

Abstract
Background:
Tacrolimus has poor solubility in water ranging from 4 to 12 μg/mL. The oral bio availabilities of tacrolimus is poor and exhibits high intra and inter-subject variability (4-89%, average 25%) in the liver and the kidney transplant recipients and in patients with renal impairment.
Aim:
The present study deals with the development and characterization of self-micro-emulsifying drug delivery system to improve the oral bioavailability of poorly soluble drug tacrolimus.
Materials and Methods:
Solubility of the tacrolimus was estimated in various oils, surfactants, and co-surfactants. Various in vitro tests such as percentage transmittance, emulsification time, cloud point, precipitation, and thermodynamic stabilities were used to find out optimized formulations. Optimized liquid self micro-emulsifying (SMEDDS) were characterized by particle size analysis and converted in solid by using the Florite RE as an adsorbent, which is further characterized by differential scanning calorimetry (DSC), scanning electron microscopy (SEM), X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), and particle size analysis.
Results:
The optimized liquid SMEDDS formulation contained 10% Lauroglycol FCC as an oil, 60% Cremophor RH, and 30% PEG (polyethylene glycol) 400 as a surfactant and co-surfactant respectively. The optimized liquid and solid SMEDDS showed higher drug release than the marketed capsule and pure API (active pharmaceutical ingredient) powder. For optimized liquid SMEDDS and solid SMEDDS, the globule sizes were found 113 nm and 209 nm respectively. The solid state characterization of solid-SMEDDS by SEM, DSC, FTIR, and XRD revealed the absence of crystalline tacrolimus in the solid-SMEDDS. Shelf-lives for liquid SMEDDS and solid SMEDDS were found to be 1.84 and 2.25 year respectively.
Conclusions:
The results indicate that liquid SMEDDS and solid SMEDDS of tacrolimus, owing to nano-sized, have potential to enhance the absorption of the drug.
Keywords: Cremophor RH, lauroglycol FCC, solid SMEDDS, tacrolimus
INTRODUCTION
In large part, the success of solid organ transplantation lies in the appropriate utilization of immunosuppressive medications. Tacrolimus, a lipophilic 23-member macrolide lactone isolated from Streptomyces tsukubaensis (molecular weight of 803.5 Da), is an important immunosuppressant widely used in the transplant patients, but with a narrow therapeutic window. The half-life of tacrolimus in human is 8.7-11.3 h.[1,2,3] Absorption of tacrolimus has been shown to be highly variable between individuals. After oral administration, the drug is generally absorbed with a mean time of peak concentrations of 1.5-2 h. However, in some patients, the drug may be absorbed over a prolonged absorption period, resulting in a more flat absorption profile.[4] The mean bioavailability is approximately 21%, although there is large inter-subject variability.
SMEDDS formulation contains oil (synthetic or natural) with hydrophilic or lipophilic surfactants and co-surfactant. This mixture is isotropic in nature and emulsifies when exposed to GI media under mild agitation and forms oil in water emulsion.[5,6] Based on the type of surfactants used and the presence or absence of oil they are classified in four different types. Among these, type-III are known to form SMEDDS that is self-micro emulsifying oil in water emulsion having globule size less than 200 nm in most cases.[7] Lipid-based formulations specially SMEDDS improve as well as normalize drug absorption, which is particularly beneficial for low therapeutic index drug like tacrolimus. These formulations enhance absorption by a number of mechanisms like improving drug solubility and maintaining drug in solution state throughout GI tract, inhibition of P-glycoprotein-mediated drug efflux and pre-absorptive metabolism by gut membrane-bound cytochrome enzymes,[8,9] the promotion of lymphatic transport, which delivers drug directly to the systemic circulation. Although avoiding hepatic first-pass metabolism,[10] and also by increasing GI membrane permeability.[11]
In recent years as an alternative to liquid SMEDDS, solid SMEDDS are developed by incorporating SMEDDS into solid inert pharmaceutical excipients. Various solidification techniques are used to prepare solid SMEDDS such as adsorption to solid carrier, spray drying, melt extrusion, nanoparticle technology, etc., Among these adsorption to solid carrier is the simplest and economical technique, which gives stable free flowing solid SMEDDS powder. It can be easily filled in hard gelatin capsule and easily disperses upon GI fluid contact.[12] Generally adsorbent used are highly porous in nature and they have the capacity to adsorb high amount of liquid. Examples of some commonly used adsorbents are Florite RE, Nusilin US2, Aerosil 200, Sylysia 350, Syloid® 244 FP (porous silicon dioxide). These adsorbent were used by some researcher to successfully develop solid SMEDDS to increase dissolution of lipophilic drug such as Curcumin and Nitredipin.[13]
In 2008 Borhade et al.[5] formulated SMEDDS of tacrolimus and reported significantly higher immunosuppressant activity in mice as compared to marketed Pangraf capsules. Authors reported that higher dissolution and improved solubility could have played an important role for improving therapeutic efficacy. Xin et al.[8] investigated the preliminary role of Schisandra sphenanthera extract on the pharmacokinetic profile of tacrolimus in healthy volunteers. Author assumed that interaction of herbal extract with Pgp (P-glycoprotein) and CYP (cytochrome) 3A4 may be the factors that might increase the oral AUC (area under curve) of tacrolimus. Recently, in 2010, Qin et al.[9] investigated in vitro and in vivo effects of Schisandra sphenanthera extract (Wuzhi Tablet) on the absorption and first-pass intestinal and hepatic metabolism of tacrolimus. Extract inhibited Pgp mediated efflux of tacrolimus. Moreover, this herbal extract was also responsible for inhibiting tacrolimus metabolism in human and rat liver microsomes. The objective of present studies was to develop a new stable liquid SMEDDS and solid SMEDDS of lipophilic immunosuppressant tacrolimus in order to increase in vitro dissolution by using the bio enhancer excipients. Bio-enhancer excipients have the ability to block Pgp and CYP 450 enzyme systems. In this study, we have selected excipients based on solubility and emulsification study; also their bio-enhancing property was considered during the selection process. Prepared batches were characterized by emulsification efficiency, globule size analysis, in vitro release profile, etc., Shelf-lives of optimized formulations were determined. Additionally, solid SMEDDS was characterized by scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), and differential scanning calorimetry (DSC). A new SMEDDS have been formulated with enhance dissolution rate and have potential to improve oral bioavailability of tacrolimus due to the bio enhancing property of excipients.
MATERIALS AND METHODS
Materials
Tacrolimus was gifted from Alembic Ltd., Pharma Vadodara, India. The Marketed Capsule of tacrolimus (Pangraf-Panacea) was purchased from a local supplier. Phosal 53 MCT and Phosal 75 SA were gifted from Lipoid, Germany. Lauroglycol FCC, Gelucire 50/13, Labrafil M2130, and Maisin 35-1 were kindly gifted from Gattefosse, Mumbai, India. Cremophor RH and EL were kindly gifted from Matrix lab, Hyderabad, India. PEG 400 and Tween were purchased from Rankem, Mumbai, India. Florite RE was gifted from Tomita Pharmaceutical Co., Ltd, Tokushima, Japan. High-performance liquid chromatography (HPLC) grade Acetonitrile was purchased from RFCL limited, New Delhi, India.
Methods
Solubility studies
The solubility of tacrolimus in various vehicles, including oils, surfactants, and co-surfactant was determined by the shake flask method.[14] An excess amount of tacrolimus was added to each vial containing 1 mL of the vehicle. After sealing, the mixture was vortexed using a vortex mixer at a maximum speed for 10 min in order to facilitate proper mixing of tacrolimus with the vehicle. Mixtures were then shaken in an orbital shaker maintained at room temperature until equilibrium (24 h). Mixtures were centrifuged at 15,000 rpm for 10 min, and the resulting supernatant was filtered through the Whatman filter paper. The filtrate was quantified by the developed HPLC method.[15] Briefly, mobile phase was acetonitrile 100% at a flow rate of 0.9 mL/min. The wavelength used for detection was 213 nm. A calibration curve was constructed from tacrolimus concentrations ranging between 5 and 250 μg/mL, which yielded a linear correlation (r2 = 0.997).
Emulsification studies
Emulsification studies were conducted to select the optimum Oil: Smix (surfactant; co-surfactant mixture) ratio. Surfactant and co-surfactant were mixed at a fixed ratio of 2:1. Oil to Smix ratios were 4:6, 3:7, 2:8, 1:9 and the mixture was homogenized with the aid of the gentle heat (30-40°C) and vortexed for 2 min in a vortex mixer. 0.5 mL formulation was added in 125 mL of water in the beaker during constant stirring on a magnetic stirrer at low speed. The resulting emulsions were observed visually for the relative turbidity. The emulsions were allowed to stand for 2 h and their % transmittance was measured at 638.2 nm by a UV-1800 spectrophotometer (Shimadzu, Japan) using the distilled water as a blank.[14]
Preparation of liquid SMEDDS
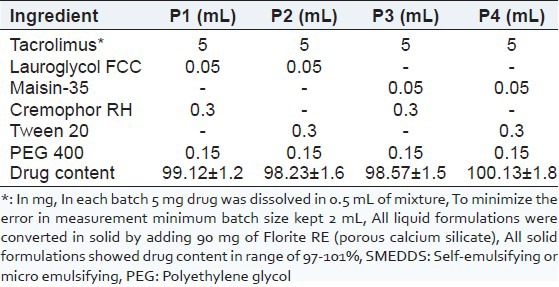
The formulations were prepared by dissolving the required amount of tacrolimus in the mixture of oil, surfactant and co-surfactant, at 40°C in a water bath. This mixture was mixed by vortexing until a transparent preparation was obtained. Then, a formulation equivalent to 5 mg tacrolimus was manually filled in a hard gelatin capsule of size “1” [Table 1].
Table 1.
Composition of liquid SMEDDS and solid SMEEDDS. Data are expressed as mean (SD [n=3])
Thermodynamic stability studies
The objective of the thermodynamic stability was to evaluate the effect of temperature variation on the SMEDDS formulations. Tacrolimus SMEDDS were centrifuged at 15,000 rpm for 15 min and the formulations were observed visually for phase separation. The formulations were subjected to freeze-thaw cycles (−5°C for 2 days followed by +40°C for 2 days). The samples were observed visually after freeze-thaw cycles.
Cloud point measurement
The formulations were compared for cloud point value. Each formulation was diluted with water in the ratio of 1:100 and placed in a water bath with a gradual increase in temperature. At the cloud point, drop in sample % transmittance was measured spectrophotometrically.[16]
Precipitation analysis
The prepared SMEDDS were diluted with 0.1N HCl up to 250 times. The diluted microemulsion was observed at 1 h and 6 h for any sign of phase separation or drug precipitation.[17]
Preparation of solid SMEDDS
Thermodynamically stable liquid SMEDD was converted to solid SMEDDS to get the advantage of the solid dosage form. The prepared liquid formulation of tacrolimus was added drop-wise on a solid carrier in glass mortar. The mixture was physically mixed until uniform free-flowing powder was obtained. This powder was screened through a 40# sieve. Then the powder was filled in a “00” size hard gelatin capsule shell.[12]
Drug content
The total amount of the drug in the formulation was analyzed by dissolving the formulation in 10 mL ACN (acetonitrile). This solution was vortexes for 10 min in vortex mixture. The mixture was centrifuged at 15,000 rpm for 10 min. Then the supernatant was filtered through the Whatman filter paper. The filtrate was analyzed by HPLC after suitable dilution [Table 1].
In vitro dissolution
Liquid and solid SMEDDS was filled in capsule shell and in vitro release profile was taken in a USP (united state pharmacopoeia) apparatus 2 at 37 ± 0.5°C, at 75 RPM in 300 mL 0.1N HCl. At pre-determined intervals, 3 mL of the medium was sampled and filtered through the Whatman filter paper. Then, 20 μL of the fitrate was analyzed by HPLC.
Globule size analysis
The globule size and size distribution were analyzed by the dynamic light scattering with a globule size apparatus (Malvern Zetasizer version 6.11). Liquid and solid SMEDDS were diluted 250-times with 0.1 N HCl at 25°C under gentle shaking. After equilibrium, the emulsions were filtered through a Whatman filter paper. The filtrates were analyzed by Zeta sizer. A laser beam at 632 nm wavelength was used and light scattering was monitored at 25°C at a 90° angle.[17]
Solid state characterization of solid SMEDDS
DSC analysis
The physical state of tacrolimus in solid SMEDDS was characterized by the DSC (DSC-60, Shimadzu Corporation, Japan). The samples (about 3.00 mg) were placed in standard aluminum pans and the air was used as an atmosphere. All samples were scanned at a temperature ramp speed of 10o C/min and the heat flow was set from 50°C to 200°C.
XRD analysis
X-ray powder scattering measurements were carried out with a D2 Phaser diffractometer at room temperature using the monochromatic CuKa-radiation at 34 mA and at 38 kV over a range of 2θ angles from 5° to 60° with an angular increment of 0.05°/s.[18]
SEM
The morphological features of solid tacrolimus SMEDDS were observed by scanning electron microscope (Model: ESEM TMP + EDAX; Make: Philips, Netherland Detector: Secondary and back scattered electron detector).
FTIR spectroscopy
The IR spectra were recorded on the FTIR (Jasco, FTIR model 6100, Japan) by using a potassium bromide pressed disc method. The scanning range was 400-4000 cm−1.
Determination of the shelf-life of the optimized formulations
Accelerated stability studies were performed for the determination of the shelf-life. The SMEDDS formulations were kept at three different temperatures and ambient humidity conditions (30 ± 0.5, 40 ± 0.5, and 50 ± 0.5°C) for 2 months. The samples were withdrawn after specified time intervals (0, 15, 30, 45, and 60 days) and the remaining drug content was measured using the HPLC method. Zero time samples were used as controls. The order of the reaction was determined. After determination of the order of the reaction, the reaction rate constant (K) for the degradation was measured from the slope of the lines at each elevated temperature using the following equation:
Slope = −K/2.303
The plot of the logarithm of K values at various elevated temperatures against the reciprocal of absolute the temperature was drawn (Arrhenius plot). From the plot, the K value at 25°C was determined and was used to calculate shelf-life by substituting in the equation:
t0.9 = 0.1052/K25
Where t0.9 is the time required for 10% degradation of the drug and is referred to as the shelf-life.[19]
RESULTS AND DISCUSSION
Solubility studies
The equilibrium solubility of tacrolimus in various oil, surfactant and co-surfactant were shown in Figure 1. From the SMEDDS point of view, solubility in various excipients is an important criterion. In SMEDDS, the drug was soluble in the oil phase and/or was present at the interface. It depended on the hydrophobicity of the drug and the HLB value of the system. Maisin 35-1 and Lauriglycol FCC showed higher solubility compare to other oils. In order to prevent the precipitation of drug on dilution it is necessary to select oil, which shows higher solubility.[20,21]
Figure 1.
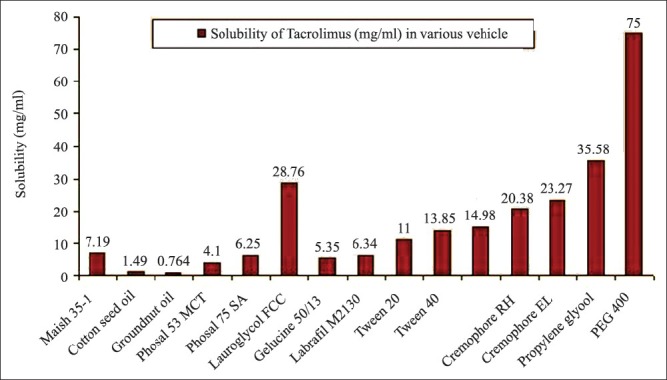
Solubility of tacrolimus in the different vehicles. Data are expressed as mean ± standard deviation (n = 3); *=10% solution
Tween 80 shows higher solubility compare to Tween 20. In our study, we selected the Tween 20 due to its higher ability to form spontaneous emulsion (data of surfactant/co-surfactant selection was not shown) having the higher value of % transmission, yet able to keep drug in solubilized state upon dilution. The same parameters were kept in mind during the selection of Cremophor RH over Cremophor EL. Drug showed the highest solubility in co-surfactant PEG 400.
Emulsification studies
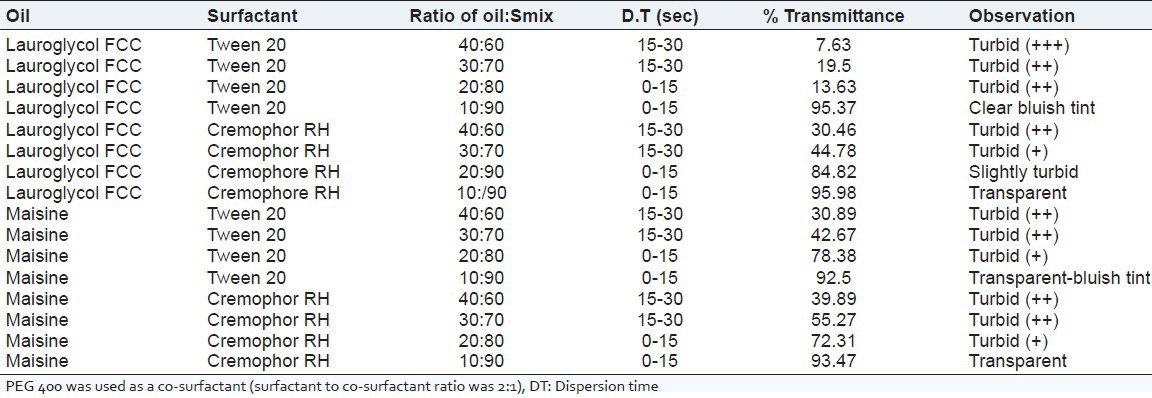
Emulsification studies were conducted in order to select proper ratio of Oil:Smix. From the data (not shown) of screening of surfactant and co-surfactant, ratio of surfactant to co-surfactant was kept 2:1 constant for all batches. Emulsification studies were performed to evaluate the ability of Smix to emulsify oil phase [Table 2]. Both Maisin 35-1 and Lauroglycol FCC oils required 90% of Smix at 2:1 ratio to form almost transparent emulsion in less than 15 s.
Table 2.
Emulsification studies for screening of excipients
Thermodynamic stability studies
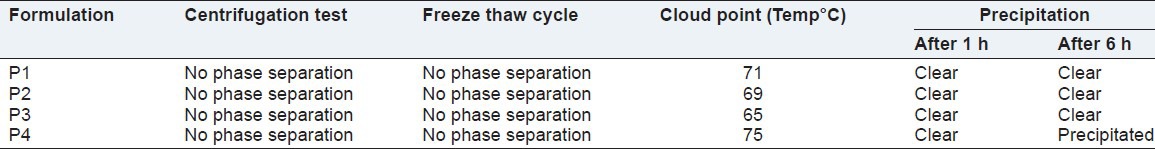
Thermodynamic stability studies were performed to observe the ability of the formulation to withstand different stress conditions. A stable SMEDDS formulation should not lose its ability of spontaneous emulsification upon dilution. All liquid formulations were found to be stable in the centrifugation test and in the freeze-thaw cycle. There was no sign of phase separation. Results are shown in Table 3.
Table 3.
Thermodynamic stability, cloud point, precipitation studies
Cloud point
The cloud point is the temperature above which the formulation clarity turns into cloudiness. At higher temperatures, phase separation can occur. Since both drug solubilisation and formulation stability will decline with this phase separation, the cloud point of the formulation should be over 37°C. In this study, the cloud points of all formulations were very high as reported in Table 3.[16]
Precipitation analysis
Out of four different formulations, the P4 was precipitated on dilution with 0.1 N HCL at 250 times after 6 h. Although there was no sign of precipitation up to 2 h in all formulations, Table 3.
Drug content
All liquid and solid SMEDDS showed drug content within 97-102%, Table 1.
In vitro dissolution
The marketed formulation [Figure 2[ showed that 71% drug was released within 120 min, whereas, the pure API powder showed that only 20% drug was released within 120 min. In vitro release profile of liquid P1 showed that 90% drug was released within 5 min. It might be due to quick emulsification properties of SMEDDS and its ability to keep drug in solubilized state upon dilution, whereas solid SMEDDS containing Florite RE as a carrier showed that 70% drug was release within 5 min. This indicates that solid SMEDDS also able to disperse spontaneously and can release drug quickly. P1 liquid and P1 solid formulations showed comparatively better dissolution profile than o8/30/2013ther SMEDDS formulations.
Figure 2.
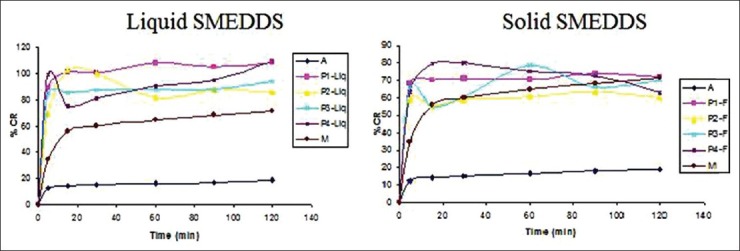
In vitro dissolution profile in 0.1 N HCL (A = pure API powder, M = marketed product)
Globule size analysis
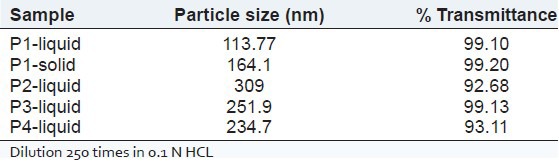
Droplet size of SMEDDS is a critical step in the pathway of enhancing drug bioavailability. For P1 liquid and P1 solid formulations the globule size was found to be 113.7 nm and 164.1 nm respectively [Figure 3]. The polydispersibility indexes were 0.349 and 0.825 for P1 liquid and P1 solid respectively. There was not much different between globule size of P1 solid and P1 liquid, indicating suitability of Florite RE as a carrier. No significant difference between the droplet sizes and % transmittance [Table 4] of liquid and solid SMEDDS suggested the capability of the lipid components of solid SMEDDS to retain its emulsification properties irrespective of physical form change.
Figure 3.
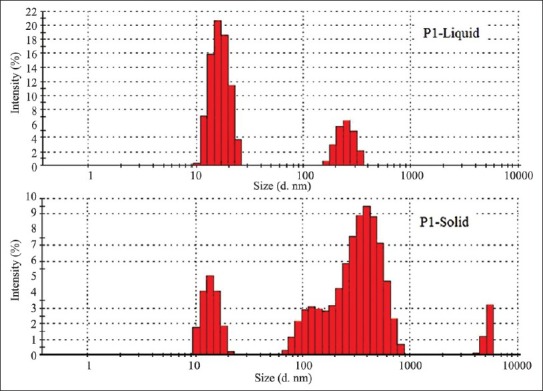
Globule size of liquid P1 and solid P1 self-microemulsifying drug delivery system
Table 4.
Globule size analysis and % transmittance
DSC analysis
DSC thermograms are shown in Figure 4. Sharp endothermic peak was observed at 129.6°C temperatures, which corresponds to melting temperature of the tacrolimus, Figure 4a. There was no endothermic peak in the Florite RE thermogram. The physical mixture shows an endothermic peak of tacrolimus. However, this endothermic peak was not found in thermogram of the solid SMEDDS which confirms the presence of drug in an amorphous form.
Figure 4.
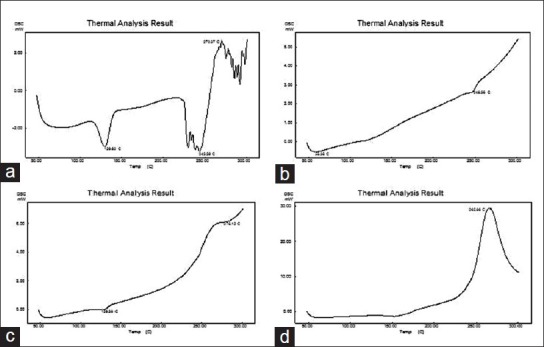
Differential scanning calorimetric thermogram of tacrolimus powder (a), Florite RE (b), physical mixture of tacrolimus and Florite RE (c) and solid self-microemulsifying drug delivery system of tacrolimus (d)
XRD analysis
The XRD pattern for tacrolimus powder is shown in Figure 5a. This shows the characteristic sharp peaks at a particular diffraction angle. Florite RE showed characteristics XRD pattern [Figure 5b]. All major characteristic peaks of Florite RE were also observed in the physical mixture [Figure 5c]. But, this crystalline pattern was not found in the XRD plot of solid SMEDDS [Figure 5d]. Florite RE showed characteristics XRD pattern [Figure 5b]. This further confirmed that the drug was present in an amorphous form.
Figure 5.
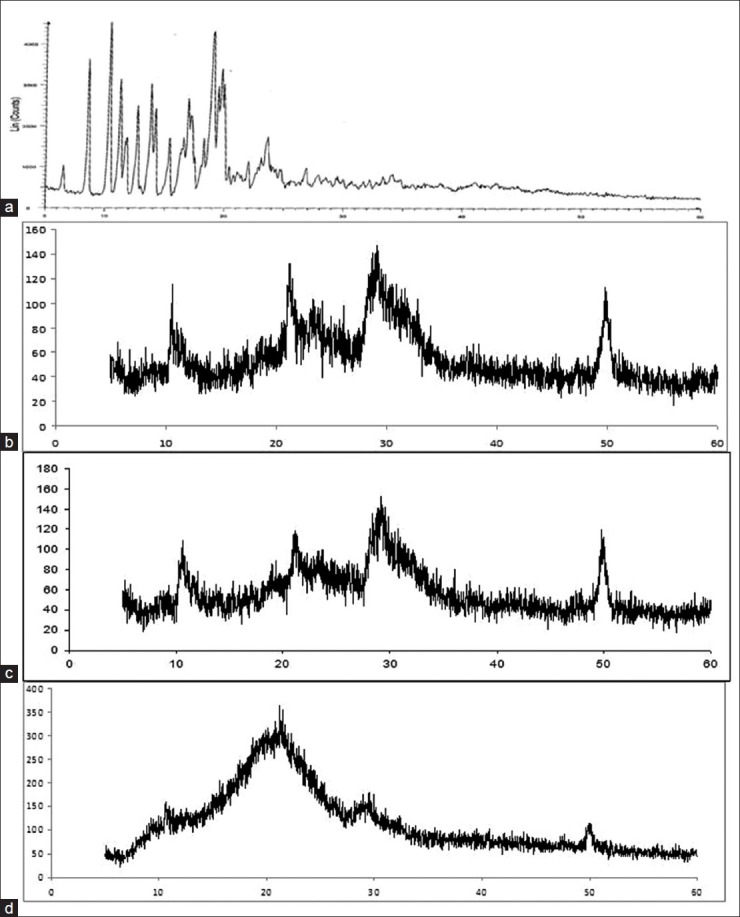
X-ray diffraction pattern of powder tacrolimus (a), Florite RE (b), physical mixture (c), solid self-microemulsifying drug delivery system of tacrolimus (d)
SEM
The scanning electron micrographs of tacrolimus powder, Florite RE and P1 solid SMEDDS are shown in Figure 6. Tacrolimus powder [Figure 6a] appeared as smooth-surfaced crystalline powder. Florite RE [Figure 6b] appeared with a rough surface with porous particles. However, the solid SMEDDS [Figure 6c] did not show any crystalline drug powder, indicating that the liquid SMEDDS is absorbed or coated inside the pores of Florite RE.
Figure 6.

Scanning electron microscopy images of (a) tacrolimus; (b) Florite RE; (c) solid self-microemulsifying drug delivery system of tacrolimus
FITR
The IR spectra of tacrolimus, Florite RE, Physical mixture and solid SMEDDS are presented in Figure 7. Tacrolimus powder shows O-H stretching vibration at 3450, cm−1, C = O (ester and ketone) stretching vibrations at 1733, and 1690 cm−1, C = O (keto-amide) and C = C stretching vibration at 1638 cm−1, C–O (ester) stretching vibration at 1184 cm−1, C–O–C (ether) stretching vibrations at 1091 cm−1. Majority of above peaks are weakened, sifted or disappeared in physical mixture and solid SMEDDS. All of the above indicated an interaction of tacrolimus with the carrier, which might be hydrogen bonds results from the dissolving of tacrolimus in SMEDDS excipients.
Figure 7.
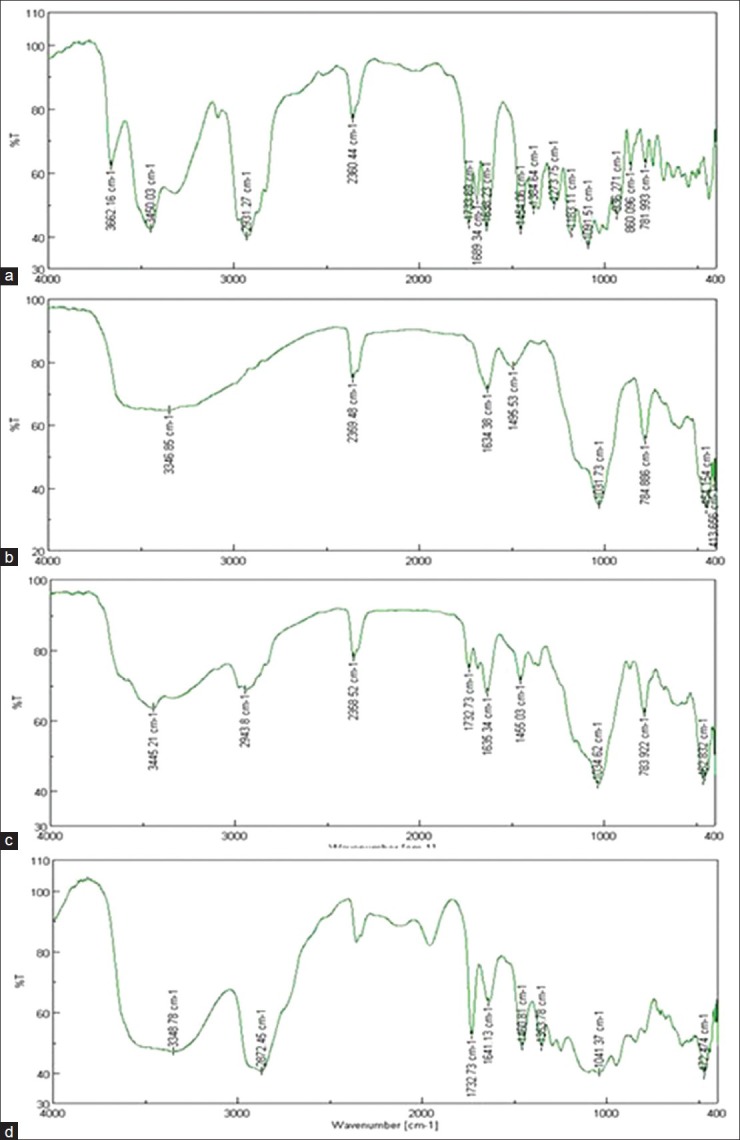
Fourier transform infrared spectroscopy spectra of powder tacrolimus (a), Florite RE (b), physical mixture (c), solid self-microemulsifying drug delivery system of the tacrolimus (d)
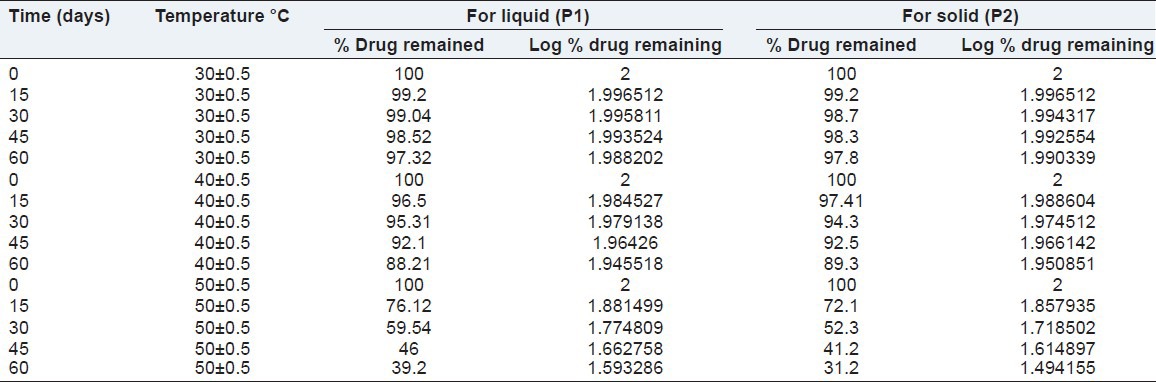
Shelf-life of optimized formulations
Accelerated stability studies were performed for the determination of the shelf-lives of the P1 liquid and P1 solid SMEDDS formulations. The amount of drug remaining undecomposed in the formulation P1 liquid and P1 solid at each time interval is shown in Table 5. It can be seen from the table that the concentration of drug remaining undecomposed in the P1 liquid formulation at the end of 60 days was 97.32%, 88.21% and 39.2% at 30 ± 0.5, 40 ± 0.5 and 50 ± 0.5°C respectively. The concentration of drug remaining undecomposed in the solid P1 formulation at the end of 60 days was 97.8%, 89.3% and 31.2% at 30 ± 0.5, 40 ± 0.5 and 50 ± 0.5°C respectively.
Table 5.
Percent drug remaining in P1 liquid and P1 solid stored at elevated temperatures (30±0.5, 40±0.5 and 50±0.5°C)
The order of degradation of tacrolimus in the SMEDDS formulations was found to follow first-order kinetics. The reaction rate constant “K” for the degradation was measured from the slope of the lines at each elevated temperature. The plot of the logarithm of the K values for liquid P1 and solid P1 SMEDDS [Table 6] at each elevated temperature against the reciprocal of the absolute temperature was drawn (Arrhenius plot) as shown in Figures 8 and 9. From the plot, the K value at 25°C was determined and was used to calculate the shelf-life. The shelf-life of the liquid P1 at room temperature was calculated to be 1.84 years and for the solid P1 formulation, the shelf-life was 2.55 years.
Table 6.
Degradation rate constant for tacrolimus in P1 liquid SMEDDS and P1 solid SMEDDS
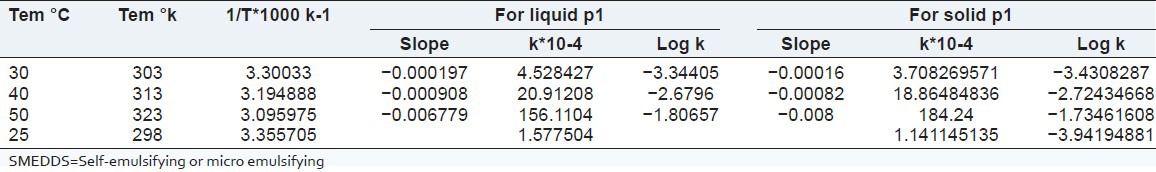
Figure 8.
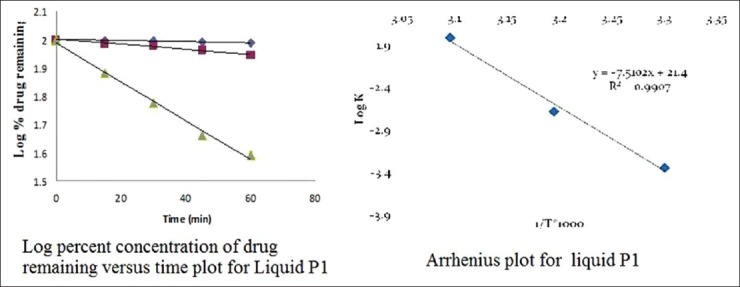
Stability data of liquid P1
Figure 9.
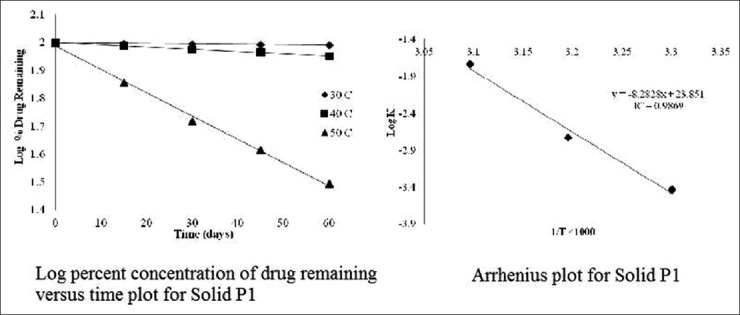
Stability data of solid P1
CONCLUSION
Liquid SMEDDS and solid SMEDDS were prepared for immunosuppressant tacrolimus. Optimized liquid SMEDDS contains 10% Lauroglycol FCC, 60% Cremophor RH and 30% PEG 400, which showed spontaneous emulsification properties and good thermodynamic stability. Liquid SMEDDS and solid SMEDDS showed a better in vitro drug release profile compared with pure API and the marketed product. SEM, IR, XRD and DSC results proved that the drug was present in an amorphous state in solid SMEDDS. The present study confirmed that the new self-microemulsifying systems containing bio-enhancer excipients are promising strategies for enhancing dissolution rate and thereby oral bioavailability of the tacrolimus. Excipients used in the formulation are known for inhibition of CYP metabolism and pgp efflux studies. The optimized formulation is able to inhibit CYP metabolism and Pgp efflux (data are not shown).
ACKNOWLEDGMENT
We are grateful for financial support provided from A. K. Raval charitable trust.
Footnotes
Source of Support: We are grateful for financial support provided from the A. K. Raval charitable trust
Conflict of Interest: None declared
REFERENCES
- 1.Spencer CM, Goa KL, Gillis JC. Tacrolimus. An update of its pharmacology and clinical efficacy in the management of organ transplantation. Drugs. 1997;54:925–75. doi: 10.2165/00003495-199754060-00009. [DOI] [PubMed] [Google Scholar]
- 2.Venkataramanan R, Swaminathan A, Prasad T, Jain A, Zuckerman S, Warty V, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet. 1995;29:404–30. doi: 10.2165/00003088-199529060-00003. [DOI] [PubMed] [Google Scholar]
- 3.Kershner RP, Fitzsimmons WE. Relationship of FK506 whole blood concentrations and efficacy and toxicity after liver and kidney transplantation. Transplantation. 1996;62:920–6. doi: 10.1097/00007890-199610150-00009. [DOI] [PubMed] [Google Scholar]
- 4.van Duijnhoven E, Christiaans M, Undre N, Stevenson P, van Hooff J. The effect of breakfast on the oral bioavailability of tacrolimus in diabetic and nondiabetic patients before transplantation. Transplant Proc. 1998;30:1268–70. doi: 10.1016/s0041-1345(98)00237-1. [DOI] [PubMed] [Google Scholar]
- 5.Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS Pharm Sci Tech. 2008;9:13–21. doi: 10.1208/s12249-007-9014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–82. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv Drug Deliv Rev. 2008;60:625–37. doi: 10.1016/j.addr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Xin HW, Wu XC, Li Q, Yu AR, Zhu M, Shen Y, et al. Effects of Schisandra sphenanthera extract on the pharmacokinetics of tacrolimus in healthy volunteers. Br J Clin Pharmacol. 2007;64:469–75. doi: 10.1111/j.1365-2125.2007.02922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin XL, Bi HC, Wang XD, Li JL, Wang Y, Xue XP, et al. Mechanistic understanding of the different effects of Wuzhi Tablet (Schisandra sphenanthera extract) on the absorption and first-pass intestinal and hepatic metabolism of Tacrolimus (FK506) Int J Pharm. 2010;389:114–21. doi: 10.1016/j.ijpharm.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 10.Charman WN. Boca Raton: CRC Press; 1992. Lipid vehicle and formulation effects on intestinal lymphatic drug transport; pp. 113–79. [Google Scholar]
- 11.Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16:237–46. doi: 10.1016/s0928-0987(02)00055-6. [DOI] [PubMed] [Google Scholar]
- 12.Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: Preparation techniques and dosage forms. Drug Discov Today. 2008;13:606–12. doi: 10.1016/j.drudis.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Sun J, Wang Y, Liu X, Liu Y, Fu Q, et al. Solid self-emulsifying nitrendipine pellets: Preparation and in vitro/in vivo evaluation. Int J Pharm. 2010;383:1–6. doi: 10.1016/j.ijpharm.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329:166–72. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 15.Patel P, Panchal S, Mehta T, Solanki S, Patel C. Reversed-phase high performance liquid chromatographic method for determination of tacrolimus in bulk and pharmaceutical formulation. Int J Pharm Pham Sci. 2011;3:220–2. [Google Scholar]
- 16.Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355:269–76. doi: 10.1016/j.ijpharm.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 17.Singh AK, Chaurasiya A, Singh M, Upadhyay SC, Mukherjee R, Khar RK. Exemestane loaded self-microemulsifying drug delivery system (SMEDDS): Development and optimization. AAPS Pharm Sci Tech. 2008;9:628–34. doi: 10.1208/s12249-008-9080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balakrishnan P, Lee BJ, Oh DH, Kim JO, Hong MJ, Jee JP, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS) Eur J Pharm Biopharm. 2009;72:539–45. doi: 10.1016/j.ejpb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Bali V, Ali M, Ali J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of ezetimibe. Colloids Surf B Biointerfaces. 2010;76:410–20. doi: 10.1016/j.colsurfb.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty S, Shukla D, Mishra B, Singh S. Lipid – An emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm. 2009;73:1–15. doi: 10.1016/j.ejpb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Nazzal S, Smalyukh II, Lavrentovich OD, Khan MA. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: Mechanism and progress of emulsion formation. Int J Pharm. 2002;235:247–65. doi: 10.1016/s0378-5173(02)00003-0. [DOI] [PubMed] [Google Scholar]