

Abstract
As the adult mammalian heart has limited potential for regeneration and repair, the loss of cardiomyocytes during injury and disease can result in heart failure and death. The cellular processes and regulatory mechanisms involved in heart growth and development can be exploited to repair the injured adult heart through ‘reawakening’ pathways that are active during embryogenesis. Heart function has been restored in rodents by reprogramming non-myocytes into cardiomyocytes, by expressing transcription factors (GATA4, HAND2, myocyte-specific enhancer factor 2C (MEF2C) and T-box 5 (TBX5)) and microRNAs (miR-1, miR-133, miR-208 and miR-499) that control cardiomyocyte identity. Stimulating cardiomyocyte dedifferentiation and proliferation by activating mitotic signalling pathways involved in embryonic heart growth represents a complementary approach for heart regeneration and repair. Recent advances in understanding the mechanistic basis of heart development offer exciting opportunities for effective therapies for heart failure.
The life of all animals, from early embryogenesis and throughout adulthood, depends on the second to second uninterrupted function of the heart. Thus, it is not surprising that congenital and acquired diseases of the heart can have catastrophic consequences and that heart disease is the most common cause of adult death worldwide1. Numerous insults, including ischaemic coronary artery disease, hypertension, genetic mutations and chemotherapy, can cause heart disease, which is associated with loss or dysfunction of cardiac muscle cells, diminished pump function, arrhythmias and eventual death2. The adult mammalian heart cannot efficiently generate new cardiac muscle cells in response to injury, and although numerous drugs and mechanical devices can temporarily improve cardiac function, such approaches do not replace lost cardiac muscle and are inevitably transient. This underscores the need for innovative strategies for heart repair.
Over the past decade, considerable effort and resources have gone into the development of stem cell-based therapies for cardiac repair3–6. Studies in animal models and humans have demonstrated that cellular replacement strategies for heart repair, in which stem cells and other cell types are injected directly into the injured heart or into the coronary circulation, can have modest beneficial effects on cardiac function7,8. However, there is little evidence that the transplanted cells are retained in injured hearts, and the mechanistic basis of cardiac improvement following stem cell delivery remains unclear. A likely possibility is that the transient improvement observed following stem cell delivery is due to paracrine effects of the grafted cells. Thus, the need remains for additional innovative strategies to enhance cardiac regeneration and replace cardiomyocytes following cardiac injury and disease9.
As the heart consists of several cell types (BOX 1), an alternative approach to cardiac repair is to reprogramme non-muscle cells in the injured heart to adopt a cardiac muscle cell fate. This approach exploits developmental gene regulatory networks that have been deciphered over the past two decades10–12 to convert non-muscle cells into cardiomyocyte-like cells. Such reprogrammed cells can potentially integrate and repopulate the injured heart to enhance cardiac function13–16. Insights into the control of cell proliferation have also catalysed new approaches for the expansion of cardiomyocyte numbers, which will be required to replenish dead or diseased cardiomyocytes from dysfunctional adult hearts17–24. This Review highlights the regulatory mechanisms involved in heart growth and development and describes how these mechanisms can be harnessed to promote heart regeneration and repair.
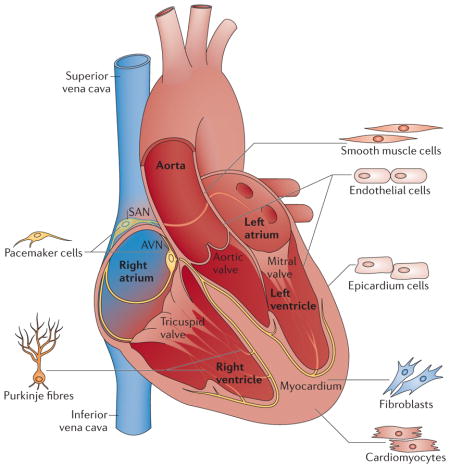
Box 1. Cell types of the heart.
The four-chambered heart consists of different cell types (see the figure). All these cell types contribute to structural, biochemical, mechanical and electrical properties of the functional heart. Atrial and ventricular cardiomyocytes form the muscular walls of the heart (that is, the myocardium). More than 50% of the cells of the heart are cardiac fibroblasts. Endothelial cells form the endocardium, the interior lining of blood vessels and cardiac valves. Smooth muscle cells contribute to the coronary arteries and inflow and outflow vasculature. The epicardium gives rise to the precursors of the coronary vasculature and cardiac fibroblasts. Pacemaker cells and Purkinje fibres in the conduction system are specialized cardiomyocytes that generate and conduct electrical impulses. The sinoatrial node (SAN), which is composed of a group of pacemaker cells, resides in the right atrium generating impulses to initiate heart contraction. The atrioventricular node (AVN) is located between the atria and ventricles, and it conducts an electrical impulse from the atria to the ventricles.
Stages of cardiogenesis
Myocardial cells are derived from the mesoderm, which emerges from the anterior region of the primitive streak during gastrulation25,26. Bone morphogenetic protein (BMP) produced by the endoderm induces the cardiomyocyte fate, whereas WNT-mediated signals from the underlying neural tube and notochord suppress cardiomyocyte specification. In the mouse embryo at embryonic day 6.5 (E6.5), cardiac progenitor cells migrate in an anterior–lateral direction under the head folds to form two groups of cells on either side of the midline where cardiac markers are first detected. The first population of cells to migrate to the heart-forming region, termed the primary heart field, further extends across the midline to form the cardiac crescent at E7.5 (BOX 2). Cells in the secondary heart field appear in the extracrescent tissue and reside medial and anterior to the primary heart field. By E8, the cardiac crescent fuses at the midline, forming a beating heart tube that contains cardiomyocytes and underlying endothelial cells. Secondary heart field cells migrate anteriorly and posteriorly to the heart tube, contributing to the future outflow tract, the right ventricle and portions of the atria. The heart tube undergoes uneven growth and remodelling to form the primitive ventricles and the atria. This is followed by looping, which brings the inflow and outflow segments to the anterior pole of the heart and also positions the future cardiac chambers for proper development10–12,27,28.
Box 2. Developmental stages and regulation of cardiogenesis.
The developmental stages of the mouse embryonic heart are shown (see the figure, parta). During mouse embryogenesis, two cardiac progenitor cell populations derived from a common progenitor at gastrulation contribute to the formation of the heart. At embryonic day 7.5 (E7.5), the cardiac crescent, which is also referred to as the primary heart field, is formed by early cardiac progenitor cells in the anterior mesoderm. The secondary heart field is derived from the pharyngeal mesoderm located medial and anterior to the cardiac crescent. By E8.0, cells from the cardiac crescent migrate to the midline to form a linear heart tube, serving as a scaffold for subsequent heart growth. The heart tube is further expanded posteriorly and anteriorly with cells migrating from the secondary heart field, giving rise to the arterial and venous poles. The linear heart tube undergoes a rightward looping (E8.5) as a consequence of uneven growth and remodelling, leading to the formation of primitive ventricles and atria. As a result, the venous pole moves anteriorly, positioning the future cardiac chambers for proper development (E10.5). Heart maturation involves septation formation in the ventricles and atria, as well as valve formation at E15. The primary heart field contributes to the left ventricle and right and left atria. The secondary heart field contributes to the right ventricle, the outflow tract and the right and left atria. Cardiac neural crest cells migrating from the dorsal neural tube into the arterial pole participate in separation of the outflow tract.
The signalling pathways and transcriptional regulation involved in cardiogenesis are fairly well established (see the figure, partb). The formation of the vertebrate heart is controlled by intrinsic and extrinsic signals, and crosstalk between molecules in the primary heart field and secondary heart field. The gene regulatory events and cellular movements that control cardiogenesis are temporally and spatially regulated by complex signalling networks. Inductive signals such as bone morphogenetic protein (BMP), Notch, WNT and sonic hedgehog (SHH) in vertebrates activate a set of genes encoding transcriptional activators that are expressed in the primary and secondary heart fields. These transcription factors are key regulators of genes that connect upstream signalling to muscle-specific genes, as well as genes that encode proteins involved in heart growth and patterning. GATA4 and NKX2.5 (NK2 homeobox 5) are central transcription factors in the primary heart field and the secondary heart field (purple shading). The transcription factor T-box 5 (TBX5) is only expressed in the primary heart field (orange shading). Genes expressed in the secondary heart field are shaded in blue. The ISL1 protein is a marker for cardiac progenitor cells in the secondary heart field. The protein product of TBX1 is a central regulator of the secondary heart field, as it controls the development of the cardiac outflow tract.
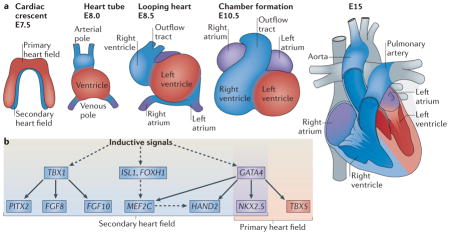
FGF, fibroblast growth factor; FOXH1, forkhead box H1; MEF2C, myocyte-specific enhancer factor 2C; PITX2, paired-like homeodomain transcription factor 2. Dashed lines indicate that additional components are involved, and solid lines indicate direct regulation.
Cardiogenesis is a precisely regulated process that is governed by signalling pathways and networks of transcription factors28 (BOX 2). Our understanding of the molecular mechanisms underlying this process has suggested strategies for inducing cardiomyocyte formation in the injured adult heart to promote cardiac repair and regeneration.
Cardiac regeneration
During embryogenesis, the mammalian heart grows through the proliferation of cardiomyocytes29. After birth, cardiomyocytes undergo another round of DNA synthesis without cytokinesis, which renders most of them binucleated. At this point, cell division ceases and postnatal heart growth is achieved primarily through the hypertrophy (enlargement) of cardiomyocytes29–31. Although the adult human heart clearly lacks sufficient regenerative potential to replace the billions of cardiomyocytes lost following myocardial infarction, there is evidence that cardiomyocytes can slowly self-renew, fostering ongoing debate regarding the extent of cardiomyocyte proliferation and turnover in the uninjured adult mammalian heart32–34.
Turnover of adult mammalian cardiomyocytes
The weight of evidence supporting adult human cardiomyocyte renewal is based on an approach that took advantage of radiocarbon (14C) that was released for a short time during atomic bomb testing (from 1955 to 1963) and incorporated into human DNA32. Using the integration of 14C to establish the age of cardiomyocytes in humans, cardiomyocytes were found to turn over at a rate of ~1% of the entire population per year in a person at the age of 25 years, and this rate declines to ~0.45% at the age of 75 years. This suggests that nearly 50% of cardiomyocytes are replenished during a normal lifespan32. In an independent analysis of tissue samples from patients with cancer between 19 and 104 years of age who had received an infusion of iododeoxyuri-dine, ~22% of cardiomyocytes were found to renew annually33. This suggests that the myocyte compartment of the adult human heart is fully replaced up to 15 times between the ages of 20 and 100 years33. The different cardiomyocyte renewal rates detected in the two studies are difficult to reconcile, but both studies indicate that cardiomyocytes in the adult human heart turn over at a measureable rate, suggesting that regenerative strategies could potentially enhance this process.
Furthermore, studies in mice using multi-isotope imaging mass spectrometry to study cardiomyocyte turnover, and to determine whether new cardiomyocytes are derived from pre-existing myocytes or from a progenitor pool, showed that cardiomyocyte renewal occurs at a low rate (~5.5% in young adults and ~2.6% in old mice) through the division of pre-existing cardiomyocytes during normal ageing34. Overall, these results suggest that the mammalian heart possesses a capacity for renewal that is insufficient for regeneration in response to injury, but that might be enhanced by therapy.
Cardiac regeneration in zebrafish
In contrast to the low level of regeneration in mammalian adult hearts, certain fish and amphibians possess a robust capacity for cardiac regeneration throughout life. For example, when up to 20% of the ventricle is amputated, adult zebrafish can fully regenerate the heart without scar formation within 2 months post surgery35. The source of new cardiomyocytes in the regenerating zebrafish heart is unclear. Initial studies concluded that cells from the epicardium (the outer layer of the heart) were activated in response to injury and gave rise to cardiomyocytes and other cell types involved in heart regeneration, such as vascular smooth muscle cells36. By contrast, subsequent lineage tracing studies, using the tamoxifen-inducible Cre–loxP recombination system to label cardiomyocytes with a regulatable GFP transgene that is controlled by the cmlc2 (cardiac-specific cardiac myosin light chain 2) promoter after birth, followed by amputation at adulthood, showed that newly formed cardiomyocytes were GFP positive37,38. This supports the notion that new myocardium is generated through dedifferentiation and proliferation of pre-existing cardiomyocytes rather than from a progenitor stem cell population37,38.
Cardiomyocytes contain highly organized sarcomeres that are required for synchronous contraction. When cardiomyocytes undergo dedifferentiation, the sarcomere structure collapses and is displaced to the cell periphery. The mechanisms responsible for sarcomere collapse and reformation are unclear, but sarcomere disassembly is probably essential for DNA synthesis and cell division to occur. Adult human cardiomyocytes are primarily binucleated with highly stable sarcomeres that probably prevent cytokinesis. By contrast, neonatal mammalian cardiomyocytes and zebrafish cardiomyocytes contain less elaborate sarcomeres, which probably accounts for their ability to proliferate. Cell cycle regulators such as Mps1 (monopolar spindle protein 1; which is a mitotic checkpoint kinase) are upregulated during zebrafish cardiac regeneration. Zebrafish hearts with a mutation in Mps1 fail to regenerate and form scars in response to injury. Together, this further suggests that the regenerative response in zebrafish is dependent on the proliferation of pre-existing cardiomyocytes35.
Cardiac regeneration in neonatal mice
Unlike zebrafish, adult mammalian hearts form fibrotic scars in response to injury. The difference in the capacity for cardiac regeneration between adult fish hearts and mammalian hearts could be due to the distinct heart anatomies and cardiomyocytes characteristics in these organisms. Mammalian hearts are four-chambered with double-circulation that allows the chambers to generate high pressure, whereas fish hearts are two-chambered with single circulation that pumps blood with a relatively lower force39. In addition, most cardiomyocytes in the zebrafish heart are mononucleated throughout life and retain significant proliferative capacity40. By contrast, ~90% of cardiomyocytes in mice become binucleated shortly after birth as a consequence of DNA replication without cell division30. The lower pressure load and mononucleated cardiomyocytes of neonatal mice resemble those of adult zebrafish.
The neonatal mouse heart retains regenerative potential for ~7 days after birth41,42. When the ventricle apex of the heart is amputated in 1 day-old mouse pups (P1), the heart can undergo full regeneration without scar formation, similar to adult zebrafish (FIG. 1). Lineage tracing of cardiomyocytes demonstrated that newly formed cardiomyocytes are derived from the proliferation of pre-existing cardiomyocytes rather than from a stem cell population. Surgical resection of the ventricle apex 7 days postnatally (P7; that is when most cardiomyocytes have withdrawn from the cell cycle) results in a loss of the regenerative capability. The heart also fully regenerates in a model of neonatal myocardial infarction in which the left anterior descending (LAD) coronary artery is ligated on day 1 after brith42. However, this regenerative capability is lost when LAD coronary artery ligation is performed at P7 or later. In the absence of regeneration after removal of the ventricle apex or LAD coronary artery ligation at P7, extensive cardiac fibrosis ensues, highlighting the reciprocal relationship between regeneration and fibrosis in the heart. The similarity in cardiac regeneration capability between zebrafish and neonatal mammals suggests that this process is mainly driven by the dedifferentiation and proliferation of existing cardiomyocytes, and not by cardiac progenitor cells. In the future, it will be of interest to identify factors that can promote cardiomyocyte proliferation to extend the neonatal regenerative response to injury.
Figure 1. Regeneration of the mammalian neonatal heart.

The neonatal mouse heart possesses regenerative capacity. In neonatal mice, the early response to cardiac injury-induced cardiomyocyte loss includes inflammatory infiltration, activation of epicardial-specific genes and angiogenesis. Global proliferation of cardiomyocytes in the injured heart replaces the scar tissue with cardiomyocytes and restores cardiac function within 3 weeks after the injury. The regenerative capacity of the mammalian neonatal heart declines with age, whereas cardiac fibrosis increases with age in response to injury. The regenerative potential of the mouse heart is lost when injury occurs 7 days postnatally or later during adulthood, when fibrotic scar tissue replaces the lost cardiomyocytes and leads to reduced cardiac function.
Regulators of cardiac proliferation
Positive cell cycle regulators, such as cyclins, cyclin-dependent kinases (CDKs) and proto-oncoproteins, are highly expressed in the embryonic heart and downregulated in the adult heart29,43. Overexpression of cell cycle activators such as cyclin A2, cyclin D2 and SV40 large T antigen has been shown to promote dedifferentiation and proliferation of post-mitotic cardiomyocytes17–19,44–48. In addition, several signalling pathways can modestly reactivate proliferation in adult cardiomyocytes, albeit relatively inefficiently.
FGF1 and inhibition of p38 increase proliferation
Differentiation and proliferation are typically opposing cellular processes, such that molecules that promote differentiation usually repress cell cycle re-entry. The MAPK p38 induces differentiation and cell cycle exit49, and its activity is inversely correlated with cardiac growth. Overexpression of p38 inhibits neonatal cardiomyocyte proliferation, whereas the inhibition of its activity or its genetic deletion in the heart increases the number of cardiomyocytes in mitosis17 (FIG. 2a). Inhibition of p38 can initiate proliferation of adult differentiated cardiomyocytes in vitro by inducing dedifferentiation48. Furthermore, the inhibition of p38 coupled with the addition of fibroblast growth factor 1 (FGF1) can block apoptosis that is induced by myocardial ischemia–reperfusion, enhance cardiomyocyte proliferation and improve cardiac function post-myocardial infarction in adult rats48,50. The addition of FGF1 alone or in combination with the inhibition of p38 also increased angiogenesis, as increased blood vessel formation was observed in the scar area. However, p38 inhibition alone did not affect capillary density but stimulated cardiomyocyte mitosis transiently by activating cell cycle genes, such as the gene encoding cyclin A17. Although the efficiency of promoting adult cardiomyocyte proliferation was low, these studies suggest that this approach could serve as a potential therapeutic option for cardiac repair.
Figure 2. Regulation of cardiomyocyte proliferation.
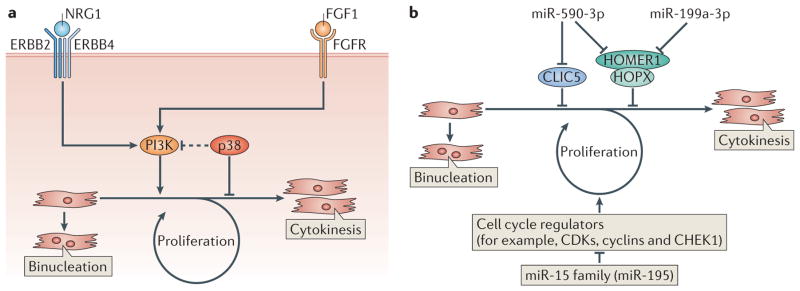
a | Regulation of cardiomyocyte proliferation by fibroblast growth factor 1 (FGF1) and neuregulin 1 (NRG1). Inhibition of the MAPK p38 in the presence of FGF1 or the activation of NGR1 signalling promotes cardiomyocyte re-entry into the cell cycle by activating PI3K, leading to DNA synthesis and cytokinesis. However, most cardiomyocytes in mice become binucleated shortly after birth as a consequence of DNA replication without cell division. b | Regulation of cardiomyocyte proliferation by microRNAs (miRNAs). miRNAs can positively (miR-590-3p and miR-199a-3p) or negatively (miR-15 family, including miR-195) regulate cardiomyocyte proliferation. miR-590-3p and miR-199a-3p promote cardiomyocyte proliferation by inhibiting the expression of genes encoding proteins that inhibit cell proliferation such as HOMER1, HOP homeobox (HOPX) and chloride intracellular channel 5 (CLIC5). The miR-15 family of miRNAs inhibits the cell cycle, and thus cardiomyocyte proliferation, by downregulating genes encoding proteins that activate the cell cycle. CDKs, cyclin-dependent kinases; CHEK1, checkpoint kinase 1; FGFR, FGF receptor.
Neuregulin 1 induces cardiomyocyte proliferation
Neuregulin 1 (NRG1) induces cardiomyocyte proliferation by activating ERBB2–ERBB4 heterodimer or ERBB4–ERBB4 homodimer receptors19 (FIG. 2a). Only differentiated, mononucleated cardiomyocytes, and not binucleated cardiomyocytes, displayed proliferative potential in the presence of NRG1. When adult cardiomyocytes undergo mitosis following NRG1 stimulation, sarcomeres disassemble to facilitate cytokinesis. This event, which was also seen in response to cardiomyocyte proliferation induced by the inhibition of p38 in the presence of FGF1, resembles the myofibrillar breakdown in proliferating embryonic cardiomyocytes. The activation of PI3K represents a common mechanism through which FGF1 and NRG1 promote cardiomyocyte proliferation. NRG1 reduced scar tissue formation and improved cardiac function when applied to adult mouse hearts two weeks post-myocardial infarction.
In adult mice, NRG1 activates the ERBB2–ERBB4 receptor to stimulate cardiomyocyte proliferation. However, during embryogenesis, NRG1 participates in signalling between the endocardium and myocardium that is required for differentiation and maturation, but not proliferation, of trabecular cardiomyocytes. This signalling is controlled by BMP10 (REF. 51). Thus, the distinct roles of NRG1 in adult and embryonic cardiomyocytes suggest that conclusions garnered from developmental studies may not always be applicable to adult disease therapy.
microRNAs regulate cardiomyocyte proliferation
The regulatory pathways that govern heart development and growth are modulated by numerous microRNAs (miRNAs)52,53. Based on the sequence of their seed region (that is, 6–8 specific nucleotides at their 5′ end), miRNAs suppress collections of mRNAs that often encode proteins participating in similar biological networks.
In a study to identify miRNAs involved in cardiomyocyte mitotic arrest, the expression patterns of miRNAs from the mouse ventricle at day 1 and day 10 were compared23. miR-195, a member of the miR-15 family, was highly upregulated at day 10 when cardiomyocytes have exited the cell cycle. Overexpression of miR-195 in the developing heart caused cardiomyocyte cell cycle arrest at G2 and led to hypoplasia and congenital heart abnormalities, such as ventricular septal defects23. It also prevented heart regeneration post-myocardial infarction at P1 (FIG. 2b). Conversely, inhibition of miR-15 family members that share the same seed sequence, using locked nucleic acid (LNA)-modified anti-miRNAs, led to an increase in mitotic cardiomyocytes in neonatal mice, promoted adult cardiomyocyte proliferation, improved cardiac function after myocardial infarction in adults and also improved contractile function after ischaemia–reperfusion injury42,54. These results suggest that the miR-15 family negatively regulates cardiomyocyte mitosis, and that upregulation of members of this miRNA family shortly after birth may contribute to cell cycle arrest23 (FIG. 2b). Among the many miR-195 target mRNAs, checkpoint kinase 1 (Chek1), which encodes a protein kinase that promotes the G2–M phase transition and mitotic progression, was downregulated in miR-195 transgenic hearts and upregulated in hearts treated with an anti-miRNA that inhibits miR-195.
To identify miRNAs that can promote neonatal cardiomyocyte proliferation, a high-throughput screen was performed using a whole genome miRNA library containing 875 miRNA mimics. Among 40 miRNAs that induced DNA synthesis and increased cytokinesis in neonatal mouse and rat cardiomyocytes, miR-590-3p and miR-199a-3p were selected for further studies of adult heart regeneration24 (FIG. 2b). Although these miRNAs contain different seed sequences, several of their predicted targets are the same. For example, miR-590-3p and miR-199a-3p target the HOMER1 mRNA, which encodes a protein that modulates Ca2+ signalling in the heart by interacting with the Ca2+ release channel ryanodine receptor (RyR). Also, both miRNAs were predicted to target the mRNA of HOP homeobox (HOPX), which encodes a homeodomain protein that inhibits cardiomyocyte proliferation. HOMER1 and HOPX were confirmed as miR-590-3p and miR-199a-3p targets using siRNAs, and additional targets specific to either miR-590-3p or miR-199a-3p were identified. For example, chloride intracellular channel 5 (CLIC5), an inhibitor of cell proliferation, was found to be a target of miR-590-3p but not miR-199a-3p. Consistent with in vitro data, overexpression of miR-590-3p and miR-199a-3p in neonatal mice increased cardiomyocyte, but not cardiac fibroblast, proliferation24. Of note, the presence of miR-590-3p and miR-199a-3p in cardiomyocytes reduced fibrotic scar size and improved cardiac function post-myocardial infarction 24. These results suggest that miRNAs could be used as therapeutic reagents to promote cardiomyocyte re-entry into the cell cycle, reduce scar formation and improve cardiac function after cardiomyocyte loss following injury.
Inactivation of Hippo signalling promotes proliferation
The Hippo signalling pathway, which was discovered in Drosophila melanogaster, is evolutionarily conserved and has a key role in regulating cell proliferation and organ size55. This pathway comprises a series of adaptors and kinases that promote the phosphorylation of the transcriptional co-activator Yes-associated protein (YAP), thereby preventing its nuclear entry. Specifically, the mammalian STE20-like protein kinase 1 (MST1) and MST2 kinases interact with the cofactor Salvador (SAV); this enables them to phosphorylate and activate a complex consisting of large tumour suppressor homologue 1 (LATS1) or LATS2 and MOB (Mps one binder), which phosphorylates and inactivates YAP (FIG. 3). Inactivation of YAP has a role in maintaining organ size during normal development, and dysregulation of this inhibition has been reported to cause cancer. YAP activates gene expression by interacting with DNA-binding transcription factors, such as TEAD (TEA domain family members), SMAD1, RUNX4 (RUNT-related transcription factor 4), T-box 5 (TBX5) and p73 (REFS 55,56).
Figure 3. Regulation of cardiomyocyte proliferation by the Hippo pathway.

Activation of the Hippo pathway, which includes the scaffold protein Salvador (SAV) and the kinases mammalian STE20-like protein kinase 1 (MST1), MST2, large tumour suppressor 1 (LATS1) and LATS2, results in the phosphorylation and nuclear exclusion of the transcriptional co-activator Yes-associated protein (YAP) to impede cardiomyocyte proliferation. Deletion of the upstream scaffold protein SAV or the inhibitory Hippo kinases LATS2, MST1 and MST2 promotes cardiomyocyte proliferation, as unphosphorylated YAP can activate the expression of genes in the WNT signalling pathway by interacting with β-catenin. Genetic deletion of YAP in the embryonic heart inhibits cardiomyocyte proliferation, leading to myocardial hypoplasia and embryonic lethality. Overexpression of YAP also promotes cardiomyocyte proliferation through the activation of pro-growth signalling pathways such as those driven by WNT and insulin-like growth factor 1 (IGF1). APC, adenomatosis polyposis coli; AXIN, axis inhibition protein; GPCR, G protein-coupled receptor; DVL, Dishevelled; GSK3β; glycogen synthase kinase 3 β; IGF1R, IGF1 receptor; LEF, lymphoid enhancer-binding factor; MOB, Mps one binder; TCF, T cell factor. Dashed icons represent components of the complex that phosphorylates β-catenin to promote its degradation, and dashed lines indicate that additional factors are involved.
Genetic loss of function of Sav in the mouse heart results in perinatal lethality owing to extreme enlargement of the heart, which is due to expansion of the trabecular and subcompact ventricular myocardial layers20. Ablation of the SAV-interacting kinases MST1 and MST2, or of the downstream kinase LATS2, causes a similar myocardial phenotype in embryonic mouse hearts20 (FIG. 3). Enlarged hearts resulting from alterations in the Hippo signalling pathway are not attributable to cardiomyocyte hypertrophy but are due to increased cardiomyocyte proliferation. Interestingly, WNT target genes are upregulated in Sav-deleted hearts. Cardiac knockout (cKO) of Sav also leads to an increase in nuclear β-catenin, which is an indicator of activated WNT signalling20. Reducing the level of β-catenin rescues the increase in cardiomyocyte proliferation that is observed in Sav-cKO hearts, suggesting that the Hippo pathway negatively regulates WNT signalling to control heart size and growth20. Among all of the components of the Hippo pathway, YAP is necessary and sufficient to regulate cardiomyocyte proliferation. Genetic deletion of Yap in the embryonic heart led to embryonic lethality, with myocardial hypoplasia due to reduced cardiomyocyte proliferation21,22. Conversely, YAP gain of function stimulates cardiomyocyte proliferation in vitro and in vivo. Thus, YAP connects Hippo signalling and other growth promoting pathways, such as insulin-like growth factor (IGF) and WNT signalling, to the control of embryonic heart growth. Whether YAP can promote the proliferation of adult cardiomyocytes and enhance adult heart regeneration in response to injury remains to be determined.
Reprogramming cells into cardiomyocytes
The conversion of various cell types into cardiomyocytes is another therapeutic approach for repairing injured hearts that is being intensely explored.
Partial reprogramming of fibroblasts into cardiomyocytes
Fibroblasts can be reprogrammed into induced pluri-potent stem (iPS) cells — cells that exhibit the properties of embryonic stem (ES) cells — through the forced expression of four pluripotency genes, namely OCT4 (also known as also known as POUF51 and OCT3), SOX2, Krüppel-like factor 4 (KLF4) and MYC (collectively referred to as OSKM factors)57,58 (FIG. 4). iPS cells generated from human fibroblasts have been differentiated into myriad cell types, including cardiomyocytes59–64. The ability to reprogramme fibroblasts of patients into beating cardiomyocytes via iPS cell intermediates offers a strategy for patient-specific disease modelling65–68.
Figure 4. Reprogramming fibroblasts into cardiomyocytes.

Several approaches have been used to reprogramme non-myocytes, such as adult fibroblasts and mouse embryonic fibroblasts (MEFs), into cardiomyocytes. In one method, the four pluripotency genes, OCT4, SOX2, Kruppel-like factor 4 (KLF4) and MYC (collectively referred to as OSKM factors), are overexpressed to reprogramme fibroblasts into induced pluripotent stem (iPS) cells, which subsequently differentiate into cardiomyocytes. A modification to this protocol is to overexpress OCT4, SOX2 and KLF4 (OSK factors), but not the oncoprotein MYC, to reduce the potential of oncogenic transformation. Transient overexpression of OSK factors epigenetically converts MEFs into plastic intermediates, without the cells becoming iPS cells. A Janus kinase (JAK) inhibitor helps prevent the formation of iPS cells from these intermediates (indicated by the dashed line) and promotes their differentiation towards cardiomyocytes. Alternatively, the forced expression of the basic helix loop helix (bHLH) transcription factor mesoderm posterior 1 (MESP1) and the transcription factor ETS2 in human dermal fibroblasts can induce their differentiation into beating cardiomyocytes via cardiac progenitors. Another approach involves the direct reprogramming of fibroblasts into cardiomyocytes, using transcription factors that specify the cardiac lineage and the differentiation of cardiomyocytes during embryogenesis. Forced expression of GATA4, myocyte-specific enhancer 2C (MEF2C) and T-box 5 (TBX5) with HAND2 (GHMT) or without HAND2 (GMT) can convert adult cardiac and skin fibroblasts into beating cardiomyocyte-like cells. GHMT or GMT factors can reprogramme non-cardiac cells into functional cardiomyocytes in vivo and improve cardiac function in response to injury. Cardiomyocytes have also been reprogrammed by introducing the microRNAs (miRNAs) miR-1, miR-133, miR-208 and miR-499 into cardiac fibroblasts. The addition of a JAK inhibitor enhanced reprogramming by these miRNAs.
It has been reported that the expression of several reprogramming factors in mouse embryonic fibroblasts (MEFs) and adult mouse fibroblasts, in combination with specific culture conditions, is sufficient to generate spontaneously contracting patches of differentiated cardiomyocytes without an intermediate pluripotent state69. The efficient generation of cardiomyocytes using this procedure only requires OSK factors, eliminating the need for the MYC oncoprotein and thus diminishing the potential for oncogenic transformation (FIG. 4). In this approach, it is assumed that the transient expression of reprogramming factors facilitates the opening of condensed chromatin, establishing a permissive state that facilitates the activation of silent genes and allows for multiple phenotypic outcomes. When provided with appropriate transcription factors and other signals that favour cardiogenesis, a high percentage of transiently pluripotent cells seem to adopt a cardiac fate over pluri-potency or other potential phenotypes. The removal from culture media of leukaemia inhibitory factor (LIF), the presence of which favours the generation and maintenance of pluripotent cells, further promotes the formation of cardiomyocytes. Moreover, the presence of inhibitors of JAK–STAT (Janus kinase–signal transducer and activator of transcription) also favours the generation of cells adopting a cardiac fate by inhibiting transition through the pluripotent state69 (FIG. 4). Inhibition of WNT signalling has also been shown to be permissive for the generation of cardiomyocytes from ES cells69.
In an alternative approach, the forced expression of the basic helix–loop–helix (bHLH) transcription factor mesoderm posterior 1 (MESP1) and the transcription factor ETS2, in mouse or human fibroblasts, generated beating cardiomyocytes with high efficiency70 (FIG. 4). In this approach, the cardiac fate seems to be established by cells passing through a cardiac progenitor intermediate state.
Although reprogramming of fibroblasts into cardiomyocytes using pluripotency genes has been successful, a major impediment to using this approach to treat heart disease in humans is the teratogenicity of iPS cells and the oncogenic potential of pluripotency genes. Other challenges include the cellular rejection of injected cells, the inefficiency of induced pluripotency to promote complete reprogramming to a cardiac cell fate and the failure of exogenously delivered cells to engraft in the injured myocardium67,68. In an effort to bypass these challenges, research is underway to directly reprogramme fibroblasts within the heart into functional cardiomyocytes, without transiting through an intermediate stem cell stage (see below).
Direct reprogramming of non-myocytes to cardiomyocytes
Cardiac fibroblasts account for ~60–70% of all heart cells71. Recent studies have focused on strategies to reprogramme cells into cardiomyocytes as a means of diminishing scar formation and generating new myocardium following myocardial infarction. Guided by the knowledge of the transcriptional circuits that specify cardiac lineage and the differentiation of cardiomyocytes during embryogenesis, different combinations of cardiac transcription factors have been tested for their ability to activate cardiac gene expression in fibroblasts in vitro. Initial results demonstrated that a combination of three transcription factors, namely GATA4, myocyte-specific enhancer factor 2C (MEF2C) and TBX5 (collectively referred to as GMT factors), could reprogramme both mice cardiac and dermal fibroblasts into induced cardiac-like myocytes (iCLMs) in vitro, with an efficiency of ~5–7%, depending on the cardiac marker assayed13 (FIG. 4). A minor fraction of these cells displayed spontaneous contractions after 4–5 weeks in culture. The efficiency of this approach is variable, and some laboratories have difficulty reproducing these results72. The variability is most likely due to the need for a precise stoichiometry of reprogramming factors to activate the cardiac gene programme, the heterogeneity of fibroblasts used in the different studies, the differences in the cell culture conditions used and other unknown variables. The inclusion of an additional bHLH transcription factor, HAND2, (in a cocktail referred to as GHMT) increased the reprogramming efficiency to ~20% (REF. 15) (FIG. 4). The newly formed iCLMs generated by these methods seem to arise from direct conversion of fibroblasts, bypassing an intermediate stem cell state. Moreover, these cells are phenotypically stable following the withdrawal of exogenous cardiogenic factors.
Fibroblasts reprogrammed by GMT or GHMT factors suppress the expression of fibroblast-specific genes and concomitantly activate a broad array of genes characteristic of cardiomyocytes13,15. Both atrial and ventricular cardiac genes are expressed in reprogrammed fibroblasts, suggesting a heterogeneous response to these factors. A small fraction of reprogrammed cells also displays spontaneous contractions on tissue culture plates, accompanied by electrical activity and Ca2+ transients13,15,73. This suggests that they are functional and similar, yet not identical, to neonatal ventricular cardiomyocytes.
Cardiomyocytes have also been generated by introducing the muscle-specific miRNAs miR-1, miR-133, miR-208 and miR-499 into cardiac fibroblasts74. Addition of a JAK1 inhibitor enhanced reprogramming by these miRNAs, consistent with studies demonstrating the cardiogenic activity of this compound in iPS cell-like cultures69. The ability of these miRNAs to induce the cardiac phenotype in fibroblasts suggests that the proteins encoded by the targets of one or more of these miRNAs repress the cardiac phenotype in fibroblasts. The identity of such proteins remains unknown, although it is interesting to note that fibroblasts repress the cardiac phenotype when artificially fused to cardiomyocytes in heterokaryons75. This suggests that the fibroblast phenotype is dominant over the cardiac phenotype.
GATA4 and TBX5 together with BAF60C (BRG1- associated factor 60C), a cardiac-restricted subunit of the SWI/SNF-like BAF chromatin remodelling complex, were reported to be sufficient to generate beating cardiomyocytes when ectopically overexpressed in the non-cardiogenic posterior mesoderm and the extra-embryonic mesoderm of the mouse amnion76. BAF60C did not display cardiogenic activity in fibroblasts or enhance the activity of GMT factors in this setting13, suggesting that its activity may require a permissive cell type or other permissive cardiogenic signals in the embryo.
The precise mechanism responsible for direct cardiac reprogramming remains to be fully defined. Various combinations of the GHMT factors have been shown to directly activate genes encoding cardiac structural proteins and to auto- and cross-regulate their own expression13–15. Presumably, when these factors achieve threshold levels of expression, they can switch on endogenous cardiac genes that gradually establish a stable cardiac phenotype.
Reprogramming fibroblasts into cardiomyocytes in vivo
The potential of cardiac regulatory genes to reprogramme fibroblasts into cardiomyocyte-like cells raises interesting possibilities for heart regeneration following ischaemic injury; such injury results in extensive cardiomyocyte death and expansion of the cardiac fibroblast pool, causing fibrosis and scar formation. Taking advantage of the fact that retroviruses only infect proliferating cells, GHMT and GMT factors have been delivered to the mouse heart after LAD ligation using retroviral expression systems14,15. Reprogrammed cells, which were identified by the presence of cardiac troponin T, functionally integrated into the heart after several weeks and formed gap junctions with neighbouring endogenous cardiomyocytes and with each other. When these iCLMs were isolated and cultured in vitro, they displayed rod-shaped morphology, a sarcomere structure, cardiomyocyte-like gene profiles, contractility and Ca2+ transients analogous to mature cardiomyocytes. Lentiviral-based expression of miR-1, miR-133, miR-208, and miR-499 also induced the formation of new cardiomyocytes in vivo post-LAD ligation74 (FIG. 4).
The expression of the cardiac reprogramming factors GHMT or GMT in vivo reduced fibrotic scar formation and led to a dramatic and sustained improvement of cardiac function post-myocardial infarction, even up to a year following this intervention14,15. The functional improvement was substantially greater than would be expected from the relatively low reprogramming efficiency observed with these factors in vitro. This suggests that the native environment of the intact heart, which contains growth factors, the extracellular matrix, other cell types and contracting neighbouring cells, may enhance reprogramming. Although fibro-blast lineage markers demonstrate that this cell type is reprogrammed into cardiomyocytes in vivo, it is also conceivable that cardiac factors may stimulate cardiac progenitor cell differentiation or facilitate the reprogramming of other cell types in the heart, such as endothelial cells, into cardiomyocytes. The improved cardiac function observed with in vivo reprogramming could also be due to improved reprogramming efficiency, enhanced cardiomyocyte division, increased blood vessel formation, increased secretion of cytokines from infiltrating immune cells and the inhibition of fibroblast proliferation. Another consideration with respect to the assessment of efficiency of in vivo reprogramming is that there is substantial heterogeneity among fibroblast populations within the heart that could display differing potential for cardiac reprogramming. In addition, blood vessel-associated cells such as mesoangioblasts, which function as a stem cell population that can differentiate into multiple mesodermal cell types, may also show selectivity for adopting a cardiac fate in response to reprogramming factors77,78. In this regard, although lineage tracing demonstrates that fibroblasts are reprogrammed into cardiomyocytes in vivo, other cell types can also be reprogrammed to adopt a cardiac fate, and therefore the reprogramming efficiency, solely based on fibroblast conversion, is likely to be underestimated.
Numerous challenges remain to be addressed before reprogramming into cardiac myoctyes in vivo can be used in humans. Human cells are difficult to reprogramme, probably because of stable epigenetic modifications that occur over time. Initial progress towards the reprogramming of human fibroblasts into cardiomyocytes in vitro has recently been reported79. Currently, direct lineage conversion gives rise to differentiated, non-proliferative cells, which limits the yield of reprogrammed cells. Therefore, providing a strategy to enhance transit through an expandable population of progenitor cells would be desirable. In addition, irregularities in cardiac architecture can cause lethal cardiac arrhythmias, so reprogramming must generate a myocardium that is seamlessly integrated with pre-existing tissue, the cardiac conduction system and the vasculature to re-establish a functional tissue.
Reprogramming into non-myocyte heart cells
In addition to cardiomyocytes, various other specialized cell types within the heart are essential for cardiac function, including certain cells of the cardiac conduction system and endothelial and smooth muscle cells that give rise to the vascular system. There has been considerable progress towards the generation of these cell types through transcriptional reprogramming.
Reprogramming cardiomyocytes into conduction system cells
The cardiac conduction system coordinates electrical impulses through the heart that mediate cardiac rhythm. The heartbeat is initiated by an impulse within the sinoatrial node (SAN), which passes through the atrium to the atrioventricular node, where impulse propagation is delayed to allow the atria to contract. Electrical impulses then progress through the ventricular conduction system, which comprises the His bundle, right and left bundle branches and Purkinje fibres, to coordinate contraction of the heart from the apex to the base. Abnormalities in the cardiac conduction system cause disturbances in cardiac rhythm and account for a high fraction of human morbidity and mortality. Current treatment for such disorders involves the implantation of costly electronic pacemakers.
In recent studies, in vivo reprogramming has been extended to the generation of cardiac conduction system cells. Forced expression of the TBX18 transcription factor in rodent ventricular cardiomyocytes, in vitro and in vivo, has been reported to confer the functional and genetic properties of pacemaker cells from the SAN without passage of cells through a pluripotent state80 (FIG. 5). Remarkably, the cells generated by this approach display characteristics of bone fide pacemaker cells such as Ca2+ cycling (the release and re-uptake of intracellular Ca2+ to control muscle contraction and relaxation) and automaticity (the ability of cells to depolarize spontaneously). They also retain their pacemaker phenotype even after exogenous TBX18 has been removed, indicating their stable conversion. Activation of Notch signalling in newborn rodent cardiomyocytes, in vitro and in vivo, has been shown to confer a ‘Purkinje-like’ phenotype81 (FIG. 5). These initial findings point towards exciting opportunities to generate biological pacemakers in vivo.
Figure 5. Reprogramming different cardiac cell types.
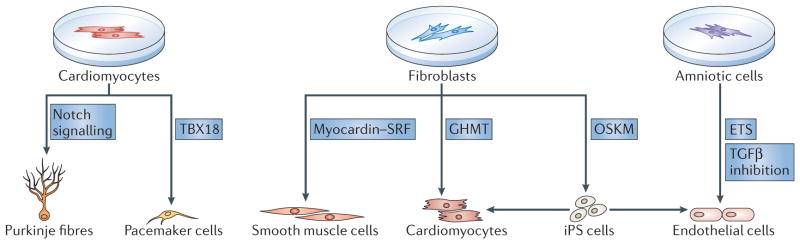
Progress has been made in using transcriptional reprogramming to generate different specialized cell types that are essential for restoring cardiac function in an injured heart. For the conduction system, pacemaker cells and Purkinje fibres can be generated by reprogramming. Overexpression of the transcription factor T-box 18 (TBX18) directly reprogrammes rodent ventricular cardiomyocytes, in vitro and in vivo, into cells resembling pacemaker cells from the sinoatrial node, without passage of cells through a pluripotent state. The activation of Notch signalling in newborn rodent cardiomyocytes,in vitro and in vivo, confers a Purkinje cell-like phenotype. For the vasculature, smooth muscle cells and endothelial cells can be produced from fibroblasts by reprogramming. Smooth muscle cells can be generated by forced expression of the cardiovascular-specific co-activator myocardin. Myocardin activates smooth muscle-specific genes through association with the transcription factor serum response factor (SRF). Adult fibroblasts can also be reprogrammed into an incomplete induced pluripotent state by transient forced overexpression of the four induced pluripotent stem (iPS) cell factors OCT4, SOX2, Kruppel-like factor 4 (KLF4) and MYC (OSKM factors); endothelial cells then form under permissive culture conditions. Endothelial cells can also be generated from amniotic cells by forced overexpression of ETS transcription factors, in combination with the inhibition of transforming growth factor-β (TGFβ) signalling, without cells transiting through an iPS cell state. Finally, cardiomyocytes can be generated by reprogramming fibroblasts indirectly via an iPS cell state that is induced by OSKM factors, or directly with the cardiac-specific transcription factors GATA4, HAND2, myocyte-specific enhancer factor 2C (MEF2C) and T-box 5 (TBX5) (collectively referred to as GHMT) (see also FIG. 4).
Reprogramming fibroblasts into smooth muscle cells
The creation of a functional myocardium requires not only cardiomyocytes, but also a functional vascular system to nourish myocardial tissue. Vascular smooth muscle cells can be generated by forced expression of the cardiovascular-specific co-activator myocardin82–84, which potently activates transcription through its association with serum response factor (SRF) (FIG. 5). DNA-binding sites for SRF are contained in nearly all smooth muscle-specific genes and in cardiac structural genes. Moreover, myocardin and SRF are necessary and sufficient for specifying the smooth muscle lineage in vivo and in vitro82,85,86.
Reprogramming non-myogenic cells into endothelial cells
Progress has been made towards reprogramming fibro-blasts into endothelial cells that can promote angiogenesis in vivo and form vascular structures on decellularized vessel scaffolds87. Exposing primary human fibroblasts to OSKM factors, but removing these factors after 4 days and before the complete pluripotent state has been reached, followed by culture under specialized conditions, allows the generation of endothelial cells that can enhance angiogenesis in infarcted hearts (FIG. 5). In a similar approach, neonatal and adult human fibroblasts have been converted into angioblast-like progenitors by transient reprogramming to pluripotency, and culturing these cells under distinct conditions allowed the generation of functional endothelial and smooth muscle cells88. Finally, forced expression of various ETS transcription factors, combined with the inhibition of transforming growth factor-β (TGFβ) signalling, efficiently reprogrammed mature amniotic cells into endothelial cells89 (FIG. 5).
The epicardium in cardiac regeneration
The epicardium, a mesothelial cell layer that encapsulates the heart, is a source of progenitor and paracrine cells that are essential for heart development, repair and regeneration. Soluble signalling molecules released by the epicardium influence the underlying myocardium90. During embryogenesis, the epicardium generates progenitor cells that differentiate into vascular smooth muscle cells, cardiac fibroblasts and possibly endothelial cells91–93. However, lineage tracing experiments suggest that the coronary artery vessels are derived from the venous plexus and not from the epicardium94. In the adult heart, the epicardium is quiescent under homeostatic conditions. However, upon injury such as myocardial infarction, activated epicardial cells show an increase in proliferation and differentiate into mesenchymal cells that express fibroblast and smooth muscle cell markers91. Identification of genetic regulatory enhancers that are activated during heart development and injury shows that activation of the epicardium is dependent on CCAAT/enhancer binding protein (C/EBP) transcription factors95. Although there is still some debate on whether the activated epicardium is a source of cardiac muscle cells, recent evidence strongly suggests that this is not the case in vivo92,96,97. In zebrafish, it is clear that epicardial cell derivatives are limited to non-myocardial cell types98. In summary, epicardial cells are activated following cardiac injury and have the potential to differentiate into various cell types. Targeting this niche with small molecules provides an attractive therapeutic strategy for regenerative medicine99.
Inflammatory response and cardiac repair
The immune system responds immediately to tissue injury, rapidly deploying inflammatory cells to the damaged site. A well-documented example of the role of the activated immune system and macrophages in tissue repair comes from skeletal muscle regeneration100–102. During ischaemic injury in the heart, cardiomyocyte death triggers an inflammatory response that is necessary to clear cellular and matrix debris and promotes scar formation. However, the inflammatory response can also perpetuate tissue damage and has long been recognized as a key mediator of the pathology and mortality associated with ischaemia–reperfusion injury, atherosclerosis and other cardiovascular diseases103,104. Recently, several studies have shed new light on both positive and negative roles for inflammation in tissue repair and regeneration. For example, a biphasic monocyte response, in which inflammatory and reparative monocyte subsets are sequentially recruited from the spleen, is necessary for appropriate healing following myocardial infarction105,106. Inhibition of C/EBP signalling in the adult epicardium greatly decreased the neutrophil influx that occurs on the epicardial surface and in the subepicardial space following ischaemic injury, leading to a reduction in fibrosis and improved cardiac function95. By contrast, new findings suggest that acute inflammation may be required for brain regeneration in adult zebrafish107 and that activation of innate immune receptors is required for efficient regenerative reprogramming strategies108. These studies force us to rethink the long-held dogma that inflammation negatively affects tissue repair and regeneration and suggests that future studies are necessary to understand how fine tuning of the immune response to cardiac injury can influence heart repair, regeneration and functional outcomes.
Looking to the future
Deciphering the signalling pathways and regulatory networks of cardiac development has led to therapeutic strategies to trigger cardiac regeneration and repair. Although progress made in promoting cardiomyocyte proliferation and in direct reprogramming of non-myocytes into cardiomyocytes and other cell types of the heart has offered new perspectives in the quest to enhance repair and regeneration of the injured adult heart, many challenges remain to be addressed.
During a heart attack, millions to billions of cardiomyocytes are lost. Generating sufficient numbers of new cardiomyocytes to replace the dead cells is especially challenging. Currently, generating new cardiomyocytes by inducing proliferation of existing cardiomyocytes or by reprogramming of non-myocytes is possible but vastly inefficient. This issue is particularly challenging, given that the cells that result from some reprogramming approaches do not proliferate, perhaps necessitating combined strategies to simultaneously promote reprogramming and proliferation. Thus, to enhance heart regeneration, it will be important to develop procedures that increase the yield and efficiency of generating new cardiomyocytes. To date, strategies to promote cardiac repair by inducing cardiomyocyte proliferation and cellular reprogramming have been tested primarily in rodents, with only a single study being reported in pigs109. Whether these approaches can be scaled up sufficiently to recreate a functional heart in humans remains to be demonstrated.
Inflammatory signals also play a key part in the response of the heart to injury. Whereas the recruitment of macrophages to sites of injury represents an essential early step to clear dead cells and extracellular debris, later aspects of the immune response can impede cardiac repair. Moreover, precise timing is necessary to therapeutically modulate the inflammatory response, as complete blockade to this process can result in cardiac rupture post-myocardial infarction105. Stimulation of inflammatory pathways, such as the Toll-like receptor pathway, may improve regeneration efficiency by modulating the expression of epigenetic regulators and thus increasing nuclear reprogramming efficiency108.
Efficient cardiac repair will probably also necessitate the delivery of reprogramming factors specifically to cardiac fibroblasts, and not to other fibroblast populations throughout the body, and molecules that promote cardiomyocyte proliferation will need to be delivered selectively to cardiomyocytes, and not to fibroblasts that develop into fibrotic scar tissue. Developing molecules that specifically affect cardiomyocytes could be technically challenging due to the complexity of the cell types in the heart.
It has been reported that cardiac arrhythmias can be induced by the delivery of immature and heterogeneous cardiomyocytes that were derived from progenitor or stem cells110,111. Currently, reprogrammed iCLMs and dedifferentiated dividing cardiomyocytes are immature and phenotypically heterogeneous, which may contribute to arrhythmogenesis. Therefore, it is crucial to promote complete cardiomyocyte differentiation after proliferation and to reprogramme cardiac fibroblasts into homogeneous, mature cardiomyocytes that can functionally integrate with existing cardiomyocytes in the adult heart to maintain contractility and conductivity.
Another concern is the safety issue associated with the viral-based delivery of factors to the injured heart. As integration of retroviruses into the genome can cause cancer, developing a strategy to enable the delivery of transcription factors without viral vectors would be important. Such methods also need to be non-invasive and yet efficient in promoting cardiomyocyte proliferation and the reprogramming of non-myocytes into cardiomyocytes and possible reprogramming of other cardiac cell types. Cardiogenic small molecules and synthetic oligonucleotides will be attractive alternatives to virus-mediated approaches in the future. Although discoveries in mouse models have provided important insights into human biology, they may not be directly applicable to human disease therapies. Preclinical trials in large animals are necessary to evaluate safety and efficacy issues.
Although beyond the scope of this Review, substantial progress has been made in the field of tissue engineering and the development of artificial matrices, such as algisyl112 and decellularized tissue scaffolds, which provide structural integrity and signals to promote the formation of heart tissue113. Combining such methods with the activation of developmental regulatory mechanisms and cellular transplantation represents an especially promising approach for successful adult heart regeneration and repair.
In summary, the regeneration of the adult human heart represents a major challenge of fundamental importance, given the magnitude of heart disease and the absence of effective long-term therapies for heart failure. Considering the complexity of the heart, which requires second to second uninterrupted function and seamless integration of cardiomyocytes with other cell types within the organ, the creation of functional heart tissue in vivo will undoubtedly necessitate a combination of approaches. The foundation of knowledge of cardiac developmental regulatory mechanisms provides a strong platform for achieving this goal.
Key points.
In response to myocardial infarction, damaged adult cardiomyocytes are replaced by activated fibroblasts that form a fibrotic scar, leading to reduced cardiac function and heart failure. As the adult heart has limited regenerative capacity, there is a need to develop innovative strategies to enhance cardiac repair and regeneration.
Cellular replacement strategies for heart repair, in which stem cells and other cell types are injected directly into the injured heart or into the coronary circulation, have shown modest beneficial effects on cardiac function. An alternative approach is to reprogramme non-muscle cells in the injured heart to adopt a cardiac fate.
Positive cell cycle regulators are highly expressed in the embryonic heart and downregulated in the adult heart. Activation of various signalling pathways in the heart can modestly reactivate proliferation in adult cardiomyocytes.
Identification of transcription factors and microRNAs that control heart formation has enabled reprogramming of non-muscle cells into cardiomyocytes and other cell types of the heart. Following injury, in vivo reprogramming of non-myocytes into cardiomyocytes has improved heart function in mice.
Epicardial cells are activated following cardiac injury and have the potential to differentiate into various cell types, offering a niche that can be targeted with small molecules. This provides an attractive approach for regenerative medicine.
The inflammatory response has a role in cardiac repair following injury. Several studies have shed light on both positive and negative roles of the inflammatory response in tissue repair and regeneration.
Acknowledgments
The authors thank G. Huang, Y.-J. Nam, A. Aurora and K. Song for constructive scientific discussions. They thank J. Cabrera for assistance with figures. Work in the author’s laboratory was supported by grants from the National Institutes of Health, the Robert A. Welch Foundation (grant I-0025), the Leducq Foundation-Transatlantic Network of Excellence in Cardiovascular Research Program, the American Heart Association-Jon Holden DeHaan Foundation and the Cancer Prevention and Research Institute of Texas (CPRIT). M.X. was supported by a Beginning Grant-in-Aid from the SouthWest Affiliate of the American Heart Association.
Glossary
- Paracrine effects
The effects of a signalling factor secreted by one cell on a nearby cell
- Cardiogenesis
The development of the embryonic heart
- Myocardial infarction
Interruption of the blood flow to the heart, causing cell death and heart damage. Also known as heart attack
- Lineage tracing
A genetic tool used to trace all progeny originating from a single cell
- Cre–loxP recombination system
Tissue-specific expression of Cre recombinase to carry out targeted gene deletion
- Ventricle apex
Anatomically, the lowest portion of the heart
- Myocardial ischaemia–reperfusion
Restoration of the blood supply to the heart tissue that is ischaemic due to a decrease in this
- Trabecular cardiomyocytes
Highly organized cardiomyocytes that form ‘projections’ into the lumen of the heart ventricles to increase surface area. They facilitate contractility of the heart
- Locked nucleic acid (LNA)-modified anti-miRNAs
Chemically modified, single-stranded RNA oligonucleotides that contain an extra bridge connecting the 2′ oxygen and 4′ carbon, which ‘locks’ the ribose in the 3′-endo conformation. This results in high stability and affinity to inactivate specific miRNAs
- Subcompact ventricular myocardial layers
The thick muscular walls of the heart ventricles
- Teratogenicity
The capability of producing congenital anomalies
- Heterokaryons
A cell that contains multiple genetically different nuclei
- Gap junctions
A specialized intercellular connection that directly connects the cytoplasm of two cells, allowing various molecules and ions to pass freely between cells
- Venous plexus
A congregation of multiple veins
Biographies
Mei Xin received her Ph.D. from the University of Texas Southwestern Medical Center, Dallas, USA. She completed her postdoctoral training in the laboratory of Eric N. Olson and is now an instructor at University of Texas Southwestern Medical Center, where she investigates microRNAs and signalling molecules regulating cardiovascular development and diseases.
Eric N. Olson is Professor and Chair of the Department of Molecular Biology at the University of Texas Southwestern Medical Center, Dallas, USA, where he holds the Robert A. Welch Distinguished Chair in Science, the Annie and Willie Nelson Professorship in Stem Cell Research and the Pogue Distinguished Chair in Research on Cardiac Birth Defects. He received his Ph.D. in biochemistry from Wake Forest University and performed his postdoctoral training at Washington University School of Medicine, St Louis, Missouri, USA. He began his scientific career at the MD Anderson Cancer Center, Houston, Texas, USA, and in 1995, he founded the Department of Molecular Biology at the University of Texas Southwestern Medical Center. He is a member of the American Academy of Arts and Sciences, the US National Academy of Sciences and the Institute of Medicine. His awards include the Basic Research Prize, the Research Achievement Award and Inaugural Distinguished Scientist Award from the American Heart Association, as well as the Pasarow Award, the Pollin Prize in Pediatric Research and the Passano Award. In 2009, the French Academy of Science awarded him the Fondation Lefoulon-Delalande Grand Prize, and in 2013, he received the March of Dimes Prize in developmental biology.
Rhonda Bassel-Duby is a professor of molecular biology at the University of Texas Southwestern Medical Center, Dallas, USA. Her research focuses on the molecular mechanisms governing heart and skeletal muscle development and the response to physiological and pathological demands. She received her Ph.D. in biochemistry from McGill University, Montreal, Canada, and trained at Harvard Medical School, Boston, Massachusetts, and the University of Texas Southwestern Medical Center.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Eric N. Olson’s homepage: http://www4.utsouthwestern.edu/olsonlab/index.html
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 3.Segers VF, Lee RT. Stem-cell therapy for cardiac disease. Nature. 2008;451:937–942. doi: 10.1038/nature06800. [DOI] [PubMed] [Google Scholar]
- 4.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–335. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wollert KC, Drexler H. Cell therapy for the treatment of coronary heart disease: a critical appraisal. Nature Rev Cardiol. 2010;7:204–215. doi: 10.1038/nrcardio.2010.1. [DOI] [PubMed] [Google Scholar]
- 6.Anversa P, Kajstura J, Rota M, Leri A. Regenerating new heart with stem cells. J Clin Invest. 2013;123:62–70. doi: 10.1172/JCI63068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Passier R, van Laake LW, Mummery CL. Stem-cell-based therapy and lessons from the heart. Nature. 2008;453:322–329. doi: 10.1038/nature07040. [DOI] [PubMed] [Google Scholar]
- 8.Ptaszek LM, Mansour M, Ruskin JN, Chien KR. Towards regenerative therapy for cardiac disease. Lancet. 2012;379:933–942. doi: 10.1016/S0140-6736(12)60075-0. [DOI] [PubMed] [Google Scholar]
- 9.Mercola M, Ruiz-Lozano P, Schneider MD. Cardiac muscle regeneration: lessons from development. Genes Dev. 2011;25:299–309. doi: 10.1101/gad.2018411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nature Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 11.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. References 10, 11 and 12 review the genetic circuits that drive the evolution and development of the heart. [DOI] [PubMed] [Google Scholar]
- 13.Ieda M, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian L, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song K, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. References 13, 14 and 15 describe in vitro and in vivo reprogramming of non-muscle cells into cardiomyocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aguirre A, Sancho-Martinez I, Izpisua Belmonte JC. Reprogramming toward heart regeneration: stem cells and beyond. Cell Stem Cell. 2013;12:275–284. doi: 10.1016/j.stem.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Engel FB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhn B, et al. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nature Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 19.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 20.Heallen T, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xin M, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Gise A, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci USA. 2012;109:2394–2399. doi: 10.1073/pnas.1116136109. References 20, 21 and 22 show that the Hippo signalling pathway governs cardiomyocyte proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porrello ER, et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109:670–679. doi: 10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eulalio A, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. Identifies miRNAs that induce cardiomyocyte proliferation. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Martinez V, Schoenwolf GC. Primitive-streak origin of the cardiovascular system in avian embryos. Dev Biol. 1993;159:706–719. doi: 10.1006/dbio.1993.1276. [DOI] [PubMed] [Google Scholar]
- 26.Tam PP, Parameswaran M, Kinder SJ, Weinberger RP. The allocation of epiblast cells to the embryonic heart and other mesodermal lineages: the role of ingression and tissue movement during gastrulation. Development. 1997;124:1631–1642. doi: 10.1242/dev.124.9.1631. [DOI] [PubMed] [Google Scholar]
- 27.Garry DJ, Olson EN. A common progenitor at the heart of development. Cell. 2006;127:1101–1104. doi: 10.1016/j.cell.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 28.Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013;5:a008292. doi: 10.1101/cshperspect.a008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–1746. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 31.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nature Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergmann O, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kajstura J, et al. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–315. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Senyo SE, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. The first study to show that the adult zebrafish heart can regenerate. [DOI] [PubMed] [Google Scholar]
- 36.Lepilina A, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127:607–619. doi: 10.1016/j.cell.2006.08.052. [DOI] [PubMed] [Google Scholar]
- 37.Jopling C, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–609. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikuchi K, et al. Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature. 2010;464:601–605. doi: 10.1038/nature08804. References 37 and 38 demonstrate that zebrafish heart regeneration involves the proliferation of existing cardiomyocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fishman MC, Olson EN. Parsing the heart: genetic modules for organ assembly. Cell. 1997;91:153–156. doi: 10.1016/s0092-8674(00)80397-9. [DOI] [PubMed] [Google Scholar]
- 40.Wills AA, Holdway JE, Major RJ, Poss KD. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development. 2008;135:183–192. doi: 10.1242/dev.010363. [DOI] [PubMed] [Google Scholar]
- 41.Porrello ER, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. The first study to show that mammalian heart regeneration can occur transiently following birth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porrello ER, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci USA. 2013;110:187–192. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–1054. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 44.Chaudhry HW, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem. 2004;279:35858–35866. doi: 10.1074/jbc.M404975200. [DOI] [PubMed] [Google Scholar]
- 45.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–118. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 46.Katz EB, et al. Cardiomyocyte proliferation in mice expressing α-cardiac myosin heavy chain–SV40 T-antigen transgenes. Am J Physiol. 1992;262:H1867–H1876. doi: 10.1152/ajpheart.1992.262.6.H1867. [DOI] [PubMed] [Google Scholar]
- 47.Kubin T, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011;9:420–432. doi: 10.1016/j.stem.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci USA. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Cuevas P, et al. Fibroblast growth factor-1 prevents myocardial apoptosis triggered by ischemia reperfusion injury. Eur J Med Res. 1997;2:465–468. [PubMed] [Google Scholar]
- 51.Grego-Bessa J, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510–525. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hullinger TG, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2012;110:71–81. doi: 10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan D. The Hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nature Cell Biol. 2011;13:877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. References 57 and 58 demonstrate that four transcription factors can induce iPS cells from fibroblasts. [DOI] [PubMed] [Google Scholar]
- 59.Marchetto MC, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee G, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 62.Liu GH, et al. Recapitulation of premature ageing with iPSCs from Hutchinson–Gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yazawa M, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carvajal-Vergara X, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Priori SG, Napolitano C, Di Pasquale E, Condorelli G. Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. J Clin Invest. 2013;123:84–91. doi: 10.1172/JCI62838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakamura K, Hirano K, Wu SM. iPS cell modeling of cardiometabolic diseases. J Cardiovasc Transl Res. 2013;6:46–53. doi: 10.1007/s12265-012-9413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bellin M, Marchetto MC, Gage FH, Mummery CL. Induced pluripotent stem cells: the new patient? Nature Rev Mol Cell Biol. 2012;13:713–726. doi: 10.1038/nrm3448. [DOI] [PubMed] [Google Scholar]
- 68.Yoshida Y, Yamanaka S. iPS cells: a source of cardiac regeneration. J Mol Cell Cardiol. 2011;50:327–332. doi: 10.1016/j.yjmcc.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 69.Efe JA, et al. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nature Cell Biol. 2011;13:215–222. doi: 10.1038/ncb2164. [DOI] [PubMed] [Google Scholar]
- 70.Islas JF, et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc Natl Acad Sci USA. 2012;109:13016–13021. doi: 10.1073/pnas.1120299109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 72.Chen JX, et al. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ Res. 2012;111:50–55. doi: 10.1161/CIRCRESAHA.112.270264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Addis RC, et al. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J Mol Cell Cardiol. 2013;60:97–106. doi: 10.1016/j.yjmcc.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jayawardena TM, et al. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. 2012;110:1465–1473. doi: 10.1161/CIRCRESAHA.112.269035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Evans SM, Tai LJ, Tan VP, Newton CB, Chien KR. Heterokaryons of cardiac myocytes and fibroblasts reveal the lack of dominance of the cardiac muscle phenotype. Mol Cell Biol. 1994;14:4269–4279. doi: 10.1128/mcb.14.6.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–711. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Minasi MG, et al. The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development. 2002;129:2773–2783. doi: 10.1242/dev.129.11.2773. [DOI] [PubMed] [Google Scholar]
- 78.Acharya A, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nam YJ, et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci USA. 2013;110:5588–5593. doi: 10.1073/pnas.1301019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kapoor N, Galang G, Marban E, Cho HC. Transcriptional suppression of connexin43 by TBX18 undermines cell–cell electrical coupling in postnatal cardiomyocytes. J Biol Chem. 2011;286:14073–14079. doi: 10.1074/jbc.M110.185298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rentschler S, et al. Myocardial Notch signaling reprograms cardiomyocytes to a conduction-like phenotype. Circulation. 2012;126:1058–1066. doi: 10.1161/CIRCULATIONAHA.112.103390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci USA. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 84.Du KL, et al. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li S, Wang DZ, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci USA. 2003;100:9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 87.Margariti A, et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc Natl Acad Sci USA. 2012;109:13793–13798. doi: 10.1073/pnas.1205526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kurian L, et al. Conversion of human fibroblasts to angioblast-like progenitor cells. Nature Methods. 2013;10:77–83. doi: 10.1038/nmeth.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ginsberg M, et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFβ suppression. Cell. 2012;151:559–575. doi: 10.1016/j.cell.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sucov HM, Gu Y, Thomas S, Li P, Pashmforoush M. Epicardial control of myocardial proliferation and morphogenesis. Pediatr Cardiol. 2009;30:617–625. doi: 10.1007/s00246-009-9391-8. [DOI] [PubMed] [Google Scholar]
- 91.Zhou B, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121:1894–1904. doi: 10.1172/JCI45529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katz TC, et al. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev Cell. 2012;22:639–650. doi: 10.1016/j.devcel.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Majesky MW. Development of coronary vessels. Curr Top Dev Biol. 2004;62:225–259. doi: 10.1016/S0070-2153(04)62008-4. [DOI] [PubMed] [Google Scholar]
- 94.Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang GN, et al. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science. 2012;338:1599–1603. doi: 10.1126/science.1229765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rinkevich Y, et al. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nature Cell Biol. 2012;14:1251–1260. doi: 10.1038/ncb2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Acharya A, Baek ST, Banfi S, Eskiocak B, Tallquist MD. Efficient inducible Cre-mediated recombination in Tcf21 cell lineages in the heart and kidney. Genesis. 2011;49:870–877. doi: 10.1002/dvg.20750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kikuchi K, et al. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development. 2011;138:2895–2902. doi: 10.1242/dev.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Russell JL, Goetsch SC, Aguilar HR, Frantz DE, Schneider JW. Targeting native adult heart progenitors with cardiogenic small molecules. ACS Chem Biol. 2012;7:1067–1076. doi: 10.1021/cb200525q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heredia JE, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kharraz Y, Guerra J, Mann CJ, Serrano AL, Munoz-Canoves P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm. 2013;2013:491497. doi: 10.1155/2013/491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Saclier M, Cuvellier S, Magnan M, Mounier R, Chazaud B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013 doi: 10.1111/febs.12166. http://dx.doi.org/10.1111/febs.12166. [DOI] [PubMed]
- 103.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58:88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kyritsis N, et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338:1353–1356. doi: 10.1126/science.1228773. [DOI] [PubMed] [Google Scholar]
- 108.Lee J, et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012;151:547–558. doi: 10.1016/j.cell.2012.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kawamura M, et al. Feasibility, safety, and therapeutic efficacy of human induced pluripotent stem cell-derived cardiomyocyte sheets in a porcine ischemic cardiomyopathy model. Circulation. 2012;126:S29–S37. doi: 10.1161/CIRCULATIONAHA.111.084343. [DOI] [PubMed] [Google Scholar]
- 110.Chen HS, Kim C, Mercola M. Electrophysiological challenges of cell-based myocardial repair. Circulation. 2009;120:2496–2508. doi: 10.1161/CIRCULATIONAHA.107.751412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang YM, Hartzell C, Narlow M, Dudley SC., Jr Stem cell-derived cardiomyocytes demonstrate arrhythmic potential. Circulation. 2002;106:1294–1299. doi: 10.1161/01.cir.0000027585.05868.67. [DOI] [PubMed] [Google Scholar]
- 112.Lee RJ, Hinson A, Helgerson S, Bauernschmitt R, Sabbah HN. Polymer-based restoration of left ventricular mechanics. Cell Transplant. 2013;22:529–533. doi: 10.3727/096368911X637461. [DOI] [PubMed] [Google Scholar]
- 113.Badylak SF, Taylor D, Uygun K. Whole-organ tissue engineering: decellularization and recellularization of three-dimensional matrix scaffolds. Annu Rev Biomed Eng. 2011;13:27–53. doi: 10.1146/annurev-bioeng-071910-124743. [DOI] [PMC free article] [PubMed] [Google Scholar]