

Abstract
Reversible, heterolytic addition of H2 across an iron-boron bond in a ferraboratrane with formal hydride transfer to the boron gives iron-borohydrido-hydride complexes. These compounds catalyze the hydrogenation of alkenes and alkynes to the respective alkanes. Notably, the boron is capable of acting as a shuttle for hydride transfer to substrates. The results are interesting in the context of heterolytic substrate addition across metal-boron bonds in metallaboratranes and related systems, as well as metal-ligand bifunctional catalysis.
Keywords: Metallaboratrane, non-noble metal hydrogenation catalysis, heterolytic dihydrogen activation
INTRODUCTION
Transition metal-catalyzed bond forming reactions often involve formal two-electron redox steps (e.g., oxidative addition and reductive elimination). Noble metal catalysts are commonly used in these reactions due, in part, to their propensity to facilitate multi-electron processes.1 There is growing interest in developing catalysts for bond forming/cleavage reactions based on earth abundant mid-to-late first-row transition metals, a goal that presents a unique set of challenges.2 First-row transition metal catalysts that can circumvent undesirable one electron processes in favor of concerted two electron reaction steps present one plausible design criterion.3
Cooperative catalysis strategies that utilize ligands that operate in tandem with a coordinated metal center to activate substrates have shown promise in addressing this issue.4,5 First-row metallaboratranes and related compounds that contain a retrodative M→B σ-interaction6 are appealing as catalysts7 because of the boron center’s ability to stabilize low valent metals.8 Akin to frustrated Lewis pairs,9 it has also been recently demonstrated that the metal-boron interaction can cooperatively facilitate the activation of H2.5,7 For instance, our lab recently reported that the diphosphine-borane nickel (MesDPBPh)Ni complex undergoes reversible oxidative addition of H2 to afford a nickel-borohydrido-hydride complex (Scheme 1).10 This nickel system is an efficient catalyst for olefin hydrogenation. Kameo and Nakazawa have also reported on the transfer hydrogenation of ketones catalyzed by a rhodium diphosphine-borane complex.11
Scheme 1.
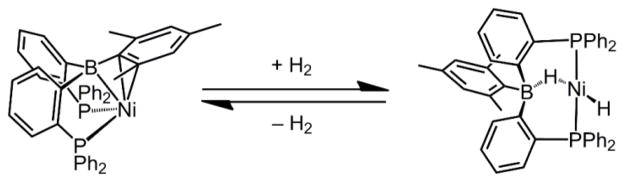
Related H2 Activation Across a Ni-B Bond
Herein we report on studies of heterolytic H-H bond cleavage at a ferraboratrane complex12 of a triphosphine-borane (TPB) ligand.13 Dihydrogen is shown to add reversibly across the Fe-B bond of (TPB)Fe(L) complexes to form corresponding iron-borohydrido-hydride complexes of the type (TPB)(μ-H)Fe(L)(H). Like the nickel system reported previously, olefin hydrogenation catalysis is accessible, albeit much slower, thereby facilitating detailed studies. As discussed below, other E-H bonds are also activated by the ferraboratanes described, including the terminal C-H bonds of arylacetylenes and the C-H bonds of formaldehyde.
RESULTS AND DISCUSSION
Reversible H2 Addition
Exposing the previously reported (TPB)Fe(N2) complex12a (1) in d6-benzene to H2 (1.2 equiv) at room temperature results in H2 addition across the Fe-B bond to give a yellow solution of the six coordinate borohydride-hydride-N2 complex (TPB)(μ-H)Fe(N2)(H) (2) (Scheme 2, Figure 1). The XRD structure of 2 shows that the Fe-B distance is significantly elongated relative to 1 (2.604(3) Å in 2 versus the calculated distance of 2.2 Å in 112b) (Figure 1). A terminal hydride ligand and a bridging hydride ligand located between the B and Fe atoms can be assigned from the electron density difference map. This structure also prevails in solution. In the 1H NMR spectrum (d6-benzene), the terminal hydride ligand on iron is observed as a triplet-of-doublets at −9.6 ppm, and the bridging hydride is observed as a broad singlet at −30.4 ppm. Replacing H2 with D2 in the reaction gives the corresponding isotopologue (TPB)(μ-D)Fe(N2)(D). Along with the expected deuteride signals in the 2H NMR spectrum, deuterium signals from the methine and terminal methyl positions of the isopropyl groups of the TPB ligand are observed. This observation establishes that facile scrambling of the hydridic ligands into the TPB isopropyl groups occurs, presumably via a reversible C-H metalation process.
Scheme 2.

H-H and E-H Bond Activations Across Fe-B Bonds
Figure 1.
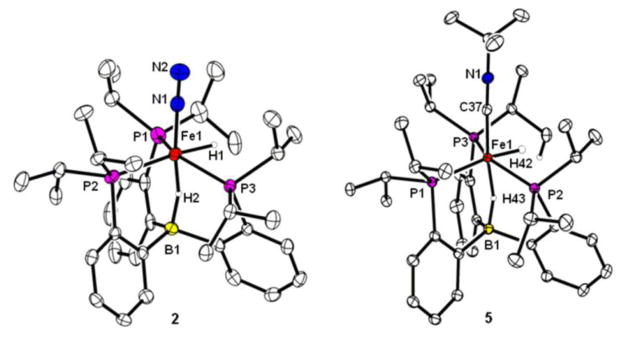
XRD structures of 2 (left) and 5 (right). Ellipsoids shown at 50% probability. Selected bond distances (Å): 2, Fe1-H1 = 1.42(2), Fe1-H2 = 1.49(2), B1-H2 = 1.17(2), Fe1-B1 = 2.604(3); 5, Fe1-H42 = 1.35(2), Fe1-H43 = 1.52(2), B1-H43 = 1.20(2), Fe1-B1 = 2.673(2).
The dinitrogen ligand (νNN = 2070 cm−1) in 2 is labile, and it can be substituted under excess H2 to give the dihydrogen analogue (TPB)(μ-H)Fe(H2)(H) (3) (Scheme 2). Exposing 1 to excess H2 (1 atm) also generates 3. Its XRD structure again confirms the presence of a bridging hydride (Figure 2). Although electron density can be located in the difference map between a widened P-Fe-P angle (136.54(2)°) and in the apical position trans to the borohydride unit, the data do not allow us to reliably distinguish between classical and non-classical hydrides. We therefore turned to NMR spectroscopy to aid in the formulation of 3.
Figure 2.
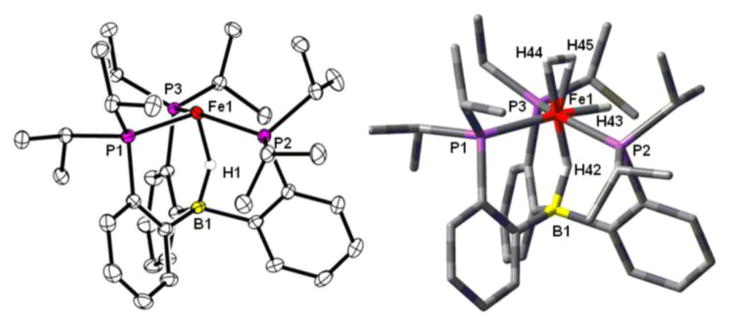
XRD (left) and DFT optimized (right) structures of 3. Ellipsoids shown at 50% probability. Selected bond distances (Å) and angles (°): XRD, Fe1-H42 = 1.56(2), B1-H42 = 1.21(2), Fe1-B1, 2.63(2), P2-Fe-P3 = 136.54(2); DFT predicted, Fe1-H42 = 1.51, Fe1-H43 = 1.50, Fe1-H44 = 1.62, Fe1-H45 = 1.59, B1-H42 = 1.24, Fe1-B1 = 2.63, H44-H45 = 0.84, P2-Fe1-P3 = 140.63.
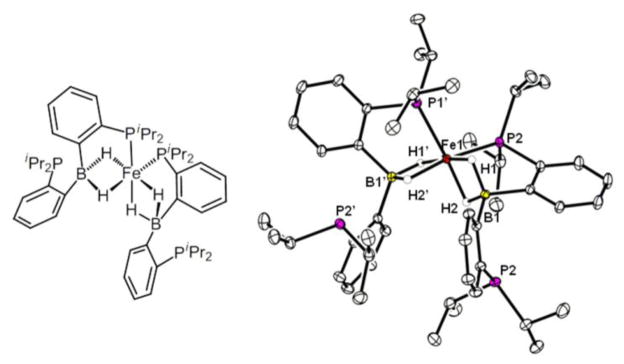
The 20 °C 1H NMR spectrum (d8-toluene) of 3 (Figure 3) shows a broad singlet resonance at −15.1 ppm, indicative of hydridic protons. A broad deuteride signal is observed in the 2H NMR spectrum when D2 is used in place of H2, and, like in 2, deuterium signals are also observed in the methyl and methine positions of the isopropyl groups of the TPB ligand. Cooling a d8-toluene solution of 3 under an H2 atmosphere to −20 °C leads to sharpening of the resonance at −15.1 ppm, which integrates to three protons (3H). A second, broad hydridic resonance integrating to one proton is also observed at −24.9 ppm, and in analogy to 2, this resonance is assigned to the bridging-borohydride (μ-H). Cooling down to −90 °C leads to broadening of the 3H resonance without reaching decoalescence, suggesting that exchange of three hydrogenic ligands is fast on the NMR timescale. Compound 3 can be heated to 50 °C before significant sample decomposition is observed (vide infra). At 50 °C, the 3H and μ-H signals both broaden into the baseline, suggesting that exchange of all four hydrogens is facile at this temperature.
Figure 3.

1H VT-NMR spectra of 3 in d8-toluene under 1 atm of H2.
To further assign the 3H unit in 3 (i.e. dihydrogen-hydride versus trihydride), we turned to minimal longitudinal relaxation (T1min) measurements.14,15 The T1min is 35 ms at −32 °C for the 3H resonance, which suggests that the 3H unit is best described as dihydrogen-hydride and implying the following assignment for 3: (TPB)(μ-H)Fe(H2)(H). This interpretation is additionally supported by DFT calculations (RB3LYP/6-31G(d)) that identify a dihydrogen-hydride structure (Figure 2 and Supporting Information) as the lowest energy isomer of 3. A stereoisomer in which the H2 ligand occupies the equatorial position and the hydride ligand is in the axial position trans to boron is calculated to be 6.1 kcal/mol higher in energy. This geometric preference parallels that of the well-characterized iron-dihydrogen-dihydride complex mer-Fe(H2)(H)2(PEt2Ph)3.16 The transition state for conversion of 3 into this higher energy stereoisomer involving H-H scission is calculated to be 6.7 kcal/mol above the most stable isomer and is in line with the observed exchange behavior on the NMR timescale.
Analogous (TPB)(μ-H)Fe(L)(H) complexes (L = CNtBu (5), CO (7)) can be synthesized (Scheme 2, Figure 1). To explore the effect of the apical ligand L on the H-H bond activation process, (TPB)Fe(L) complexes (L = CNtBu, 4; L = CO, 6) were prepared. The previously reported carbonyl complex 6 displays an η3-interaction with a P-CAr-CAr unit of the TPB ligand12a whereas isocyanide adduct 4, whose structure has been determined, does not: its Fe center is rigorously 5-coordinate. Complex 6 is diamagnetic whereas 4 gives rise to a solution magnetic susceptibility (μeff = 1.7 μB) at room temperature in C6D6. The temperature dependence of the solution susceptibility suggests an S = 0 ground state with a thermally accessible S = 1 state. No reaction occurs between (TPB)Fe(CNtBu) (4, μeff = 1.7 μB) or the previously reported (TPB)Fe(CO)12a (6) with H2 (1 atm) at room temperature over a period hours. Compound 4 is fully consumed by H2 (1 atm) over the course of 3 days at 40 °C to generate 5, while compound 6 is fully consumed by H2 (1 atm) over the course of 5 days at 80 °C to give 7. The increase in temperature and reaction time compared to the facile room temperature reaction between 1 and H2 is consistent with a scenario in which H2 substitution for L occurs prior to H2 addition across the Fe-B bond. Complex 7 can be alternatively synthesized from 1 or 2 and formaldehyde (Scheme 2).
Dihydrogen addition across the Fe-B bond of 1 is reversible. Conversion of 3 to 2 and subsequently back to 1 can be effected by exposing 3 to dynamic vacuum and then N2, or by repeated freeze-pump-thaw-N2 cycles. Reformation of the Fe-B bond can also occur through hydride transfer to unsaturated substrates (vide infra). Dihydrogen elimination from 5 and 7 does not occur when treated similarly.
Worth underscoring is that cleavage of the Fe-B bond in the present ferraboratrane system is distinct from the H2 chemistry observed for a structurally related (SiPiPr3)Fe silatrane system (SiPiPr3= [Si(o-C6H4PiPr2)3]−,) we have introduced elsewhere.17,18,19 For instance, the reaction between (SiPiPr3)Fe(N2) and H2 affords (SiPiPr3)Fe(H2), and that between [(SiPiPr3)Fe(N2)]+ and H2 affords [(SiPiPr3)Fe(H2)]+. No disruption of the Fe-Si bond is observed in either case, even if for instance isolated [(SiPiPr3)Fe(N2)]+ or [(SiPiPr3)Fe(H2)]+ is exposed to excess H2. Ligand substitution instead occurs. This sharply contrasts isoelectronic (TPB)Fe(N2) 1, where H2 addition readily affords the cleavage product 2 or 3. We also find (this report, Scheme 3) that hydrogenolysis of the iron(II) methyl complex (SiPiPr3)Fe(Me) occurs slowly at 60 °C to give an H2/H product, but once again without disruption of the Fe-Si bond. Addition of exogenous donor ligands such as H2, N2 and CO effects substitution of the coordinated H2 ligand, but the Fe-Si bond is maintained. One factor contributing to the difference between the two systems is likely the more flexible Fe-B bond in the (TPB)Fe system, as reflected in the variable Fe-B bond distances (varying by ca. 0.5 Å)12b versus more rigid Fe-Si bond distances (varying by ca. 0.2 Å)17,18,19 in (SiPiPr3)Fe that have been observed over several formal iron oxidation states. The rigidity of the Fe-Si interaction presumably reflects an appreciably stronger Fe-Si bond relative to Fe-B. One can additionally consider the relative Lewis acidity of the Ar3B versus the Ar3Si+ subunit18 in these respective systems, and their propensity to serve as H− acceptors, but one might then predict the Ar3Si+ to be the better acceptor, in contrast to the experimental observations. Indeed, this latter point may be the reason the Fe-Si interaction is stronger than the Fe-B interaction.
Scheme 3.

Chemistry of the Related Silatrane System
Reaction with Unsaturated Substrates
The ability of the (TPB)Fe scaffold to reversibly cleave H2 prompted us to study if the transfer of hydrogen from (TPB)(μ-H)Fe(L)(H) to substrates is possible. The reaction of 1 with ethylene, styrene and arylacetylenes was probed. A degassed d6-benzene solution of 1 reacts with ethylene to give a light brown solution of the paramagnetic iron-ethylene adduct (TPB)Fe(C2H4) (8; μeff = 3.2 μB, S = 1) (Scheme 4). Brown XRD quality crystals of 8 can be grown under an atmosphere of ethylene at 0 °C. Two molecules of 8 are found in the asymmetric unit cell. The iron center is bound η2 to ethylene ((Fe-C)avg = 2.108(1) Å) and lies above the plane defined by the phosphine donors by an average distance of 0.641 Å, with a corresponding elongation of the average Fe-B distance to 2.491(2) Å (compared to 2.2 Å in 1). The average C-C bond distance ((C-C)avg = 1.397(2) Å) of the η2-coordinated ethylene molecule is significantly elongated from that in free ethylene (1.337 Å). 20 The data are consistent with π-backbonding from iron to ethylene, which confers significant ferracyclopropane character to 8.21 Storing 8 for two days under an atmosphere of N2 fully regenerates 1.
Scheme 4.
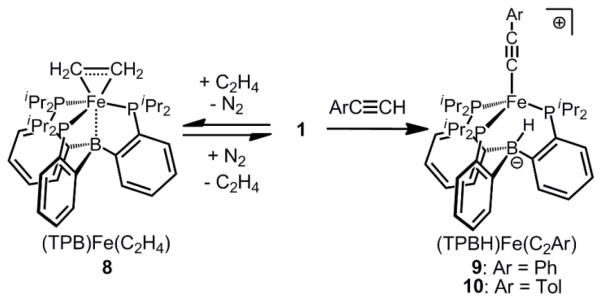
Ethylene Coordination and Arylacetylene C-H Bond Activation by 1
Compound 1 does not afford a detectable styrene adduct but reacts with both phenyl- and tolylacetylene with formal hydride transfer from the terminal C(sp)-H of the arylacetylene to the boron, forming S = 2 iron-borohydrido-arylacetylide complexes (TPBH)Fe(C2Ar) (Ar = Ph, 9, μeff = 5.1 μB; Ar = Tol, 10, μeff = 5.2 μB) (Scheme 4). Two molecules of 10 are found in the asymmetric unit, and the XRD structure shows the presence of a tolylacetylide ligand coordinated to a pyramidalized iron center (Figure 4). While the hydride on the boron cannot be reliably located by XRD, the IR spectra for both 9 and 10 show B-H stretches at 2490 cm−1 and 2500 cm−1, respectively, most consistent with a non-bridging B-H unit. The vibrational bands shift to 1826 cm−1 (predicted 1834 cm−1) for 9 and 1824 cm−1 (predicted 1841 cm−1) for 10 upon labeling with the monodeuterated arylacetylene (ArC≡CD).
Figure 4.
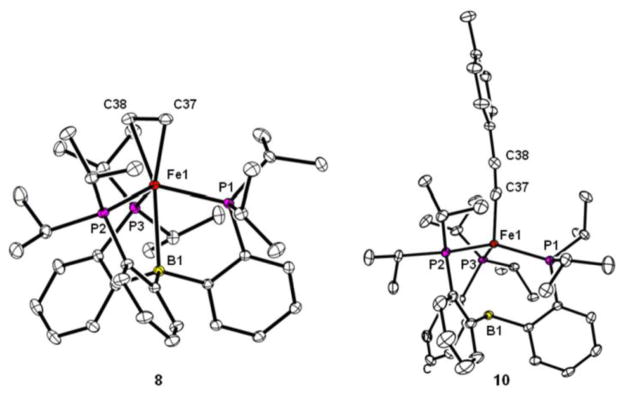
XRD structures of 8 (left) and 10 (right). Ellipsoids shown at 50% probability. Selected bond distances (Å) and angles (°): 8 (average for two molecules in the asymmetric unit cell), Fe1-B1 = 2.491(1), Fe1-C37 = 2.103(1), Fe1-C38 = 2.113(1), C37-C38 = 1.397(2), Σ(P-Fe-P) = 338.76(3); 10 (average for two molecules in the asymmetric unit cell), Fe1-B1 = 2.761(2), Fe1-C37 = 1.918(2) C37-C38 = 1.169(3), Σ(P-Fe-P) = 345.07(2).
The activation of the arylacetylene C(sp)-H bond by 1 is reversible. Mixing a d6-benzene solution of 9 with tolylacetylene (4 equiv) and, conversely, mixing a d6-benzene solution of 10 with phenylacetylene (4 equiv) both result in a mixture of 9 and 10 (Scheme 5A). The corresponding exchange reactions with B-D labelled isotopologues of 9 or 10 ((TPBD)Fe(C2Ar)) and a different all-protio arylacetylene (Ar’C≡CH) result in the exclusive formation of free ArC≡CD, indicating that the arylacetylene unit is reductively eliminated from the iron-borohydrido-arylacetylide complexes prior to activation of an incoming acetylene substrate, presumably by reversible hydride transfer from the boron to the arylacetylide to form intermediate π-adducts akin to 8 (Scheme 5B).
Scheme 5.
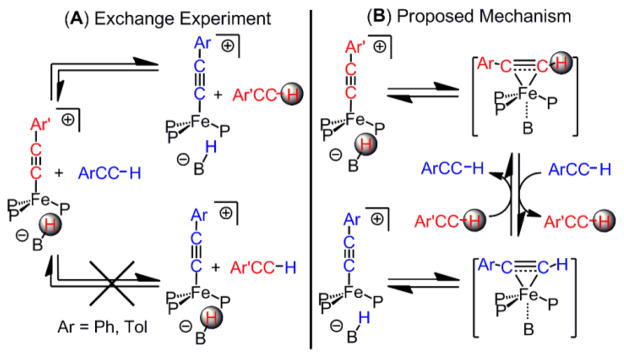
Reversible Arylacetylene C-H Bond Activation
Stoichiometric Hydrogenations
We also explored whether the transfer of hydrogen from (TPB)(μ-H)Fe(L)(H) species to unsaturated substrates might be possible. Exposing 8 to excess H2 (1 atm) results in complete conversion of ethylene to ethane and 3 as the iron-containing product (Scheme 6-i) in less than 12 h. A paramagnetic intermediate (A) and 8 can be observed by in situ 1H NMR spectroscopy. The same paramagnetic intermediate A and 8 can also be observed by in situ 1H NMR spectroscopy if the same reaction is run under ethylene (1 atm) and H2 (1 atm). The IR spectrum of the reaction mixture shows a diagnostic terminal B-H vibration at 2470 cm−1 that is attributed to A. Akin to the iron-borohydrido-alkynyl complexes 9 and 10, A is assigned to the iron-borohydrido-ethyl complex (Scheme 6-i).22 Using D2 in place of H2 in the ethylene hydrogenation reaction and monitoring leads to the observation of both B-D and B-H stretches in the IR spectrum, which is consistent with facile insertion/β-hydride elimination processes prior to ethane elimination from A. We note the similarity of A (and 9 and 10) to the well-characterized zwitterionic, tris(phosphino)borate-iron-ethyl complex (PhBP3)Fe(Et) that was observed as an intermediate in ethylene hydrogenation with the previously reported iron (PhBP3)Fe system.3e
Scheme 6.

Stoichiometric Hydrogenation Reactions
Complexes 1, 2, and 3 hydrogenate both phenylacetylene (1 equiv) and styrene (1 equiv) to ethylbenzene (1 atm H2) with 3 as the observable iron-containing product. The in situ 1H NMR spectrum of styrene hydrogenation reactions using 1, 2, or 3 show styrene, ethylbenzene and 3 in solution during the reaction course. Furthermore, for styrene hydrogenation with D2 the 2H NMR spectrum and GC-MS data of the reaction mixture show incorporation of deuterium onto both olefinic carbon atoms of free styrene, indicating that styrene coordination to the iron-center and insertion/β-hydride elimination processes are reversible.
Under stoichiometric conditions, the addition of 1 equiv of styrene to a solution of 2 in d6-benzene and under N2 (1 atm) cleanly generates 1 equiv of ethylbenzene and 1 (Scheme 6-ii). In contrast, running the same reaction under a static vacuum yields a mixture of styrene, ethylbenzene, 1, and 2 (Scheme 6-iii). These observations suggest that excess H2 or N2 is required for the hydrogenations to proceed to completion. Moreover, the bridging monohydride appears competent for transfer to a substrate.
Substrates including trans-stilbene, N-benzylideneaniline, acetone, and acetophenone were not hydrogenated under similar conditions. Compounds 5 and 7 also do not hydrogenate ethylene, styrene, or phenylacetylene under the same conditions.
Catalytic Hydrogenations
Under the catalytic conditions of 0.01 M 1, 1 atm of H2, and 30 equiv of the substrate in d6-benzene at room temperature, ethylene, styrene, and phenylacetylene are hydrogenated to ethane and ethylbenzene, respectively (Table 1). Compounds 2 and 3 can also be used as precatalysts. Ambient laboratory light does not affect the reaction and the catalysis is not inhibited by elemental mercury; it thus appears to be a homogeneous process. Norbornene is hydrogenated to norbornane, and with an atmosphere of D2 in place of H2 the cis-addition product exo,exo-2,3-d2-norbornane23 is exclusively observed, indicating the syn-addition of hydrogen and arguing against radical processes.
Table 1.
Catalytic Hydrogenations by 1 with H2
| Precatalyst | Substrate | Product | TOF (h−1) |
|---|---|---|---|
| 1 | Ethylenea | Ethane | 15 |
| 1 | Styrenea | Ethylbenzene | 0.27 |
| 1 | Phenylacetyleneb | Ethylbenzene | 0.16 |
Conditions: Room temperature, 0.01 M 1, 1 atm H2, and 0.01 M ferrocene as an internal integration standard in d6-benzene.
0.3 M substrate,
0.29 M substrate.
As determined by 1H NMR spectroscopy at > 95% product.
The hydrogenation catalysis was monitored by 1H NMR spectroscopy with ferrocene as an internal integration standard. As with stoichiometric ethylene hydrogenation, the in situ 1H NMR spectra of the catalytic ethylene hydrogenation reaction indicate the presence of ethylene adduct 8 and the putative ethyl-borohydride intermediate A as the iron-containing species during the reaction course. Complex 3 is the iron-containing product at the completion of the reaction. For styrene hydrogenation, styrene, ethylbenzene, and 3 are observed during catalysis, and scrambling of deuterium into the vinylic positions of styrene is observed under D2. In contrast, for phenylacetylene hydrogenation complex 9 is the only observed iron-species early in the reaction when the phenylacetylene concentration is high. As phenylacetylene is consumed, styrene and 3 form, and ethylbenzene begins to develop slowly thereafter.
Attempting to increase the rate of catalysis by elevating the reaction temperature results in catalyst decomposition. The decomposition product (11) can be synthesized independently in near quantitative yields by heating 3 (80 °C) under H2 (1 atm) for 2 h. The XRD structure of 11 indicates that a B-CAr bond is cleaved from the TPB ligand fragment (Figure 5). This result offers the cautionary note that B-CAr bond cleavage to give metal-borohydride products is a viable catalyst decomposition pathway.
Figure 5.
Chemical line representation and XRD structure of 11. Ellipsoids shown at 50% probability. Selected bond distances (Å): Fe1-H1 = 1.59(1), Fe1-H2 = 1.63(1), Fe1-B1 = 2.0900(6), Fe1-P1 = 2.2481(2).
Based on the results from the stoichiometric and catalytic experiments, we propose a plausible mechanistic scenario to account for the observed catalytic styrene hydrogenation by 1 (Scheme 7A), and in doing so underscore interesting aspects of the mechanism that remain unanswered. Starting from precatalyst 1, addition of H2 generates 3, a species that can be observed by 1H NMR spectroscopy during catalytic runs (resting state). Subsequent substitution of the apical H2 ligand for styrene forms the unobserved iron-styrene-hydride-borohydride species B, and insertion into the terminal hydride affords the iron-alkyl intermediate A′. Intermediate A′ is analogous to the ethyl species A (Scheme 6-i), and is also related to the structurally characterized acetylide-borohydride complex 10 (Scheme 4). Olefin coordination and insertion appears to be reversible, as labeling studies show deuterium is exchanged into the vinylic positions of free styrene under a D2 atmosphere. Elimination of ethylbenzene in the presence of H2 regenerates the catalyst resting state.
Scheme 7.

Mechanism and Alkane Elimination Pathways
The conversion of 1 to 3 likely proceeds via the H2-adduct intermediate C depicted in Scheme 7B by H2-for-N2 ligand exchange. While we have not detected such a species in the present (TPB)Fe system, its cobalt analogue (TPB)Co(H2) can be isolated and has been thoroughly characterized,24 as has the isoelectronic iron complex [(SiPiPr3)Fe(H2)]+.17 N2/H2 exchange is facile in these well-defined Co and Fe systems, and by extension we infer it would also be facile for (TPB)Fe(N2) 1 to afford (TPB)Fe(H2) C before additional reactions ensue. Since H2 reacts with 1, but not with 5 and 7 at room temperature, we think that facile H2 substitution for N2 most likely occurs prior to H-H bond cleavage. We appreciate that while this scenario is consistent with the data available, it is not demanded by the available data. For instance, it is alternatively possible that styrene substitution for the N2 ligand in 1 precedes H2 addition to form intermediate B. No direct evidence rules out this possibility. We prefer suggesting that the H2/dihydride species 3 precedes styrene binding because of the observation that H2 addition/activation by other (TPB)Fe(L) adducts, for example 4 and 6, is very slow, and also because N2, and presumably therefore also H2, displaces ethylene from 8 in solution (regenerating 1 or (TPB)Fe(H2)).
The final ethylbenzene elimination step can be envisioned to occur through two plausible routes (Scheme 7B). One such pathway involves a reductive elimination step where hydride transfer directly from the borohydride subunit generates the alkane product to form 3, likely via the dihydrogen adduct intermediate C. The other pathway proceeds through alkane elimination by hydrogenolysis of the phenylethyl group without hydride transfer from the borohydride subunit.
While the available data do not firmly distinguish between the two product elimination pathways shown in Scheme 7B, the stoichiometric hydrogenation studies described above show that 1 equiv of styrene is completely hydrogenated to ethylbenzene by 2 under an N2 atm (Scheme 6-ii). This observation implies that 2 can serve as the source of the two H-atom equivalents delivered to styrene. From complex 2, substitution of the apical N2 ligand for styrene in 2 would generate the styrene adduct intermediate B. Subsequent insertion of the bound styrene into the cis Fe-H would afford intermediate A′, which in the absence of H2 at least, eliminates ethylbenzene concomitant with N2 binding to reform 1. The presence of exogenous N2 (or alternatively H2) facilitates the generation of ethylbenzene. As noted in Scheme 6-iii, the stoichiometric reaction under static vacuum between 2 and styrene is quite slow compared with the same reaction under N2 (Scheme 6-ii). This observation can be explained by presuming the conversion of intermediate A′ to 1 requires N2 association prior to elimination to generate ethylbenzene.
CONCLUSIONS
In summary, we have demonstrated that the Fe-B bond in ferraboratranes (TPB)Fe(L) (where L = N2, 1; CNtBu, 4; CO, 6) can facilitate heterolytic cleavage of H2 and of the C(sp)-H and C(sp2)-H bonds of arylacetylenes and formaldehyde, respectively, resulting in Fe-B bond rupture and formal hydride transfer to the boron of the ligand scaffold. The formal hydride transfer from the C(sp)-H of arylacetylenes to give iron-acetylide complexes is distinct from traditional syntheses of metal-acetylide complexes in that a hydride equivalent is formally abstracted by the Lewis acidic borane unit in 1 from a C(sp)-H hydrogen.25
Dihydrogen addition across the Fe-B bond is reversible, and the boron is also capable of shuttling the hydride equivalent derived from H2 to unsaturated substrates under stoichiometric hydrogenation conditions. The hydrogen chemistry of this (TPB)Fe(L) system contrasts with the related nickel26 and cobalt24 complexes of TPB, where the metal-boron bond remains intact under an H2 atmosphere.
An understanding of the factors that govern metal-boron bond cleavage will aid in the development of cooperative catalytic reactions in metallaboratranes. The direct role, if any, of the borane ligand in assisting the H2 cleavage step is an interesting question in this context for the present iron and recently reported diphosphine-borane-iron systems,27 and also conceptually related to the nickel system (Scheme 1).10 Determining whether there is a cooperative interaction between the coordinated H2 ligand, the iron center, and the borane subunit en route to H-H cleavage (and its microscopic reverse), akin to H-H cleavage by frustrated Lewis pairs,9 calls for detailed theoretical studies that are the subject of ongoing research.
EXPERIMENTAL
General Considerations
All manipulations were carried out using standard glovebox or Schlenk techniques under an N2 atmosphere. Unless otherwise noted, solvents were deoxygenated and dried by thoroughly sparging with N2 gas followed by passage through an activated alumina column in the solvent purification system by SG Water, USA LLC. Deuterated solvents and D2 gas were purchased from Cambridge Isotope Laboratories, INC. The deuterated solvents were degassed and dried over activated 3 Å sieves prior to use. Unless otherwise noted, all compounds were purchased commercially and used without further purification. TPB,13 (SiPiPr3)Fe(Me),19 (SiPiPr3)Fe(N2)(H),17 and monodeuterated phenyl- and tolylacetylene (PhC2D and TolC2D)28 were synthesized by literature procedures. Elemental analyses were performed by Midwest Microlab, LLC., Indianapolis, IN.
NMR spectra were recorded on Varian 300 MHz, 400 MHz, and 500MHz spectrometers. 1H and 13C chemical shifts are reported in ppm relative to residual solvent as internal standards. 31P and 11B chemical shifts are reported in ppm relative to 85% aqueous H3PO4 and BF3·Et2O, respectively. Multiplicities are indicated by br (broad), s (singlet), d (doublet), t (triplet), quart (quart), quin (quintet), multiplet (m), d-d (doublet-of-doublets), and t-d (triplet-of-doublets).
FT-IR measurements were obtained on samples prepared as KBr pellets or in solution using a Bio-Rad Excalibur FTS 300 spectrometer with Varian Resolutions Pro software at 4 cm−1 resolution. The ATR-IR measurements were measured on a thin film of the complex obtained from evaporating a drop of the solution on the surface of a Bruker APLHA ATR-IR Spectrometer probe (Platinum Sampling Module, diamond, OPUS software package) at 2 cm−1 resolution. IR intensities indicated by s (strong), m (medium), and w (weak).
X-ray Crystallography
X-ray diffraction was measured on the Bruker Kappa Apex II diffractometer with Mo Kα radiation. Structures were solved using the SHELXS software and refined against F2 on all data sets by full matrix least squares with SHELXL. The crystals were mounted on a glass fiber with Paratone oil.
Computational Methods
Geometry optimizations were performed using the Gaussian03 package. B3LYP exchange-correlation functional was employed with a 6-31G(d) basis set. The GDIIS algorithm was used. A full frequency calculation was performed on each structure to ensure that they were the true minima. A single negative vibrational frequency was observed for the transition state between 3 and its equatorial-H2 isomer, confirming that this structure was the transition state. See SI for full a description of the computational method.
HD Gas Generation
D2O (1mL) was added to an evacuated, cooled sample (−78 °C) of solid lithium aluminum hydride (316 mg, 8.2 mmol) in a Schlenk flask. An evacuated Schlenk line was filled with the resulting HD gas (ca. 1 atm) as the Schlenk flask was warmed to room temperature. A J-Young NMR tube containing a freeze-pump-thawed solution was the respective complex was exposed to the HD gas.
Synthesis of (TPB)(μ-H)Fe(N2)(H) (2)
A J-Young NMR tube containing a brown-red solution 1 (20.3 mg, 31.1 mmol) in C6H6 (0.8 mL) was freeze-pump-thawed (3x) and, with the J-Young tube frozen with liquid nitrogen, exposed to H2 (1.2 equiv). The solution was thawed and mixed, giving a yellow solution. An atmosphere of N2 was subsequently introduced and the reaction was mixed for 2 h to yield 2 (100% yield by 1H NMR spectroscopy with a ferrocene integration standard). Alternatively, 2 could be synthesized from 3 by degassing a solution of 3 of free H2 by freeze-pump-thaw (3x), exposing it to an N2 atmosphere, and mixing the solution overnight (100% yield by 1H NMR spectroscopy with a ferrocene integration standard). Yields could not be determined by mass because 2 was unstable to prolonged exposure to dynamic vacuum. Yellow-orange XRD quality crystals were grown in a concentrated solution of pentane:THF (10:1) at −30 °C. 1H NMR (C6D6, 300 MHz): δ 7.9 (2H, d, 3JH-H = 6 Hz, Ar-H), δ 7.7 (1H, br s, Ar-H), δ 7.3 (3H, d, 3JH-H = 6 Hz, Ar-H), δ 7.0 (3H, t, 3JH-H = 9 Hz, Ar-H), δ 2.7 (4H, d, 2JP-H = 18 Hz, PCH), δ 2.4 (2H, br s, PCH), δ 1.4 (6H, d,3JH-H = 6 Hz, CH3), δ 1.3 (12H, br s, CH3), δ 1.1 (6H, d-d, 3JP-H = 15 Hz, 3JH-H = 6 Hz, CH3), δ 0.8 (6H, d, 3JP-H = 9 Hz, CH3), δ − 9.6 (1H, d-t, 2JH-Pcis = 81 Hz, 2JH-Ptrans = 36 Hz, Fe-H), δ −30.4 (1H, s, Fe-(μ-H)-B). 2H NMR (C6H6/C6D6, 76 MHz): δ −9.5 (1D, br s), δ −30.3 (1D, br s). 31P NMR (C6D6, 121 MHz): δ 73.6 (2P, s), δ 64.2 (1P, s). 13C NMR (C6D6, 125 MHz): δ 161.9 (s, CAr), δ 143.5 (s, CAr), δ 141.0 (s, CAr), δ 132.3 (s, CAr), δ 131.8 (s, CAr), δ 131.3 (s, CAr), δ 130.3 (s, CAr), δ 124.6 (s, CAr), δ 124.0 (s, CAr), δ 32.0 (s, PCH), δ 29.4 (s, PCH), δ 28.5 (s, PCH), δ 22.8 (s, CH3), δ 20.1 (s, CH3), δ 19.7 (s, CH3), δ 18.9 (s, CH3). 11B NMR (C6D6, 128 MHz): δ 8.2 (br). IR (KBr, cm−1): 2071 (s, N≡N), 1960 (w) 1934 (w). UV-vis (THF, nm {M−1 cm−1}): 328 {shoulder, 500}, 280 {shoulder, 11250}. Anal.: Elemental analysis could not be obtained because of the instability of the compound under dynamic vacuum.
Synthesis of (TPB)(μ-H)Fe(H2)(H) (3)
A J-Young NMR tube containing a brown-red solution 1 (21 mg, 31.1 mmol) in C6H6 (0.8 mL) was freeze-pump-thawed (3x). Upon warming to room temperature, the sample was exposed to H2 (1 atm), resulting in a clear yellow solution. The reaction was mixed for 24 h to give 3 (100% by 1H NMR spectroscopy with a ferrocene integration standard). The yield could not be determined by weight because 3 was unstable to prolonged exposure to dynamic vacuum. Yellow-orange XRD quality crystals were grown under 1 atm of H2 in a concentrated solution of pentane:THF (10:1) at −78 °C. 1H NMR (C6D6, 300 MHz): δ 8.0 (3H, d, 3JH-H = 6 Hz, Ar-H), δ 7.3 (3H, t, 3JH-H = 9 Hz, Ar-H), δ 7.2 (3H, t, 3JH-H = 9 Hz, Ar-H), δ 7.0 (3H, d, 3JH-H = 6 Hz, Ar-H), δ 4.47 (s, free H2), δ 2.3 (6H, m, PCH), δ 1.0 (18H, d-d, 3JH-P = 15 Hz, 3JH-H = 6 Hz, CH3), δ 0.8 (18H, d-d, 3JH-P = 15 Hz, 3JH-H = 6 Hz, CH3), δ −15.1 (br s, 2H). T1min (d8-toluene): 35 ms (δ −15.1, −32 °C). 2H NMR (C6H6/C6D6, 76 MHz): δ −15.4 (1D, br s). 31P NMR (C6D6, 121 MHz): δ 90.0 (3P, s). 13C NMR (C6D6, 125 MHz): δ 163.5 (s, CAr), δ 144.6 (s, CAr), δ 144.1 (s, CAr), δ 130.9 (d, JP-C = 23 Hz, CAr), δ 124.5 (s, CAr), δ 123.9 (s, CAr), δ 28.6 (s, PCH), δ 21.2 (s, CH3), δ 19.9 (s, CH3). 11B NMR (C6D6, 128 MHz): δ 7.5 (br). IR (KBr, cm−1): 2278 (w), 19618 (w), 1845 (w). UV-vis (THF, nm {M−1 cm−1}): 377 {shoulder, 1532}, 275 {14532}. Anal.: Elemental analysis could not be obtained because of the instability of the compound under dynamic vacuum.
Synthesis of (TPB)Fe(CNtBu) (4)
Tert-butyl isocyanide (20 mg, 0.24 mmol) was added to a brown solution of (TPB)Fe(N2) (40 mg, 59 μmol) in benzene (2 mL), causing an instantaneous darkening upon gentle shaking. The volatiles were removed by lyophilization and the residue was extracted with tetramethylsilane (2 mL). The resulting dark brown solution was slowly concentrated down to ca. 0.2 mL by vapor diffusion into hexamethyldisiloxane. Removal of the mother liquor by decantation, washing with cold tetramethylsilane (2 × 0.1 mL) and drying in vacuo afforded (TPB)Fe(CNtBu) as brown crystals (33 mg, 77%). 1H NMR (C6D6, 300 MHz): δ 11.2 (3H), δ 9.2 (3H), δ 8.6 (3H), δ 8.5 (9H), δ 6.4 (3H), δ 5.2 (9H), δ 3.7 (12H), δ 2.9 (9H), δ −1.5 (9H), δ −2.3 (3H). IR (KBr, cm−1): 1972 (C≡N). UV-Vis (THF, nm {cm−1M−1}): 600 {shoulder, 428}, 910 {70}. μeff (C6D6, method of Evans, 20 °C): 1.7 μB. Anal.: Calc’d for C41H64BFeNP3: C 67.50, H 8.70, N 1.92; found: C 67.20, H 8.54, N 1.72.
Synthesis of (TPB)(μ-H)Fe(CNtBu)(H) (5)
A heavy-walled Schlenk tube containing a yellow-brown solution of 4 (16.4 mg, 22.4 mmol) in C6H6 (10 mL) was freeze-pump-thawed (3x). Upon warming to room temperature, the sample was exposed to H2 (1 atm) for a few minutes. The Schlenk tube was sealed and heated under vigorous mixing at 40 °C for 85 h. Removal of the solvent in vacuo, extraction with C6H6, and lyophilization yielded a solid of 5 (17.3 mg, 98 %). Room temperature evaporation of a solution of 5 in a diethyl ether:pentane (2 mL to 1 mL) mixture yielded yellow crystals suitable for XRD analysis. 1H NMR (C6D6, 300 MHz): δ 8.1 (1H, d, 3JH-H = 9 Hz, Ar-H), δ 7.4 (3H, d, 3JH-H = 9 Hz, Ar-H), δ 7.2 (3H, d, 3JH-H = 6 Hz, Ar-H), δ 7.1 (3H, d, 3JH-H = 9 Hz, Ar-H), δ 2.7 (2H, br s, PCH), δ 2.5 (2H, br s, PCH), δ 2.2 (2H, t, 2JH-P = 9 Hz, PCH), δ 1.3 (16H, m, CH3), δ 1.1 (9H, s, C(CH3)3), δ 1.0 (6H, d, 3JH-H = 6 Hz, CH3), δ 0.8 (6H, d-d, 3JH-P = 15 Hz, 3JH-H = 6 Hz, CH3), δ 0.7 (6H, br s, CH3), δ −11.7 (1H, t-d, 2JH-Pcis = 87 Hz, 2JH-Ptrans = 27 Hz, Fe-H), δ −23.9 (1H, br s, Fe-(μ-H)-B). 13C NMR (C6D6, 125 MHz): δ 176.9 (quart, 2JC-P = 8 Hz, CNtBu), δ 163.5 (br s, CAr), δ 163.0 (br s, CAr), δ 144.9 (m, CAr), δ 143.0 (d, JC-P = 20 Hz, CAr), δ 131.9 (d, 2JC-P = 7.5 Hz, CAr), δ 130.6 (d, JC-P = 3.2 H, CAr), δ 130.5 (d, JC-P = 2.5 H, CAr), δ 128.6 (s, CAr), δ 127.5 (s, CAr), δ 124.2 (s, CAr), δ 123.6 (s, CAr), δ 55.3 (s, C(CH3)3), δ 31.6 (s, PCH), δ 30.9 (s, PCH), δ 29.2 (m, PCH), δ 28.5 (d, 2JC-P = 7.5 Hz, PCH), δ 23.8 (s, CH3), δ 20.4 (s, CH3), δ 20.3 (m, CH3), δ 20.2 (s, CH3), δ 19.9 (s, CH3), δ 19.7 (s, CH3). 31P NMR (C6D6, 121 MHz): δ 81.3 (2P, d, 2JP-P = 63 Hz), δ 72.4 (1P, s). IR (KBr, cm−1): 2027 (s, C≡N), 1942 (w, Fe-H). UV-Vis (THF, nm {cm−1 M−1}): 205 {5530}, 224 {15437}, 245 {17142}, 255 {16635}, 285 {4117}, 335 {shoulder, 2166}, 400 {1830}. Anal: Calc’d for C41H66BFeNP3: C 67.32, H 8.96, N 1.91; found: C 66.59, H 8.61, N 1.30.
Synthesis of (TPB)(μ-H)Fe(CO)(H) (7) from 6 and H2
In a J-Young NMR tube, 6 (6.0 mg, 8.9 μmol) was dissolved in C6H6 (0.7 mL) to give a brown-red solution. The solution was freeze-pump-thawed (3x) and subsequently exposed to H2 (1 atm) for ca. 5 min. The reaction was then heated at 80 °C for 5 days, during which time a clear yellow solution developed. Removal of the solvent in vacuo, extraction with C6H6, and lyophilization yielded a yellow solid of 7 (5.9 mg, 98 %). Room temperature evaporation of a solution of 7 in a diethyl ether:pentane (1 mL to 0.5 mL) mixture yielded yellow analytically pure 7. 1H NMR (C6D6, 300 MHz): δ 8.1 (2H, d, 3JH-H = 6 Hz, Ar-H), δ 7.9 (1H, d, 2JP-H = 9 Hz, Ar-H), δ 7.2 (4H, m, Ar-H), δ 7.0 (4H, m, Ar-H), δ 2.6 (2H, t, 2JH-P = 3 Hz, PCH), δ 2.4 (2H, q, 2JH-P = 6 Hz, PCH), δ 2.2 (2H, t, 2JH-P = 6 Hz, PCH), δ 1.4 (6H, d, 3JH-P = 6 Hz, CH3), δ 1.2 (12H, m, CH3), δ 0.9 (6H, d, 3JH-P = 6 Hz, CH3), δ 0.8 (6H, d-d, 3JH-P = 8 Hz, 3JH-H = 6 Hz, CH3), δ 0.6 (6H, d, 3JH-P = 6 Hz, CH3). δ −11.6 (1H, t-d, 2JH-Pcis = 81 Hz, 2JH-Ptrans= 21 Hz, Fe-H), δ −20.0 (1H, br s, Fe-(μ-H)-B). 2H NMR (C6H6, 76 Hz): δ −12.3 (1D, t, 2JP-D = 10 Hz), δ −20.8 (1D, br s). 13C NMR (THF with 1 drop of C6D6, 125 MHz): δ 222.7 (br s, CO), δ 161.8 (br s, CAr), δ 142.9 (br s, 1JC-P = 19 Hz, CAr), δ 140.8 (br s, 2JC-P = 16 Hz, CAr), δ 131.0 (s, CAr), δ 129.8 (s, CAr), δ 128.5 (s, CAr), δ 128.0 (s, CAr), δ 124.0 (s, CAr), δ 123.4 (s, CAr), δ 30.8 (s, PCH), δ 28.3 (s, PCH), δ 27.7 (s, PCH), δ 22.3 (s, CH3), δ 19.1 (s, CH3), δ 18.8 (s, CH3), δ 18.1 (s, CH3). 31P NMR (C6D6, 121 MHz): δ 83.4 (2P, d, 2JP-H = 21 Hz), δ 72.8 (1P, s). IR (KBr, cm−1): 1898 (s, C≡O), 1967 (w, Fe-H). UV-Vis (THF, nm {cm−1M−1}): 270 {4333}, 280 {4111}, 390 {1400}. Anal.: Calc’d for C37H56BFeOP3: C 65.70, H 8.34; found: C 65.64, H 8.08.
Synthesis of (TPB)(μ-H)Fe(CO)(H) (7) from Formaldehyde
Compound 1 (8 mg, 11.8 μmol) or 2 (6 mg, 8.9 μmol) was mixed with excess paraformaldehyde in C6H6 for 3 h to give a turbid, light yellow solution. The excess paraformaldehyde was filtered away and the solution was pumped down to give 7 as a yellow solid (from 1, 8 mg, 100%; from 2, 7 mg, 100%). Spectroscopic data is identical to those listed above.
Synthesis of (TPB)Fe(C2H4) (8)
A J-Young NMR tube containing an orange solution of 3 (8.4 mg, 12.5 μmol) in C6H6 (0.8 mL) was freeze-pump-thawed (3x) and exposed to ethylene gas (1 atm) for ca. 1 minute. Mixing immediately gave a brown solution of 8. Removal of solvent in vacuo yielded a brown solid (8.3 mg, 99%) of 8. Dissolution of this solid under N2 atmosphere gave mostly 8 and small amounts of 1. Over time, 8 in solution converted to 1. Crystals suitable for XRD were grown in a saturated, cold pentane:diethyl ether (2:1) solution under an ethylene atmosphere. 1H NMR (C6D6, 300 MHz): δ 33.1 (1H), δ 28.8 (1H), δ 18.7 (4H), δ 13.3 (1H), δ 5.25 (s, free C2H4), δ 4.9 (3H), δ 4.1 (1H), δ 1.9 (2H), δ −3.0 (10H), δ −6.2 (11H), δ −9.2 (11H), δ −10.0 (3H). UV-Vis (THF, nm {cm−1M−1}): 309 {shoulder, 6032}, 553 {1804}, 938 {418}. μeff (C6D6, method of Evans, 20 °C): 3.2 μB (S = 1). Anal.: Elemental analysis could not be obtained because of the instability of the compound under dynamic vacuum.
Synthesis of (TPBH)Fe(C2Ph) (9)
Phenylacetylene (40.8 mg, 400 μmol) was added to a C6H6 solution (5 mL) of 3 (9.0 mg, 13 μmol), immediately giving a gray solution. Removal of the solvent in vacuo yielded a black powder of 9 (10 mg, 100%). 1H NMR (C6D6, 300 MHz): δ 28.8 (3H), δ 13.1 (4H), δ 4.7 (18H), δ 2.6 (2H), δ 2.3 (4H), δ 1.2 (2H), δ −29.3 (1H), δ −30.9 (5H). μeff (C6D6, method of Evans, 20 °C): 5.1 μB (S = 2). UV-Vis (THF, nm {cm−1M−1}): 325 {20670}, 439 {1051}, 479 {971}, 522 {955}, 601 (br abs extending from 400 to 700 nm, 930}, 883 {1466}. IR (KBr, cm−1): 2040 (s, C≡C), 2490 (m, B-H). Anal.: Calc’d for C44H60FeP3B: C, 70.60; H, 8.08. Found: C, 70.39; H, 7.89.
Synthesis of (TPBD)Fe(C2Ph)
The B-D labeled complex of 9 was generated by the same method described for 9, except that PhC2H was replaced with PhC2D. The 1H NMR spectrum was identical to 10. IR (thin film, cm−1): 1826 (br m, B-D; predicted 1832).
Synthesis of (TPBH)Fe(C2Tol) (10)
Tolylacetylene (16.1 mg, 138 μmol) was added to a C6H6 solution (5 mL) of 3 (22.9 mg, 33.0 μmol), immediately giving a gray solution. Removal of the solvent in vacuo yielded a black powder of 10 (24.0 mg 100%). Black XRD quality crystals of 10 were grown by diffusion of hexamethyldisiloxane into a concentrated THF solution of 10. 1H NMR (C6D6, 300 MHz): δ 44.1 (1H), δ 29.4 (1H), δ 13.3 (1H), δ 4.6 (2H), δ 3.3 (1H), δ 2.7 (2H), δ 2.3 (4H), δ 1.8 (2H), δ −32.3 (1H). μeff (C6D6, method of Evans, 20 °C): 5.2 μB (S = 2). UV-Vis (THF, nm {cm−1M−1}): 326 {24476}, 444 {1243}, 486 {1138}, 527 {shoulder, 975}, 624 (915}, 887 {1575}. IR (KBr, cm−1): 2039 (s, C≡C), 2500 (m, B-H). Anal.: Calc’d for C45H62FeP3B: C, 70.88; H, 8.20. Found: C, 70.44; H, 7.80.
Synthesis of (TPBD)Fe(C2Tol)
The B-D labeled complex of 10 was generated by the same method described for 10, except that TolC2H was replaced with TolC2D. The 1H NMR spectrum was identical to 10. IR (thin film, cm−1): 1824 (br m, B-D; predicted 1841).
Synthesis of 11
A yellow, C6H6 solution of 1 (18.2 mg, 27 μmol) was heated in a J-Young NMR tube under H2 (1 atm) at 80 °C for 2 h, giving a turbid red-purple solution. The solvent was removed in vacuo, and the dark crude material was redissolved in hexamethyldisiloxane (3 mL) and filtered through a glass frit to remove a black solid (presumably iron metal). Removal of the solvent in vacuo gave a purple solid that is a mixture of 11 (1 equiv) and diisopropyl-phosphino-benzene (iPr2PPh, 1 equiv). Orange XRD quality crystals of 11 can be grown from slow evaporation of a concentrated hexamethyldisiloxane solution of 11 at room temperature (5.1 mg, 18%). Dissolution of these crystals by heating in benzene or THF results in decomposition. Therefore, spectral data is reported on the mixture of 11 with iPr2PPh. Compound 11 appears to be fluxional at RT. 1H NMR (C6D6, 300 MHz): δ 8.4 (4H, s, Ar-H), δ 7.6 (4H, s, Ar-H), δ 7.5 (3H, d, 2JH-P = 6 Hz, Ar-H), δ 2.7 (1H, d, 2JH-P = 6 Hz, PCH), δ 2.5 (1H, s, PCH), δ 1.9 (1H, quart, 3JH-H = 6 Hz, PCH), δ 1.3 (6H, d-d, 3JP-H = 4 Hz, 3JH-H = 2 Hz, CH3), δ 1.2 (6H, d, 3JH-H = 3 Hz, CH3), δ 1.1 (6H, quart, 3JP-H = 4 Hz, 3JH-H = 2 Hz, CH3), 0.9 (12H, m, CH3), −17.0 (0.25H, t, 2JH-P = 36 Hz, B-H). 13C NMR (C6D6, 125 MHz): δ 157.4 (br s, CAr), δ 144.2 (br s, CAr), δ 134.9 (d, 2JC-P = 19 Hz, CAr), δ 130.5 (s, CAr), δ 129.3 (s, CAr), δ 125.7 (d, 2JC-P = 23 Hz, CAr), δ 25.5 (br s, PCH), δ 24.9 (br s, PCH), δ 24.4 (br s, PCH), δ 23.7 (br s, PCH), δ 23.1 (br s, PCH), δ 20.0 (m, CH3), δ 19.4 (m, CH3), δ 18.2 (m, CH3). 31P NMR (C6D6, 121 MHz): δ 99.1 (4P, s), δ 9.5 (2P, s). 11B NMR (C6D6, 128 MHz): δ 41.0 (br). UV-Vis (THF, nm {cm−1M−1}): 264 {shoulder, 31650}, 520 {826}. IR (KBr, cm−1): 2088 (s, B-H), 1838 (m, B-H). Anal.: Calc’d for C51H86FeP4B2Si (i.e., 11 + Me3SiH): C, 65.96; H, 9.33. Found: C, 65.70; H, 9.16.
Synthesis of (SiPiPr3)Fe(H2)(H)
In a 100 mL Schlenk tube a red solution of (SiPiPr3)Fe(Me) (1.05 g, 1.547 mmol) in C6H6 (50 mL) was degassed by freeze-pump-thaw (3x). H2 gas (1 atm) was charged into the reaction mixture. The reaction was heated at 60 °C for over a week. The reaction solution was then quickly filtered through Celite and volatiles were removed in vacuo to give a light yellow powder. The solid was collected on a glass-frit and washed with pentane (3 mL x 2). The resulting product (SiPiPr3)Fe(H2)(H) (950 mg, 1.425 mmol, 92%) was obtained as a light yellow powder after drying under vacuum. 1H NMR (C6D6, 300 MHz): δ 8.3 (3H, d, 3JH-P = 6.8 Hz, Ar-H), δ 7.3 (3H, m, Ar-H), δ 7.2 (3H, t, 3JH-H = 7.2 Hz, Ar-H), δ 7.1 (3H, t, 3JH-H = 7 Hz, Ar-H), δ 4.5 (s, free H2), δ 2.2 (6H, m, PCH), δ 1.0 (18H, m, CH3), δ 0.8 (18H, br s, CH3), δ 0.16 (s, free CH4), δ −10.0 (3H, quin, 2JP-H = 18.4 Hz, Fe-H). T1min (d8-toluene): 32 ms (δ −10.0, −30 °C). 31P NMR (C6D6, 121 MHz): δ 100 (br); (d8-Tol, 300 MHz, −80 °C): δ 117.9 (d, 3JP-P = 43.5 Hz), δ 94.7 (bs), δ 84.7 (d, 3JP-P = 43.5 Hz). 13C NMR (THF with 1 drop of C6D6, 125 MHz): δ 157.3 (d, JC-P = 22.5 Hz, CAr), δ 150.5 (d, JC-P = 21.3 Hz, CAr), δ 130.1 (d, JC-P = 9.3 Hz, CAr), δ 128.6 (s, CAr), δ 127.4 (s CAr), δ 126.0 (d, JC-P = 2.5 Hz, CAr), δ 29.0 (br s, PCH), δ 20.4 (br s, CH3), δ 19.2 (br s, CH3), UV-Vis (THF, nm {cm−1M−1}): 353 {3040}. IR (KBr pellet; cm−1): 1941 (w, Fe-H). Anal.: Elemental analysis could not be obtained because of the instability of the compound under prolonged exposure to N2.
Isotopomers of (SiPiPr3)Fe(H2)(H); (SiPiPr3)Fe(H2)(H), (SiPiPr3)Fe(HD)(H), (SiPiPr3)Fe(H2)(D), (SiPiPr3)Fe(D2)(H), (SiPiPr3)Fe(HD)(D), and (SiPiPr3)Fe(D2)(D)
HD gas was generated by the method described above and charged into a J-young tube containing a degassed solution of SiPiPr3)Fe(Me) in C6D6. The solution was heated at 60 °C for over a week. Monitoring the progress of the reaction by 1H NMR revealed the gradual disappearance of 1 and formation of diamagnetic isotopomers (SiPiPr3)Fe(H2)(H). Spectroscopic features in the 1H NMR spectrum were identical to (SiPiPr3)Fe(H2)(H) except for the hydridic proton resonances, where isotopologues were observed. 1H{31P} NMR (C6D6, 300 MHz): δ −10.0 (3H, quart, 2JP-H = 18.4 Hz, H3), δ −10.0 (t, 1JH-D = 9.5 Hz, H2D), δ −10.23 (quin, 1JH-D = 9.3 Hz, HD2).
Synthesis of (SiPiPr3)Fe(CO)(H)
In a 50 mL Schlenk tube a yellow solution of (SiPiPr3)Fe(H2)(H) (330 mg, 0.495 mmol) in C6H6 (20 mL) was freeze-pump-thawed (3x). The solution was charged with CO (1 atm). The reaction was mixed overnight at RT and then for 1 h at 60 °C, resulting in a light yellow solution. After completion, the solution was degassed by freeze-pump-thaw (3x). The solution was filtered through Celite, and the volatiles were removed in vacuo to give a light yellow powder. The solid was collected on a glass-frit and washed with pentane (5 mL x 3). Removal of the solvent in vacuo yields (SiPiPr3)Fe(CO)(H) (266 mg, 0.384 mmol, 78%) as a light yellow powder. Crystals suitable for X-ray diffraction were obtained by RT evaporation of a pentane solution of (SiPiPr3)Fe(CO)(H). 1H NMR (d8-toluene, 300 MHz): δ 8.2 (2H, d, 3JH-H = 7.2 Hz), δ 8.1 (1H, d, 3JH-H = 7.2 Hz), δ 7.3 (3H, m), δ 7. 2 (3H, m), δ 7.1 (3H, m), δ 2.7 (2H, m), δ 2.4 (2H, m), δ 2.2 (2H, m), δ 1.5 (6H, d-d, 3JH-P = 15.2 Hz, 3JH-P = 6.8 Hz), δ 1.3 (6H, m), δ 1.1 (6H, d-d, 3JH-P = 12.6 Hz, 3JH-H = 6.6 Hz), δ 0.8 (12H, m), δ 0.5 (6H, s), δ −14.9 (1H, t-d, 3JH-Pcis = 81.0 Hz, 3JH-Ptrans = 14.1 Hz). 31P NMR (C6D6, 121 MHz, RT): δ 88.0 (br), δ 90.0 (s); (d8-toluene, 121 MHz, −80 °C): δ 109.9 (t, 3JP-P = 65 Hz, 3JP-H = 80 Hz), δ 90.0 (s), δ 77.9 (t, 3JP-P = 65 Hz, 3JP-H = 80 Hz). 13C NMR (THF with 1 drop of C6D6, 125 MHz): δ 223.0 (d, 2JC-P = 6.3 Hz, CO), δ 157.2 (d, JC-P = 8.8 Hz, CAr), δ 155.2 (d, JC-P = 10.6 Hz, CAr), δ 150.5 (d, JC-P = 11.3 Hz, CAr), δ 150.2 (d, JC-P = 8.8 Hz, CAr), δ 148.6 (d, JC-P = 18.1 Hz, CAr), δ 132.4 (d, JC-P = 9.4 Hz, CAr), δ 131.9 (d, JC-P = 8.8 Hz, CAr), δ 130.9 (s, CAr), δ 128.3 (s, CAr), δ 128.5 (s, CAr), δ 127.1 (s, CAr), δ 126.8 (s, CAr), δ 126.4 (s, CAr), δ 125.8 (s, CAr), δ 124.7 (s, CAr), δ 32.4 (s, PCH), δ 30.7 (s, PCH), δ 30.0 (s, PCH), δ 29.6 (s, PCH), δ 28.7 (s, PCH), δ 22.8 (s, CH3), δ 22.1 (s, CH3), δ 20.2 (s, CH3), δ 19.5 (s, CH3), δ 19.2 (s, CH3), δ 19.1 (s, CH3), δ 18.9 (s, CH3), δ 18.0 (s, CH3). UV-Vis (THF, nm {cm−1M−1}): 340 {2,050}, 400 {1,500}. IR (KBr pellet; cm−1): 1882 (s, C≡O), 1944 (m, Fe-H). Anal.: Calc’d for C37H57FeOP3Si: C, 64.16; H, 8.00. Found: C, 64.15; H, 8.13.
Synthesis of (SiPiPr3)Fe(13CO)(H)
In a J-Young NMR tube an orange C6D6 solution of (SiPiPr3)Fe(H2)(H) was degassed by three freeze-pump-thaw (3x). Subsequently, 13CO (1 atm) was added and the reaction was allowed to mix overnight at RT. The 1H NMR spectrum was identical to (SiPiPr3)Fe(CO)(H). IR (KBr; cm1): 1836 (s, 13C≡O).
Generation of (TPBH)Fe(Et) (A)
Compound A was observed as an intermediate of catalytic ethylene hydrogenation under the reaction conditions described in section “Catalytic Hydrogenation Studies.” We were also able to generate A starting from complex 8. This procedure described below is more amendable to observing A spectroscopically. A dark yellow C6D6 solution (0.5 mL) of 8 (2.8 mg, 4.2 μmol) under ethylene (1 atm) in a J-Young tube was frozen (−196 °C) and H2 was added (1 atm). The reaction was thawed and quickly mixed only immediately prior to measuring the 1H NMR spectrum, revealing a mixture of 8 and A. Further mixing of the solution for ca. 45 min yielded a purple solution of A with a small residual amount of 8 by 1H NMR spectroscopy. Free C2H4, C2H6, and H2 were also observed in the 1H NMR spectrum. Compound A could not be isolated as a solid due its instability. For example, an ATR IR spectrum of a thin film of the reaction mixture obtained by solvent evaporation under an N2 atmosphere over a period less than 30 sec gave vibrational bands diagnostic of 1, 2, and A. 1H NMR (C6D6, 300 MHz): δ 17.3 (1H), δ 6.6 (1H), δ 5.26 (s, free C2H4), δ 4.4 (1H), δ 4.47 (br s, free H2), δ 3.3 (2H), 0.80 (s, free C2H6), δ −1.9 (8H), δ −5.5 (6H). IR (thin film; cm−1): 2470 (br s, B-H of A), 2069 (m, N≡N of (TPB)(μ-H)Fe(N2)(H) 2), 2009 (s, N≡N of (TPB)Fe(N2) 1). UV-Vis, obtained 45 min after exposing A under 1 atm of ethylene to 1 atm of H2 (THF, nm {cm−1M−1}): 325 {20670}, 439 {1051}, 479 {971}, 522 {955}, 601 (br abs extending from 400 to 600 nm, 930}, 883 {1466}. Magnetic data could not be obtained due to residual 10 in the reaction mixture. Anal.: Elemental analysis could not be obtained because of the instability of the compound under dynamic vacuum.
Catalytic Hydrogenation Studies
Compound 1 (0.045 g, 0.07 mmol) and ferrocene (0.012 g, 0.07 mmol) were dissolved in 1.5 mL of C6D6, giving a 0.045 M precatalyst stock solution. Ferrocene was used as an internal 1H NMR integration standard and did not affect the rates of hydrogenation. For a catalytic run, 0.1 mL of the stock solution was taken and mixed with 0.35 mL of C6D6, and 30 equiv of substrate in a J-Young NMR tube (3.2 mL capacity). For styrene hydrogenation, this equates to 0.01 M 1 and 0.3 M styrene. For phenylacetylene hydrogenation, this equates to 0.01 M 1 and 0.29 M phenylacetylene. For ethylene hydrogenation, this equates to 0.01 M 1 and 0.30 M ethylene. The sample in the J-Young NMR tube was subsequently degassed by freeze-pump-thaw (3x) and backfilled with 1 atm H2 (0.11 mmol). The J-Young NMR tube was continually inverted (12 min−1) to ensure adequate mass transfer. The tube was periodically refilled with H2 to maintain 1 atm of H2. All reactions were monitored periodically by 1H NMR spectroscopy until >95% completion. All reactions resulted in clean conversion of the substrate to the corresponding product. Catalytic runs in the presence of a drop of mercury or in the absence of ambient laboratory light had no affect on the reactions. Catalytic hydrogenations could also be cleanly effected by pre-generating 2 or 3 before the addition of the substrate.
Supplementary Material
Acknowledgments
We acknowledge the Gordon and Betty Moore Foundation and the NIH (GM070757) for funding. M.-E.M acknowledges a Fellowship for Advanced Researchers from the Swiss National Science Foundation. Dr. Charlene Tsay and Lawrence Henling are acknowledged for assistance with X-ray crystallography, and Dr. David VanderVelde is acknowledged for assistance with NMR spectroscopy. We acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis BRP at Caltech for their generous support of the Molecular Observatory.
Footnotes
The authors declare no competing financial interests.
Supporting Information. Spectral data, catalysis data, computational details, solid state structures (4 and (SiPiPr3)Fe(CO)(H)), and CIF files (for 2, 3, 4, 5, 8, 10, 11, and (SiPiPr3)Fe(CO)(H)). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; 2010. [Google Scholar]
- 2.Bullock RM, editor. Catalysis Without Precious Metals. Wiley-VCH; 2010. [Google Scholar]
- 3.For representative examples of mid-to-late first row transition metal catalysts that perform two-electron transformations see: Bolm C, Legros J, Le Paih J, Zani L. Chem Rev. 2004;104:6217–6254. doi: 10.1021/cr040664h.Hu X. Chem Sci. 2011;2:1867–1886.Knijnenburg Q, Horton AD, van der Heijden H, Kooistra TM, Hetterscheid DGH, Smits JMM, de Briun B, Budzelaar PHM, Gal AW. J Mol Catal A. 2005;232:151–159.Tondreau AM, Atienza CCH, Weller KJ, Nye SA, Lewis KM, Delis JGP, Chirik PJ. Science. 2012;335:567–570. doi: 10.1126/science.1214451.Daida EJ, Peters JC. Inorg Chem. 2004;43:7474–7585. doi: 10.1021/ic0488583.Federsel C, Boddien A, Jackstell R, Jennerjahn R, Dyson PJ, Scopelliti R, Laurenczy G, Beller M. Angew Chem, Int Ed. 2010;49:9777–9780. doi: 10.1002/anie.201004263.
- 4.(a) Noyori R, Yamakawa M, Hashiguchi S. J Org Chem. 2001;66:7931–7944. doi: 10.1021/jo010721w. [DOI] [PubMed] [Google Scholar]; (b) Morris RH. Chem Soc Rev. 2009;38:2282–2291. doi: 10.1039/b806837m. [DOI] [PubMed] [Google Scholar]; (c) Chakraborty S, Guan H. Dalton Trans. 2010;39:7427–7436. doi: 10.1039/c002942d. [DOI] [PubMed] [Google Scholar]; (d) Casey CP, Guan H. J Am Chem Soc. 2007;129:5816–5817. doi: 10.1021/ja071159f. [DOI] [PubMed] [Google Scholar]; (e) Sui-Seng C, Freutel F, Lough AJ, Morris RH. Angew Chem, Int Ed. 2008;47:940–943. doi: 10.1002/anie.200705115. [DOI] [PubMed] [Google Scholar]; (f) Langer R, Leitus G, Ben-David Y, Milstein D. Angew Chem, Int Ed. 2011;50:2120–2124. doi: 10.1002/anie.201007406. [DOI] [PubMed] [Google Scholar]
- 5.Tsoureas N, Kuo YY, Haddow MF, Owen GR. Chem Comm. 2011;47:484–486. doi: 10.1039/c0cc02245d. [DOI] [PubMed] [Google Scholar]
- 6.For discussions on the M-B bonding situation: Sircoglou M, Bontemps S, Mercy M, Saffon N, Takahashi M, Bouhadir G, Maron L, Bourissou D. Angew Chem, Int Ed. 2007;46:8583–8586. doi: 10.1002/anie.200703518.Amgoune A, Bourissou D. Chem Comm. 2011;47:859–871. doi: 10.1039/c0cc04109b.Hill AF. Organometallics. 2006;25:4741–4743.Parkin G. Organometallics. 2006;25:4744–4747.
- 7.(a) Owen GR. Chem Soc Rev. 2012;41:3535–3546. doi: 10.1039/c2cs15346g. [DOI] [PubMed] [Google Scholar]; (b) Tsoureas N, Owen GR, Hamilton A, Orpen AG. Dalton Trans. 2008:6039–6044. doi: 10.1039/b809247h. [DOI] [PubMed] [Google Scholar]
- 8.(a) Crossley IR, Foreman MRSt-J, Hill AF, Owen GR, White AJP, Williams DJ, Willis AC. Organometallics. 2008;27:381–386. [Google Scholar]; (b) Crossley IR, Foreman MRSt–J, Hill AF, White AJ, Williams D. Chem Comm. 2005:221–223. doi: 10.1039/b413305f. [DOI] [PubMed] [Google Scholar]; (c) Crossley IR, Hill AF, Willis AC. Organometallics. 2007;26:3891–3895. [Google Scholar]; (d) Crossley IR, Hill AF. Dalton Trans. 2008:201–203. doi: 10.1039/b712058c. [DOI] [PubMed] [Google Scholar]; (e) Crossley IR, Hill AF, Willis AC. Organometallics. 2008;27:312–315. [Google Scholar]; (f) Hill AF, Smith MK, Wagler J. Organometallics. 2008;27:2137–2140. [Google Scholar]; (g) Crossley IR, Hill AF, Willis AC. Organometallics. 2009;29:326–336. [Google Scholar]
- 9.Stephan DW, Erker G. Angew Chem, Int Ed. 2009;49:46–76. doi: 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]
- 10.Harman WH, Peters JC. J Am Chem Soc. 2012;134:5080–5082. doi: 10.1021/ja211419t. [DOI] [PubMed] [Google Scholar]
- 11.Kameo H, Nakazawa H. Organometallics. 2012;31:7476–7484. [Google Scholar]
- 12.(a) Moret ME, Peters JC. Angew Chem, Int Ed. 2011;50:2063–2067. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moret ME, Peters JC. J Am Chem Soc. 2011;133:18118–18121. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bontemps S, Bouhadir G, Dyer PW, Miqueu K, Bourissou D. Inorg Chem. 2007;46:5149–5151. doi: 10.1021/ic7006556. [DOI] [PubMed] [Google Scholar]
- 14.The H-D coupling constant (JH-D) is also in principle an excellent distinguishing characteristic between the η2-dihydrogen and dihydride ligands.15 Unfortunately, H-D coupling cannot be resolved in 3, likely due to fast exchange within the HnD3-n unit and H/D scrambling into the methine and terminal methyl positons on the isopropyl groups of the TPB ligand.
- 15.(a) Desrosiers PJ, Cai L, Lin Z, Richards R, Halpern J. J Am Chem Soc. 1991;113:4173–4184. [Google Scholar]; (b) Kubas GJ. Chem Rev. 2007;107:4152–4205. doi: 10.1021/cr050197j. [DOI] [PubMed] [Google Scholar]; (c) Morris RH. Coord Chem Rev. 2008;252:2381–2394. [Google Scholar]
- 16.Van der Sluys LS, Eckert J, Eisenstein O, Hall JH, Huffman JC, Jackson SA, Koetzle TF, Kubas GJ, Vergamini PJ, Caulton KG. J Am Chem Soc. 1990;112:4831–4841. [Google Scholar]
- 17.Lee Y, Kinney RA, Hoffman BM, Peters JC. J Am Chem Soc. 2011;133:16366–16369. doi: 10.1021/ja207003m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee Y, Peters JC. J Am Chem Soc. 2011;133:4438–4446. doi: 10.1021/ja109678y. [DOI] [PubMed] [Google Scholar]
- 19.Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartell LS, Roth EA, Hollowell CD, Kuchitsu K, Young JE. J Chem Phys. 1965;42:2683–2683. [Google Scholar]
- 21.Select iron-ethylene complexes of ferracyclopropane character: Zenneck U, Frank W. Angew Chem, Int Ed. 1986;25:831–833.Schneider JJ, Czap N, Blaser D, Boese R. J Am Chem Soc. 1999;121:1409–1410.Bennett MA, Ditzel EJ, Hunter AD, Khan K, Kopp MR, Neumann H, Robertson GB, Zeh H. J Chem Soc, Dalton Trans. 2000:1733–1741.Fürstner A, Martin R, Krause H, Seidel G, Goddard R, Lehmann CW. J Am Chem Soc. 2008;130:8773–8787. doi: 10.1021/ja801466t.
- 22.A similar paramagnetic (S = 2) iron ethyl-hydro-tris(pyrazolyl)borate complex: Shirasawa N, Nguyet TT, Hikichi S, Moro-oka Y, Akita M. Organometallics. 2001;20:3582–3598.
- 23.Marchand AP, Marchand NW. Tetrahedron Lett. 1971;12:1365–1368. [Google Scholar]
- 24.Suess DLM, Tsay C, Peters JC. J Am Chem Soc. 2012;134:14158–14164. doi: 10.1021/ja305248f. [DOI] [PubMed] [Google Scholar]
- 25.(a) Reinhard N. Coord Chem Rev. 1982;47:89–124. [Google Scholar]; (b) Bianchini C, Laschi F, Masi D, Ottaviani FM, Pastor A, Peruzzini M, Zanello P, Zanobini F. J Am Chem Soc. 1993;115:2723–2730. [Google Scholar]; (c) Field LD, George AV, Malouf EY, Slip IHM, Hambley TW. Organometallics. 1991;10:3842–3848. [Google Scholar]
- 26.Tsay C, Peters JC. Chem Sci. 2012;3:1313–1318. [Google Scholar]
- 27.Suess DLM, Peters JC. J Am Chem Soc. 2013 doi: 10.1021/ja400836u. [Online early access] [DOI] [Google Scholar]
- 28.Rubina M, Gevorgyan V. J Am Chem Soc. 2001;123:11107–11108. doi: 10.1021/ja016934k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.