

Abstract
Mutations in mitochondrial DNA cause a number of neurological diseases with defined neuropathology; however, mutations in this genome have also been found to be important in a number of more common neurodegenerative diseases. In this review, the authors discuss the importance of mitochondrial DNA mutations in a number of different diseases and speculate how such mutations could lead to cell loss. Increasing our understanding of how mitochondrial DNA mutations affect mitochondrial metabolism and subsequently result in neurodegenerative disease will prove vital to the development of targeted therapies and treatments.
Keywords: mitochondria, mitochondrial DNA, neurodegeneration, neuropathology, cell death
Understanding the molecular basis of neurodegenerative diseases is a major challenge yet critically important due to the prevalence of these disorders in the aging population. Neurodegenerative disorders are diverse, encompassing Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease, multiple sclerosis (MS), and amylotrophic lateral sclerosis (ALS), with each disease displaying a selective involvement of different brain regions and abnormalities involving distinct proteins.
One unifying aspect of all of these neurodegenerative disorders is the detection of mitochondrial abnormalities in brain tissue. The underlying question in this area of research, however, is whether the mitochondrial defect is the primary cause of the cell loss or whether this occurs secondary to other disease processes. This review will explore the role that mitochondrial DNA (mtDNA) defects play in neural dysfunction and degeneration. We highlight the evidence of severe neurodegeneration in patients harboring primary defects of mtDNA and then examine the role mtDNA defects play in other neurodegenerative diseases.
Mitochondrial Biology and Genetics
Mitochondria are double-membraned organelles that were originally primitive, autonomous bacteria-like organisms. During evolution, they were engulfed by a eukaryotic cell, which provided protection in exchange for a supply of energy. The relationship was so successful that we now rely on these organelles for 80% to 90% of our cellular energy in the form of ATP. This endosymbiotic relationship resulted in the origin of the mitochondrial genome, which has been maintained throughout evolution (Margulis 1971).
Among the many functions of mitochondria, generation of ATP is arguably the most important. These organelles are highly efficient in their ability to utilize molecular oxygen and metabolic substrates to generate cellular ATP. Mitochondria consist of 2 membranes: an intermembrane space and an inner matrix. The molecular machinery necessary for the generation of ATP resides within the inner mitochondrial membrane and consists of 5 protein multisubunit complexes, termed the electron transport chain. This chain of complexes functions to transfer electrons across the membrane, driven by the extrusion of protons, creating a proton-motive force for ATP generation at the final complex, ATP synthase. This process is known as oxidative phosphorylation.
Mitochondria are very dynamic organelles that form a reticulum within the cell. There is constant fission and fusion of mitochondria (Fig. 1). Mitochondria have other important cellular functions; among these are regulation of calcium homeostasis and programmed cell death. Mitochondria are also the main site of reactive oxygen species (ROS) generation as a by-product of oxidative phosphorylation. Electrons leaked from complexes I and III react with molecular oxygen to form hydrogen peroxide, superoxide, and hydroxyl radicals. Generation of these radicals has been shown to be harmful to the cell with damage to proteins, lipids, and DNA (Camello-Almaraz and others 2006).
Figure 1.
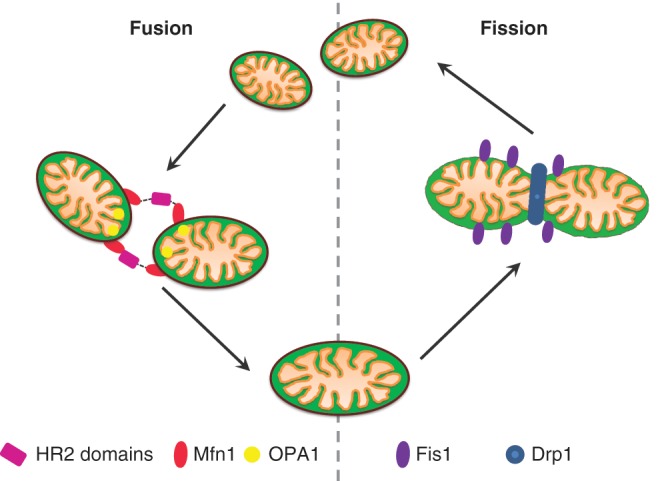
Mitochondrial dynamics. Mitochondria are dynamic organelles that constantly fuse and divide. This figure highlights the molecular machinery necessary for these processes to occur.
The mitochondrion is especially complex as it is the only organelle under the dual control of both the nuclear genome and its own mtDNA. MtDNA is a double-stranded, circular genome of 16,569 bp (Fig. 2; Anderson and others 1981). It is a highly compact genome that lacks introns and only one major noncoding region (the displacement or D-loop). The 37 genes code for 22 transfer RNAs (tRNAs), 2 ribosomal RNAs, and 13 polypeptide subunits that help form the oxidative phosphorylation system. Due to a combined lack of protective histones, ROS generation in the inner membrane, and limited repair mechanisms, mtDNA is particularly susceptible to damage and has a mutation rate estimated to be 10 to 20 times higher than that of nuclear DNA (Brown and others 1979).
Figure 2.
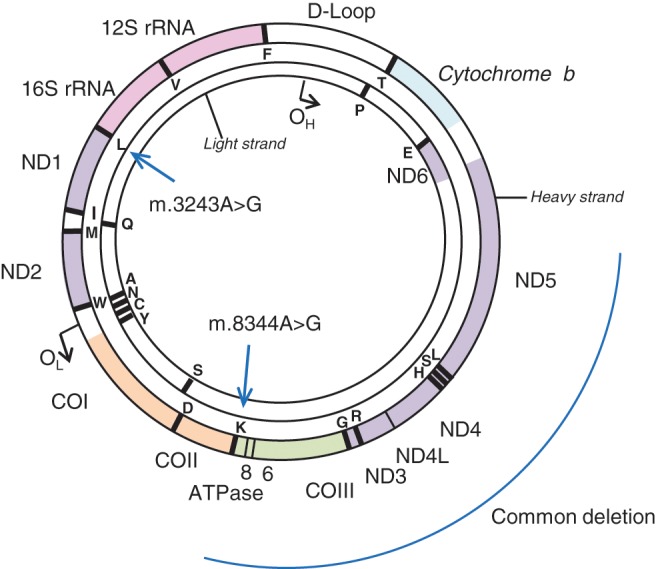
Mitochondrial genome. The mitochondrial genome exists as a closed, double-stranded circular genome of 16,569 base-pairs in length. This encodes for 37 genes, including 13 polypeptide components of the electron transport system. This figure highlights some of the mutations associated with mitochondrial disease including m.3243A>G, m.8344A>G, and the site of the common mtDNA deletion.
The unique features of mitochondrial genetics are crucial to our understanding of disorders resulting from mitochondrial dysfunction. The first feature of mtDNA is maternal inheritance, which means that mtDNA may be passed on only from the mother to the offspring (Giles and others 1980). The paternal mitochondria are destroyed after entry of the sperm into the oocyte at time of fertilization (Sutovsky and others 1999). MtDNA exists in multiple copies within a cell, which can range from hundreds to several thousands of copies. Mutations residing within the mtDNA can exist in a homoplasmic state, where all the mtDNA molecules are either wild type or mutated, or in a heteroplasmic state, where varying levels of mutated and wild-type mtDNA can coexist in a single cell. The vast majority of mtDNA mutations are functionally recessive, and therefore, high levels of mutated mtDNA are necessary before a phenotype is observed in cells. The threshold effect is likely to result from a loss of wild-type mtDNA molecules, and below a critical level, a functional defect within a cell is likely to result.
MtDNA replicates independently of the cell cycle, and this has important consequences when considering the effects of mtDNA mutations in neurological diseases. The independent replication of mtDNA even in postmitotic cells results in adjacent cells having very different mutation loads. This results in the observation of mosaic deficiency of respiratory function in tissues. This segregation is thought to be random, but the respiratory drive can lead to high copy number of mtDNA molecules (characterized in muscle as ragged red muscle fibers). The replication of mtDNA in neurons and other postmitotic cells also leads to the observation of clonal expansion of somatic mtDNA mutations. MtDNA mutations, which may be generated by oxidative stress, can clonally expand to high levels within individual cells. The mechanism of clonal expansion is still under debate, but the most likely explanation is that this occurs by random intracellular drift (Elson and others 2001).
Neurodegeneration Caused by Primary Mitochondrial DNA Abnormalities
MtDNA disorders were originally thought to be rare causes of neurological disease. However, recent studies have shown that as many as 1 in 200 individuals carry potentially pathogenic mtDNA mutations, although the number clinically affected is much lower. It is difficult to estimate the true prevalence of mtDNA disease because of the clinical and genetic heterogeneity, as well as the likely underdiagnosis in clinical practice. Current prevalence figures for the northeast of England are a minimum of 9.2 per 100,000 of the population (Schaefer and others 2008).
MtDNA defects take the form of large-scale rearrangements (usually deletions) or point mutations. Hundreds of different mutations have been described, and new defects are constantly being identified. Some of these defects are inherited maternally and can lead to a large number of affected individuals within a family. Because the CNS has intense metabolic requirements and is particularly ATP dependent, any deficit in energy generation can have catastrophic effects on neural functioning. Patients harboring mutations in mtDNA are often severely neurologically impaired due to neurodegeneration in the CNS. Generally, the degree and pattern of neurodegeneration within patients with mitochondrial disease can vary quite dramatically depending on the type of mutation and additionally the segregation of mutation load throughout brain regions and, indeed, individual neurons. Identification of the selective involvement of particular groups of neurons in patients harboring specific mutations has allowed us to explore mechanisms underlying some of the pathological changes. The insight we can gain from studying postmortem tissues from patients harboring these defects is important in our understanding of the mechanisms occurring in other neurodegenerative diseases and aging.
Specific Mitochondrial Defects
There are a number of mtDNA defects that have been extensively characterized from a neuropathological perspective. These either take the form of mtDNA point mutations or large-scale single deletions. It is of considerable interest that each genetic defect can present with not only different clinical features but also different degrees of neurodegeneration affecting different brain regions.
Point Mutations
m.3243A>G MTTL
This is the most common mtDNA point mutation and can present with a variety of different phenotypes depending in part on the level of heteroplasmy (Goto and others 1990). The best characterized neurological phenotype is the syndrome called MELAS (mitochondrial encephalopathy lactic acidosis with stroke-like episodes). Patients harboring this mutation generally manifest with a multisystem disorder, with symptoms such as stroke-like episodes, deafness, diabetes, migraines, gut immobility, myopathy, and seizures. Neuropathological examination of postmortem brain tissues from these patients often reveals the presence of multiple infarcts in gray matter cortex, which are often observed in posterior brain regions such as the temporal, parietal, and occipital lobes. These changes are thought to underlie the stroke-like episodes; however, they are generally not restricted to a particular vascular territory and are often accompanied by profound neuronal cell loss, neuronal eosinophilia, and astrogliosis (Tanji and others 2001). In addition, there is evidence of calcification of the basal ganglia, which is particularly prominent in the vasculature and can be detected during brain imaging scans (Sue and others 1998). There is evidence of cerebellar degeneration, which typically shows abnormalities and loss of Purkinje cells with the formation of abnormally thickened dendrites containing an accumulation of mitochondria.
m.8344A>G MTTK
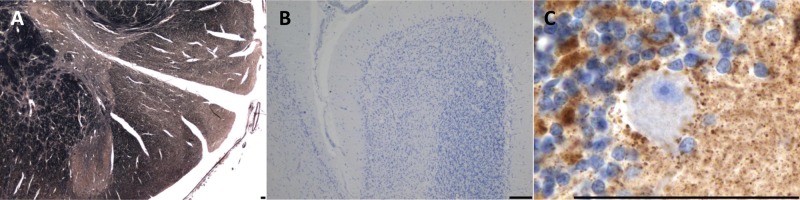
Another common point mutation is m.8344A>G in the tRNALys, which is responsible for a collection of mitochondrial diseases known as myoclonic epilepsy ragged red fibers (MERRF). Although additional mutations have been described to cause MERRF, the m.8344A>G mutation is responsible for more than 90% of cases (Shoffner and others 1990). The main presenting feature is often the myoclonus; however, other symptoms, such as epilepsy, seizures, ataxia, neuropathy, and cardiac abnormalities, develop. The main neuropathological findings describe degenerative features within the olivocerebellar pathway, with severe neuron loss from the inferior olivary nucleus (Fig. 3), Purkinje cells, and dentate nucleus. There are often high levels of respiratory chain deficiency in surviving neurons (Fig. 3) and a strong proliferation of astrocytes. Studies have described the presence of enlarged mitochondria containing inclusions within surviving neurons within the cerebellum (Fukuhara 1991). There is additional evidence of neurodegeneration in the gracile and cuneate nuclei and evidence of demyelination of the spinal column (Sparaco and others 1993). A study examining the mutant mtDNA heteroplasmy levels in neurons residing within the cerebellum failed to show a clear correlation between mutation load and the extent of neurodegeneration (Zhou and others 1997). This suggests that other factors may be important for determining neuronal cell dysfunction.
Figure 3.

Inferior olivary degeneration in a patient harboring the m.8344A>G mutation. (A) Severe neuronal cell loss from the inferior olivary nucleus (hematoxylin and eosin stain). (B) High mitochondrial density within remaining inferior olivary neurons (antiporin immunohistochemistry). (C) Complex I–deficient neuron (arrow) evident in the inferior olivary nucleus (anti–complex I subunit 20 kDa immunohistochemistry). (D) Absence of complex IV subunit I expression in the inferior olivary neurons (anti-COXI immunohistochemistry). Scale bar represents 100 µm.
Leber’s hereditary optic neuropathy
Leber’s hereditary optic neuropathy (LHON) was first described by Theodore Leber in 1871 (Leber 1871). The salient clinical feature of LHON is the almost exclusive involvement of the optic nerve. Patients typically present with a subacute or acute, painless loss of central vision usually between the ages of 20 and 40 years, with a strong predilection for males. The first mtDNA point mutation recognized as being pathogenic in LHON was m.11778G>A (Wallace and others 1988); however, additional point mutations m.3460G>A and m.14484T>C have subsequently been identified. It has been shown that more than 95% of LHON pedigrees are known to harbor one of these mtDNA point mutations, and these mutations all involve genes encoding for complex I subunits of the electron transport chain (Mackey 1996).
Given the cell-specific nature of ocular pathology in LHON, the neurodegeneration appears to be limited to the retinal ganglion cell (RGC) layer, with sparing of the retinal pigment epithelium and photoreceptors. Within the RGC layer, there is pronounced cell body and axonal degeneration, with associated demyelination and atrophy observed from the optic nerves to the lateral geniculate bodies. Previous studies provide evidence of impaired glutamate transport, increased mitochondrial ROS, and evidence of apoptotic cell death in the RGCs (Carelli and others 2004).
Single large-scale mtDNA deletions
Large-scale deletions or rearrangements of the mtDNA are responsible for the clinical phenotype Kearns Sayre syndrome (KSS), which usually has an onset before 20 years of age, with clinical symptoms consisting of opthalmoplegia, retinopathy pigmentosa, complete heart block, cerebellar ataxia, and an elevated cerebrospinal fluid protein level. As this multisystem disorder has an early onset of disease, life span is quite often reduced (Zeviani and others 1988).
The neuropathological hallmark of KSS is severe spongiform degeneration of the white matter regions of the brain, including the cerebrum, cerebellum, thalamus, basal ganglia, brain stem, and spinal cord. The severity of these lesions can vary from slight myelin pallor to mild spongy change to striking vacuolation of the white matter (Sparaco and others 1993). Ultrastructural analysis of affected white matter has revealed the presence of myelin splits at the level of intraperiod line. This has been attributed to a specific vulnerability of the myelin-producing glial cells, oligodendrocytes, to the single mtDNA deletion removing important genes encoding for OXPHOS components (Oldfors and others 1990).
Mutations in Nuclear Encodes Genes Involved in mtDNA Maintenance and Expression
As the mitochondrial genome is under dual genetic control, there have been an increasing number of mutations described in nuclear genes involved in mtDNA maintenance and expression. The most important of these is the mitochondrial polymerase gamma (POLG1), which is the sole polymerase responsible for the synthesis and repair of the mitochondrial genome. Mutations within POLG1 result in secondary damage to mtDNA, including mtDNA deletions and depletion. This genetic defect therefore provides a useful model for the damage to mtDNA observed in aging and other neurodegenerative diseases. Patients harboring mutations within POLG1 present with a diverse spectrum of clinical symptoms, such as autosomal dominant and autosomal recessive familial external ophthalmoplegia (Lamantea and others 2002; Van Goethem and others 2001, 2003b), autosomal recessive sensory ataxia neuropathy with dysarthria and ophthalmoplegia (Van Goethem and others 2003a), adult-onset mitochondrial spinocerebellar ataxia and epilepsy, parkinsonism, and Alpers’ hepatocerebral syndrome (Huttenlocher and others 1976). Although the clinical symptoms do vary, mutations in POLG1 generally result in the development of a severe encephalopathy, with several common features including the selective involvement of the CNS and liver with rapidly progressive disturbances of consciousness, severe epilepsy, and the development of acute cerebral lesions with a propensity toward greater posterior brain involvement.
Neuropathologically, patients harboring mutations within the POLG1 gene show evidence of severe degeneration of the olivopontocerebellar pathways, with profound neuronal cell loss in the inferior medullary olives and cerebellum. It is likely that the degeneration of these structures is likely to be responsible for the clinical manifestation of ataxia. In addition, degeneration of the posterior spinal cord columns is often described (as seen in Fig. 4A; Cottrell and others 2000). Recently, postmortem evaluation of a patient harboring multiple deletions due to two heterozygous mutations in POLG1 revealed a loss of pigmented neurons within the substantia nigra with formation of α-synuclein–positive Lewy bodies. Furthermore, neuronal cell loss from the cerebellar Purkinje cells (Fig. 4B), dentate nucleus, and gracile nucleus is similar to the degenerative features observed in other patients harboring POLG1 defects. High levels of neuronal respiratory chain deficiency, particularly complex I, are frequently observed (Fig. 4C). High levels of mtDNA deletion (>65%) were detected in remaining substantia nigra neurons in association with COX deficiency (Betts-Henderson and others 2009). In addition, neurodegeneration due to mitochondrial inherited recessive ataxia syndrome mainly occurs in the spinal cord, brain stem, and cerebellum with severe deficiencies of complex I in surviving neuronal populations (Hakonen and others 2008).
Figure 4.
Neurodegeneration in a patient harboring two heterozygous, p.A467T and p.W748S, mutations in POLG1 and secondary multiple mtDNA deletions (A) Prominent demyelination of the spinal posterior columns (Loyez stain). (B) Atrophy of the cerebellum with severe Purkinje cell and granular cell loss in the cerebellar cortex (Cresyl fast violet stain). (C) Profound complex I deficiency in remaining Purkinje cell population (anti–complex I subunit 30 kDa immunohistochemistry). Scale bar represents 100 µm.
Mitochondrial DNA Mutations in Other Neurodegenerative Diseases
Mutations within the mitochondrial genome have been implicated as playing a role in a number of neurodegenerative disorders including PD, MS, and AD (Bender and others 2006; Horton and others 1995; Lin and others 2002). The mitochondrial dysfunction that may be caused by high levels of these mutations has also been suggested to have an important role in the pathogenesis of these disorders. However, mtDNA mutations are also thought to play an important part in normal human aging. MtDNA mutations increase with advancing age in a number of tissues, including muscle, colon, and brain (Bender and others 2006; Kraytsberg and others 2006; Taylor and others 2003). At high levels, these mutations cause mitochondrial dysfunction, which can be detected within the laboratory as a reduction in activity of complex IV (cytochrome c oxidase) of the electron transport chain. Both mtDNA point mutations and large-scale deletions have been shown to occur with aging, although the type of mutation that occurs seems to be tissue specific; for example, in the substantia nigra, mitochondrial dysfunction is caused by deletions, whereas in the colon, it is point mutations that are responsible (Bender and others 2006; Taylor and others 2003). Recent studies have examined these mtDNA defects in substantia nigra neurons in both aging and PD. These studies have shown a remarkably high level of mtDNA deletions with normal aging (about 50% of all mtDNA molecules) and a slight, nonsignificant increase in patients with PD (Bender and others 2006). mtDNA point mutations are thought not to be important in these neurons (Reeve and others 2009). The significance of these high levels of mtDNA deletions in the pathogenesis of PD is still under investigation, but it is of interest that patients with POLG1 mutations and secondary mtDNA defects also develop symptoms of Parkinsonism.
Mitochondria have recently been brought to the forefront of research into the pathogenesis of PD. Studies investigating early-onset, familial forms of this disease have shown that the mutations responsible for some of these cases occur within genes that have been shown to associate with mitochondria and that may have a role in mitochondrial function. Pink1 and Parkin, for example, have been shown to associate with each other and with mitochondria to affect mitochondrial dynamics and degradation (Matsuda and others 2010). DJ-1 has also recently been shown to be important for mitochondrial dynamics as well as protecting cells from oxidative stress–induced cell death (Irrcher and others 2010). A deficiency in activity and protein levels of complex I of the respiratory chain has also been reported in PD. This deficiency has been reported to occur not only in the substantia nigra of patients but also in their platelets and muscle (Schapira and others 1989, 1990).
Respiratory chain deficiency caused by mutations in mtDNA has been shown to also occur in AD and recently in MS (Cottrell and others 2001). Other mitochondrial changes such as redistribution of axonal mitochondria in MS and alterations in morphology and biochemistry in AD have also been reported (Andrews and others 2005; Mancuso and others 2010). Mitochondrial dysfunction has also been reported in ALS; 20% of familial ALS cases are caused by mutations in the SOD1 gene, and mutant SOD1 has been shown to associate with mitochondria, affecting morphology and bioenergetics. It has also recently been reported that mutant SOD1 can also affect mitochondrial axonal transport, which subsequently affects mitochondrial dynamics (Shi and others 2010).
Mouse and Cell Culture Models
Mouse models
It is difficult to pinpoint in many diseases whether mitochondrial dysfunction is causal for the development of a neurodegenerative phenotype. However, the recent development of transgenic animal models and cell culture systems has proven to be invaluable in exploring some of the challenging questions posed by research in this area. In 2004, a mutator mouse was developed that accumulates high levels of mtDNA point mutations and deletions due to a proofreading deficiency of the polymerase γ (POLG1) gene (Trifunovic and others 2004). This mouse displays a phenotype consistent with premature aging (at about 25 weeks), kyphosis (curvature of the spine), weight loss, alopecia, and reduced life span (they die at about 48 weeks). Subsequently, another group produced a similar mouse model that possessed very similar phenotypes (Kujoth and others 2005). To date, no detailed neuropathological study has been carried out on this model, but neurological changes would be interesting to investigate.
By disrupting TFAM, a high-mobility group box protein that binds to mtDNA and packages it into nucleoids, in dopaminergic neurons, Ekstrand and others (2007) created a mouse with a primary mitochondrial defect, as shown by decreased mtDNA copy number as well as increased numbers of respiratory deficient neurons, that displays some of the characteristic features of PD. These mice exhibit progressive motor function impairment, with an adult onset and a loss of dopaminergic neurons with the formation of protein inclusions. These inclusions were negative for alpha synuclein staining and were associated with mitochondria. The same group has also created a mouse model in which defects in TFAM primarily affect neocortex and hippocampal neurons. This mouse (the MILON mouse) shows an age-related increase in respiratory deficiency in large forebrain neurons and neurodegeneration. By creating chimeras between these MILON and wild-type mice, it is possible to create mice with varying degrees of respiratory deficiency in forebrain neurons. Results from these studies have shown that respiratory deficiencies greater than 20% can cause clinical symptoms, whereas levels greater than 60% to 80% result in the death of these animals (Dufour and others 2008). Most interestingly, this model also shows transneuronal degeneration whereby neurons with a deficiency negatively affect neighboring neurons (Dufour and others 2008). Studies such as this highlight that mitochondrial deficiencies are an important cause of neurodegeneration, both in terms of the cells harboring the deficiencies and also their neighbors, and this degeneration may then ultimately lead to death.
Cell culture models
Although cells from patients with primary mitochondrial defects, including fibroblasts and myoblasts, are grown regularly, growing neurons from patients with neurodegenerative diseases is virtually impossible. Finding ways to model mitochondrial defects in neuronal cells is therefore a challenge. Recent studies have used mouse embryonic stem (ES) cell lines, which contain varying levels of mitochondrial deficiencies. These cell lines were created as transmitochondrial cybrids. This involves treating parental ES cells with rhodamine 6G to remove all mtDNA and then fusing these cells with enucleated fibroblasts containing differing levels of mitochondrial DNA defects. The resulting cells then have defects of mitochondrial complex I and complex IV deficiency. Because of the pluripotent nature of these cells, they can then be differentiated into neuronal cells. These cell lines were used for several of the studies described below, particularly those of Trevelyan and others (2010) and Abramov and others (2010).
Mechanism of Neuronal Degeneration in Relation to mtDNA Mutations
As previously mentioned, it is well documented that high levels of mtDNA mutation cause respiratory dysfunction in mitochondria and that primary and secondary defects of mtDNA can lead to profound neurodegeneration. What is not understood is what the effect of this mitochondrial dysfunction has on neurons and their survival. We review some potential mechanisms for the degeneration of neurons in disease based on mitochondrial dysfunction caused by mtDNA mutations (Fig. 6).
Figure 6.

Mitochondrial DNA mutations and neuronal cell death. Mitochondrial DNA mutations at high levels cause mitochondrial dysfunction, which will have consequences on ATP levels and other cellular processes. This mitochondrial dysfunction may then be the cause of neuronal loss in a number of diseases. This figure postulates how this might occur.
Mitochondria are abundant at regions of neurons with high energy demand
Mitochondria are vital for neuronal function. The majority of the ATP generated in neurons is consumed through maintenance of the Na+/K+ gradients required for impulse transmission. There are regions of the neuron, however, that require large amounts of energy; these include the nodes of Ranvier and synapses (Berthold and others 1993; Bristow and others 2002; Kageyama and Wong-Riley 1982; Fig. 5). Mitochondria cluster at these regions to provide ATP to meet the energy demand. One example of where degenerative disease can lead to changes in this organization of mitochondria is in MS (Andrews and others 2005). The demyelination seen in this disease is associated with changes in the localization of the mitochondria. The mitochondria proliferate and redistribute along the length of the axon. Despite these changes, free radical damage accumulates, which will affect mitochondrial function and may lead to axonal loss. The presence of mtDNA mutations in the mitochondria of these axons will compound these effects and may accelerate the damage to these axons. Recent evidence has suggested that mtDNA mutations, and in particular deletions, are important in MS with significant respiratory deficiency caused by mtDNA deletions in patient gray matter (Campbell and others 2010).
Figure 5.

Mitochondria and neuronal function. Mitochondria are essential for neuronal function and supply the majority of the energy required for cellular processes. There are a number of regions of the cell where mitochondria accumulate, where a number of energy-demanding processes take place. This figure reviews some of the important cellular regions where mitochondria may be important.
Mitochondrial oxidative metabolism and calcium handling
Recently, the effects of mtDNA mutations in neurons differentiated from embryonic stem cells have been examined, and two important discoveries have come to light. First, the calcium handling within neurons that harbor mitochondrial DNA mutations is altered, and in particular it is the response to repeated stimuli, which seems to be particularly affected (Trevelyan and others 2010). Second, in neurons containing a complex I defect, mitochondrial membrane is affected and in fact is increased due to reversal of the F1F0ATPase. Thus, impaired respiratory chain function and membrane potential changes cause oxidative stress, which may then lead to cell death. These studies give direct evidence that a primary mitochondrial defect can affect two very important processes required for the normal functioning of neurons (Abramov and others 2010).
Mitochondrial dynamics
The fission and fusion of mitochondria in cells serve important functions to maintain mitochondrial network integrity (an overview of this process is shown in Fig. 2). The fusion of mitochondria via their inner and outer membranes leads to a mixing of the organelle contents, and this may be beneficial if one of the fusion partners carries a high level of mitochondrial DNA mutation and hence is respiratory deficient. By fusing with a wild-type mitochondrion, the level of mutation can be reduced. These dynamic processes require a number of proteins, several of which have been found to be important in human degenerative disease, for example, mfn2 in Charcot-Marie-Tooth type A and OPA1 in dominant optic atrophy (Delettre and others 2002; Zuchner and others 2004). Changes in this process can have implications for mitochondria themselves but also for the whole neuron. A loss of mitochondrial membrane potential has been proposed to cause a fission event; if the membrane potential cannot be restored, then the mitochondria loses OPA1, an essential fusion protein, and is targeted for degradation through the autophagy pathway (Twig and others 2008). Fission and fusion have been recently shown to be important in a number of other neurodegenerative diseases such as PD, and changes in these processes have been reported in relation to AD and ALS.
Mutations in genes such as PINK1, parkin, and DJ-1, which cause familial forms of PD, have been shown to cause changes in mitochondrial dynamics. Mutations in parkin and PINK1 in drosophila lead to enlarged and swollen mitochondria, suggesting a defect in mitochondrial fission (Clark and others 2006; Greene and others 2003). Studies investigating this effect on mitochondrial dynamics in more detail have shown through either overexpression of DRP1 (a fission protein) or by loss of function mutations in OPA1 and mfn2 that it seems likely that mutations in these genes may even inhibit mitochondrial fusion (Park and others 2009). More recently, mutations in DJ-1 have also been shown to affect mitochondrial dynamics, although in this case, it was shown that a DJ-1 deficiency led to a fragmented mitochondrial network, suggesting a role in fusion (Irrcher and others 2010). In AD, is has been shown that amyloid-β (Aβ) can fragment mitochondrial networks by inducing fission (Wang and others 2008). It has also been shown that increased levels of ROS can lead to mitochondrial fission (Andres-Mateos and others 2007); therefore, the increase in ROS levels associated with normal aging as well as with neurodegenerative diseases may lead to the fragmentation of the mitochondrial network and hence neuronal dysfunction leading to cell death. Increased levels of ROS may also be associated with high levels of mitochondrial DNA mutations leading to respiratory dysfunction.
Taken together, these studies inform us that mitochondrial dynamics are important for neuronal function and that alterations in mitochondrial dynamics may have detrimental consequences. Although the effect of high levels of mtDNA mutations on the mitochondrial membrane potential is still debated, it seems likely that changes in these processes would occur, perhaps leading to increased fission of the mitochondrial network.
mtDNA Mutations and Cell Death
There are at least two distinct pathways by which neurons might undergo cell death: apoptosis and necrosis. Each of these processes is morphologically and functionally distinct. In terms of mitochondria, however, apoptosis is the most important. Apoptosis is an evolutionarily conserved mechanism that exists to modulate tissue development and homeostasis throughout life and can be bought about by two distinct pathways: the mitochondrial pathway and the death receptor pathway. Initiation of the apoptotic pathways is thought to occur in a diverse range of neurodegenerative diseases. It is a highly regulated process that is morphologically defined by cell shrinkage, membrane blebbing, formation of apoptotic bodies, and condensation and fragmentation of nuclear DNA (Saraste 1999). Mitochondria are known to play a key role in triggering or increasing the process of apoptosis. The mitochondrial pathway is characterized by a release of SMAC, AIF, and cytochrome c from the mitochondria into the cytosol. Cytochrome c interacts with apoptotic protease activating factor-1 and pro–caspase 9 to form the apoptosome (Li and others 1997). This triggers a downstream cascade of events, which ultimately ends in the demise of a cell. Apoptosis has previously been explored in patients harboring mtDNA mutations with some variation in findings. ROS-induced apoptosis is also thought to be important. A study using the mutator mouse has recently shown in muscle that mitochondrial DNA mutations can lead to a reduction in mitochondrial complex protein levels, impaired mitochondrial bioenergetics, and a reduction in mitochondrial membrane potential. Interestingly, despite all these changes within the mitochondria, these data did not show an increase in oxidative damage or ROS but did show evidence of increased apoptosis (Hiona and others 2010). Thus, it seems that mitochondrial dysfunction due to mtDNA mutations can lead to cell death in postmitotic cells without involving oxidative stress.
Autophagy is the main pathway through which intact mitochondria can be degraded within the cell. It is a complicated and tightly regulated process. Essentially, autophagy involves the sequestering of proteins and organelles to be degraded within a double membrane termed the autophagosome; this structure then fuses with a lysosome, the lysosomal hydrolases degrade the contents of the autophagosomes, and the products of this degradation are then returned to the cytosol. The components of this pathway are then recycled. An accumulation of autophagosomes within a cell may be associated with cell death (Mehrpour and others 2010; Ravikumar and others 2009). Central to the control of this pathway is mTOR. This protein negatively regulates autophagy; inhibition stimulates autophagy while its activation inhibits the pathway. mTOR can respond to a number of different cellular stimuli, including amino acid levels (starvation), changes in ATP and calcium levels, and ROS. This would seem an ideal garbage disposal system for the cell, removing dysfunctional organelles and proteins to prevent the damage of other components (Dennis and others 2001; Mehrpour and others 2010). Recent evidence has shown that the targeting of mitochondria to autophagy depends on a loss of membrane potential and the association of parkin with pink1 on the outer mitochondrial membrane (Narendra and others 2010). It is clear, however, from the data showing that mitochondrial dysfunction accumulates with age and in disease that mitochondria harboring high levels of mutation are not degraded and may persist within the cell. Therefore, mtDNA mutations may not lead to cell death through an autophagy-related pathway.
Research investigating the role of autophagy in AD pathogenesis is more limited than for PD, but this field is expanding. Autophagy is a prominent process within AD neurons, and previous studies have shown that the mtDNA copy number is increased in neurons undergoing oxidative stress in both neuronal cytoplasm and also in vacuoles associated with lipofuscin. Increased levels of cytochrome c oxidase subunit–I (COXI) were also reported, and this increased expression was associated with mitochondria undergoing phagocytosis (Moreira and others 2007). These data suggested that neurons in AD showed upregulated mitochondrial degradation and turnover. Autophagic vacuoles, also found to be common in neurons in AD, have been shown to accumulate Aβ protein precursor, suggesting that these vacuoles may play a central role in the formation of the protein inclusions associated with this disease (Moreira and others 2007).
Links between mtDNA Mutations and Protein Accumulation
The accumulation of damaged protein into intracellular inclusions as well as extracellular structures is common in many neurodegenerative disorders. Mitochondrial dysfunction has been suggested to be important in the processes leading to this aggregation by a number of studies. As mentioned previously, mitochondria are the primary source of ROS within the cell. These damaging species are generated during the generation of ATP, in particular through electron transfer in complexes I and III of the respiratory chain. These molecules are known to be damaging to cellular components including DNA, protein, and lipids. Interestingly, a number of these proteins, for example, Aβ and α-synuclein, have been shown to directly interact with mitochondria and/or mitochondrial proteins (Cole and others 2008; Devi and Anandatheerthavarada 2010; Hashimoto and others 2003; Muirhead and others 2010; Shavali and others 2008). The interaction of these proteins with mitochondria has been proposed to contribute not only to their accumulation, particularly due to the damaging nature of ROS, primarily produced in the mitochondria, but also to the mitochondrial dysfunction seen in this disease.
Alpha-synuclein, for example, has the potential to bind to lipids and so may interact with mitochondrial membranes. It is also thought that α-synuclein fibrils have the potential to form pores in membranes, which may then affect mitochondrial membrane potential. The release of proteins such as cytochrome c and BAD is involved in the signaling pathways involved in apoptosis, and there is evidence to suggest that α-synuclein may also interact with these molecules (Hashimoto and others 2003; Spillantini and others 1997; Waxman and Giasson 2009). The unfolded nature of α-synuclein means that it may also be taken up by the TOM/TIM protein transporters within the mitochondrial membranes. Alpha-synuclein may then affect mitochondrial function from within. Similar potential effects are also reported for Aβ; for example, Aβ might enter mitochondria through the protein translocases TOM/TIM, and once inside, they may lead to mitochondrial dysfunction. Aβ has also been shown to interact with α-synuclein, which may then cause mitochondrial dysfunction. Aβ has also been reported to produce free radicals in vitro. This would enhance oxidative stress and may disrupt calcium homeostasis (Devi and Anandatheerthavarada 2010; Devi and others 2006; Reddy 2009). All the effects mentioned above may be compounded if mitochondrial dysfunction, caused by mtDNA mutations, is already present.
It is important to note, however, that protein accumulation into inclusions are not typically seen in mitochondrial disease, suggesting therefore that the mtDNA mutations and the mitochondrial dysfunction they cause are not sufficient to cause the accumulation of proteins within neurons. Although the accumulation of protein in neurodegenerative diseases is the primary pathological hallmark, it is still debated whether the protein accumulation is damaging to the cell or protective. For example, in the case of α-synuclein accumulation into Lewy bodies, we are yet unable to decide whether it is beneficial for the cell to accumulate toxic forms of damaged α-synuclein into Lewy bodies or if by doing so the cell becomes damaged and undergoes apoptosis.
Conclusions
MtDNA mutations are indeed important new players in neuronal degeneration. Understanding their role is complex due to the complicated mitochondrial genetics and the numerous roles mitochondria play within neurons. Important insights are given by studying patients with primary mtDNA defects both from a clinical and neuropathological perspective. These patients often have marked neurodegeneration with progressive features leading to severe disability and death. However, studies in these patients are inevitably limited by tissue availability. Studies in mouse models and cell culture have also given important insights into the relationship of the mtDNA defect and cell dysfunction and loss.
The major unanswered question remains the potential importance of mtDNA defects in the common neurodegenerative diseases such as AD and PD. In these patients, there can be high levels of mtDNA mutations and the presence of respiratory deficient cells, indicating a potential role. However, in these diseases, pathogenic mechanisms are still uncertain, and interactions between mitochondrial function and other neuropathological features require much more research. It would be naïve, in our view, to think that mtDNA abnormalities play a primary role, but the accumulation of respiratory-deficient cells is likely to have a detrimental effect, contributing to disease progression.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: The authors’ work is supported by Newcastle University Centre for Brain Ageing and Vitality supported by BBSRC, EPSRC, ESRC, and MRC (G0700718); the Wellcome Trust (074454/Z/04/Z); Marriott Research Fund; and UK NIHR Biomedical Research Centre for Ageing and Age-Related Disease.
References
- Abramov AY, Smulders-Srinivasan TK, Kirby DM, Acin-Perez R, Enriquez JA, Lightowlers RN. and others. 2010. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain 133:797–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J. and others. 1981. Sequence and organization of the human mitochondrial genome. Nature 290:457–65 [DOI] [PubMed] [Google Scholar]
- Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B. and others. 2007. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A 104:14807–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews HE, Nichols PP, Bates D, Turnbull DM. 2005. Mitochondrial dysfunction plays a key role in progressive axonal loss in multiple sclerosis. Med Hypotheses 64:669–77 [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH. and others. 2006. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–7 [DOI] [PubMed] [Google Scholar]
- Berthold CH, Fabricius C, Rydmark M, Andersen B. 1993. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol 22:925–40 [DOI] [PubMed] [Google Scholar]
- Betts-Henderson J, Jaros E, Krishnan KJ, Perry RH, Reeve AK, Schaefer AM. and others. 2009. Alpha-synuclein pathology and Parkinsonism associated with POLG1 mutations and multiple mitochondrial DNA deletions. Neuropathol Appl Neurobiol 35:120–4 [DOI] [PubMed] [Google Scholar]
- Bristow EA, Griffiths PG, Andrews RM, Johnson MA, Turnbull DM. 2002. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol 120: 791–6 [DOI] [PubMed] [Google Scholar]
- Brown WM, George M, Jr, Wilson AC. 1979. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A 76: 1967–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. 2006. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol 291:C1082–8 [DOI] [PubMed] [Google Scholar]
- Campbell G, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O. and others. 2010. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol Available from http://dx.doi.org/10.1002/ana.2210910.1002/ana.22109 10.1002/ana.22109 [DOI] [PMC free article] [PubMed]
- Carelli V, Rugolo M, Sgarbi G, Ghelli A, Zanna C, Baracca A. and others. 2004. Bioenergetics shapes cellular death pathways in Leber’s hereditary optic neuropathy: a model of mitochondrial neurodegeneration. Biochim Biophys Acta 1658:172–9 [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH. and others. 2006. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–6 [DOI] [PubMed] [Google Scholar]
- Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. 2008. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res 314:2076–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell DA, Blakely EL, Johnson MA, Ince PG, Borthwick GM, Turnbull DM. 2001. Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol Aging 22:265–72 [DOI] [PubMed] [Google Scholar]
- Cottrell DA, Ince PG, Blakely EL, Johnson MA, Chinnery PF, Hanna M. and others. 2000. Neuropathological and histochemical changes in a multiple mitochondrial DNA deletion disorder. J Neuropathol Exp Neurol 59:621–7 [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Pelloquin L, Belenguer P, Hamel CP. 2002. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol Genet Metab 75:97–107 [DOI] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. 2001. Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–5 [DOI] [PubMed] [Google Scholar]
- Devi L, Anandatheerthavarada HK. 2010. Mitochondrial trafficking of APP and alpha synuclein: relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim Biophys Acta 1802:11–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. 2006. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26:9057–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour E, Terzioglu M, Sterky FH, Sorensen L, Galter D, Olson L. and others. 2008. Age-associated mosaic respiratory chain deficiency causes trans-neuronal degeneration. Hum Mol Genet 17:1418–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E. and others. 2007. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A 104:1325–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson JL, Samuels DC, Turnbull DM, Chinnery PF. 2001. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet 68:802–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara N. 1991. MERRF: a clinicopathological study. Relationships between myoclonus epilepsies and mitochondrial myopathies. Rev Neurol (Paris) 147:476–9 [PubMed] [Google Scholar]
- Giles RE, Blanc H, Cann HM, Wallace DC. 1980. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A 77:6715–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S. 1990. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348:651–3 [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. 2003. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 100:4078–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonen AH, Goffart S, Marjavaara S, Paetau A, Cooper H, Mattila K. and others. 2008. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet 17:3822–35 [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Rockenstein E, Crews L, Masliah E. 2003. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med 4:21–36 [DOI] [PubMed] [Google Scholar]
- Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T. and others. 2010. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 5:e11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton TM, Graham BH, Corral-Debrinski M, Shoffner JM, Kaufman AE, Beal MF. and others. 1995. Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 45:1879–83 [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, Solitare GB, Adams G. 1976. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol 33:186–92 [DOI] [PubMed] [Google Scholar]
- Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M. and others. 2010. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 19:3734–46 [DOI] [PubMed] [Google Scholar]
- Kageyama GH, Wong-Riley MT. 1982. Histochemical localization of cytochrome oxidase in the hippocampus: correlation with specific neuronal types and afferent pathways. Neuroscience 7:2337–61 [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. 2006. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:518–20 [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE. and others. 2005. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–4 [DOI] [PubMed] [Google Scholar]
- Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S. and others. 2002. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol 52:211–9 [DOI] [PubMed] [Google Scholar]
- Leber T. 1871. Ueber hereditäre und congenital angelegte Sehnervenleiden. Graefes Arch Clin Exp Ophthalmol 17:249-291 [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES. and others. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–89 [DOI] [PubMed] [Google Scholar]
- Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. 2002. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet 11:133–45 [DOI] [PubMed] [Google Scholar]
- Mackey DA, Oostra RJ, Rosenberg T, Nikoskelainen E, Bronte-Stewart J, Poulton J. and others. 1996. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet 59:481–5 [PMC free article] [PubMed] [Google Scholar]
- Mancuso M, Orsucci D, LoGerfo A, Calsolaro V, Siciliano G. 2010. Clinical features and pathogenesis of Alzheimer’s disease: involvement of mitochondria and mitochondrial DNA. Adv Exp Med Biol 685:34–44 [DOI] [PubMed] [Google Scholar]
- Margulis L. 1971. Symbiosis and evolution. Sci Am 225:48–57 [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA. and others. 2010. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrpour M, Esclatine A, Beau I, Codogno P. 2010. Overview of macroautophagy regulation in mammalian cells. Cell Res 20:748–62 [DOI] [PubMed] [Google Scholar]
- Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M. and others. 2007. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 3:614–5 [DOI] [PubMed] [Google Scholar]
- Muirhead KE, Borger E, Aitken L, Conway SJ, Gunn-Moore FJ. 2010. The consequences of mitochondrial amyloid beta-peptide in Alzheimer’s disease. Biochem J 426:255–70 [DOI] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J. and others. 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfors A, Fyhr IM, Holme E, Larsson NG, Tulinius M. 1990. Neuropathology in Kearns-Sayre syndrome. Acta Neuropathol 80:541–6 [DOI] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. 2009. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun 378:518–23 [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Futter M, Jahreiss L, Korolchuk VI, Lichtenberg M, Luo S. and others. 2009. Mammalian macroautophagy at a glance. J Cell Sci 122:1707–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH. 2009. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol 218: 286–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Taylor G, Elson JL, Bender A, Taylor RW. and others. 2009. The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell 8:496–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M. 1999. Oxidative phosphorylation at the fin de siecle. Science 283:1488–93 [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW. and others. 2008. Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63:35–9 [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. 1990. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–7 [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. 1989. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1:1269. [DOI] [PubMed] [Google Scholar]
- Shavali S, Brown-Borg HM, Ebadi M, Porter J. 2008. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett 439:125–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Wei Y, Zhang J, Gal J, Zhu H. 2010. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J Alzheimers Dis 20 Suppl 2:S311–24 [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. 1990. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 61:931–7 [DOI] [PubMed] [Google Scholar]
- Sparaco M, Bonilla E, DiMauro S, Powers JM. 1993. Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defects. J Neuropathol Exp Neurol 52:1–10 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. 1997. Alpha-synuclein in Lewy bodies. Nature 388:839–40 [DOI] [PubMed] [Google Scholar]
- Sue CM, Crimmins DS, Soo YS, Pamphlett R, Presgrave CM, Kotsimbos N. and others. 1998. Neuroradiological features of six kindreds with MELAS tRNA(Leu) A2343G point mutation: implications for pathogenesis. J Neurol Neurosurg Psychiatr 65:233–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. 1999. Ubiquitin tag for sperm mitochondria. Nature 402:371–2 [DOI] [PubMed] [Google Scholar]
- Tanji K, Kunimatsu T, Vu TH, Bonilla E. 2001. Neuropathological features of mitochondrial disorders. Semin Cell Dev Biol 12:429–39 [DOI] [PubMed] [Google Scholar]
- Taylor RW, Barron MJ, Borthwick GM, Gospel A, Chinnery PF, Samuels DC. and others. 2003. Mitochondrial mutations in human colonic crypt stem cells. J Clin Invest 112:1351–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Kirby DM, Smulders-Srinivasan TK, Nooteboom M, Acin-Perez R, Enriquez JA. and others. 2010. Mitochondrial DNA mutations affect calcium handling in differentiated neurons. Brain 133:787–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE. and others. 2004. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–23 [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G. and others. 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. 2001. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 28:211–2 [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Martin JJ, Dermaut B, Lofgren A, Wibail A, Ververken D, | others. 2003a. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord 13:133–42 [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Schwartz M, Lofgren A, Dermaut B, Van Broeckhoven C, Vissing J. 2003b. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet 11:547–9 [DOI] [PubMed] [Google Scholar]
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM. and others. 1988. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242:1427–30 [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y. and others. 2008. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105:19318–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. 2009. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta 1792:616–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, Schon EA. and others. 1988. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 38: 1339–46 [DOI] [PubMed] [Google Scholar]
- Zhou L, Chomyn A, Attardi G, Miller CA. 1997. Myoclonic epilepsy and ragged red fibers (MERRF) syndrome: selective vulnerability of CNS neurons does not correlate with the level of mitochondrial tRNAlys mutation in individual neuronal isolates. J Neurosci 17: 7746–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL. and others. 2004. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36:449–51 [DOI] [PubMed] [Google Scholar]