity in BD subjects and controls was performed by means of indicator-dilution type techniques that have been described in detail elsewhere.[15] We estimated the single pass transpulmonary utilization of the radiolabelled synthetic, hemodynamically inactive, and specific for ACE substrate 3H-benzoyl-Phe-Ala-Pro (3H-BPAP) by PCEB-ACE under first-order reaction conditions and calculated related kinetic parameters. Briefly, a 1.3 mL bolus of normal saline solution containing 17 μCi of 3H-BPAP; (22.2 Ci/mmole) was injected through the distal port of a central vein catheter and was distributed through the pulmonary vascular bed. Simultaneously, effluent arterial blood was withdrawn via a radial artery catheter by means of a peristaltic pump into a fraction-collector (1.2 mL blood/tube, 16 tubes). Blood was collected into 1.75 mL of normal saline containing 5 mM EDTA and 6.8 mM 8-hydroxyquinoline 5-sulfonic acid to prevent further activity of ACE in blood, and heparin 1,000 IU/L (“stop” solution). Four additional tubes containing 1.75 mL of “stop” solution, 1.2 mL of blood withdrawn before isotope injection, and 0.02 mL of the isotope mixture were used to calculate the amount of administered radioactivity.
After centrifugation of the blood samples (3,000 revolutions/minute for 10 minutes), 0.5 mL of the supernatant was transferred into a scintillation vial, and total 3H radioactivity was measured in 5 mL Ecoscint (National Diagnostics, Atlanta, Ga., USA). For determination of the radioactivity associated with metabolites, another 0.5 mL of the supernatant was transferred into a separate vial containing 2.5 mL HCL (0.12 N). Three mL of Toluene Scintillator (Packard, Meriden, Ct., USA) was added, samples were mixed, and radioactivity was measured 48 hours later. In this way, ~70% of the 3H BPAP metabolite 3H-benzoyl-Phe and < 10% of the parent 3H-BPAP were extracted in the organic phase of the mixture (toluene). The precise values were calculated by identically processing separate tubes containing substrate or previously synthesized product.[15,16]
Estimates of PCEB-ACE activity included single pass 3H-BPAP percent metabolism (%M), transpulmonary hydrolysis (v), and functional capillary surface area (FCSA), originally termed Amax/Km.[15]
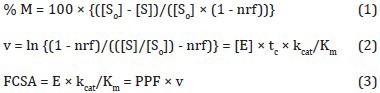
with [So] and [S] being the initial and final substrate concentrations, respectively, in the effluent arterial plasma in dpm/mL, nrf being the nonreactive fraction of cis-BPAP (7%), and [E], tc, kcat, Km, E and PPF being the enzyme concentration available for reaction, capillary transit time (i.e., enzyme-substrate reaction time), catalytic rate constant, Henri-Michaelis-Menten constant, total enzyme mass available for reaction, and pulmonary plasma flow, respectively.[15,17,18] Cardiac output (CO) was calculated as previously described, and PPF was calculated as = CO × (1 - Hematocrit).[18] Substrate hydrolysis (v) and %M reflect ACE activity per capillary, whereas FCSA (Amax/Km) reflects ACE activity per vascular bed.[17,18,19]
Statistical analysis
Group data are presented as mean ± SEM. For normally distributed data the Student's t-test was used. For non-normally distributed data, comparisons were performed using the Mann-Whitney U-test. Fisher's exact test was used for gender comparison. Spearman's rank correlation coefficients (rs) were calculated to describe the relationships between the quantitative variables. In all analyses, two-tailed P-values < 0.05 were considered significant.
RESULTS
Patients’ characteristics
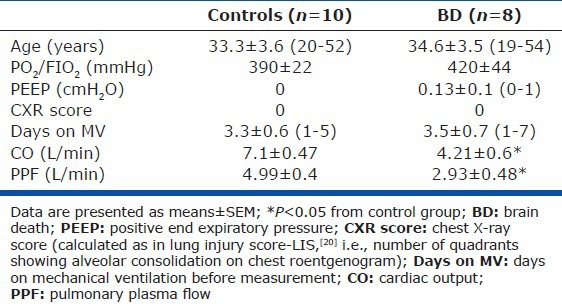
Patient demographics, as well as clinical and laboratory data obtained on the day of PCEB-ACE activity determination, are shown in Table 1. Eight patients (six men) had developed BD and 10 patients (nine men) served as controls. All 18 patients had been mechanically ventilated for similar time periods, and with the same mode (assist control ventilation) and tidal volumes (8 mL/Kg). The diagnoses of BD were: Spontaneous (n = 2) or traumatic (n = 3) subarachnoid hemorrhage and spontaneous (n = 2) or traumatic (n = 1) intracerebral hemorrhage. Controls suffered brain trauma without deterioration to BD. There were no statistical differences between the groups in sex, age, or time on mechanical ventilation. No patient suffered from ALI or other overt lung pathology as denoted by PO2/FIO2 values and their chest X-rays (Table 1).
Table 1.
Demographic and clinical data of the study patients
BD induces pulmonary endothelial ACE activity reduction
ACE substrate utilization was reduced in BD patients: BPAP %M (35 ± 6.8%) and v (0.49 ± 0.13) were decreased in BD subjects, as compared to controls (55.9 ± 4.9, P = 0.033 and 0.9 ± 0.15, P = 0.033, respectively; Figures 1A and B).
Figure 1.
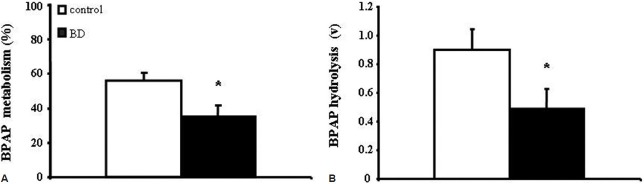
Brain death is associated with pulmonary endothelial ACE activity reduction, as compared to brain-injured controls. Substrate (BPAP) percent metabolism (%M; Panel A) and transpulmonary hydrolysis (v; Panel B), both reflecting enzyme activity per capillary, were decreased in BD subjects (n= 8), as compared to controls (n= 10). Data are presented as means ± SEM, *P< 0.05 from controls
Functional capillary surface area is decreased in BD patients
FCSA available for BPAP hydrolysis was decreased in BD patients (1,563 ± 562 mL/min) as compared to controls (4,235 ± 559 mL/min, P = 0.003; Fig. 2).
Figure 2.
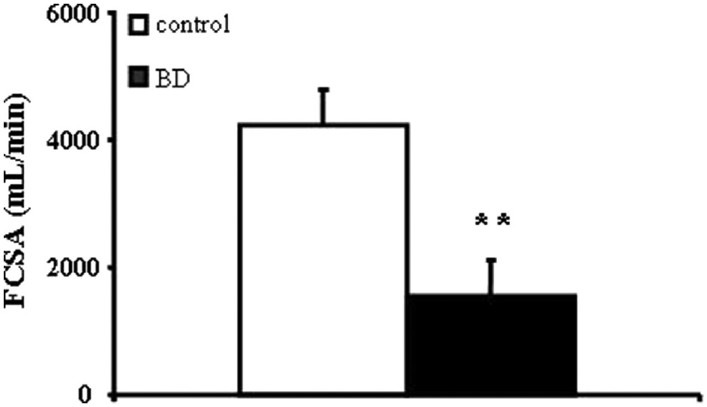
Functional capillary surface area (FCSA), a reflection of enzyme activity per vascular bed, is decreased in BD patients (n= 8) as compared to brain-injured controls (n= 10). Data are presented as means ± SEM, **P< 0.01 from controls
BD is associated with increased circulating inflammatory parameters
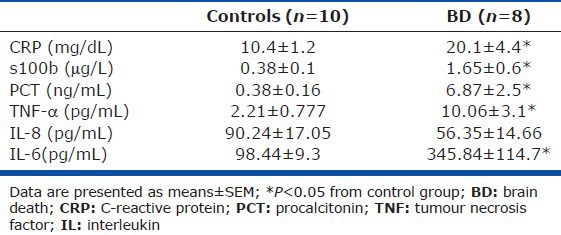
Circulating CRP, S-100b, and PCT were higher in plasma of BD subjects as compared to controls (Table 2). In addition, plasma levels of cytokines TNF-α and IL-6 were increased in BD as compared to controls, while no difference was noted in plasma IL-8 levels between the two groups (Table 2). In our population, neither substrate %M or v, nor FCSA were correlated with any of the aforementioned inflammatory indices.
Table 2.
Laboratory data of the study patients
DISCUSSION
Lung or lung-heart transplantation may be the life-saving intervention for patients with end-stage lung disease, including subjects with pulmonary arterial hypertension that deteriorate despite adequate specific treatment. Severe early allograft dysfunction is a major complication that may be related, among others, to existing preclinical injury in the donor lung. In that respect, there is evidence that lung injury sustained very early following BD may compromise post-transplant graft function.[2] It is thus necessary to further improve our means of detecting the presence of such pathology. To the best of our knowledge, this study provides the first direct in vivo evidence, obtained at patients’ bedsides, that pulmonary endothelial dysfunction is present in BD subjects with no overt lung pathology.
Endothelial integrity in peripheral organs of brain-dead donors has more recently gained considerable attention in relation to potential harmful effects of BD on donor organ quality. Animal and human studies have shown that under conditions of hemodynamic instability, such as the ones that follow BD, vascular endothelial cells are susceptible to shifts in mechanical forces and shear stress,[24] or systemic inflammatory insults.[25] Endothelial dysfunction is among the earliest features occurring under and contributing to lung injury pathogenesis, as evidenced among others by the altered expression of circulating endothelial-specific proteins, soluble adhesion molecules, and endothelial barrier alterations.[8,9] It should be noted, however, that the aforementioned indices of pulmonary endothelial injury are either surrogate or have been estimated in ex vivo systems. In contrast, estimating PCEB-ACE activity by means of indicator-dilution type technique provides a direct, sensitive, and quantifiable means of assessing pulmonary endothelial function at the bedside in humans.[8,9,14,15,16]
PCEB-ACE is an ectoenzyme uniformly distributed along the luminar pulmonary endothelial surface with its catalytic site exposed to the blood stream, thus allowing interactions with bloodborne substrates and inhibitors without requiring the time and energy expense that would be needed by interactions with a cytosolic enzyme. Due to the very high concentrations in the capillaries, estimations of pulmonary endothelial ACE activity by the indicator-dilution technique is in practical terms equal to monitoring PCEB-ACE activity;[9] it has been additionally shown that the contribution of plasma ACE to the single pass transpulmonary utilization of the substrate is minimal and that PCEB-ACE from alveolar capillaries with a diameter less than 20 μm are primarily responsible for the majority of the hydrolysis.[26]
This method may additionally distinguish between abnormalities secondary to endothelial dysfunction per se (expressed by reduced %M and v) and decreased functional capillary surface area.[15,17] PCEB-ACE activity reduction has been among the earliest signs of ALI in animal models, preceding acid-base imbalance, gas exchange and hemodynamic abnormalities, barrier dysfunction, and morphologic changes at the light and electron microscopy level.[9] Assessment of PCEB-ACE activity in critically ill patients revealed that both transpulmonary hydrolysis and FCSA (alias Amax/Km) decrease early during the ALI/ARDS continuum,[16] while PCEB-ACE activity is also reduced in humans with pulmonary hypertension of diverse etiologies.[14,17]
In the present study and in the absence of ALI or other apparent lung pathology, we detected significant reductions of both substrate %M and v in BD subjects as compared to brain-injured controls (Figures 1A and B). These data reflect reduced enzyme activity per capillary and may be related to (1) capillary endothelial dysfunction associated with alterations of either the PCEB-ACE microvascular concentration, the enzyme kinetic constants, or both (Equation 2); and (2) a decrease in capillary transit time (Equation 2). However, pulmonary capillary flow does not appear to change over a wide CO range,[14,17,27,28] thus resulting in unchanged transit times. In addition, our BD patients had lower CO than the controls (Table 1) implying that, if anything, capillary flows would decrease resulting in higher capillary transit times and thus higher substrate utilization as per Equation 2 (i.e., the opposite rather than the observed phenomenon). Thus, our BD patients exhibit true pulmonary capillary endothelial dysfunction, as compared to the brain-injured controls.
In addition, BD subjects exhibited significant decreases in FCSA as compared to brain-injured controls (Fig. 2). FCSA is proportional to the enzyme mass available for reaction (perfused capillary surface bed multiplied by the enzyme mass expressed on the endothelial surface) and the enzyme kinetic constants (Equation 3). Taken thus together, the observed decreases in substrate utilization and in FCSA, the latter should be related to both enzyme mass and/or kinetic constant reductions (capillary endothelial dysfunction), and to the lower CO and pulmonary plasma flows observed in the BD patients (Equation 3).
Could the decreased CO values observed in BD subjects, per se, cause the decreased enzyme activity parameters as compared to controls? As previously analyzed, this should indeed be partly the case for the lower FCSA observed in our BD subjects. We have, however, shown that substrate hydrolysis by PCEB-ACE in humans and animals does not change over a wide range of pulmonary blood flows.[14,28] Capillary transit times could be increased under extremely low capillary flows,[27] but such a phenomenon would have produced instead increased v and %M. Thus a true, non pulmonary blood flow-related, reduction in PCEB-ACE activity appears to be present in our BD patients as compared to brain-injured controls.
An additional important issue is related to the potential systemic effects of low CO and if such effects could indirectly affect PCEB-ACE activity. In this respect, low CO is expected to lead to increased circulating natural ACE substrate angiotensin I through induction of the rennin angiotensin system (RAS). If such an increase could lead to pulmonary ACE saturation, the estimated PCEB-ACE activity would be expected to decrease. It has, however, been shown that circulating angiotensin I concentrations are significantly lower than the Km of its interaction with ACE (~0.1 nM versus ~33 μM, respectively)[29] meaning that PCEB-ACE saturation by angiotensin I is not possible. In a similar respect, an effect of the low CO via the inflammatory milieu cannot be excluded. It should be noted, however, that patients suffering from idiopathic pulmonary arterial hypertension exhibiting long-term low CO (lower than those observed in our BD group) did not show decreases in either BPAP %M or v compared with related controls despite the significantly higher CO of the latter,[14] thus making such a possibility rather unlikely.
Several reports link systemic inflammation to pulmonary endothelial dysfunction.[8] In an effort to investigate if the observed PCEB-ACE activity reductions coexist with the presence of systemic inflammation, we additionally measured circulating inflammatory and brain injury-related biomarkers. A more prominent inflammatory process was detected in BD patients as compared to controls (Table 2); however, no significant relationship was observed among any inflammatory compound measured and the pulmonary endothelial ACE activity indices. Circulating CRP, PCT, and S100b in BD were all higher compared to controls (Table 2). Serum PCT levels have been found increased after BD, predicting early graft failure, especially in cardiac donors.[30] Yet, a relationship of the above phenomenon to infection or systemic inflammatory changes in the brain-dead donor has not been established.[31] The higher levels of the brain-specific marker S-100b observed in our BD patients reflect the presence and the severity of cerebral damage,[32,33] but might also denote the major inflammatory impact of BD on peripheral organ dysfunction.[34,35,36]
Most experimental and clinical studies confirm the presence of increased circulating proinflammatory cytokines shortly after BD occurs[25,33,37,38,39,40] due to brain tissue ischemia itself and/or to α-adrenergic derangements.[25,41] A massive inflammatory response is triggered following BD, characterized among others by increased serum levels of proinflammatory cytokines such as IL-6 and TNF-α as well as by upregulation of their receptors in peripheral organs; these phenomena appear associated with inferior donor organ viability.[34,39,42,43,44,45] TNF-α and IL-6 participate in the early inflammatory response following BD by activating, among others, endothelial cells to express adhesion molecules (intercellular adhesion molecule-1 and E- and P-selectins);[25] they additionally regulate the production of the potent neutrophil activator and chemoattractant IL-8 by endothelial and epithelial cells of the capillary alveolar membrane.[6,36,46,47] Accordingly, our BD subjects exhibited increased plasma levels of both IL-6 and TNF-α (Table 2). However, despite the well-established effects of IL-6 and TNF-α on the pulmonary endothelium,[8,9] our study does not provide direct evidence on a cause-effect phenomenon between these cytokines and the observed PCEB-ACE activity reduction in the BD patients.
Neutrophil infiltration in apparently healthy lungs of brain-dead donors has been shown to correlate with IL-8 levels in patients’ bronchoalveolar lavage fluid (BALF), while elevated lung IL-8 expression was associated with graft failure post-transplantation.[38] In contrast with the aforementioned seminal investigation, in our cohort lung and BALF IL-8 levels were not measured, not allowing for direct comparisons. In our study, no significant differences in plasma IL-8 levels were noted between BD patients and brain-injured controls, implying that this chemokine might not have contributed to the observed reductions seen in PCEB-ACE activity in the former.
In summary, this is the first study to demonstrate that subtle pulmonary endothelial dysfunction, as assessed by PCEB-ACE activity reduction, is present in BD patients in the absence of ALI or other overt lung pathology. Assessing pulmonary endothelial ACE activity at the bedside by means of indicator-dilution type techniques provides a direct and quantifiable index of pulmonary endothelial dysfunction that may reveal the existence of preclinical lung pathology in potential BD lung donors. Future studies should investigate the impact of such subtle pulmonary endothelial dysfunction in the outcome of lung transplantation.
ACKNOWLEDGMENTS
The authors would like to thank Dr. P. Kopterides for proof editing the manuscript. Drs. Constantinos Glynos and Chariclea Athanasiou contributed equally to the study and are considered co-first authors.
Footnotes
Source of Support: This study was supported by the Thorax Foundation.
Conflict of Interest: None declared.
REFERENCES
- 1.Hosenpud JD, Bennett LE, Keck BM, Fiol B, Novick RJ. The Registry of the International Society for Heart and Lung Transplantation: Fourteenth official report--1997. J Heart Lung Transplant. 1997;16:691–712. [PubMed] [Google Scholar]
- 2.Fisher AJ, Donnelly SC, Hirani N, Burdick MD, Strieter RM, Dark JH, et al. Enhanced pulmonary inflammation in organ donors following fatal non-traumatic brain injury. Lancet. 1999;353:1412–3. doi: 10.1016/S0140-6736(99)00494-8. [DOI] [PubMed] [Google Scholar]
- 3.Cooper DK, Basker M. Physiologic changes following brain death. Transplant Proc. 1999;31:1001–2. doi: 10.1016/s0041-1345(98)01876-4. [DOI] [PubMed] [Google Scholar]
- 4.Pratschke J, Wilhelm MJ, Kusaka M, Basker M, Cooper DK, Hancock WW, et al. Brain death and its influence on donor organ quality and outcome after transplantation. Transplantation. 1999;67:343–8. doi: 10.1097/00007890-199902150-00001. [DOI] [PubMed] [Google Scholar]
- 5.Fisher AJ, Dark JH, Corris PA. Improving donor lung evaluation: A new approach to increase organ supply for lung transplantation. Thorax. 1998;53:818–20. doi: 10.1136/thx.53.10.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avlonitis VS, Fisher AJ, Kirby JA, Dark JH. Pulmonary transplantation: The role of brain death in donor lung injury. Transplantation. 2003;75:1928–33. doi: 10.1097/01.TP.0000066351.87480.9E. [DOI] [PubMed] [Google Scholar]
- 7.McKeating EG, Andrews PJ, Signorini DF, Mascia L. Transcranial cytokine gradients in patients requiring intensive care after acute brain injury. Br J Anaesth. 1997;78:520–3. doi: 10.1093/bja/78.5.520. [DOI] [PubMed] [Google Scholar]
- 8.Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49:119–33. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: From basic science to the critically ill. Intensive Care Med. 2004;30:1702–14. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- 10.Bao X, Lu C, Frangos JA. Temporal gradient in shear but not steady shear stress induces PDGF-A and MCP-1 expression in endothelial cells: Role of NO, NF kappa B, and egr-1. Arterioscler Thromb Vasc Biol. 1999;19:996–1003. doi: 10.1161/01.atv.19.4.996. [DOI] [PubMed] [Google Scholar]
- 11.Chiu JJ, Wung BS, Shyy JY, Hsieh HJ, Wang DL. Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arterioscler Thromb Vasc Biol. 1997;17:3570–7. doi: 10.1161/01.atv.17.12.3570. [DOI] [PubMed] [Google Scholar]
- 12.Chiu JJ, Wang DL, Chien S, Skalak R, Usami S. Effects of disturbed flow on endothelial cells. J Biomech Eng. 1998;120:2–8. doi: 10.1115/1.2834303. [DOI] [PubMed] [Google Scholar]
- 13.Yoshisue H, Suzuki K, Kawabata A, Ohya T, Zhao H, Sakurada K, et al. Large scale isolation of non-uniform shear stress-responsive genes from cultured human endothelial cells through the preparation of a subtracted cDNA library. Atherosclerosis. 2002;162:323–34. doi: 10.1016/s0021-9150(01)00735-3. [DOI] [PubMed] [Google Scholar]
- 14.Langleben D, Orfanos SE, Giovinazzo M, Hirsch A, Baron M, Senécal JL, et al. Pulmonary capillary endothelial metabolic dysfunction: Severity in pulmonary arterial hypertension related to connective tissue disease versus idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2008;58:1156–64. doi: 10.1002/art.23405. [DOI] [PubMed] [Google Scholar]
- 15.Orfanos SE, Langleben D, Khoury J, Schlesinger RD, Dragatakis L, Roussos C, et al. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in humans. Circulation. 1999;99:1593–9. doi: 10.1161/01.cir.99.12.1593. [DOI] [PubMed] [Google Scholar]
- 16.Orfanos SE, Armaganidis A, Glynos C, Psevdi E, Kaltsas P, Sarafidou P, et al. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation. 2000;102:2011–8. doi: 10.1161/01.cir.102.16.2011. [DOI] [PubMed] [Google Scholar]
- 17.Orfanos SE, Hirsch AM, Giovinazzo M, Armaganidis A, Catravas JD, Langleben D. Pulmonary capillary endothelial metabolic function in chronic thromboembolic pulmonary hypertension. J Thromb Haemost. 2008;6:1275–80. doi: 10.1111/j.1538-7836.2008.03046.x. [DOI] [PubMed] [Google Scholar]
- 18.Catravas JD, White RE. Kinetics of pulmonary angiotensin-converting enzyme and 5’-nucleotidase in vivo. J Appl Physiol. 1984;57:1173–81. doi: 10.1152/jappl.1984.57.4.1173. [DOI] [PubMed] [Google Scholar]
- 19.Douzinas EE, Orfanos SE, Livaditi O, Augustatou K, Villiotou V, Kavantzas N, et al. Hypoxemic resuscitation prevents pulmonary capillary endothelial dysfunction induced by normoxemic resuscitation from hemorrhagic shock. Crit Care Med. 2009;37:869–75. doi: 10.1097/CCM.0b013e31819b81ec. [DOI] [PubMed] [Google Scholar]
- 20.Murray JF, Matthay MA, Luce JM, Flick MR. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:720–3. doi: 10.1164/ajrccm/138.3.720. [DOI] [PubMed] [Google Scholar]
- 21.Wijdicks EF. The diagnosis of brain death. N Engl J Med. 2001;344:1215–21. doi: 10.1056/NEJM200104193441606. [DOI] [PubMed] [Google Scholar]
- 22.Livaditi O, Kotanidou A, Psarra A, Dimopoulou I, Sotiropoulou C, Augustatou K, et al. Neutrophil CD64 expression and serum IL-8: Sensitive early markers of severity and outcome in sepsis. Cytokine. 2006;36:283–90. doi: 10.1016/j.cyto.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Dimopoulou I, Tsagarakis S, Korfias S, Zervakis D, Douka E, Thalassinos N, et al. Relationship of thyroid function to post-traumatic S-100b serum levels in survivors of severe head injury: Preliminary results. Intensive Care Med. 2004;30:298–301. doi: 10.1007/s00134-003-2058-7. [DOI] [PubMed] [Google Scholar]
- 24.Ali MH, Schumacker PT. Endothelial responses to mechanical stress: Where is the mechanosensor? Crit Care Med. 2002;30(5 Suppl):S198–206. doi: 10.1097/00003246-200205001-00005. [DOI] [PubMed] [Google Scholar]
- 25.Barklin A. Systemic inflammation in the brain-dead organ donor. Acta Anaesthesiol Scand. 2009;53:425–35. doi: 10.1111/j.1399-6576.2008.01879.x. [DOI] [PubMed] [Google Scholar]
- 26.Catravas JD, Orfanos SE. Pathophysiologic functions of endothelial angiotensin-converting enzyme. In: Born GVR, Schwartz CJ, editors. Vascular Endothelium. Physiology, Pathology and Therapeutic Opportunities. Stuttgart: Schattauer; 1997. pp. 193–204. [Google Scholar]
- 27.Orfanos SE, Chen XL, Ryan JW, Chung AY, Burch SE, Catravas JD. Assay of pulmonary microvascular endothelial angiotensin-converting enzyme in vivo: Comparison of three probes. Toxicol Appl Pharmacol. 1994;124:99–111. doi: 10.1006/taap.1994.1013. [DOI] [PubMed] [Google Scholar]
- 28.Orfanos SE, Ehrhart IC, Barman S, Hofman WF, Catravas JD. Endothelial ectoenzyme assays estimate perfused capillary surface area in the dog lung. Microvasc Res. 1997;54:145–55. doi: 10.1006/mvre.1997.2031. [DOI] [PubMed] [Google Scholar]
- 29.Ryan JW. Processing of endogenous polypeptides by the lungs. Annu Rev Physiol. 1982;44:241–55. doi: 10.1146/annurev.ph.44.030182.001325. [DOI] [PubMed] [Google Scholar]
- 30.Wagner FD, Jonitz B, Potapov EV, Qedra N, Wegscheider K, Abraham K, et al. Procalcitonin, a donor-specific predictor of early graft failure-related mortality after heart transplantation. Circulation. 2001;104(12 Suppl 1):I192–6. doi: 10.1161/hc37t1.094836. [DOI] [PubMed] [Google Scholar]
- 31.Rangeard O, Audibert G, Perrier JF, Loos-Ayav C, Lalot JM, Agavriloaie M, et al. Relationship between procalcitonin values and infection in brain-dead organ donors. Transplant Proc. 2007;39:2970–4. doi: 10.1016/j.transproceed.2007.02.101. [DOI] [PubMed] [Google Scholar]
- 32.Ingebrigtsen T, Romner B. Biochemical serum markers of traumatic brain injury. J Trauma. 2002;52:798–808. doi: 10.1097/00005373-200204000-00038. [DOI] [PubMed] [Google Scholar]
- 33.Townend W, Ingebrigtsen T. Head injury outcome prediction: A role for protein S-100B? Injury. 2006;37:1098–108. doi: 10.1016/j.injury.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Birks EJ, Burton PB, Owen V, Mullen AJ, Hunt D, Banner NR, et al. Elevated tumor necrosis factor-alpha and interleukin-6 in myocardium and serum of malfunctioning donor hearts. Circulation. 2000;102(19 Suppl 3):III352–8. doi: 10.1161/01.cir.102.suppl_3.iii-352. [DOI] [PubMed] [Google Scholar]
- 35.Birks EJ, Yacoub MH, Burton PS, Owen V, Pomerance A, O’Halloran A, et al. Activation of apoptotic and inflammatory pathways in dysfunctional donor hearts. Transplantation. 2000;70:1498–506. doi: 10.1097/00007890-200011270-00018. [DOI] [PubMed] [Google Scholar]
- 36.Takada M, Nadeau KC, Hancock WW, Mackenzie HS, Shaw GD, Waaga AM, et al. Effects of explosive brain death on cytokine activation of peripheral organs in the rat. Transplantation. 1998;65:1533–42. doi: 10.1097/00007890-199806270-00001. [DOI] [PubMed] [Google Scholar]
- 37.Birks EJ, Owen VJ, Burton PB, Bishop AE, Banner NR, Khaghani A, et al. Tumor necrosis factor-alpha is expressed in donor heart and predicts right ventricular failure after human heart transplantation. Circulation. 2000;102:326–31. doi: 10.1161/01.cir.102.3.326. [DOI] [PubMed] [Google Scholar]
- 38.Fisher AJ, Donnelly SC, Hirani N, Haslett C, Strieter RM, Dark JH, et al. Elevated levels of interleukin-8 in donor lungs is associated with early graft failure after lung transplantation. Am J Respir Crit Care Med. 2001;163:259–65. doi: 10.1164/ajrccm.163.1.2005093. [DOI] [PubMed] [Google Scholar]
- 39.Murugan R, Venkataraman R, Wahed AS, Elder M, Hergenroeder G, Carter M, et al. Increased plasma interleukin-6 in donors is associated with lower recipient hospital-free survival after cadaveric organ transplantation. Crit Care Med. 2008;36:1810–6. doi: 10.1097/CCM.0b013e318174d89f. [DOI] [PubMed] [Google Scholar]
- 40.Nijboer WN, Schuurs TA, van der Hoeven JA, Fekken S, Wiersema-Buist J, Leuvenink HG, et al. Effect of brain death on gene expression and tissue activation in human donor kidneys. Transplantation. 2004;78:978–86. doi: 10.1097/01.tp.0000135565.49535.60. [DOI] [PubMed] [Google Scholar]
- 41.Mertes PM, el Abassi K, Jaboin Y, Burtin P, Pinelli G, Carteaux JP, et al. Changes in hemodynamic and metabolic parameters following induced brain death in the pig. Transplantation. 1994;58:414–8. doi: 10.1097/00007890-199408270-00004. [DOI] [PubMed] [Google Scholar]
- 42.Deng MC, Erren M, Kammerling L, Gunther F, Kerber S, Fahrenkamp A, et al. The relation of interleukin-6, tumor necrosis factor-alpha, IL-2, and IL-2 receptor levels to cellular rejection, allograft dysfunction, and clinical events early after cardiac transplantation. Transplantation. 1995;60:1118–24. doi: 10.1097/00007890-199511270-00011. [DOI] [PubMed] [Google Scholar]
- 43.Lopau K, Mark J, Schramm L, Heidbreder E, Wanner C. Hormonal changes in brain death and immune activation in the donor. Transpl Int. 2000;13(Suppl 1):S282–5. doi: 10.1007/s001470050342. [DOI] [PubMed] [Google Scholar]
- 44.Plenz G, Eschert H, Erren M, Wichter T, Böhm M, Flesch M, et al. The interleukin-6/interleukin-6-receptor system is activated in donor hearts. J Am Coll Cardiol. 2002;39:1508–12. doi: 10.1016/s0735-1097(02)01791-6. [DOI] [PubMed] [Google Scholar]
- 45.Weiss S, Kotsch K, Francuski M, Reutzel-Selke A, Mantouvalou L, Klemz R, et al. Brain death activates donor organs and is associated with a worse I/R injury after liver transplantation. Am J Transplant. 2007;7:1584–93. doi: 10.1111/j.1600-6143.2007.01799.x. [DOI] [PubMed] [Google Scholar]
- 46.Lentsch AB, Ward PA. Regulation of experimental lung inflammation. Respir Physiol. 2001;128:17–22. doi: 10.1016/s0034-5687(01)00260-2. [DOI] [PubMed] [Google Scholar]
- 47.Strieter RM, Kunkel SL. Acute lung injury: The role of cytokines in the elicitation of neutrophils. J Investig Med. 1994;42:640–51. [PubMed] [Google Scholar]