

Abstract
Members of the Cbl protein family (Cbl, Cbl-b, and Cbl-c) are E3 ubiquitin ligases that have emerged as critical negative regulators of protein tyrosine kinase (PTK) signaling. This function reflects their ability to directly interact with activated PTKs and to target them as well as their associated signaling components for ubiquitination. Given the critical roles of PTK signaling in driving oncogenesis, recent studies in animal models and genetic analyses in human cancer have firmly established that Cbl proteins function as tumor suppressors. Missense mutations or small in-frame deletions within the regions of Cbl protein that are essential for its E3 activity have been identified in nearly 5% of leukemia patients with myelodysplastic/myeloproliferative disorders. Based on evidence from cell culture studies, in vivo models and clinical data, we discuss the potential signaling mechanisms of mutant Cbl-driven oncogenesis. Mechanistic insights into oncogenic Cbl mutants and associated animal models are likely to enhance our understanding of normal hematopoietic stem cell homeostasis and provide avenues for targeted therapy of mutant Cbl-driven cancers.
Keywords: Cbl, E3 ubiquitin ligase, MDS/MPN, Leukemia, Tyrosine kinase, Cellular signaling, Hematopoietic
Introduction to Cbl Proteins
Cbl (Casitas B-lineage lymphoma) proteins are an evolutionarily-conserved family of multi-domain proteins that serve as regulators of cellular signaling based on their protein tyrosine kinase (PTK)-directed E3 ubiquitin ligase function. While simpler organisms such as C. elegans and Drosophila possess a single Cbl gene, mammalian genomes encode three distinct Cbl-family proteins: Cbl (also known as c-Cbl), Cbl-b, and Cbl-c (also known as Cbl-3 or Cbl-SL) [1–6].
All Cbl-family proteins, including the three mammalian members of the protein family, share a highly homologous N-terminal region that serves as the structural platform for direct binding to phosphopeptide motifs in activated tyrosine kinases and is accordingly known as the tyrosine kinase-binding (TKB) domain. This domain is in fact a unique assembly of a four-helical bundle (4H), a variant SH2 domain and an EF hand domain, only found in Cbl-family proteins [7]. The TKB domain is followed by a highly conserved linker helical region (LHR) and a RING (Really Interesting New Gene) finger domain; these two regions together form the structural platform for binding to an ubiquitin conjugating enzyme (E2) and this interaction is essential for E3 ligase activity of Cbl proteins. The three N-terminal domains (TKB, linker and RING finger) are necessary as well as sufficient for activated tyrosine kinase-directed E3 ligase activity [8,9]. Among mammalian Cbl proteins, Cbl and Cbl-b contain extensive additional C-terminal regions that serve to mediate protein-protein interactions. These include a proline-rich region that mediates interactions of Cbl proteins with SH3 domain-containing proteins including Src-family kinases, Grb2, Cin-85 and others; several tyrosine residues whose phosphorylation generates binding motifs for interaction with SH2 domain-containing signaling proteins including Vav-family guanine nucleotide exchange factors (GEFs) for Rho-family GTPases (human Cbl Y700), p85 subunit of PI 3-kinase (human Cbl Y731) and Crk-family adaptor proteins that link Cbl proteins to C3G, a GEF for Ras-related small GTPase Rap1 (human Cbl Y774). The distal C-terminal region encodes a leucine zipper (LZ)-like region that is known to facilitate dimerization under certain experimental conditions [10,11]; this region also functions as a ubiquitin-associating domain (UBA) capable of binding to ubiquitin chains [12]. However, the functional roles of the LZ/UBA domain remain unclear. The shorter Cbl-c lacks all of the C-terminal regions except for a short proline-rich region; the domain organization of Cbl-c is more similar to the C. elegans Cbl (SLI-1) and the shorter alternatively-spliced form of Cbl in Drosophila [13]. A visual representation of Cbl protein domains and structures from a number of species can be found in Figure 1.
Figure 1.

Conservation of domain structure and primary sequence of Cbl family proteins through evolution. Structure and domains of Cbl proteins from various indicated species are shown. Amino acid (AA) sequence identity within the indicated domains relative to human Cbl are shown in the table to the right. The protein designations are indicated on left. TKB – tyrosine kinase binding; 4H – 4-helical bundle; EF – calcium-binding EF hand, SH2 – phosphotyrosine-binding Src Homology 2 domain; LHR – linker helical region; RF – RING finger domain; P – proline-rich region; UBA – ubiquitin-associating domain.
A large body of biochemical and cell biological work [1–3,14] has led to our current paradigms that Cbl-family E3 ubiquitin ligases serve as negative regulators of PTK signaling. In essentially every mammalian cell system, Cbl proteins are recruited to PTK-coupled cell surface receptors (either receptor tyrosine kinases [RTKs] or receptors non-covalently associated with PTKs, such as antigen and cytokine receptors) upon engagement of these receptors with appropriate ligands. An essential component of the recruitment of Cbl proteins to PTK-coupled receptors involves the generation of phosphotyrosine-containing motifs, either on receptor cytoplasmic tails themselves (as with many RTKs) or on associated intermediate proteins (as with certain RTKs or antigen receptors), which serve as docking sites for the TKB domains of Cbl proteins [15]. Binding of Cbl with adaptors such as Grb2 has also been shown to recruit Cbl to activated RTKs [16]. The PTK association allows Cbl proteins to ubiquitinate both the PTKs and associated receptors (for non-covalently associated PTKs). The Cbl protein-PTK association also leads to phosphorylation of C-terminal motifs in Cbl and association of SH2 domain-containing signaling proteins. It is clear that proteins that are in complex with activated PTK-associated Cbl proteins also undergo ubiquitination [17]. Generally, ubiquitination of targets by Cbl proteins occurs as either mono- or poly-ubiquitination [2]. Cbl-dependent mono-ubiquitination and K63-linked poly-ubiquitination appears to be a feature of activated RTKs [18], while other targets, such as ZAP70/Syk and Src-family kinases, appear to undergo poly-ubiquitination [19–21]. In most cases, these modifications direct ubiquitinated targets of Cbl proteins toward degradation. However, degradation can occur through two distinct pathways: mono- or K63-linked poly-ubiquitination of activated cell surface receptors enables their sorting to the lysosomes through recognition of the ubiquitin chains by Endosomal Sorting Complex for Transport (ESCRT) machinery, which facilitates the entry of ubiquitin-tagged receptors into inner vesicles of the multivesicular body (MVB) compartment; in contrast, many poly-ubiquitinated targets of Cbl proteins undergo proteasomal degradation. Interestingly, degradation-independent negative regulation by Cbl proteins, potentially by regulating subcellular localization alone, has been suggested in selected cases such as the PI 3-kinase [22].
The negative regulatory role of Cbl proteins indicated by biochemical and cell biological analyses has led to studies of animal models that have provided a number of novel insights into both their physiological functions and the pathological consequences of their defective expression or function. Germline deletion of Cbl is associated with a series of mild to moderate phenotypes during development. Cbl-null mice exhibit mild hyper-cellularity of a number of lymphoid organs, including the bone marrow and spleen with increased numbers of megakaryocytes [23], expansion of hematopoietic stem/progenitor cells in the bone marrow [24], mild increase in mammary ductal branching [23], altered positive selection of T-cells in the thymus [25], reduced male fertility [26], lean muscles due to elevated insulin action [27] and other phenotypes; however, these mice have a relatively normal life span. In contrast, Cbl-b deletion does not produce any apparent developmental abnormalities in mice; however, Cbl-b-null mice exhibit hyperactive T cell responsiveness to antigens leading to autoimmune disease, a failure to induce energy in T cells [28,29], and reduced muscle atrophy in response to unloading [30]; however, if infectious challenges are avoided, these mice also have a relatively normal life span. In contrast to individual knockouts, germ line deletion of Cbl and Cbl-b together leads to early embryonic lethality, indicating their redundant but essential functions during embryogenesis. Importantly, use of a conditional (floxed) allele of Cbl on a Cbl-b-null background has allowed for tissue-selective deletion of Cbl and Cbl-b, demonstrating that these two proteins function redundantly to regulate key physiological functions. Cbl plus Cbl-b deletion in T cells using T cell-specific Lck-Cre led to a systemic autoimmune/inflammatory disease and dramatic hyper-responsiveness of T cells to antigen stimulation [31]. Cbl/Cbl-b double deletion in the B-cell compartment in mice led to loss of peripheral tolerance [32]. Finally, deletion of Cbl and Cbl-b in hematopoietic stem cells led to a myeloproliferative disease that was fatal within 8 weeks of birth [33]. Notably, deletion of the predominantly epithelial tissue-expressed Cbl-c gene is without a phenotype [34]; however, a combined deletion of Cbl-c with Cbl or Cbl-b has not yet been examined. Collectively, the knockout animal models suggest that Cbl and Cbl-b proteins serve selective physiological roles, perhaps due to their relative levels of expression in various tissues. More importantly, however, the models show that these two members function cooperatively during animal development as well as in maintenance of normal tissue homeostasis in adults.
Oncogenic Mutations of Cbl in Human Leukemia
A potential role for alterations of Cbl-family protein function in oncogenesis was implied by very early observations. The founding family member Cbl was identified as the cellular homolog of the viral oncogene v-Cbl, the transforming gene of a murine retrovirus that causes hematological malignancies in mice [35]. The v-Cbl oncogene represents a fusion of the viral gag protein with the N-terminal 357 amino acids of Cbl, which we now know functions as the TKB domain, deleting the remaining sequences that confer the essential E3 ligase activity on Cbl [36]. Both Cbl-1-357, corresponding to v-Cbl sequences without the viral gag protein, as well as a 17 amino acid internal deletion of Cbl cloned from the murine transformed B cell line 70Z/3 (and now commonly known as Cbl-70Z mutant) were dominantly transforming in rodent fibroblasts [37]. Analyses of Cbl-1-357 versus Cbl70Z and other engineered mutants of the helical linker demonstrated that oncogenic versions of Cbl with an intact C-terminal region were more transforming [37]. The differential transforming activity correlated with the degree of elevated PTK signaling induced by these oncogenes and resulted in the formulation of dominant-negative inhibition of Cbl-b (via the TKB domain) and potential gain of function (via the C-terminal regions) mechanisms of Cbl oncogenesis in the rodent transformation system [38]. These early studies provided a strong basis for an expectation that aberrations of Cbl proteins might occur in human cancer but it was only recently that this prediction was indeed confirmed.
Beginning with initial reports in 2007, a large number of studies have now established that mutations clustered in the linker helical region and RING finger domain of Cbl (and rarely Cbl-b) are the features of myeloid leukemias, predominantly in the category of myelodysplastic/myeloproliferative disorders (MDS/MPD) [39–59]. These disorders include juvenile myelomonocytic leukemia (JMML), chronic myelomonocytic leukemia (CMML), and a typical chronic myeloid leukemia (aCML). At present, over 3,500 patients have been collectively analyzed and mutations have been observed in about 4% of cases (based on current compilation of data in COSMIC, or Catalogue of Somatic Mutations In Cancer, maintained by the Sanger Institute) (http://www.sanger.ac.uk/genetics/CGP/cosmic/). Clustering of the cancer-associated Cbl mutations in the linker and RING finger regions (Figure 2) strongly argues for the loss of ubiquitin ligase activity as a critical feature of conversion to oncogenes. This argument is further strengthened by the predominance of mutations on highly conserved residues in the RING finger (e.g., the Zn-coordinating cysteine and histidine residues as well as known E2 contact residues) as well as the helical linker [60–62]. While not all the mutations have been directly studied in cell culture models, those that have been examined have confirmed the lack of E3 ligase function [40,43,47]. Of note, Cbl mutations are sometimes found in the germline of a small subset of patients which then undergo somatic duplication to initiate leukemogenesis. This is particularly the situation in patients with JMML in which Cbl mutations account for nearly 15% of all cases [53,54].
Figure 2.
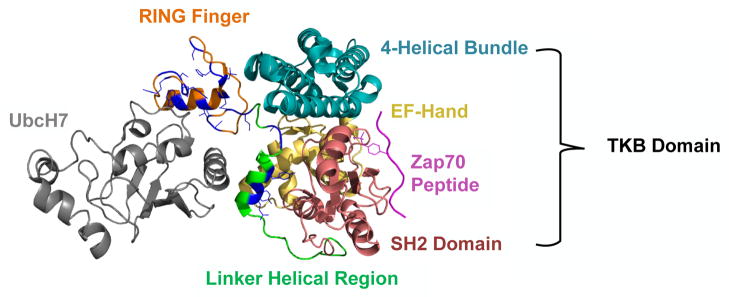
Location of human Cbl mutations in the context of known structure of RING finger and helical linker region. The N-terminus of Cbl containing the TKB (4H – blue, EF – yellow, SH2 – red), linker helical region (green), and RING finger domains (orange) is shown in complex with a Zap70 peptide (purple) and the E2 ubiquitin-conjugating enzyme UbcH7 (grey). Residues that have been found to be mutated in human leukemia/myelodysplastic syndromes are shown in blue. Note that the mutations cluster in and around the binding pocket responsible for E2 interactions. (Modeled using pyMol software [http://www.pymol.org/]. 1FBV PDB file [Zheng et al. 2000] was downloaded from NCSB Protein Data Bank [http://www.rcsb.org/pdb]).
Mechanisms of Mutant Cbl-Induced Oncogenesis
Notable features associated with Cbl mutations in patients strongly suggest their role as oncogenesis-driver mutations. In nearly all patients harboring Cbl mutations, the mutant allele is found duplicated together with loss of the wild type allele in a process known as acquired uniparental disomy (aUPD). The resulting loss of both wildtype alleles may provide one mechanism of oncogenesis, via a complete loss of function of a tumor suppressor. A mere deletion of the wildtype allele with a hemizygous mutant allele is rarely observed in patients, suggesting that the a UPD to produce a duplication of the mutant Cbl allele is a critical oncogenic event. However, overexpression of oncogenic mutants of Cbl (such as Cbl 70Z) in rodent fibroblasts is dominantly transforming even when endogenous Cbl and Cbl-b are expressed [37,38]. This is consistent with a dominant-negative mechanism in which the mutant protein would compete with the remaining copy of wildtype Cbl (as seen in a minority of human patients) and Cbl-b (retained in nearly all cases) for binding to targets that are negatively regulated by wildtype Cbl proteins. Compelling evidence for this hypothesis comes from the fact that neither Cbl nor Cbl-b knockouts have overt phenotypes, while double deletion in the hematopoietic compartment results in a myeloproliferative disease remarkably similar to the human disease associated with Cbl mutants [33]. Furthermore, Cbl RING finger mutant knock-in mice with a wildtype Cbl allele do not present with MPD while a hemizygous Cbl RING finger knock-in mutant (together with a Cbl-null allele) produces MPD [63]. Introduction of a point mutation (G306E) in the TKB domain of the transforming Cbl-1-357 truncation negated the ability of the TKB domain to bind to PTK targets. Consistent with the competitive dominant-negative inhibition model in rodent fibroblasts, this resulted in a loss of Cbl 1-357’s transforming ability [38]. The aUPD of the mutant Cbl locus is also expected to increase the relative level of mutant versus the wildtype Cbl proteins and thereby allow a more effective dominant negative inhibition of Cbl-b. Studies carried out by Sanada et al. [47] support this possibility as the ectopic expression of mutant Cbl proteins promoted a higher degree of proliferation in a Cbl-null background versus a wildtype background in murine hematopoietic stem cells [47].
However, duplication of the mutant Cbl allele in leukemia patients also suggests the possibility of a gain of function mechanism in addition to dominant-negative inhibition of endogenous Cbl proteins. It is noteworthy that no instances of bi-allelic loss of Cbl without an oncogenic mutation has been reported in patients, indicating that dominant-negative inhibition of Cbl-b and/or the gain of function phenotype of mutants is essential. In favor of a gain of function mechanism, it is of note that none of the patients have revealed truncations akin to v-Cbl oncogene that could function as relatively pure dominant-negative mutants by competing with the wildtype protein for the target PTK binding. Studies in genetically tractable models will be needed to unambiguously define the contributions of the three mechanisms discussed above.
Signaling pathways of mutant Cbl-driven oncogenesis
A key question connected to how mutant Cbl proteins drive oncogenesis involves which upstream cell surface receptors (either RTKs or receptors associated with PTKs) are aberrantly active in leukemic cells carrying Cbl mutations. An equally critical question is which downstream signaling pathways mediate oncogenesis. Answers to these questions will be critical in designing therapeutic approaches for patients whose disease is driven by Cbl mutations. Based on the known roles of Cbl proteins as negative regulators of PTKs, together with their ability to directly complex with a subset of downstream mediators of PTK signals (indicated under Introduction to Cbl proteins), it is likely that a number of signaling pathways are concurrently elevated in leukemic cells. However, it is probable that only some of these are essential for oncogenesis and deciphering these will be important.
Because of the extensive protein-protein interaction domains within the C-terminal region of Cbl, multiple signaling pathways are likely to be involved in mutant Cbl-driven oncogenesis. The hypothesized gain of function mechanism of mutant Cbl proteins involve the role of these proteins to act as multi-domain scaffolds to aid in the formation of complexes that paradoxically result in hyperactivation rather than attenuation of signaling through Cbl’s normal physiological interacting partners. While normal Cbl protein attenuates the signaling functions of its partners by promoting their ubiquitination, mutant Cbl (lacking E3 ligase activity) instead brings its bound partners into close proximity with activated PTKs (bound at the TKB domain of mutant Cbl and not subjected to ubiquitination-dependent degradation), resulting in a synergistic increase in signaling from these supra-molecular complexes (Figure 3).
Figure 3.

Potential mechanisms of mutant Cbl-induced leukemogenesis. (left) Mutant Cbl proteins retain the ability to bind to activated protein tyrosine kinases (shown is a generic receptor tyrosine kinase), but cannot promote their ubiquitination. Mutant Cbl (indicated by the red “no” symbol; typically expressed in cancers from duplicated mutant alleles) interacts with activated target tyrosine kinases in competition with remaining wild-type Cbl and/or Cbl-b (dominant-negative inhibition); the ineffective degradation and downregulation of activated protein tyrosine kinases results in enhanced signaling to promote leukemogenesis. (right) Mutant Cbl proteins bound to activated protein tyrosine kinases juxtapose downstream signaling effectors (that interact with Cbl mutants via intact C-terminal regions) to activated tyrosine kinases. These stabilized and hyperactive macromolecular signaling complexes enhance signaling (gain of function) through the downstream signaling pathways that are negatively regulated by WT Cbl under physiological conditions and possibly by novel pathways. (middle) These mechanisms contrast with the normal case, whereby Cbl binds to activated protein tyrosine kinases and also downstream signaling effectors, ubiquitinating both and resulting in downregulation of signaling.
A potentially important C-terminal domain that may be involved in the gain of function of mutant Cbl is the proline-rich region, which has been shown to interact with SH3 domain-containing proteins such as Src-family kinases (SFKs) and Grb2 [64,65]. SFKs are involved in the propagation of signaling from a variety of cell surface receptors, including cytokine receptors, RTKs, integrins and G-protein coupled receptors [66]. Given the availability of clinically-active SFK inhibitors, definitive analyses of the role of SFKs are urgently needed.
Clinical analyses have revealed that Cbl mutations in patients, especially those with JMML, are mutually exclusive with other mutations known to drive oncogenesis by finally activating the Ras signaling pathways [44,52, 55]. Notably, in vitro analyses have shown elevated Erk signaling in hematopoietic stem cells from animal models of mutant Cbl oncogenesis [33,63]. These findings suggest that mutant Cbl proteins may indeed promote oncogenesis via the Ras-MAPK signaling pathway. In addition to general upregulation of this pathway due to misregulated PTK signaling, it is also possible that activation of this pathway may occur due to Cbl’s Grb2 interactions or Cbl’s known interactions via the TKB domain with Sprouty2, a negative regulator of Ras-Erk signaling [67].
The tyrosine phosphorylation sites within Cbl’s C-terminus are also likely to play a prominent role in mutant Cbl’s gain of function. Each one of the three major tyrosine phosphorylation sites on Cbl (Y700, Y731, and Y774) have been well-characterized and mediate direct interactions with key signaling pathways. Cbl Y700 is known to interact with Vav-family proteins, which are guanine nucleotide exchange factors for Rho/Rac family GTPases [17]. Rho/Rac GTPases along with Vav GEFs have been implicated in oncogenesis, owing to their ability to participate in actin cytoskeletal reorganization, migration, and transcriptional regulation [68]. Furthermore, Rho GTPases have been established as regulators of hematopoietic stem cell programs and play key roles in oncogenesis by other leukemia oncogenes [69]. Cbl Y731 has been shown to bind the p85 regulatory subunit of PI3-kinase, a key regulator of proliferation and survival signaling via Akt and other pathways in HSCs and leukemogenesis [65, 70]. Interestingly, hematopoietic cells in the Cbl RING finger mutant knock-in mouse model of myeloproliferative disease show a higher level of Akt activation compared to Cbl-null mice; the Akt hyperactivation was accompanied by increased Y731 phosphorylation [63].
Cbl Y774 is a known interaction site of Crk adaptor proteins [71]. Crk adaptors play a major role in the regulation of a number of effectors such as Rap GTPases through interaction with the GEF C3G, and these pathways have been linked to regulation of cell adhesion, migration, and oncogenic signaling [72]. A multitude of recent evidence has indicated that the overexpression of Crk proteins, which are prominent interaction partners of Cbl [73, 74,2], is commonly associated with human cancers [75].
Studies of clinical samples as well as mouse models of leukemia associated with mutant Cbl have also revealed the elevation of pSTAT5 and other signaling intermediates whose direct link to Cbl remains to be explored. The general increase in signaling downstream of PTKs is expected since their post-activation degradation is expected to be reduced in mutant Cbl-expressing cells. While studies thus far have clearly raised the possible involvement of a number of signaling pathways in mutant Cbl-driven oncogenesis, establishing a direct causative role of these pathways will require further analyses.
Related to upstream receptors and PTKs that function aberrantly in mutant Cbl-dependent leukemogenesis, both patient-derived leukemic cells and cells derived from animal models have revealed hypersensitivity to a number of growth factors/cytokines, including many, such as GM/CSF, SCF, FLT3 ligand, thrombopoietin, and IL-3, that are known to regulate hematopoietic stem/progenitor cell proliferation [33,47]. Recent studies using a genetic cross between the RING finger knock-in and FLT3 ligand-null mice suggested that FLT3 signaling is important in promoting leukemogenesis [63]. More genetic studies together with biochemical analyses using cell line and animal models will be needed to dissect out the key receptors and PTKs involved in mutant Cbl-driven oncogenesis.
Clearly, a systematic analysis of the various protein-protein interaction motifs is needed in order to narrow down the critical binding partners and signaling pathways contributing to mutant Cbl-dependent leukemogenesis. These analyses will greatly benefit from being performed in a variety of backgrounds of Cbl, including one that is deficient of all endogenous Cbl proteins (such as a Cbl/Cbl-b double knockout [Cbl-c is primarily restricted to epithelial cells]), which will allow for differentiation of dominant negative and gain of function mechanisms.
Conclusions
Cbl proteins have now been established as tumor suppressors and mutations of Cbl are known to drive the MDS/MPN subset of hematological neoplasms. These advances have led to a need to better understand the function of Cbl proteins and how their mutant forms contribute to oncogenesis. Acquired uniparental disomy causes duplication of mutant Cbl genes and loss of wildtype Cbl in most patients, suggesting that a combination of the loss-of-function of a tumor suppressor, dominant-negative inhibition of Cbl-b and a gain of function mechanism all lead to the development of oncogenesis. Understanding the contributions of these mechanisms and delineating the essential roles of signaling pathways that are rendered hyperactive in leukemic cells should facilitate the development of novel therapies targeting mutant Cbl-induced and related malignancies.
Acknowledgments
This work was supported by the NIH grants CA87986, CA105489, CA99163, CA116552 and NCI 5U01CA151806-02 to H.B; the NIH grants CA96844 and CA144027 and Department of Defense grants W81XWH-07-1-0351 and W81XWH-11-1-0171 to V.B; Department of Defense grant W81 XWH-10-1-0740 and Nebraska DHHS Stem Cell grant to MN; the NIH grant CA127239 to A.N.; and the NCI Core Support Grant to UNMC-Eppley Cancer Center. N.P was a postdoctoral fellow and S.N. was a predoctoral trainee under the NCI Institutional Cancer Biology Training Grant (CA009476). L.D. was an exchange scholar of the China Scholarship Council. D.F. was a recipient of a scholarship from the China Medical University Shenyang, China.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Lupher ML, Jr, Rao N, Eck MJ, Band H. The Cbl protooncoprotein: A negative regulator of immune receptor signal transduction. Immunol Today. 1999;20:375–382. doi: 10.1016/s0167-5699(99)01484-x. [DOI] [PubMed] [Google Scholar]
- 2.Duan L, Reddi AL, Ghosh A, Dimri M, Band H. The Cbl family and other ubiquitin ligases: Destructive forces in control of antigen receptor signaling. Immunity. 2004;21:7–17. doi: 10.1016/j.immuni.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Thien CB, Langdon WY. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signaling responses. Biochem J. 2005;391:153–166. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kales SC, Ryan PE, Nau MM, Lipkowitz S. Cbl and human myeloid neoplasms: the cbl oncogene comes of age. Cancer Res. 2010;70:4789–4794. doi: 10.1158/0008-5472.CAN-10-0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naramura M, Nadeau S, Mohapatra B, Ahmad G, Mukhopadhyay C, et al. Mutant Cbl proteins as oncogenic drivers in myeloproliferative disorders. Oncotarget. 2011;2:245–250. doi: 10.18632/oncotarget.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naramura M, Band V, Band H. Indispensable roles of mammalian Cbl family proteins as negative regulators of protein tyrosine kinase signaling: Insights from in vivo models. Commun Integr Biol. 2011;4:159–162. doi: 10.4161/cib.4.2.14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 8.Ota S, Hazeki K, Rao N, Lupher ML, Jr, Andoniou CE, et al. The RING finger domain of Cbl is essential for negative regulation of the Syk tyrosine kinase. J Biol Chem. 2000;275:414–422. doi: 10.1074/jbc.275.1.414. [DOI] [PubMed] [Google Scholar]
- 9.Lill NL, Douillard P, Awwad RA, Ota S, Lupher ML, Jr, et al. The evolutionarily conserved N-terminal region of Cbl is sufficient to enhance down-regulation of the epidermal growth factor receptor. J Biol Chem. 2000;275:367–377. doi: 10.1074/jbc.275.1.367. [DOI] [PubMed] [Google Scholar]
- 10.Bartkiewicz M, Houghton A, Baron R. Leucine zipper-mediated homodimerization of the adaptor protein c-Cbl. A role in c-Cbl’s tyrosine phosphorylation and its association with epidermal growth factor receptor. J Biol Chem. 1999;274:30887–30895. doi: 10.1074/jbc.274.43.30887. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, DeYoung SM, Hwang JB, O’Leary EE, Saltiel AR. The roles of Cbl-b and c-Cbl in insulin-stimulated glucose transport. J Biol Chem. 2003;278:36754–36762. doi: 10.1074/jbc.M300664200. [DOI] [PubMed] [Google Scholar]
- 12.Davies GC, Ettenberg SA, Coats AO, Mussante M, Ravichandran S, et al. Cbl-b interacts with ubiquitinated proteins; differential functions of the UBA domains of c-Cbl and Cbl-b. Oncogene. 2004;23:7104–7115. doi: 10.1038/sj.onc.1207952. [DOI] [PubMed] [Google Scholar]
- 13.Keane MM, Ettenberg SA, Nau MM, Banerjee P, Cuello M, et al. Cbl-3: A new mammalian cbl family protein. Oncogene. 1999;18:3365–3375. doi: 10.1038/sj.onc.1202753. [DOI] [PubMed] [Google Scholar]
- 14.Swaminathan G, Tsygankov AY. The cbl family proteins: Ring leaders in regulation of cell signaling. J Cell Physiol. 2006;209:21–43. doi: 10.1002/jcp.20694. [DOI] [PubMed] [Google Scholar]
- 15.Wang HY, Altman Y, Fang D, Elly C, Dai Y, et al. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of zap-70. J Biol Chem. 2001;276:26004–26011. doi: 10.1074/jbc.M010738200. [DOI] [PubMed] [Google Scholar]
- 16.Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, et al. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002;21:303–313. doi: 10.1093/emboj/21.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura-Shimura Y, Duan L, Rao NL, Reddi AL, Shimura H, et al. Cbl-mediated ubiquitinylation and negative regulation of Vav. J Biol Chem. 2003;278:38495–38504. doi: 10.1074/jbc.M305656200. [DOI] [PubMed] [Google Scholar]
- 18.Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Rao N, Ghosh AK, Ota S, Zhou P, Reddi AL, et al. The non-receptor tyrosine kinase Syk is a target of Cbl-mediated ubiquitylation upon B-cell receptor stimulation. EMBO J. 2001;20:7085–7095. doi: 10.1093/emboj/20.24.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao N, Miyake S, Reddi AL, Douillard P, Ghosh AK, et al. Negative regulation of Lck by Cbl ubiquitin ligase. Proc Natl Acad Sci U S A. 2002;99:3794–3799. doi: 10.1073/pnas.062055999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao N, Ghosh AK, Douillard P, Andoniou CE, Zhou P, et al. An essential role of ubiquitination in Cbl-mediated negative regulation of the Src-family kinase Fyn. Signal Transduct. 2002;2:29–39. doi: 10.1002/1615-4061(200205)2:1/2<29::AID-SITA29>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol. 2001;2:870–875. doi: 10.1038/ni0901-870. [DOI] [PubMed] [Google Scholar]
- 23.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008;22:992–997. doi: 10.1101/gad.1651408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naramura M, Kole HK, Hu RJ, Gu H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci U S A. 1998;95:15547–15552. doi: 10.1073/pnas.95.26.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El Chami N, Ikhlef F, Kaszas K, Yakoub S, Tabone E, et al. Androgen-dependent apoptosis in male germ cells is regulated through the protooncoprotein Cbl. J Cell Biol. 2005;171:651–661. doi: 10.1083/jcb.200507076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molero JC, Jensen TE, Withers PC, Couzens M, Herzog H, et al. c-Cbl-deficient mice have reduced adiposity, higher energy expenditure, and improved peripheral insulin action. J Clin Invest. 2004;114:1326–1333. doi: 10.1172/JCI21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 29.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 30.Nakao R, Hirasaka K, Goto J, Ishidoh K, Yamada C, et al. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol Cell Biol. 2009;29:4798–4811. doi: 10.1128/MCB.01347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naramura M, Jang IK, Kole H, Huang F, Haines D, et al. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–1199. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- 32.Kitaura Y, Jang IK, Wang Y, Han YC, Inazu T, et al. Control of the B cell-intrinsic tolerance programs by ubiquitin ligases Cbl and Cbl-b. Immunity. 2007;26:567–578. doi: 10.1016/j.immuni.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naramura M, Nandwani N, Gu H, Band V, Band H. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci U S A. 2010;107:16274–16279. doi: 10.1073/pnas.1007575107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffiths EK, Sanchez O, Mill P, Krawczyk C, Hojilla CV, et al. Cbl-3-deficient mice exhibit normal epithelial development. Mol Cell Biol. 2003;23:7708–7718. doi: 10.1128/MCB.23.21.7708-7718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langdon WY, Hartley JW, Klinken SP, Ruscetti SK, Morse HC., 3rd v-cbl, an oncogene from a dual-recombinant murine retrovirus that induces early B-lineage lymphomas. Proc Natl Acad Sci U S A. 1989;86:1168–1172. doi: 10.1073/pnas.86.4.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blake TJ, Shapiro M, Morse HC, 3rd, Langdon WY. The sequences of the human and mouse c-cbl proto-oncogenes show v-cbl was generated by a large truncation encompassing a proline-rich domain and a leucine zipper-like motif. Oncogene. 1991;6:653–667. [PubMed] [Google Scholar]
- 37.Andoniou CE, Thien CB, Langdon WY. Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO J. 1994;13:4515–4523. doi: 10.1002/j.1460-2075.1994.tb06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonita DP, Miyake S, Lupher ML, Jr, Langdon WY, Band H. Phosphotyrosine binding domain-dependent upregulation of the platelet-derived growth factor receptor alpha signaling cascade by transforming mutants of Cbl: Implications for Cbl’s function and oncogenicity. Mol Cell Biol. 1997;17:4597–4610. doi: 10.1128/mcb.17.8.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, et al. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007;110:1022–1024. doi: 10.1182/blood-2006-12-061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood. 2007;110:1004–1012. doi: 10.1182/blood-2007-01-066076. [DOI] [PubMed] [Google Scholar]
- 41.Abbas S, Rotmans G, Lowenberg B, Valk PJ. Exon 8 splice site mutations in the gene encoding the E3-ligase CBL are associated with core binding factor acute myeloid leukemias. Haematologica. 2008;93:1595–1597. doi: 10.3324/haematol.13187. [DOI] [PubMed] [Google Scholar]
- 42.Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–10357. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113:6182–6192. doi: 10.1182/blood-2008-12-194548. [DOI] [PubMed] [Google Scholar]
- 44.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114:1859–1863. doi: 10.1182/blood-2009-01-198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009;27:6109–6116. doi: 10.1200/JCO.2009.23.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reindl C, Quentmeier H, Petropoulos K, Greif PA, Benthaus T, et al. CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/myelodysplastic syndrome subtypes. Clin Cancer Res. 2009;15:2238–2247. doi: 10.1158/1078-0432.CCR-08-1325. [DOI] [PubMed] [Google Scholar]
- 47.Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460:904–908. doi: 10.1038/nature08240. [DOI] [PubMed] [Google Scholar]
- 48.Barresi V, Palumbo GA, Musso N, Consoli C, Capizzi C, et al. Clonal selection of 11q CN-LOH and CBL gene mutation in a serially studied patient during MDS progression to AML. Leuk Res. 2010;34:1539–1542. doi: 10.1016/j.leukres.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 49.Fernandes MS, Reddy MM, Croteau NJ, Walz C, Weisbach H, et al. Novel oncogenic mutations of CBL in human acute myeloid leukemia that activate growth and survival pathways depend on increased metabolism. J Biol Chem. 2010;285:32596–32605. doi: 10.1074/jbc.M110.106161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gelsi-Boyer V, Trouplin V, Roquain J, Adelaide J, Carbuccia N, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:365–375. doi: 10.1111/j.1365-2141.2010.08381.x. [DOI] [PubMed] [Google Scholar]
- 51.Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28:3858–3865. doi: 10.1200/JCO.2009.27.1361. [DOI] [PubMed] [Google Scholar]
- 52.Muramatsu H, Makishima H, Jankowska AM, Cazzolli H, O’Keefe C, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood. 2010;115:1969–1975. doi: 10.1182/blood-2009-06-226340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42:794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet. 2010;47:686–691. doi: 10.1136/jmg.2010.076836. [DOI] [PubMed] [Google Scholar]
- 55.Perez B, Kosmider O, Cassinat B, Renneville A, Lachenaud J, et al. Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:460–468. doi: 10.1111/j.1365-2141.2010.08393.x. [DOI] [PubMed] [Google Scholar]
- 56.Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer. 2010;10:401. doi: 10.1186/1471-2407-10-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shiba N, Kato M, Park MJ, Sanada M, Ito E, et al. CBL mutations in juvenile myelomonocytic leukemia and pediatric myelodysplastic syndrome. Leukemia. 2010;24:1090–1092. doi: 10.1038/leu.2010.49. [DOI] [PubMed] [Google Scholar]
- 58.Makishima H, Jankowska AM, McDevitt MA, O’Keefe C, Dujardin S, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117:e198–e206. doi: 10.1182/blood-2010-06-292433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shiba N, Taki T, Park MJ, Nagasawa M, Kanazawa T, et al. CBL mutation in childhood therapy-related leukemia. Leukemia. 2011;25:1356–1358. doi: 10.1038/leu.2011.75. [DOI] [PubMed] [Google Scholar]
- 60.Kobashigawa Y, Tomitaka A, Kumeta H, Noda NN, Yamaguchi M, et al. Autoinhibition and phosphorylation-induced activation mechanisms of human cancer and autoimmune disease-related E3 protein Cbl-b. Proc Natl Acad Sci U S A. 2011;108:20579–20584. doi: 10.1073/pnas.1110712108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dou H, Buetow L, Hock A, Sibbet GJ, Vousden KH, et al. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat Struct Mol Biol. 2012;19:184–192. doi: 10.1038/nsmb.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kales SC, Ryan PE, Lipkowitz S. Cbl exposes its RING finger. Nat Mol Biol. 2012;19:131–133. doi: 10.1038/nsmb.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rathinam C, Thien CB, Flavell RA, Langdon WY. Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell. 2010;18:341–352. doi: 10.1016/j.ccr.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 64.Meisner H, Conway BR, Hartley D, Czech MP. Interactions of Cbl with Grb2 and phosphatidylinositol 3′-kinase in activated Jurkat cells. Mol Cell Biol. 1995;15:3571–3578. doi: 10.1128/mcb.15.7.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Panchamoorthy G, Fukazawa T, Miyake S, Soltoff S, Reedquist K, et al. p120cbl is a major substrate of tyrosine phosphorylation upon B cell antigen receptor stimulation and interacts in vivo with Fyn and Syk tyrosine kinases, Grb2 and Shc adaptors, and the p85 subunit of phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:3187–3194. doi: 10.1074/jbc.271.6.3187. [DOI] [PubMed] [Google Scholar]
- 66.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 67.Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: Modified modulators, matchmakers or missing links? J Endocrinol. 2009;203:191–202. doi: 10.1677/JOE-09-0110. [DOI] [PubMed] [Google Scholar]
- 68.Lazer G, Katzav S. Guanine nucleotide exchange factors for RhoGTPases: Good therapeutic targets for cancer therapy? Cell Signal. 2011;23:969–979. doi: 10.1016/j.cellsig.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 69.Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, et al. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936–947. doi: 10.1182/blood-2009-09-198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686–707. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- 71.Ribon V, Hubbell S, Herrera R, Saltiel AR. The product of the cbl oncogene forms stable complexes in vivo with endogenous Crk in a tyrosine phosphorylation-dependent manner. Mol Cell Biol. 16:45–52. doi: 10.1128/mcb.16.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 73.Reedquist KA, Fukazawa T, Panchamoorthy G, Langdon WY, Shoelson SE, et al. Stimulation through the T cell receptor induces Cbl association with Crk proteins and the guanine nucleotide exchange protein C3G. J Biol Chem. 1996;271:8435–8442. doi: 10.1074/jbc.271.14.8435. [DOI] [PubMed] [Google Scholar]
- 74.Sawasdikosol S, Chang JH, Pratt JC, Wolf G, Shoelson SE, et al. Tyrosine-phosphorylated Cbl binds to Crk after T cell activation. J Immunol. 1996;157:110–116. [PubMed] [Google Scholar]
- 75.Sriram G, Birge RB. Emerging roles for Crk in human cancer. Genes Cancer. 2010;1:1132–1139. doi: 10.1177/1947601910397188. [DOI] [PMC free article] [PubMed] [Google Scholar]