

Abstract
Background. Since the mapping of the human genome in 2003, the development of biomarker targeted therapy and clinical adoption of “personalized medicine” has accelerated. Models for insurance subsidy of biomarker/test/drug packages (“codependent technologies” or technologies that work better together) are not well developed. Our aim was to create a framework to assess the safety, effectiveness, and cost-effectiveness of these technologies for a national coverage or reimbursement decision. Methods. We extracted information from assessments of recent Australian reimbursement applications that concerned genetic tests and treatments to identify items and evidence gaps considered important to the decision-making process. Relevant international regulatory and reimbursement guidance documents were also reviewed. Items addressing causality theory were included to help explain the relationship between biomarker and treatment. The framework was reviewed by policy makers and technical experts, prior to a public consultation process. Results. The framework consists of 5 components—context, clinical benefit, evidence translation, cost-effectiveness, and financial impact—and a checklist of 79 items. To determine whether the biomarker test, the drug, both, or neither should be subsidized, we considered it crucial to identify whether the biomarker is a treatment effect modifier or a prognostic factor. To aid in this determination, the framework explicitly allows the linkage of different types of evidence to examine whether targeting the biomarker varies the likely clinical benefit of the drug, and if so, to what extent. Conclusions. The first national framework to assess personalized medicine for coverage or reimbursement decisions has been developed and introduced and may be a suitable model for other health systems.
Keywords: personalized medicine, drug approval process, genetic testing, reimbursement mechanisms
Until recently, health professionals have had limited information about the likely response of a patient to therapy. Treatment strategies were generally based on aggregated information and subsequently modified according to individual response. With increased understanding of genetics, it is now possible to personalize medicine so that the risk profile of a patient can be determined prospectively to guide treatment so that it is more effective from initiation and is used only in those who will respond and/or with fewer side effects.1–4
Several drugs, particularly for cancer, have been developed and marketed with a “companion diagnostic,” a test to determine whether a patient has a biomarker that will predict response to a drug.5,6 Examples include trastuzumab and HER2 testing for breast cancer, cetuximab and K-RAS mutation testing for metastatic colorectal cancer, and gefitinib and EGFR testing for lung cancer.7–10 Such treatment is potentially more clinically and cost-effective as it targets only patients likely to respond.11–13
The US Federal Drug Administration has made preliminary efforts to provide guidance on prospective, scientifically robust codevelopment of a drug and companion diagnostic,5 and there is growing international discussion investigating ways of dealing with these codependent technologies from an assessment and reimbursement perspective.14–16 However, there is also growing frustration from industry and health professionals that personalized medicine is not living up to its promise,17 partly because the current models of assessment internationally are inadequate to inform coverage or reimbursement decisions regarding these distinctive technologies.18
Both the treatment and companion diagnostic in a personalized medicine need to be assessed for performance in order to make a coverage or reimbursement decision. This is not a straightforward process.16,19 To determine what factors influence these decisions, Meckley and Neumann18 selected 6 personalized medicine case studies and extracted data on the quality of evidence supporting each case study, type of regulatory oversight each received, whether clinical guidelines supported the technology, and whether the technology had been found to be cost-effective. They noted there was poor evidentiary support—in the form of randomized controlled trials assessing the direct impact of testing on health outcomes—for most of these technologies and that the key factor influencing a positive reimbursement decision appeared to be the strength of the evidence base.
The recent Review of Health Technology Assessment in Australia similarly recognized that codependent technologies (or technologies that work better together), such as personalized medicines, are problematic to assess for reimbursement decisions.20 As a consequence, research was undertaken to develop an assessment framework to assist policy makers to make evidence-based decisions about subsidized access to these emerging technologies.
Three objectives were formulated to ensure that the assessment framework was feasible:
to identify the different decision-making scenarios that would apply specifically to a personalized medicine (i.e., targeting drug therapy on the basis of a biomarker),
to identify the criteria needed to inform an assessment of these technologies, and
to formulate an approach that recognizes the scarcity of direct evidence, that is, randomized trials assessing the impact on health outcomes of testing versus no testing for the biomarker to guide treatment with the new drug.
Methods
First Stage
Five codependent technologies that had previously been assessed for coverage or reimbursement decisions were reviewed (Table 1):
Table 1.
Case Studies of Pharmacogenetic Codependent Technologies
| Case Study (Biomarker/Therapy) | Decision-Making Body [Therapeutic Purpose] | Evidence Quality | Evidence Gaps (n = 67 Information Items)a | Test Reimbursed?b | Drug Reimbursed?b |
|---|---|---|---|---|---|
| EGFR/gefitinib for non–small-cell lung cancer (2nd line) | PBAC [targeted treatment] | No direct evidence Linked evidence, moderate quality |
8/67 (12%) | Not considered | Yes |
| K-RAS/cetuximab for metastatic colorectal cancer (1st line) | PBAC [targeted treatment] | No direct evidence Linked evidence, poor quality |
32/67 (48%) | Not considered | No |
| K-RAS/panitumumab for metastatic colorectal cancer (2nd line) | PBAC [targeted treatment] | No direct evidence Linked evidence, poor quality |
21/67 (31%) | Not considered | No |
| PDGFR rearrangements/imatinib for primary or secondary clonal eosinophiliac | MSAC [targeted treatment] | Direct evidence, poor quality Plus Linked evidence, moderate quality |
3/67 (4%) | Yes | Yes |
| KIT D816V/imatinib for aggressive systemic mast cell disease without eosinophilia (2nd line) | MSAC [rule out imatinib treatment] | Direct evidence, poor quality Plus Linked evidence, moderate quality |
4/67 (6%) | Nod | Yes |
Note: PBAC = Pharmaceutical Benefits Advisory Committee; MSAC = Medical Services Advisory Committee.
Sixty-seven information items (denominator) were collated from submissions at the completion of stage 1. Evidence gaps (numerator) were defined as a complete absence of information in the submission; however, please note that frequently, the information items were only partially/inadequately addressed and in some instances items were not applicable.
Decision at the time the framework was being developed.
Systemic mast cell disease, hypereosinophilic syndrome, and chronic eosinophilic leukemia.
PDGFR rearrangements and the KIT D816V mutation are mutually exclusive so, as the PDGFR test was funded, there was no need to fund the KIT D816V test.
EGFR/gefitinib for non–small-cell lung cancer,
K-RAS/cetuximab for metastatic colorectal cancer,
K-RAS/panitumumab for metastatic colorectal cancer,
PDGFR rearrangements/imatinib for primary or secondary clonal eosinophilia (systemic mast cell disease, hypereosinophilic syndrome, and chronic eosinophilic leukemia),21 and
KIT D816V/imatinib for aggressive systemic mast cell disease without eosinophilia.21
These case studies were selected as they were the most recent codependent technologies to be assessed for a reimbursement decision by our national committees (either for the test or drug). In all cases, the drug was considered for reimbursement prior to consideration of the biomarker test. Three of the 5 source documents were available only as commercial-in-confidence.
The information provided in each independent assessment report on these 5 technology applications was categorized and tabulated. Sixty-seven information items were identified as being present in at least 1 of the 5 applications. A gap analysis was conducted for each personalized medicine across the 67 items to determine what key information was considered absent on the basis of 1) matters raised within the assessment report (mentioned in the independent assessment report (commentary) of an applicant’s submission undertaken on behalf of the Pharmaceutical Benefits Advisory Committee [PBAC] or discussed in the independent assessment report undertaken on behalf of the Medical Services Advisory Committee [MSAC]) and 2) matters raised during the appraisal and decision-making process (relevant MSAC or PBAC meeting minutes or formal advice from the Economics Subcommittee of PBAC. Each of the 5 personalized medicines was independently rated by 3 experienced evaluators of reimbursement applications, in terms of whether the 67 information items were provided in the application (yes, no, partially) and whether or not the information was needed (yes, no, not applicable). A free-text column was used to comment on whether difficulties were likely to arise when reviewing an item.
It was noted that reimbursement was more likely when there were fewer evidence gaps present in the application and when the evidence was of better quality (Table 1). This latter finding is consistent with Meckley and Neumann.18 However, given that in both Meckley and Neumann’s case studies and in our case studies there was a lack of direct randomized controlled trial evidence of the biomarker test impact on patient health outcomes, it was thought that a framework that allowed the linkage of different types of evidence to support a claim for reimbursement might provide policy makers with fewer evidence gaps and thus reduce decision-making uncertainty.
Second Stage
To ensure that the linkage of evidence was done rigorously, the relationship between biomarker status (as identified by a test) and drug treatment outcomes needed to be adequately explained. The collated list of items was cross-checked against Bradford Hill causality theory22 to ensure that there were multiple opportunities to explain the association between biomarker and drug treatment outcomes, even in the absence of generally accepted experimental evidence. The Bradford Hill criteria, namely, strength, specificity, and temporality of the association between the biomarker and drug treatment on health outcomes; consistency and coherence of effect; biological plausability and gradient (e.g., dose response); and producing the effect upon experimentation or by analogy were addressed, and 5 additional items were included in the checklist. The list of items was then structured in a format consistent with that used for assessing pharmaceuticals for reimbursement decisions in Australia.23
Currently available international guidance documents for the appraisal of technologies or appraisal of applications for test/drug reimbursement were reviewed to determine whether any further items would be relevant to the framework’s development. To identify this literature, Embase and Medline were canvassed, along with Internet searches of regulatory and reimbursement agency Web sites and the health technology assessment database (http://www.crd.york.ac.uk). No new items were identified in the international literature, although some of the documents provided detail that was considered useful as explanatory material in the framework.
Third Stage
Feedback was sought on the framework from a Steering Committee comprising chairs and members of the 2 committees responsible for funding decisions for new technologies (MSAC and PBAC) in Australia, as well as representatives of the funder (government).
The structure of the framework was considered by the Steering Committee to be consistent with the information needed to make reimbursement decisions. Committee members chose not to prioritize any of the 72 items, as all were considered important. The following amendments were suggested by committee members and incorporated into the framework: a more precise definition of the biomarker; explanatory detail regarding the proposed test, including a new checklist item on the proposed Medicare descriptor for the test; a new item on the need for testing for new somatic mutations following treatment; a new item on the method and timing of specimen retrieval; a new item concerning the analytic validity of the test; reordering items and in some cases collapsing items that contained similar concepts or splitting items that contained multiple concepts; and modifying wording of items or explanations to make it clearer regarding the scope or purpose of some of the items.
Fourth Stage
On the basis of feedback from the Steering Committee, a finalized assessment framework containing a checklist of 79 items and explanatory material was developed and released for public consultation between 16 September and 17 December 2010 (http://www.health.gov.au/internet/hta/publishing.nsf/Content/whats-new).
Twelve submissions were received through public consultation. Suggestions were made to extend the scope of the framework in the future and to clarify the government administrative processes for codependent technology applications. The key concern specific to the framework was whether the requested evidence could be feasibly provided, particularly for codependent technologies targeting rare diseases. In response to this feedback, examples were included in the framework (see Additional File 2) to make it more explicit that the linkage of different types of relevant evidence was encouraged when there were deficiencies in an experimental evidence base. Similarly, guidelines are being produced that will further explain each of the concepts in the framework, and a government process for case managing codependent technology applications has been developed (summarized at http://www.health.gov.au/internet/hta/publishing.nsf/Content/co-1).
Results
Decision-Making Scenarios
In Australia, both clinical and cost-effectiveness (i.e., value for money) are considered as part of reimbursement decision making. Regardless of whether decision making occurs on the basis of clinical effectiveness or cost-effectiveness, 4 distinct scenarios can arise when assessing a codependent technology:
The drug is (cost-)effective in an untested population but (cost-)ineffective when conditioned on biomarker status as identified by the test. This might occur if the biomarker does not explain the variation in treatment effect, other prognostic factors are more important in terms of the drug’s effect than the identified biomarker, or if the test for the biomarker is not accurate. In this scenario, the drug is reimbursed but not the test.
The drug is (cost-)effective in an untested population but more (cost-)effective when conditioned on the biomarker identified by the test. In this scenario, the drug is reimbursed but the decision to reimburse the test will depend on the level of uncertainty surrounding the relationship between biomarker and the treatment effect of the drug;
The drug is not (cost-)effective in an untested population but is (cost-)effective when conditioned on biomarker status as identified by the test. In this scenario, reimbursement of both test and drug will depend on the level of uncertainty surrounding the relationship between the biomarker and the treatment effect of the drug.
The drug is not (cost-)effective in either an untested or tested population. In this scenario, neither the drug nor the test is subsidized.
Possible Applications of the Assessment Framework
The framework was developed to assess a new personalized medicine in the first instance (prototype situation). However, it was recognized that reimbursement of a drug and its companion test may not occur contiguously, nor would the test and drug be necessarily submitted for funding by the same applicant, in which case the framework needed to be sufficiently flexible to address different reimbursement situations (Table 2). These situations are described in more detail below.
Table 2.
Reimbursement Situations Requiring Different Applications of the Assessment Framework
| Reimbursement Situation | Biomarkera | Test | Drug |
|---|---|---|---|
| Prototype situation (see Additional file 1) | Probable new marker | New reimbursement application | New reimbursement application |
| Situation I | Valid marker | Currently reimbursed | New reimbursement application |
| Situation II | Valid marker | New reimbursement application | Currently reimbursed |
| Situation III | Valid marker | New reimbursement application | New reimbursement application |
| Situation IV | Group of markers | Currently reimbursed ± new reimbursement application | Currently reimbursed ± new reimbursement application |
Prototype Situation
The framework for assessing personalized medicines consists of 5 domains and a checklist of 79 items (see Additional File 1). Section A provides the rationale for the codependent relationship between biomarker test and drug, section B provides the supporting evidence of clinical benefit (in a manner that allows the linkage of different types of evidence when direct evidence is not available, see also Additional File 2), section C outlines how the evidence of clinical benefit can be translated to the local setting, section D provides the economic model incorporating clinical and cost data for the biomarker test and drug and for the drug without use of the test, and section E describes the financial or budgetary impact of funding both test and drug.
Extensions of the Framework
In addition to the situation in which a new test and drug are being submitted for coverage or reimbursement in the context of an as yet unproven biomarker, 4 other situations were identified.
Situation I
When a new drug is submitted for reimbursement for targeting a previously established (valid)24,25 biomarker using a test that is currently reimbursed, the aim is to discriminate the superior (or noninferior) treatment effect of the drug alone. It would be inefficient to address all of the 79 items for this new drug, so only items that address specific areas of uncertainty would require assessment. This would mean that some basic information regarding the previous codependent technology assessment would need to be in the public domain.
Situation II
When a drug and companion test for an established biomarker have been accepted as cost-effective, evaluation of a new test, for the same biomarker, would require an assessment only of the comparative accuracy of the new and old test. If the spectrum of disease identified by the new test in the patient population does not change, supporting evidence of treatment effectiveness would not be required.
Situation III
When the biomarker has been previously assessed but both the proposed test and the proposed drug are new, it is likely that the majority of the checklist items would need to be assessed, although the biomarker’s prognostic or predictive impact may not need review.
Situation IV
When a new biomarker (or group of biomarkers) is identified as part of a new application, the aim is to gauge whether this new biomarker(s), when targeted by the drug, results in further improved patient health outcomes. This scenario could encompass the possibility of a new or currently listed drug, as well as a new or currently listed test (a complex scenario). Thus, all of the checklist items would need to be assessed.
Discussion
Key Considerations when Developing the Framework
Examination of the clinical effectiveness of a codependent technology requires an innovative approach to assessment. Data are needed to support the claim of a relationship between biomarker status and the treatment effect of the drug, primarily because this directly informs the decision to reimburse the test, the drug, both, or neither. The biological plausibility of the relationship is essential. Specifically, the causal pathway could suggest that the test is identifying a biomarker that is an independent prognostic factor (prognostic test), treatment effect modifier (predictive test), or both (see example in Additional File 3) or the relationship is unknown.26
A prognostic factor is a risk factor that affects the likely progress of patients regardless of the particular treatment they are given.27 If, for example, a biomarker in a tumor sample acts as an independent prognostic factor for an early death from metastatic colorectal cancer, then regardless of the treatment given (i.e., the new drug A or the old drug B), these patients will have a worse prognosis than those without the biomarker. In a reimbursement/policy framework, this indicates that the 2 health technologies (prognostic and therapeutic) have a low level of codependency. By identifying those patients with a better (or worse) prognosis, irrespective of treatment, the test may be used to provide more cost-effective targeting of the new drug, but all possible comparator treatments would need to be considered in making a reimbursement decision as they are also likely to be more cost-effective in the identified subgroup. These health technologies may include established treatments that, following a reimbursement decision, are retrospectively targeted to certain patient groups in which there will be an optimal effect in terms of toxicity, uptake, effectiveness, and cost-effectiveness. Prognostic impact can be distinguished using the study designs described in Figure 1, Figure 3, and to a lesser extent Figure 4, in Additional File 1.
Figure 1.
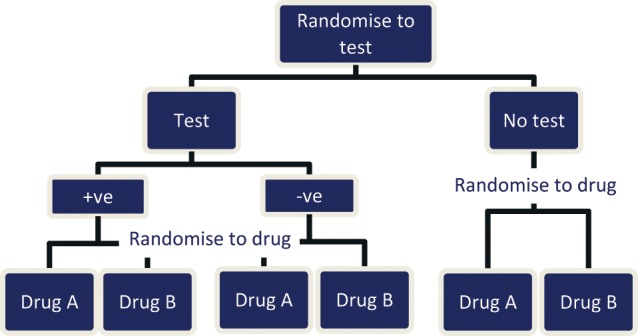
Double-randomized controlled trial.
Figure 3.
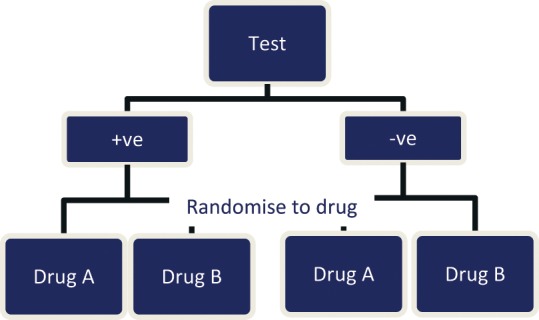
Biomarker-stratified design.
Note: At drug randomization (assuming a reasonable sample size), all variables other than biomarker status should be fairly evenly distributed in drug A and drug B groups. This explains the likely test (biomarker)–drug relationship but not the incremental benefit of the test, that is there may be uncertainty as to whether the biomarker +ve/-ve is responsible for the differential treatment effect or some other unmeasured variable.
Figure 4.
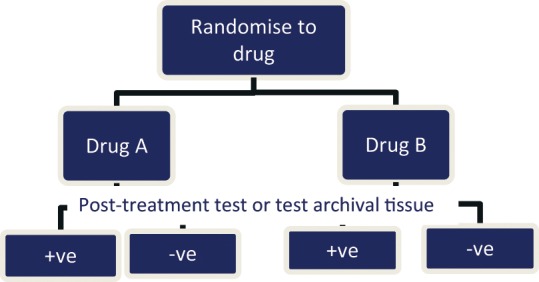
Biomarker-stratified design via subgroup analysis.
When treatment effect varies according to biomarker status, the drug and test are considered highly codependent. Drug A may have been developed specifically to target a biomarker to produce a clinical benefit to the patient (e.g., survival, quality of life, reduced complications). (In some cases, the development of the test and the treatment is a joint enterprise.) If this successfully predicts a favorable treatment effect, then among patients with the biomarker, those receiving drug A in addition to drug B would have better health outcomes than patients receiving drug B alone, whereas patients without the biomarker will receive the same clinical benefit regardless of whether drug A is used in addition to drug B. If drug A replaces drug B, then patients without the biomarker would be effectively untreated.
An adequately powered randomized trial that prospectively assesses comparative treatment effect on patient-relevant health outcomes according to subgroups delineated by the biomarker would be ideal to determine whether an effect modification is occurring. As prediction of treatment effect variation suggests that there is a unique relationship between the biomarker and drug, reimbursement of both technologies would be considered, particularly if this predicts a qualitative difference (i.e., better rather than worse or noninferior) rather than a quantitative difference (i.e., the extent of effect is improved). Treatment effect modification can be distinguished using the study designs described in Figure 1, Figure 3, and to a lesser extent Figure 4, in Additional File 1.
A comparison with a “no testing” arm similarly allows the incremental benefit of the biomarker test to be determined, that is, receiving drug A when biomarker positive and drug B when biomarker negative versus drug A being administered to everyone (see Figure 1 and Figure 2, Additional File 1). This may assist when there is uncertainty as to whether the biomarker explains the differential treatment effect between drug A and drug B or whether some other unmeasured variable is responsible.
Figure 2.

Single-randomized controlled trial (targeted treatment)
Note: This design cannot explain test (biomarker)-drug relationship. Additional evidence would need to be provided to show whether the biomarker is a treatment-effect modifier or a prognostic factor.
It is helpful to envision the ideal randomized controlled trial evidence that would be needed to answer the decision makers’ question as to whether the biomarker test and/or drug should be subsidised.28 In this case, a double-randomized controlled trial (Figure 1, Additional File 1) may be considered ideal evidence as it addresses each of the biomarker-drug relationship issues described previously. However, the practicalities are such that trials of this design are rarely, if ever, going to be conducted. As the aim of biomarker targeting is to maximize the therapeutic effect of a drug, and it is more efficient (both financially and logistically, i.e., in terms of sample size requirements) to measure this effect in a biomarker-enriched population (particularly when the biomarker is uncommon), it is unlikely that double-randomized controlled trials would be conducted. Similarly, if the new drug therapy is meant to replace an existing therapy in patients who are biomarker positive rather than to be used in combination with an existing therapy, then it could be considered unethical to conduct a trial where there is a chance that biomarker-negative patients would be receiving a new drug that has no effect on them, apart from perhaps an increased risk of adverse events, and be forgoing a known effective treatment.
Given these practical limitations with double randomized controlled trials, it was considered reasonable to take a pragmatic approach and allow an applicant for a codependent technology to build a chain of argument through the linkage of different types of evidence (“linked evidence approach”; see Additional File 2 and Option 2, Section B, in Additional File 1). The key is to present this linkage so that decision makers can see that obvious uncertainties have been addressed, that data are defensibly transferrable across different parts of the linkage, and that available evidence for the linkage has been gathered systematically and transparently and has been executed in an internally valid manner (i.e., the data are not selectively used or affected by bias and confounding).
Other factors need to be considered when assessing a personalized medicine, including whether the test or drug is additional to the current tests or treatment being received or replaces them; if, when using a linked evidence approach, there is a reference standard for the biomarker test or whether the test itself is proposed as the reference standard21,29; whether testing can be conducted on biopsied tumor samples taken at diagnosis or after first-line treatment; or whether the method of sample preservation, storage, or previous treatment or instability of the biomarker state over time will affect the accuracy of the test results. Some biomarkers are identified only through the use of multiple tests, and the positive and negative predictive value of these tests will vary according to the prevalence of a biomarker state in the population being tested. Each of these factors has been identified as requiring an answer in the assessment framework.
International Context
The implications of poor primary research and/or poor assessment frameworks to address personalized medicines include 1) fragmented or poor decision making as a consequence of considering the drug and biomarker test independently, rather than as an integrated package (see Table 1); 2) poor guidance to trialists and industry regarding appropriate trial design and thus wasted resources in producing and presenting suboptimal evidence to funding agencies; and 3) poor health outcomes for patients as a consequence of receiving ineffective or potentially harmful treatment if a personalized medicine has not been assessed rigorously and yet is reimbursed.
Most countries that ascribe to the evidence-based assessment of technologies to make resource and policy decisions have guidance available on evaluating single interventions, such as drugs.23 Guidance on the assessment of tests has been produced only recently (2008–2011) in the United States,30–32 England,33 and Europe.34 Australia developed its own guidance for the assessment of diagnostic tests for reimbursement purposes in 2005,28,35 proposing a linked evidence approach when assessing tests, which has subsequently been recommended in each of the international guidance documents mentioned above. Although there have been recent developments in regulatory policy in the United States to allow joint approval of a codependent test and drug,32 to our knowledge, no authority to date has developed a system to evaluate a package of codependent test and drug technologies for reimbursement purposes. As can be seen from the framework that has been developed, there are many domains where both the biomarker test and drug need to be considered together when evaluating their clinical benefit and cost-effectiveness.
Strengths and Limitations of the Framework
The assessment framework that has been developed is novel as it tackles the concept of personalized medicine within a coverage or reimbursement context. A formal assessment framework provides clarity for industry, with regard to policy makers’ expectations, and can drive research aimed at addressing these expectations. It also facilitates consistency in decision making and helps to identify areas of uncertainty for a reimbursement decision. The framework recognizes that often the “ideal” clinical evidence to address decision makers’ questions is not available. This is both a strength and limitation of the framework. It is a strength in that this pragmatic approach allows potentially beneficial medicines to be subsidized, despite deficiencies in the supporting evidence.36 However, linking evidence from different studies conducted in different populations can never provide evidence about the impact of a new biomarker test and new drug on patient outcomes with the same strength and quality as a double-randomized controlled trial. A trial would capture the entire causal pathway, including the unexpected and unknown effects.28 The linkage of individual pieces of evidence to estimate the effect of a trial must therefore be applied and interpreted with caution. Identifiable uncertainties or assumptions concerning linkages in the pathway can be explored using decision analytic modeling, but modeling itself may be prone to oversimplification and potential bias.
An area of economic uncertainty for decision makers is how to allocate value to the components in a codependent technology package.37 Australia assesses value across a number of technologies, including diagnostics, but in current practice, the Australian system is quite passive. Suppliers of the technologies are allowed to set a price for each component reflective of the supplier’s notion of value, and then decision makers judge whether the value of the package as a whole is acceptable in terms of incremental cost-effectiveness. This might be problematic when the supplier differs for each of the technologies in a package or for health systems that actively allocate value but are inflexible in revising this value when 2 technologies become linked.
Another potential limitation of the assessment framework is that it has yet to be evaluated over the long-term or empirically assessed as to its utility. Applications for personalized medicines have been accepted in Australia using the framework since late 2010 via a newly created Health Technology Assessment Access Point (HTAAP). This process case manages a personalized medicine to ensure that each codependent technology is appraised by the relevant decision-making committee and that coordinated advice is provided for a reimbursement decision. Applicants to the HTAAP are encouraged to use the framework (described as the Draft Information Requests for Assessing Co-dependent Technologies on the HTAAP site, http://www.health.gov.au/internet/hta/publishing.nsf/Content/co-1, and in the HTAAP information pack for applicants) as it is the backbone upon which more detailed guidelines are being produced on codependent technology evaluation. It is also conceptually consistent with the current Australian guidelines for evaluating diagnostic tests and drugs.23,35
Australia is a small market in global terms so, currently, 4 applications have been evaluated since the codependent technology framework was drafted, with another 4 commencing the submission process. The rate of applicants seeking reimbursement of these technologies has increased rapidly, although it is unclear whether this is because there is now a recognized method outlining the type of evidence that policy makers expect to see or because there have been more personalized medicines getting regulatory approval. Either way, reports suggest that the framework has assisted in providing valuable guidance to the decision maker, facilitating efficient processing of a reimbursement decision for both biomarker test and drug.
Acknowledgments
Our thanks go to Brian Richards, Graeme Suthers, Brian K. Harrison, Robyn Ward, and Lloyd Sansom for providing input on the items described in the framework.
Footnotes
Financial support for this project was provided in part by a contract with the Australian Government Department of Health and Ageing. The funding agreement ensured the authors’ independence in designing the framework, interpreting the data, writing and publishing the report. The following author is employed by the sponsor: Andrew Mitchell.
Supplementary material for this article (additional files 1, 2, and 3) is available on the Medical Decision Making Web site at http//mdm.sagepub.com/supplemental.
References
- 1. Finkelstein Y, Bournissen FG, Hutson JR, Shannon M. Polymorphism of the ADRB2 gene and response to inhaled beta- agonists in children with asthma: a meta-analysis. J Asthma. 2009;46(9):900–5 [DOI] [PubMed] [Google Scholar]
- 2. Roden DM, Altman RB, Benowitz MD, et al. Pharmacogenomics: challenges and opportunities. Ann Intern Med. 2006;145:749–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang B, Wang J, Huang SQ, Su HH, Zhou SF. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr Drug Metab. 2009;10(7):781–834 [DOI] [PubMed] [Google Scholar]
- 4. Xie H-G, Frueh FW. Pharmacogenomics steps toward personalized medicine. Future Med. 2005;2(4):325–37 [DOI] [PubMed] [Google Scholar]
- 5. Food and Drug Administration, Department of Health and Human Services Drug-diagnostic co-development: concept paper. 2005
- 6. Scartozzi M, Bearzi I, Mandolesi A, et al. Epidermal growth factor receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer. 2009;9:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65 [DOI] [PubMed] [Google Scholar]
- 8. Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–10 [DOI] [PubMed] [Google Scholar]
- 9. Shak S. Overview of the trastuzumab (Herceptin) anti-HER2 monoclonal antibody clinical program in HER2-overexpressing metastatic breast cancer. Herceptin Multinational Investigator Study Group. Semin Oncol. 1999;26(4 Suppl 12):71–7 [PubMed] [Google Scholar]
- 10. Tamura K, Okamoto I, Kashii T, et al. Multicentre prospective phase II trial of gefitinib for advanced non-small cell lung cancer with epidermal growth factor receptor mutations: results of the West Japan Thoracic Oncology Group trial (WJTOG0403). Br J Cancer. 2008;98(5):907–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Essers BA, Seferina SC, Tjan-Heijnen VC, et al. Transferability of model-based economic evaluations: the case of trastuzumab for the adjuvant treatment of HER2-positive early breast cancer in the Netherlands. Value Health. 2010;13(4):375–80 [DOI] [PubMed] [Google Scholar]
- 12. Meckley LM, Gudgeon JM, Anderson JL, Williams MS, Veenstra DL. A policy model to evaluate the benefits, risks and costs of warfarin pharmacogenomic testing. Pharmacoeconomics. 2010;28(1):61–74 [DOI] [PubMed] [Google Scholar]
- 13. Wong W, Carlson J, Thariani R, Veenstra D. Cost effectiveness of pharmacogenomics: a critical and systematic review. Pharmacoeconomics. 2010;28(11):1001–13 [DOI] [PubMed] [Google Scholar]
- 14. Conti R, Veenstra DL, Armstrong K, Lesko LJ, Grosse SD. Personalized medicine and genomics: challenges and opportunities in assessing effectiveness, cost-effectiveness, and future research priorities. Med Decis Making. 2010;30(3):328–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parliamentary Office of Science and Technology Personalised medicine. Postnote. 2009;329 [Google Scholar]
- 16. Terasawa T, Dahabreh I, Castaldi P, Trikalinos T. Systematic reviews on selected pharmacogenetic tests for cancer treatment: CYP2D6 for tamoxifen in breast cancer, KRAS for anti-EGFR antibodies in colorectal cancer, and BCR-ABL1 for tyrosine kinase inhibitors in chronic myeloid leukemia. Draft. Rockville, MD: Agency for Healthcare Research and Quality; 2009 [PubMed] [Google Scholar]
- 17. Laksman Z, Detsky AS. Personalized medicine: understanding probabilities and managing expectations. J Gen Intern Med. 2011;26(2):204–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meckley LM, Neumann PJ. Personalized medicine: factors influencing reimbursement. Health Policy. 2010;94(2):91–100 [DOI] [PubMed] [Google Scholar]
- 19. Schmitt F. HER2+ breast cancer: how to evaluate? Adv Ther. 2009;26(Suppl 1):S1–8 [DOI] [PubMed] [Google Scholar]
- 20. Australian Government Department of Health and Ageing Review of Health Technology Assessment in Australia: A Discussion Paper. Canberra, Australis: Commonwealth of Australia; 2009 [Google Scholar]
- 21. Buckley E, Merlin T. Molecular testing for the diagnosis of systematic mast cell disease, hypereosinophilic syndromes and chronic eosinophilic leukaemia. MSAC Application 1125b. Canberra, Australia: Australian Government; 2010 [Google Scholar]
- 22. Bradford Hill A. The environment and disease: association or causation? Proceedings of the Royal Society of Medicine. 1965;58:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Australian Government Department of Health and Ageing Guidelines for Preparing Submissions to the Pharmaceutical Benefits Advisory Committee. Canberra, Australia: Commonwealth of Australia; 2008 [Google Scholar]
- 24. US Department of Health and Human Services Food and Drug Administration Guidance for Industry: Pharmacogenomic Data Submissions. Procedural. Rockville, MD: Food and Drug Administration; 2005 [Google Scholar]
- 25. Food and Drug Administration Expert Working Group (Efficacy) of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Guidance for Industry: E15 Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories. US Department of Health and Human Services [Google Scholar]
- 26. Lee CK, Lord SJ, Coates AS, Simes RJ. Molecular biomarkers to individualise treatment: assessing the evidence. Med J Aust. 2009;190(11):631–6 [DOI] [PubMed] [Google Scholar]
- 27. Clark GM, Zborowski DM, Culbertson JL, et al. Clinical utility of epidermal growth factor receptor expression for selecting patients with advanced non-small cell lung cancer for treatment with erlotinib. J Thorac Oncol. 2006;1:837–46 [PubMed] [Google Scholar]
- 28. Lord SJ, Irwig L, Bossuyt PM. Using the principles of randomized controlled trial design to guide test evaluation. Med Decis Making. 2009;29(5):E1–E12 [DOI] [PubMed] [Google Scholar]
- 29. Merlin T, Weston A, Tooher R. Extending an evidence hierarchy to include topics other than treatment: revising the Australian “levels of evidence.” BMC Med Res Methodol. 2009;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agency for Healthcare Research and Quality Methods Guide for Medical Test Reviews. Rockville, MD: Agency for Healthcare Research and Quality, US Department of Health and Human Services; 2010 [Google Scholar]
- 31. Centers for Disease Control and Prevention, Office of Public Health Genomics ACCE Model List of 44 Targeted Questions Aimed at a Comprehensive Review of Genetic Testing 2010 [cited]. Available from: URL: http://www.cdc.gov/genomics/gtesting/ACCE/acce_proj.htm
- 32. Food and Drug Administration In vitro companion diagnostic devices: draft guidance for industry and Food and Drug Administration staff. In: US Department of Health and Human Services, ed. Rockville, MD: US Department of Health and Human Services; 2011 [Google Scholar]
- 33. NICE Interim Methods Statement (Pilot). Version 8. London: National Institute of Health and Clinical Excellence Centre for Health Technology Evaluation, Diagnostics Assessment Programme; 2010 [Google Scholar]
- 34. EUnetHTA HTA Core Model for Diagnostic Technologies. Work Package 4: European Network for Health Technology Assessment; 2008 [Google Scholar]
- 35. Medical Services Advisory Committee Guidelines for the Assessment of Diagnostic Technologies. Canberra, Australia: Commonwealth of Australia; 2005 [Google Scholar]
- 36. Steinberg EP, Tunis S, Shapiro D. Insurance coverage for experimental technologies. Health Affairs. 1995;14:143–58 [DOI] [PubMed] [Google Scholar]
- 37. Garrison LP, Austin MJF. The economics of personalized medicine: a model of incentives for value creation and capture. Drug Information Journal. 2007;41:501–9 [Google Scholar]