

Abstract
Owing to their antimicrobial properties, silver nanoparticles (NPs) are the most commonly used engineered nanomaterial for use in a wide array of consumer and medical applications. Many discussions are currently ongoing as to whether or not exposure of silver NPs to the ecosystem (i.e. plants and animals) may be conceived as harmful or not. Metallic silver, if released into the environment, can undergo chemical and biochemical conversion which strongly influence its availability towards any biological system. During this process, in the presence of moisture, silver can be oxidized resulting in the release of silver ions. To date, it is still debatable as to whether any biological impact of nanosized silver is relative to either its size, or to its ionic constitution. The aim of this review therefore is to provide a comprehensive, interdisciplinary overview—for biologists, chemists, toxicologists as well as physicists—regarding the production of silver NPs, its (as well as in their ionic form) chemical and biochemical behaviours towards/within a multitude of relative and realistic biological environments and also how such interactions may be correlated across a plethora of different biological organisms.
Keywords: silver nanoparticles, silver ions, biological impact
1. Introduction
Over the past 20 years the increase in nanoparticle (NP) research has been concomitant with the overwhelming increase in the level of nanotechnology-related products being produced [1,2]. This new industrial revolution promises to provide an advantageous basis for numerous applications, including medicine, consumer products (such as cosmetics and sporting equipment), environmental remediation and information technology [2]. For the basis of this review, the following definition for an NP will be used; i.e. a nano-object with all three external dimensions in the nanoscale (1–100 nm) [3]. Common within all engineered NPs is their ability to exhibit significantly novel and distinct physical, chemical and biological properties because of their nanoscale size.
Silver (Ag) NPs are an example of the numerous different types of NPs being engineered for use in a wide array of consumer, industrial and technological applications. Ag is a (precious) metal with the chemical symbol Ag, an abbreviation of the Latin word for silver, i.e. argentum. Ag NPs are clusters of Ag atoms that range in diameter from 1 to 100 nm and are attracting interest since their most innovative characteristics are associated with their increased antimicrobial properties [4–6]. Owing to these antimicrobial properties, Ag NPs are currently the most commonly used NPs in consumer and industrial applications (www.wilsoncenter.org). The use of Ag as an antimicrobial agent, however, is not a new concept. Ag pots and coins were used in ancient times to keep water sterile and in particular Ag-nitrate (AgNO3) has been used since the seventeenth century as an essential multipurpose medicinal product [7,8]. Examples of recent consumer applications using Ag as an antimicrobial agent consist of food supplements, materials for food packaging, coatings on medical devices, water disinfectants, air filters, electronic appliances, odour-resistant textile fabrics and cosmetic products such as deodorants [9,10].
Despite the prominent advantages of using Ag NPs in such applications, the possible health as well as environmental effects of Ag and of NPs in general [11,12] have raised concerns as to their mass use and production without having a clear understanding of their specific behaviour in aquatic and biological systems. Such concerns have been the basis for increased attention towards gaining an insight into the potential human health and environmental effects following exposure to Ag NPs. Human exposure to Ag has been described to occur orally, dermally or by inhalation, and occupational exposure is one primary concern. Patients that inhale or ingest Ag in large quantities and over a long period of time develop a disease called ‘argyria’ which means that their skin develops a blue or grey discoloration [13]. It is, however, not described if the patients have been exposed to nanosized Ag or to the bulk form of this material.
Environmental concerns are heightened due to the use of Ag NPs in washing machines and similar applications which can cause direct NP release into environmental systems and thus exposure of aquatic or terrestrial organisms to Ag NPs. Owing to the initial production of Ag NPs by humans, occupational exposure is of primary concern. Although toxic effects of Ag NPs to a number of organisms have been demonstrated [14], many questions concerning the specific interactions of Ag NPs with organisms at the biochemical and cellular level remain open. This is highlighted by the disparity whether the effects of Ag NPs are in fact a direct result of the NPs themselves, or rather due to the interaction with Ag ions [15] that are released in aqueous suspensions [16]. In the presence of moisture, metallic Ag NPs are oxidized, which results in the release of Ag ions (Ag+). Ag oxidation is typically a slow reaction which strongly depends both on the Ag NP properties, such as size and coating, and on the environmental conditions [13]. Several studies illustrate this contradicting picture and further highlight other aspects that may contribute towards the environmental and biological impact of Ag [14].
As soon as Ag NPs are released into the environment, such as for instance in the air or water, in experimental media or in biological fluids they start to transform which modifies their properties influencing their transport, fate and possible toxicity [17]. It is found in elemental form and in various ores such as argentite (silver sulfide, Ag2S). Ag is insoluble in water, whereas many Ag salts, such as e.g. AgNO3 are water soluble [18–20]. Ag occurs naturally in several oxidation states, usually as metallic Ag and Ag+; other possible oxidation states are Ag2+ and Ag3+ [21,22], these two latter forms are, however, not of relevance for aqueous solutions and under environmental or biological conditions.
The aim of this review is to describe the chemical and biochemical transformations of Ag NPs, which determine their bioavailability to bacteria, algae and higher organisms, as well as to consider uptake mechanisms of Ag NPs and of Ag+ at the cellular level (figure 1). These considerations provide then a basis for understanding possible adverse effects.
Figure 1.

Ag NPs released into the environment can either interact with the environment as metallic particles or be oxidized to Ag+. Both species can then undergo further interactions with aquatic and/or biological fluids and interact with proteins or chloride or thiols (a). All Ag species (underlined with a grey box) can then interact with aquatic systems such as (b) algae, (c) bacteria or (d,d’) mammalian cells. (b) Represents a dark field scanning electron microscope (STEM) image of a section through an algae cell Chlamydomonas reinhardtii (courtesy by E. Müller & S. Handschin, EMEZ, ETH Zürich) evidencing the cell wall surrounding the cell. Clearly visible are the photosynthetic thylakoid membranes of the chloroplast which occupy most of the cellular space. For Ag NPs to be effective in inhibiting photosynthesis, particles need to pass through the cell wall, cell membrane and the chloroplast envelope. Conventional transmission electron micrograph image of a bacteria Salmonella typhimurium (c) shows the surrounding of the bacteria with a cell membrane that encloses all essential components of the cytoplasma. Note that bacteria do not have membrane-bound compartments. (d) Represents a TEM picture from an epithelial lung cell in vitro showing the typical polarization of an epithelium with the microvilli's at the apical surface (arrow). At higher magnification (d’) the typical membrane-bound intracellular compartments like cell nucleus (n) and mitochondria (m) can be identified.
2. Production and characteristics of Ag nanoparticles
There are numerous reports on the synthesis of Ag NPs [18,20,23]. In general, there are two strategies to produce metal NPs. One is the so-called bottom-up method, where nanostructures are obtained through assembly of smaller, basic units into larger structures. The other is the top-down method, which starts from bulk material, whose size is decreased during the process. Common top-down techniques include e.g. photo-lithography and electron beam lithography [21,24].
A comprehensive coverage of all methods would be beyond the scope of this review. Instead, this review will describe the most commonly used bottom-up techniques that are used in the current literature.
The most extensively studied method for synthesizing Ag NPs is the controlled reduction of Ag salts [25,26]. This reduction is often accomplished using a chemical reducing agent [27–29] but also via electrochemical [23,30] or photochemical reduction [31,32].
This wet-chemical method, which involves Ag salts reduced (in one or more steps) to metal Ag, can be steered by many different parameters, such as the choice of the reducing agent, the concentrations of reagents, temperature, pH, mixing rate and reaction time [20,31]. In order to tailor the particles' properties, many studies have focused on the generation of different shapes, such as rods, wires, cubes, thin plates or bipyramids [33–35]. In addition, substantial effort has been devoted to the construction of core–shell structures, such as e.g. Ag@Au (silver core particles with a gold shell) or Ag@SiO2 NPs [23,36].
In general, strong reducing agents result in the formation of smaller NPs, whereas weaker reducing agents usually lead to larger Ag particles, as shown by Hoonacker et al., who studied the influence of borohydride and citrate on particle size distribution and colloidal stability [24,33]. Borohydride or dimethyl formamide are examples of strong reducing agents used to produce Ag NPs. Pastoriza-Santos and Liz-Marzan have shown the impact of temperature on the reaction rate and consequently on particle formation [25,37]. The so-called polyol-process uses ethylene glycol as both reducing agent and solvent, the reaction is carried out at 160°C using Ag nitrate as the Ag source [28,38]. Sun and Xia showed in 2002 how this reaction can be tuned by adapting the reaction parameters. They produced single-crystalline Ag nanocubes with controllable dimensions [23,39].
Another method makes use of a Tollens process to produce Ag NPs in a one-step method [31]. This reaction has been long used in the electroless deposition of Ag to generate reflective mirrors on solid supports and combines reducing sugars and Ag ammoniacal solutions [31]. The obtained particle size distributions are generally narrow. As described previously, the appropriate choice of reaction conditions is crucial also in this process. The Tollens method is an example of a green synthesis of Ag NPs. According to Raveendran et al. [33], a green synthetic approach includes the (i) selection of solvent medium, (ii) selection of environmentally benign reducing agents, and (iii) selection of non-toxic particle stabilizers. A recent review by Sharma et al. [36] summarizes current green chemistry approaches to synthesize Ag NPs focusing on the above-mentioned Tollens method, the polysaccharaide method (using polysaccharides [33] as capping and/or reducing agents), irradiation methods (using e.g. laser irradiation [37] of an aqueous solution of Ag salts in combination with a surfactant), polyoxometalates, which are water soluble and can undergo stepwise, multi-electron redox reactions [38] and biological methods [39]. The latter rely on the reduction of Ag ions using extracts from bio-organisms, which may act both as reducing and capping agents.
For any application of NPs, it is crucial to develop techniques to control their dispersion/aggregation. Usually, capping agents are used to enhance the particles' colloidal stability and the choice of capping material used to protect or passivate the NP surface is absolutely crucial, since it impacts on size and morphologies and even the targeted application or particle cell interactions in general [33]. Citrate is a commonly used reducing agent and stabilizer in the synthesis of metal colloids and has long been applied to controllably synthesize Ag NPs [40]. Another very popular stabilizer used in the synthesis of colloidal Ag is 3-aminopropyl-trimethoxysilane [41]. Variation of typical parameters showed that by adapting the reaction temperature either silica spheres with adsorbed Ag NPs or Ag particles with thin silica shells could be obtained. Long chain thiols have been studied extensively to coat and stabilize metal NPs [20] and the impact of chain length and ligating head groups were investigated with regards to particle size distribution and stability [42]. Liu et al. [43] studied the impact of multidentate thiol ligands on capping efficiency. They could demonstrate that due to multiple binding sites on the NP surface, multidentate capping agents yield smaller and more stable NPs. In addition to such small molecules, a wide variety of synthetic and natural polymers have been investigated as Ag NP stabilizers. Poly(vinyl pyrrolidone) (PVP) was first used more than 30 years ago [44] and has been used to prepare metal NPs ever since. Other generally used polymers include polyacrylates, poly(vinyl alcohol) and polyacrylamide [20]. In addition to linear polymers, branched macromolecules have been studied as stabilizers for Ag NPs in the past. Luo et al. [45] produced small Ag NPs (20–40 nm) using a third-generation poly(propylene imine) dendrimer AgNO3 solution without the introduction of additional reducing reagents and protective reagents in a microwave-assisted synthesis.
3. Behaviour of Ag nanoparticles in fresh waters and experimental media and biological fluids
The behaviour of Ag NPs in aqueous solutions will be considered with a special emphasis on the composition of freshwater (lakes or rivers), of wastewater and of similar synthetic solutions, in particular of culture media for aquatic organisms and cells. The most important processes for the bioavailability of Ag NPs and effects to aquatic organisms include agglomeration or aggregation of NPs to form larger particles, oxidation of Ag(0) to Ag+, subsequent dissolution to dissolved Ag+ species, speciation and solubility of Ag+ in solution and reactions modifying the reactivity of Ag(0)-NP [46–48]. The most important Ag+ solid phases to be considered for solubility are AgCl(s) and Ag2S(s). The solubility of these two solid phases must be carefully considered in experimental media for biological studies of Ag NP and of Ag+ effects, as well as for their behaviour in natural systems and is therefore discussed here in some detail.
To evaluate the fate of Ag NPs in aqueous media, an essential question is their stability with regards to their size and colloidal dispersion [49]. Very small NPs are likely to be transported with the water phase and to easily interact with organisms. If the size of Ag NPs is increasing by agglomeration processes, they become less mobile, will tend to be deposited to the sediments and therefore become less available to organisms in the water column. This, in turn, may then affect deposit feeders and other benthic organisms. Stable Ag NP suspensions, as produced in the laboratory, are stabilized by using various surface coatings, as described in §2, which confer stability by building up an electrical charge at the surfaces of Ag NPs, or by steric stabilization [50]. The surface charge of Ag NPs is most often negative at neutral pH, if weak acids are used as coatings and is strongly pH-dependent. If the surface charge of Ag NPs is neutralized as a function of pH, agglomeration occurs, as shown for example for carbonate-coated Ag NPs and for citrate-coated Ag NPs at low pH [51,52]. Polymer-coated Ag NPs, especially with PVP, appear to be more stable as a function of pH and of media composition, as they are sterically stabilized [52,53].
A very important parameter for colloidal stability is the ionic strength (I). At higher ionic strength, Ag NP suspensions are destabilized, in a similar way as other colloids. Calcium (Ca2+) concentrations in the millimolar range, as they occur in freshwaters, have been shown to cause agglomeration of Ag NPs [51,54]. In seawater with I = 0.7 and 10 mM Ca2+, Ag NPs are expected to strongly agglomerate. Synthetic culture media for aquatic organisms often contain divalent cations in the millimolar concentration range, or have higher ionic strength. The OECD medium for algal tests contains 0.12 mM Ca2+ and 0.12 mM Mg2+, and its ionic strength is I = 0.002 [55]. Ag NP suspensions may be expected to be stable in this medium, but the stability will depend on the particular coating properties [16,53]. The most commonly used growth medium for fish cells is Liebovitz's L-15. It contains relatively low Ca2+(1.26 mM), but has a high ionic strength containing 0.14 M of NaCl. Moreover, the Leibovitz's L-15 medium is supplemented with several amino acids, including cysteine and methionine at millimolar concentration. Ag NPs are thus expected to agglomerate in this medium, and dissolved Ag+ will be mostly either complexed to Cl− or bound to cysteine and methionine, so that free Ag+ will only be present at very low levels.
The dissolution of Ag NPs implies an oxidation reaction at the NP surface, from the elemental Ag(0) to Ag+, and possibly a subsequent binding of Ag+ to a ligand, according to the general scheme:
| 3.1 |
and
| 3.2 |
where the oxidant is oxygen or a reactive oxygen species, or another strong oxidant, and L is a ligand forming a complex with Ag+ [46]. These reactions are of high relevance for possible toxic effects of Ag NPs, as released Ag+ appear in many cases to determine their toxicity, or at least substantially contribute to the adverse biological effects observed [48,56,57].
Dissolution of Ag NPs is expected to be dependent on the presence of oxygen [58,59]. The presence of ligands for Ag+ may increase the dissolution rate of Ag NPs and lead to increased dissolution by formation of Ag+ complexes [60]. Thiol ligands such as cysteine strongly bind Ag+ [61]. Dissolution of Ag NPs was strongly increased in the presence of a high concentration of cysteine (400 μM) [60]. Experiments in our laboratory with lower cysteine concentrations (1–5 μM) indicated increased dissolution at short reaction times, and decreased dissolution at longer times, probably due to reduction of Ag(I) by cysteine (Sigg & Lindauer 2013, unpublished data). However, some strong Ag(I) ligands may also strongly bind to the Ag NP surface and prevent further dissolution reactions. Interactions with proteins as ligands may also increase the dissolution of Ag NPs [62]. In the presence of algae, some biotic ligands are released, which may affect the dissolution of Ag NPs and the bioavailability of Ag+ [46]. Furthermore, the size of Ag NPs affects the extent and kinetics of Ag NP dissolution, with the smallest NPs being dissolved faster and to a larger extent [63,64]. The nature of the coating however, appears to be less significant than the size effect [64].
Ag NP dissolution is expected to significantly depend on pH and to be more efficient at low pH [16]. In this study, release of Ag+ in the presence of oxygen increased by a factor of 5 if the pH was lowered from pH 7 to 4. In Ag NP dissolution experiments using various natural water samples, dissolved Ag+ increased by about a factor 4 in the case of lake water with pH 6.4 and low ionic strength, in comparison with lake water at pH 8 and higher ionic strength (Odzak & Sigg 2013, unpublished data). These results should be considered in view of pH in natural waters in the typical range of 6–8.5, and in experimental media for aquatic organisms pH 7.0–7.5. However, pH in gastric fluids may be as low as pH 1.6 (human gastric fluid) [64] and pH 0.8 in fish guts [65]. Extensive and rapid Ag NP dissolution would be expected at this low pH.
Ag+ in solution will interact with various ions and molecules that are present in aqueous media. Important ligands to be considered for Ag+ are chloride, sulfide and organic ligands with thiol groups [61], as well as carboxylic acids which are used as Ag NP coatings (e.g. citrate, lactate). Binding by natural organic ligands (fulvic and humic acids, other low molecular weight organic acids) is an important process in natural waters [66]. Chloride is an ubiquitous component of natural waters and is usually present in freshwaters in the concentration range of 0.1–1 mM, in wastewaters 2–20 mM, with higher values in industrial wastewaters, and in seawater with 0.5 M. Synthetic media for algae or Daphnia contain chloride in the range of 0.2–4 mM, media for fish cells or for mammalian cells in the range of 140–200 mM.
Figure 2 illustrates the solubility of AgCl(s) as a function of chloride concentrations in the range of 1 × 10−5–0.16 M (calculated with VMinteq). It shows that the sum of the dissolved Ag-species is limited to about 0.6 μM in the typical millimolar concentration range for chloride, but that the dissolved species increase at higher chloride concentrations. However, the speciation of Ag+ is different at low and high Cl− concentrations, as the concentration of free Ag ions [Ag+] steadily decreases with increasing Cl−, and dissolved Ag at higher Cl− concentrations is mostly composed of AgCl complexes, with AgCl0(aq), an uncharged species and AgCl2−, a negatively charged species. In culture media with high chloride concentrations, Ag(I) may thus be predominantly present as neutral or negatively charged species. This calculation was done for pH 7.5, but the results will be similar at lower pH. Precipitation of AgCl(s) is a process of concern in toxicological studies carried out under laboratory conditions at high Cl− concentrations and high total Ag concentrations [63]. However, at expected nanomolar Ag concentrations in natural systems, AgCl(s) solubility will not be exceeded.
Figure 2.
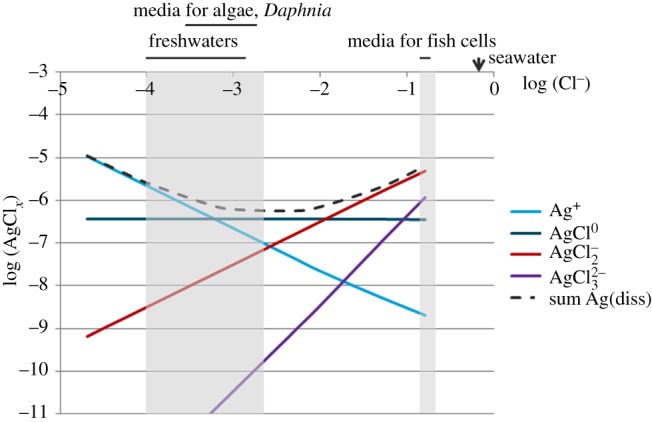
Dissolved Ag-species concentrations (M) at equilibrium with AgCl(s) over a range of (Cl−) (M) concentrations (pH 7.5). The dashed line represents the sum of all dissolved species, including Ag+, AgCl0(aq), AgCl2−, AgCl32−. The shaded areas represent the chloride concentration ranges of freshwaters and media for freshwater algae and invertebrates, and of media for fish and mammalian cells.
It has been suggested that the uncharged species AgCl0(aq) may play a role in Ag+ uptake in algae [67]. However, this interpretation has been challenged by further studies on Ag+ uptake by algae in the presence of Cl−, which concluded on the influence of Cl− on diffusion limitation of Ag+ uptake [68].
The solubility of Ag2S is extremely low, as illustrated in figure 3. Total dissolved Ag at equilibrium with Ag2S is mostly lower than 10−10 M, and Ag+ are at extremely low levels. Dissolved Ag+ in the presence of sulfide is only present as Ag–HS complexes. Small traces of HS− in a medium may thus lead to reactions with Ag+ and to the formation of Ag2S-precipitates at the surface of Ag NPs, or to precipitation of new Ag2S particles. This process has been observed for Ag NPs added in a pilot wastewater treatment plant, in which one tank was not aerated and contained HS− [69]. Typical dissolved sulfide concentrations in wastewaters with a partly anaerobic treatment may be in the micromolar to millimolar range [70]. Dissolved sulfide is expected to be very low in aerated freshwaters, as it would be rapidly oxidized. However, nanomolar concentrations of sulfide have been measured in some aerobic surface waters [71]. The very strong decrease of Ag+ solubility in contact with HS− appears to lead to decreased toxicity of Ag NPs [58,72].
Figure 3.
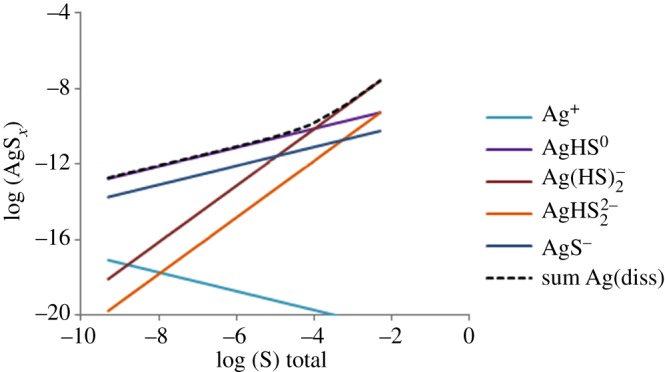
Dissolved Ag-species concentrations (M) at equilibrium with Ag2S(s) over a range of total S(-II) concentrations (M, pH 7.5). The dashed line represents the sum of dissolved Ag-species, including Ag+, AgHS0(aq), Ag(HS)2−, AgHS22−, AgS−.
Ag+ has a very high affinity to thiol ligands, such as cysteine, mercaptoethanol, thiourea and similar compounds [61]. In the presence of an excess concentration of one of these ligands over Ag+, the concentration of free Ag+ will be very low. As an example, in the presence of an excess of cysteine over total Ag of only 20%, free Ag+ decreases from 1 × 10−6 M to less than 1 × 10−10 M. Ag+ also forms very stable complexes with glutathione and prevents its oxidation [73]. Cysteine has been used to complex Ag+ in toxicity experiments with Ag NPs and has been shown to suppress toxicity to algae of Ag NPs with various coatings [46]. In a similar way, N-acetylcysteine has been used to study the toxicity of Ag NPs to nematodes and also suppressed toxicity in this case [57]. The presence of cysteine and other amino acids in the fish cell medium (Leibovitz's L-15) is likely to influence the toxic effects of Ag NPs, if experiments are carried out with the full medium including serum. Also proteins typically contain thiol groups, to which Ag+ may strongly bind and possibly replace essential metal ions [74,75]. Binding of Ag(I) to natural organic ligands was shown to be very strong at low Ag concentrations and was assumed to be due to the presence of thiol ligands [66].
By contrast, carboxylic acids such as citrate or lactate, which are used for Ag NP coatings weakly bind Ag+ [61,76]. Using the complex stability constant for an Ag+–citrate complex, as given by Yang et al. [76], and a 100-fold excess of citrate over Ag+ (Ag(tot) = 1 × 10−7 M), the calculation results in only 10% of Ag+ complexed with citrate. In suspensions containing these molecules as coatings or as dissolved ligands, Ag+ will thus be to a large part available to algae or to other cells [57]. However, the coatings may serve as ion buffers, which partly bind Ag+ and then release it to the cells. Binding of Ag+ by humic acids also appears to be weak, if Ag+ is mostly bound by carboxylic groups rather than by thiol groups [77].
From these various processes and chemical reactions, some of the behaviour of Ag NPs in various types of ecosystems may be predicted. In terms of colloidal stability, Ag NPs are expected to be most stable in freshwater at low ionic strength, such as in crystalline rock areas, and to be increasingly destabilized in freshwater with higher ionic strength (in carbonate rock areas), as in estuarine waters and in seawater. Dissolution, which is of primary importance for Ag bioavailability, will be most efficient at low pH (less than 7) and in the presence of strong binding ligands for Ag+. Bioavailability of Ag+ is decreased in the presence of strong binding ligands, including inorganic sulfide and organic thiol ligands.
Cell culture and biological media are very complex due to their high protein content and ligands as well as possible chelators for Ag+ in the solution. The chemical composition of cell culture media essentially consists of four groups of components: inorganic salts, carbohydrates, amino acids and various supplements (i.e. vitamins, fatty acids, lipids and growth factors). As each cell culture medium contains a defined amount of salts, vitamins, pH-buffering substances and nutrients, the addition of fetal bovine serum (FBS), which is commonly used in cell culture media as a primary growth supplement, is difficult to characterize. A high variety between different batches is likely even from a single manufacturer as FBS is produced out of pooled fetal blood collected in slaughterhouses. However, the undefined complex mixture in biological and cell culture media leads to transformations of Ag NPs and complexation of Ag+.
Interactions of dissolved Ag+ with ions present in experimental media are expected to influence also Ag bioavailability to exposed organisms. Despite the absence of systematic studies, several observations on ameliorating effects of Cl− on Ag toxicity to fish [78–80], of cysteine to algae [45] and N-acetylcysteine to nematodes [74] suggest that Ag–chloro complexes and Ag–cysteine (or Ag-N-acetylcysteine) complexes are not internalized in cells. Moreover, uptake experiments with algae that make use of cysteine to remove Ag adsorbed to cell surfaces indicate that Ag–cysteine complexes are not taken up by algae [46]. Nevertheless, more studies addressing the bioavailability of various Ag complexes will be needed to help understand the effects of Ag+ complexation upon Ag toxicity.
4. Nanoparticle transformation at biological interfaces
Transformation processes similar to those that affect the fate of Ag NPs in aqueous solutions are expected to occur once particles interact with biological surfaces and fluids or even within cells and specific compartments. One open question relevant to understanding how Ag NPs are taken up in organisms relates to the chemical composition of the NPs that reach a cell membrane. Currently, no studies with aquatic organisms have addressed this issue while an increasing body of literature demonstrating protein NP interactions, also suggests that it is the identity of biomolecules adsorbed to NPs rather than the original NP that might relate to the observed biological effects [81]. Interestingly, it is the characteristic behaviour of NPs to interact with biological molecules that is being exploited in nanotechnology (i.e. nanomedicine) developments and applications [82]. While transformation might occur in all cellular compartments where NPs are trafficked to after their uptake, the focus of this part of the review is upon the primary fluid to which Ag NPs become exposed.
4.1. Aquatic organisms
NPs at the interface with aquatic organisms will interact with an extraordinary number of diverse biological surfaces. These include skin, gills or gut tissues that have in common epithelial cellular structures in fish and invertebrates, as well as cell walls, which enclose the plasma membrane in microorganisms. These structures are covered with a film of biopolymers produced by the organisms for a variety of functions mostly referred to as mucus (in the case of fish and invertebrates) or extrapolymeric substances (EPS; in the case of microorganisms). Besides being of primary importance in conferring protection from chemical, physical and biological stressors, production of biopolymers by aquatic organisms is used for adhesion to substrates, locomotion, feeding or protection against predators or grazers [83,84]. Both the chemical composition and the amount of polymers composing mucus and EPS are expected to modify the physico-chemical features of NPs before they reach epithelial surfaces.
Mucus in fish and invertebrates is mainly composed of high molecular weight, filamentous, highly glycosylated glycoproteins, enzymes and other proteins and peptides, lipids and polysaccharides. Composition and abundance are highly variable and differ among species. Furthermore, the compositions of mucus are influenced by biological factors as gender, developmental stage or pathogens, and by environmental conditions such as pH or anthropogenic chemicals [85]. Similarly, EPS of autotrophic and heterotrophic communities is composed of enzymes for nutrient acquisition, other proteins, polysaccharides and small organic acids that remain to be identified [86]. Also the composition of EPS is influenced by the species making up the community, their growth state as well as nutrient availability or chemical conditions. Nevertheless, the concentrations of biomolecules in mucus or EPS in the range of micromolar to millimolar correspond to a number of molecules with the potential to completely coat the NP surface. Thus, a protein concentration of 70 g L–1 as found in the skin mucus of rockfish [87], corresponds to a number of molecules in high excess compared with the total surface area available for adsorption onto 10 μM Ag in the form of 20 nm NPs. This concentration would result in estimate of 8.4 × 107 protein/NP, whereas an NP with a diameter of 20 nm may be covered with almost 100 proteins with a radius of 2 nm (assuming an Mr of 20 kDa: http://www.calctool.org/CALC/prof/bio/protein_size). High densities of mucus are also expected to influence diffusion of NPs to epithelial cells [88]. Correspondingly, EPS bacterial biofilms showed to be important in controlling diffusion of various NPs, including Ag NPs [89].
While particle modification eventually influences their bioavailability, Ag NPs and Ag+ may in turn impair protective functions of mucus or EPS. Studies with fish have observed increased mucus production in rainbow trout exposed to titanium dioxide NPs [90], single-walled carbon nanotubes [91] and more recently in perch exposed to Ag NPs and dissolved Ag [92]. Increased mucus production in fish is a general response observed upon exposure to anthropogenic chemicals to decrease their bioavailability to epithelial tissues. Interaction of Ag NPs and Ag+ with mucus enzymes might also occur, leading to indirect effects as increased susceptibility to infections in fish and invertebrates. Similarly, interactions with EPS enzymes may result in damaged nutrient acquisition or increased grazing susceptibility in communities of microorganisms.
Interestingly, lectins and lysozyme, which are components of mucus and EPS [84], also are a focus of interest for nanomedical applications. Lectins, which are proteins capable of binding carbohydrates, have also been used conjugated to NPs to increase the retention of NPs in the gastrointestinal tract. Because of their property to adhere to the intestinal epithelium and generally to mucosal surfaces, lectins are exploited in medical applications to increase cellular internalization of drugs or their delivery across mucosal surfaces [93]. Other studies indicate lysozyme to confer exceptional colloidal stability, controlled self-assembly ability and biocompatible surface to lysozyme monolayer-stabilized Au [94].
No information is currently available on the interactions between Ag NPs and Ag+ with biomolecules in mucus or EPS produced by various aquatic organisms. Whether Ag NPs and Ag+ will interact with components of mucus or EPS will depend on binding affinities and concentration ratios as well as reaction rate constants. Addressing these types of questions is expected to progress the understanding of particle bioavailability, uptake and toxicity in aquatic organisms.
4.2. Biological fluids
In a synthetic human gastrointestinal model, Ag NPs were estimated to be capable of reaching the intestinal wall in their initial size and composition and, following intestinal digestion of AgNO3 in the presence of proteins, particle formation (20–30 nm) composed of Ag, sulfur and chlorine, may occur [95]. Not only is the gastrointestinal tract covered with mucus but also the epithelium of the airways, where a low viscous mucus layer is covered with a thick layer of high viscous mucus. This heterogeneity also influences particle mobility as shown for various NPs [96], but the chemical reactivity of mucus components with Ag NPs and/or Ag+ is only rarely described so far. A recent study has shown that ingested Ag NPs undergo accelerated oxidative dissolution in gastric acid resulting in an interaction with reduced selenium species [97], however, further studies are needed to reveal more details on these complex mechanisms.
In physiological media or in vivo, the bioavailability of Ag NP in internal compartments will be strongly influenced by the local environment: charge and pH, dissolved solutes, proteins, lipids and other potential adsorbing compounds will all have an effect on the aggregation state (agglomeration) and the dissolution of Ag NPs.
In the blood circulation, Ag NPs or Ag+ encounter approximately 7000 proteins and isoforms [98,99], which can interact with them. Many studies have been performed to assess bioavailability and toxicokinetics of Ag NPs administered into the bloodstream [100,101], however, to the best of our knowledge, we have not found any studies.
5. Molecular mechanisms of Ag uptake
In order to understand the possible toxicology of Ag NPs, the mechanisms of cellular homeostasis of ionic Ag should be considered. The molecular mechanisms that enable Ag to be taken up and excreted by a cell have long been ignored; and even though some evidence is now available, the overall mechanism remains largely unknown. The role of Ag in biology has not been thoroughly investigated probably owing to the fact that Ag is a non-essential metal, and therefore research focused mainly on toxicity effects rather than more subtle cellular homeostatic mechanisms. In fact, what is known of Ag cellular homeostasis has mainly to do with its interaction with the essential metal copper (Cu). Both excess and deficiency of Cu is deleterious for the cell, thus Cu intracellular concentration is tightly regulated by Cu transporter proteins [102]. Notably, some of these proteins have been shown to transport Ag as well as Cu [103,104].
The strong inhibition by Ag+ in Cu uptake experiments suggests that Ag+, which are isoelectric to Cu+, can be taken up by the same high-affinity Cu transporter (CTR1) [103]. Inhibition of Cu uptake by Ag has been shown in bacteria [105], plant [106], fish [107] and humans [103,108]. Moreover, recently, it has been demonstrated that CTR1 is an important Ag transporter. Cells over-expressing Ctr1 accumulated Ag and cells lacking Ctr1 had a 50% reduction in Ag content compared with wild-type [104]. In addition, the evidence that Ag+ inhibit Cu uptake suggests that monovalent Cu is the favourite substrate for CTR1. This evidence is also confirmed by an enhanced uptake of Cu when cells are pre-exposed to the reducing agent ascorbate [103,109]. From yeast [110] to mammals [111,112], uptake of Cu (and Iron) has been shown to be dependent on reductase proteins located at the plasma membrane. In Cu uptake studies, inhibition of Cu uptake by Ag is used as a marker of CTR1-mediated transport [108]. However, it should also be mentioned that Ctr1-deficent cells (Ctr1 knock out embryonic cells) still accumulated some Cu or Ag, approximately 30 [113] or 50% [104], respectively, showing that there are alternative Ctr1-independent systems. Ctr1-deficient cells express residual Cu transport activity that saturates and is characterized by a Km for Cu of 10 μM (rather than 1–5 μM of CTR1) [113]. Unlike CTR1-mediated Cu uptake, the CTR1-independent Cu transport activity appears to be a Cu(II) transporter, as suggested by the lack of stimulated Cu uptake by ascorbate and the absence of competition by Ag+ [113]. The principal candidate for CTR1-independent Cu uptake is the divalent metal transporter (DMT1), however other unknown transporters cannot be excluded. DMT1 has been demonstrated to mediate proton-coupled transport of a broad group of divalent ions, primarily iron [114], but also Cu with lower efficiency [115]. On the other hand, even though the name of this transporter suggests transport of Cu(II), Cu(I) was shown to be the species transported by DMT1 [98,116]. This observation could suggest a role for DMT1 in Ag+ uptake however it has not yet been shown.
Another example to CTR1-independent Ag and Cu uptake is well known from the fish physiology literature [117,118]. Freshwater fish constantly lose electrolytes from their concentrated extracellular fluids by diffusion to the dilute environment. The key transporters for the compensation of lost electrolytes are the epithelial sodium channels (ENaC), which are located at the apical membrane of polarized epithelial cells including the gill and facilitate passive uptake of Na+ from the dilute environment and the Na+/K+-ATPase that is located at the basolateral membrane of epithelial cells [107,118]. It is hypothesized that Ag and Cu can inhibit Na uptake through competition for entry across the apical ENaC [118] and also through inhibition of the basolateral Na+/K+-ATPase [107]. This uptake route for Cu and Ag is likely to occur only in the freshwater fish, where external Na+ is only a few millimolar or less [117]. Furthermore, a similar disruption of Na homeostasis was also described in Daphnia magna [119]. However, even though the existence of the apical ENaC has been claimed more than a decade ago, its molecular characterization is still lacking so its existence needs to be confirmed.
Interestingly, inhibition of Na uptake was recently shown in rainbow trout exposed to dialysed Ag NPs while exposure to its measured dissolved fraction did not result in any Na uptake inhibition, suggesting an NP-specific effect [120].
In addition to this ‘Na-dependent’ uptake route, in fish, similar to all other vertebrate a Cu uptake specific transporter (CTR1), which is Na-independent, has been shown [121,122]. Furthermore, the Cu efflux pumps or Cu-ATPases (ATP7A and ATP7B) have also been shown in fish [123]. In addition to Cu-ATPases at the basolateral membrane, a role for a putative Cu : Cl− symporter has been proposed by Handy et al. [124] based on observations of DIDS (anion transport inhibitor) sensitivity and correlation between the rate of Cu appearance in basolateral solutions and apical Cl− concentrations. However, the role of these transporters in the response to excess Ag+ or Ag NPs in fish still needs to be confirmed.
In summary, a number of assertions can be made regarding the correlation of the biological impact of Ag NPs and their possible interaction with metals transporters, in particular Cu transporters. First of all because of their size, Ag NPs could not be transported across Cu transporters. For example, CTR1 functions as a trimer forming a membrane ‘pore’ and the centre of the pore was estimated to be 9 Å (0.9 nm) [125]. Therefore, any possible interaction of CTR1 with Ag NPs would be with Ag+ dissolved from the NP possibly inside the acidic environment of endosomes or lysosomes (figure 4). For this reason, the most likely mechanism of uptake of Ag NPs is by endocytosis as described for many other NPs [126]. Notably, there are some studies reporting electron microscopy data suggesting that Ag NPs are taken up by the epithelial cells possibly by endocytosis [127,128]. Moreover, following waterborne Ag NPs exposure Ag accumulated in the gill, but also liver [127,129,130] of fish showing that Ag NPs are efficiently transported across gill epithelial cells and reach the liver via the blood. Understanding how Ag NPs interact with serum proteins (e.g. albumin) is of critical importance to determine hepatic Ag NPs uptake.
Figure 4.

Proposed model for Ag NP uptake and interactions with Cu transporters in a putative gill cell. While Ag NPs might enter the gill cell only through endocytosis, Ag+ dissolving from the particle surface could enter the cell via the sodium channel (ENaC) or an unknown transporter (?) or possibly sharing Cu transporters routes via DMT1 or CTR1. However, CTR1 is more likely to be involved in intracellular transport of Ag from endosomes or other vesicles. Cu enters via CTR1 at the basolateral membrane or via DMT1, endocytosis or other unidentified transporters at the apical membrane in enterocytes. Previous entry via CTR1 and DMT1 Cu is reduced by the metalloreductases. After entry Cu is bound to the chaperone ATOX1 which delivers Cu to the Cu-ATPases located at the trans golgi network (TGN). The Cu-ATPases ATP7A and ATP7B may then deliver Cu to cuproenzymes and secretory Cu-proteins or excrete Cu once Cu levels are high. A possible route of excretion for intracellular Ag+ would be through the same pathway (CTR1-ATOX1-Cu-ATPases), however, this hypothesis remains to be demonstrated. Ag NPs or Ag+ dissolving from their surfaces can also interact and inhibit the Na/K-ATPase located at the basolateral membrane. (Adapted from Minghetti et al. [123].)
6. Uptake of Ag nanoparticle in aquatic organisms
During the last 5 years a number of studies have dealt with questions on the fate and ecotoxicity of Ag NPs in aquatic systems. Published ecotoxicity experiments have informed on concentrations leading to lethal or sublethal effects in a variety of organisms. So far there is no evidence of a particularly high adverse effect, which would be predictable from increased reactivity of materials displaying nano dimensions. Despite this, there is still little knowledge on how Ag NPs interact with aquatic organisms and major questions remain to be examined. For instance, it is generally not known whether assessed toxicity results from internalization of particles in cells of exposed organisms. While Ag+ have been shown to explain toxicity to bacteria and algae and partly to other organisms too [46,76,131], other studies show that exposures to ionic and particulate Ag result in distinct transcriptional responses [132,133]. Nevertheless, conclusive evidence for direct effects of particles is still missing. Moreover, as discussed earlier, while environmental studies indicate the susceptibility of Ag NPs to be transformed as a function of pH, ionic strength and chemicals occurring in the environment, no attention has been given to the biological transformation of Ag NPs and their influence on NP uptake and toxicity once particles come in contact with aquatic organisms, or after their internalization. The important role of protein–NP interaction is recognized in nanomedicine and nanomaterials, where it is exploited for the development of new applications such as targeted drug delivery [81,82]. At this stage, progress in understanding how Ag NPs exert toxicity to aquatic organisms would benefit from focusing research priorities on understanding particle transformation and its influence on uptake and distribution in organisms.
Determining the bioavailability, uptake and intracellular accumulation of Ag NPs in aquatic organisms is essential for the evaluation of their toxicity to aquatic life and transfer along the food chain. While unicellular organisms do absorb contaminants directly through cell walls and cell membranes, multicellular organisms as invertebrates and fish can absorb contaminants directly from the water and via food ingestion. Only a limited number of studies have reported so far on cellular internalization of Ag NPs in aquatic organisms. Combining dark field single particle optical microscopy and spectroscopy, Ag NPs were detected in the chorion of zebra fish embryos [134]. However, it is still uncertain whether and to which extent Ag NPs are taken up by fish. For instance, no Ag particles were detectable in perch upon analysis of several organs by dark field microscopy [92]. Larger aggregates of Ag NPs were found to be internalized in the nematodes Caenorhabditis elegans upon particle ingestion [135]. Using transmission electron microscopy (TEM) together with energy dispersive X-ray analysis Ag NPs were also detected in the gut epithelium of the deposit feeder Nereis diversicolor feeding on sediments contaminated by Ag NPs [136]. Intracellularly, Ag NPs were found to be localized mainly in endosomes as well as in lysosomes, where they occur as aggregates. In the same study, the finding that Ag from exposures to AgNO3 was mainly found to be associated with metallothionein and was taken as indirect evidence that detected particles correspond to the Ag NPs to which the organisms were exposed. Individual electron dense particles were also observed in the freshwater algae Ochromonas danica, but their elemental composition was not examined [137]. Since formation of insoluble inorganic granules represent one way of detoxifying metals through their immobilization [138], identifying the metal of interest is essential [139]. Other studies have used inductively coupled plasma mass spectrometry (ICP-MS) to quantify Ag NP uptake in some aquatic organisms. Thus, increased Ag levels upon exposure to Ag NPs were measured in gill tissue of rainbow trout and zebra fish [127,132], as well as in soft tissues of clams [140,141]. Differently, low levels of Ag detected in the green algae Chlamydomonas reinhardtii exposed to Ag NPs were explained by uptake of dissolved Ag+. Moreover, no Ag NP uptake was detected in a cell wall-free mutant of the same algae indicating that not only the cell wall of algae but also the cell membrane constitute a barrier for particle internalization in this algae [47,57].
These studies confirm the potential of Ag NPs to be taken up and distributed in various aquatic organisms, but the toxicological significance of these findings is uncertain because of the general lack of quantitative data. TEM analysis is labour intensive to prepare samples and analysis is time consuming limiting high throughput for a large number of samples [142]. On the other hand, since ICP-MS measurements do not allow sources of Ag to be distinguished, uptake studies require appropriate controls with an Ag salt because Ag NP suspensions might include a variable concentration of Ag+ which derive from particle synthesis or dissolution. The internalized Ag that derives from particles is then determined by the difference to the Ag level found to be accumulated upon exposure to the Ag salt concentration corresponding to the dissolved concentration in the Ag NP treatment. Determining whether Ag NPs or Ag+ have been internalized in cells also requires particular consideration of sample preparation. Metal ions display considerable binding affinity to a variety of biological ligands and part of the metal at biological interfaces will inevitably interact with potential binding sites before being taken up by cells. Moreover, particles may merely be physically trapped in biological samples because of structural characteristics, as for instance gill tissues in fish or molluscs, which display an extraordinary large surface area. Sample preparations including centrifugation steps might also non-specifically entrap particles. Thus, depending on sample characteristics and preparation, Ag detected by ICP-MS might also include Ag that has not been taken up in cells. One way to operationally define intracellular Ag in biological material consists of removing not internalized particles and Ag+ by repeated washing in presence of a strong Ag ligand such as cysteine (figure 5). Experiments with the green algae Chlamydomonas reinhardtii showed that the Ag detected in algae upon exposure to Ag NPs decreased by more than 95% upon subsequent re-suspension and centrifugation of the cells in Ag-free medium in presence of cysteine, whereas more than 50% Ag was removed after 1 min of washing algae with cysteine while remaining constant upon subsequent washes [47].
Figure 5.
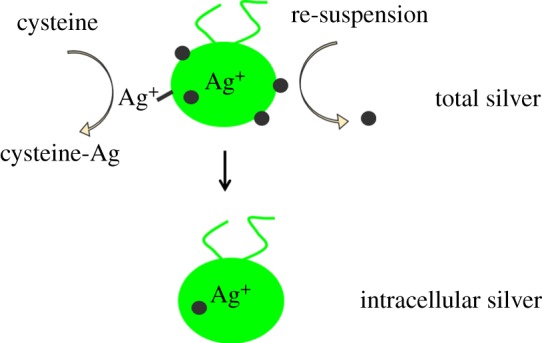
Schematic of Ag distribution in algae after exposure to Ag NPs. Total Ag determined by ICP-MS includes Ag NPs and Ag+ adsorbed to algal surfaces as well as internalized Ag in the form of particles or dissolved Ag. Assessing the intracellular concentration of Ag requires re-suspending algae in an Ag-free medium to remove adsorbed particles and washing with cysteine, which as a strong Ag ligand removes adsorbed Ag+. Black dots represent Ag NPs. (Online version in colour.)
Therefore, depending on the procedure adopted to prepare samples the level of intracellular Ag internalization determined by ICP-MS may vary considerably. Currently, the available data on Ag NPs uptake in aquatic organisms are scarce and there is urgent need for more quantitative data which are a requisite to examine how observed toxic effects of Ag NPs relate to Ag accumulated in cells. Moreover, data on particle uptake and accumulation will be important to evaluate potential transfer of Ag NPs along aquatic food chains.
7. Bacterial interactions of Ag nanoparticles
Owing to their simplistic structure and also culturing requirements, bacteria, both Gram-negative and Gram-positive, have been used extensively during the last decade as a model to determine the potential risk posed by Ag NPs, most notably in the field of nano-ecotoxicology [143]. In addition to the plethora of literature highlighting the possible risks posed by nano Ag, a select number of studies have taken the additional step and assessed whether or not Ag NPs are internalized within bacteria; an essential requirement when determining any NP–bacteria interaction [144].
In a recent study by Chamakura et al. [145], Ag NPs within a size range of 10–50 nm [146] were not found to be present inside Gram-negative Escherichia coli after exposure for 2 h at a particle concentration of up to 40 µg ml–1 using conventional TEM combined with elemental analysis; a further necessity when assessing/confirming the entry and intracellular localization of spherical NPs, which elicit a similar size to intracellular organelles (i.e. ribosomes) [139]. This finding was in conjunction with a negative finding for mutagenicity, as determined by a colony formation assay, within the Escherichia coli strain after 2 h exposure at 0.6–10.0 ppm, which was associated with the lack of internalization of Ag NPs [145]. Dror-Ehre et al. [147] also studied the interaction of Ag NPs with Escherichia coli using conventional TEM. In this study, Dror-Ehre and colleagues reported that Ag NPs were present inside the bacteria as well as being attached to the outer membrane. Although the two-dimensional images shown by Dror-Ehre and colleagues may be suggestive of such a conclusion, the authors did not incorporate elemental analysis nor did they perform electron tomography (three-dimensional TEM) in order to confirm the presence of the Ag NPs within the Gram-negative bacteria, thus making the known presence within the bacteria questionable. Dror-Ehre et al. [147] did, however, suggest the notion that due to an aggregation observed of the proposed internalized Ag NPs (15 nm inside bacterium versus 6 nm outside bacterium). While it has been reported that nano-objects may agglomerate/aggregate when present within a membrane-bound vesicle within eukaryotic cells [148], it is debatable whether or not the status of aggregation/agglomeration of the NP sample can be a determining factor that proves cellular internalization or not. From these two studies, it can be suggested that Ag NPs do not easily penetrate into Gram-negative bacteria. However, a study by Yuan et al. [149] showed a limited number (although no quantification of the internalized fraction was performed) of Ag NPs to be internalized by Gram-negative Nitrosomonas europaea after 3 h exposure at a particle concentration of up to 10 mg ml−1. Similar to Chamakura et al. [145], Yuan et al. [149] used conventional TEM analysis combined with elemental analysis, suggesting that the ability of Ag NPs to gain access beyond the lipid bilayer and enter Gram-negative bacteria is specific to the bacterial strain used and not a function of the methodology used by the different authors. However, the Ag NPs used by Yuan et al. [149] were between 4 and 10 nm in size; a factor of five times smaller than Ag NPs studied by Chamakura et al. [145]. This difference could further suggest that particle size is a contributing factor regarding the ability for Ag NPs to enter Gram-negative bacteria. This hypothesis however, is counteracted by the study of Dror-Ehre et al. [147], who used Ag NPs ranging from 4 to 20 nm in diameter (with varying surfactants and charges on the NP surfaces). The findings from this single study, alternatively suggest therefore, that perhaps neither size nor surface characteristics of the Ag NPs play a significant role in the internalization of Ag NPs by Gram-negative bacteria.
Although such an equivocal stance is paramount within the field since the majority of studies have assessed the Ag NP–bacterial interaction using Gram-negative bacteria, in a recent study by Mirzajani et al. [150] the Gram-positive bacterial strain Staphylococcus aureus was used. Ag NPs (diameter 1–5 nm) were reported to be present within bacteria after 24 h exposure using conventional TEM. Similar to the study of Dror-Ehre et al. [147], Mirzajani et al. [150] did not use further elemental or tomography analysis of the bacterial samples, instead Mirzajani et al. [150] proposed that the presence of Ag NPs within the bacteria was due to a change in the specific electron density with the bacteria. Such assessment of the internalization of Ag NPs is debatable however, owing to the extensive sample preparation procedure in which contamination material (e.g. uranyl acetate staining) can be easily observed as electron dense substances, possibly indicative of nanomaterials [139]. It is therefore not clear whether or not the Ag NPs were internalized by Staphylococcus aureus over the 24 h exposure period.
Although it has been shown by Yuan et al. [149] that Ag NPs may enter bacteria, although at a limited number, it can be suggested that Ag NPs do not readily enter either Gram-negative or positive bacteria. This notion, while possible, is difficult to comprehend based on the extensive literature database that eludes to the fact that Ag NPs can elicit both an adverse (cytotoxic) and positive (antimicrobial) response [143]. The lack of any feasible and conceptual proof and understanding if Ag NPs can enter bacteria provides strength to the hypothesis that the (adverse and positive) biochemical effects observed within biological systems following exposure Ag NPs are caused by Ag+ and not the NPs themselves [151]. However, if Ag+ drive the biochemical effects monitored, then do they enter the bacteria themselves or not? If the Ag+ can enter bacteria, then how is this achieved? It is perceived that through many different ion channels, Ag+ are able to gain entry into bacteria and subsequently interact with the inner components (i.e. nucleoid region).
In summary therefore, there is a large knowledge gap within the field as regards the ability for Ag NPs to enter bacteria, and if they possibly are engulfed, by what mechanisms. Furthermore, Ag+ seem to present the most notable possibility for the passage of Ag into bacteria, yet it is only hypothesized how their passage into bacteria may occur. In order to understand these aspects of the Ag NP–bacterial interaction, it is therefore essential for future studies to also investigate the localization of Ag NPs in association with bacteria and if a direct or indirect interaction occurs, and how this relates to any response observed.
8. Uptake of Ag nanoparticles in mammals
At present, there are few references related to the bioavailability of Ag NPs to terrestrial animals. Most of the information comes from laboratory studies using highly controlled exposure methods. From these studies, and the few that have looked at environmentally relevant exposures, it is possible to determine a rough picture of the bioavailability of Ag NPs to terrestrial organisms. However, many questions remain essentially unknown, especially with respect to realistic exposure models.
Once terrestrial animals come into contact with Ag NPs, the physiological route of exposure also needs to be considered as each pathway will confer different bioavailability on the host. Uptake of Ag NPs to terrestrial organisms can occur via air, soil and water, and these uptake routes all confer potentially different bioavailability. The primary route of uptake into terrestrial animals is likely to be via the lungs or through the gastrointestinal epithelia, with trans-dermal transport being a minor route. In addition, if retention of the NPs occurs in the animal, potential sources of transfer of NPs to offspring may occur through lactation or could be passed through the birth or the placenta. However, only very few studies have actually looked at bioavailability of Ag NPs to terrestrial animals with environmentally realistic exposure scenarios. One study has shown that once into contact with terrestrial organisms such as earthworms, Ag NPs were shown to bioaccumulate 5.1 ± 0.5% and 11.0 ± 0.3% of the Ag NPs and Ag+ (respectively) from the contents of their guts; after 48 h, the corresponding values were 0.38 ± 0.03% and 2.3 ± 0.1%, Ag NPs and Ag+ respectively [152].
By far, the best sources of information regarding the bioavailability of Ag NPs comes from controlled laboratory studies, primarily performed on rats or mice and generally using either i.v., instillation or inhalation as means of controlled exposure. Some of these studies are summarized in review form by Kruszewski et al. [153]. The bioavailability of orally administered Ag NPs was 1.2% in rats treated with 1 mg kg−1 Ag NPs and 4.2% in animals treated with 10 mg kg−1 Ag NPs [101]. Ag was found to be present in all examined organs of rats exposed to Ag NPs, with the highest levels in the liver and spleen for all Ag treatments [154]. In this study, Ag concentrations in the organs were highly correlated to the amount of Ag+ in the Ag NP suspension, indicating that mainly Ag+, and to a much lesser extent Ag NPs, passed the intestines in exposed rats [154]. In addition, it has been shown that Ag NPs injected in rats can be translocated from the blood to all the main organs and that the concentration of Ag in tissues was significantly higher in rats treated with 20 nm Ag NPs when compared with 200 nm Ag NPs [100]. The accumulation of Ag was also observed in all examined rabbit organs, including liver, kidney, spleen, lung, brain, testis and thymus up to 28 days following a single intravenous dose [155].
An uptake of Ag NPs into mammalian cells such as lung epithelial cells could be shown in vitro by TEM using an air–liquid exposure cell system to avoid particle dissolution (and aggregation) in cell culture media. The Ag NPs were endocytosed and highly aggregated inside vesicular structures, but they did not cause cytotoxicity or induce the release and expression of oxidative stress and (pro)-inflammatory markers. For equal AgNO3 exposure concentrations, similar effects were observed, and it has been concluded that no acute cytotoxic and (pro)-inflammatory effects for Ag at a realistic exposure dose can be assumed [156]. This is in agreement with another comprehensive review recently published by Chernousova & Epple [157], who showed that the risks for humans and the environment seem to be limited.
However, the fate of intracellular Ag NPs or Ag+ cannot be answered so far. The problem is that the majority of cell culture studies are done under suspension, and as already discussed it is then difficult to differentiate between the particle and ionic effects. One possibility to overcome the issue with suspension experiments is to use air–liquid exposure systems, if lung cells are considered in the study [156]. Using such sophisticated systems, the direct exposure of a defined solution with either Ag NPs or Ag+ onto the cell surface can be controlled, and therefore the possible observed effects can directly be linked to a particle or ion effect.
To the present, the only side effects to humans exposed to Ag+ are Ag deposits in colocalization with sulfur and selenium as found in patients with ‘argyria’ an irreversible discoloration of the skin [13,158]. However, the exact chemical reaction that leads to Ag deposition is still unclear.
9. Conclusions
In order to understand the potential toxicity of Ag NPs, it is essential to understand their chemical and biochemical behaviour in aquatic and biological systems. Dissolution, which is of primary importance for Ag bioavailability, is very much influenced by the pH and the presence of strong binding ligands, which is completely different in the various environments. In addition, the routes for exposure and the varied transformation processes in environmental and physiological compartments make the prediction of Ag NPs bioavailability difficult. One important point is that each study with Ag NPs has to be related in more detail to the chemical and biochemical conversion in the medium, where the experiments have been performed, only this will allow for a more comprehensive understanding of the potential of Ag NPs for further applications.
For further experimental research, we suggest in detail the following points:
— determination of the release of ions from the Ag NPs in the medium/system used in order to distinguish between effects from the particle and the ionic form;
— further studies on uptake of Ag NPs in various types of cells and of organisms, however, considering the different systems when data are compared, i.e. prokaryotic and eukarytic cells do not behave similar upon one stimulus;
— use of realistic exposure systems for both aquatic and biological systems, i.e. use of realistic dose and exposure scenarios;
— understanding the role of protein binding to the surface of Ag NPs; and
— revealing relevant mechanisms of endocytotic uptake of Ag NPs and the importance of intracellular NP quantification, this information will help to understand how intracellular Ag NP number and Ag+ concentration relate to possible toxic effects.
Acknowledgements
The authors thank Ekin Oeztuerk Ilgue for her assistance with the reference library.
Funding statement
This work was financed by the Federal Office of Public Health, Switzerland, Swiss National Science Foundation, the National Research Program 64, the German Research Foundation (SPP1313), the Swiss Nanoscience Institute (SNI) within the National Center of Research (NCCR) in Nanoscale Science and the Adolphe Merkle Foundation. The authors acknowledge the support of the Electron Microscopy Centre EMEZ of the Swiss Federal Institute of Technology ETHZ for the algae imaging work.
References
- 1.Gwinn MR, Vallyathan V. 2006. Nanoparticles: health effects—pros and cons. Environ. Health Perspect. 114, 1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maynard A, Aitken R. 2007. Assessing exposure to airborne nanomaterials: current abilities and future requirements. Nanotoxicology 1, 26–41. ( 10.1080/17435390701314720) [DOI] [Google Scholar]
- 3.ISO/TS 27687: 2008.
- 4.Edwards-Jones V. 2009. The benefits of silver in hygiene, personal care and healthcare. Lett. Appl. Microbiol. 49, 147–152. ( 10.1111/j.1472-765X.2009.02648.x) [DOI] [PubMed] [Google Scholar]
- 5.Eckhardt S, et al. 2013. Nanobio silver: its interactions with peptides and bacteria, and its uses in medicine. Chem. Rev. ( 10.1021/cr300288v) [DOI] [PubMed] [Google Scholar]
- 6.Morones JR, Elechiguerra JL, Camacho A, Holt K, Kouri JB, RamÃrez JT, Yacaman MJ. 2005. The bactericidal effect of silver nanoparticles. Nanotechnology 16, 2346–2353. ( 10.1088/0957-4484/16/10/059) [DOI] [PubMed] [Google Scholar]
- 7.Klasen H. 2000. Historical review of the use of silver in the treatment of burns. I. Early uses. Burns 26, 117–130. ( 10.1016/S0305-4179(99)00108-4) [DOI] [PubMed] [Google Scholar]
- 8.Maillard JY, Hartemann P. 2012. Silver as an antimicrobial: facts and gaps in knowledge. Crit. Rev. Microbiol. ( 10.3109/1040841X.2012.713323) [DOI] [PubMed] [Google Scholar]
- 9.Hagen Mikkelsen S, et al. 2011. Survey on basic knowledge about exposure and potential environmental and health risks for selected nanomaterials. Danish Ministry of the Environment–Environmental Protection Agency. Environmental Project 1370. See http://www.nfp64.ch/SiteCollectionDocuments/nfp64_publikationen_mikkelsen.pdf.
- 10.Rai M, Yadav A, Gade A. 2009. Silver nanoparticles as a new generation of antimicrobials. Biotechnol. Adv. 27, 76–83. ( 10.1016/j.biotechadv.2008.09.002) [DOI] [PubMed] [Google Scholar]
- 11.Oberdorster G, Stone V, Donaldson K. 2007. Toxicology of nanoparticles: a historical perspective. Nanotoxicology 1, 2–25. ( 10.1080/17435390701314761) [DOI] [Google Scholar]
- 12.Oberdorster G, Oberdorster E, Oberdorster J. 2005. Nanotoxicology, an emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 113, 823–839. ( 10.1289/ehp.7339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albers I. 1816. Observations on a change of colour in the skin, produced by the internal use of the nitrate of silver. Med. Chir. Trans. 7, 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Lima R, Seabra AB, Duran N. 2012. Silver nanoparticles: a brief review of cytotoxicity and genotoxicity of chemically and biogenically synthesized nanoparticles. J. Appl. Toxicol. 32, 867–879. ( 10.1002/jat.2780) [DOI] [PubMed] [Google Scholar]
- 15.Lubick N. 2008. Nanosilver toxicity: ions, nanoparticles—or both? Environ. Sci. Technol. 42, 8617 ( 10.1021/es8026314) [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Hurt RH. 2010. Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environ. Sci. Technol. 44, 2169–2175. ( 10.1021/es9035557) [DOI] [PubMed] [Google Scholar]
- 17.Levard C, Hotze EM, Lowry GV, Brown GE. 2012. Environmental transformations of silver nanoparticles: impact on stability and toxicity. Environ. Sci. Technol. 46, 6900–6914. ( 10.1021/es2037405) [DOI] [PubMed] [Google Scholar]
- 18.Panáček A, et al. 2006. Silver colloid nanoparticles: synthesis, characterization, and their antibacterial activity. J. Phys. Chem. B 110, 16 248–16 253. ( 10.1021/jp063826h) [DOI] [PubMed] [Google Scholar]
- 19.Howe P, Dobson S. 2002. Silver and silver compounds. Geneva, Switzerland: World Health Organization. [Google Scholar]
- 20.Nair LS, Laurencin CT. 2007. Silver nanoparticles: synthesis and therapeutic applications. J. Biomed. Nanotechnol. 3, 301–316. ( 10.1166/jbn.2007.041) [DOI] [Google Scholar]
- 21.Eustis S, Krylova G, Smirnova N, Eremenko A, Tabor C, Huang W, El-Sayed MA. 2006. Using silica films and powders modified with benzophenone to photoreduce silver nanoparticles. J. Photochem. Photobiol. A Chem. 181, 385–393. ( 10.1016/j.jphotochem.2005.12.024) [DOI] [Google Scholar]
- 22.Roper WL. 1990. Toxicological profile for silver. Atlanta, GA: Agency for Toxic Substances and Disease Registry U.S. Public Health Service; See http://www.atsdr.cdc.gov/toxprofiles/tp146.pdf. [PubMed] [Google Scholar]
- 23.Sun Y, Xia Y. 2002. Shape-controlled synthesis of gold and silver nanoparticles. Science 298, 2176–2179. ( 10.1126/science.1077229) [DOI] [PubMed] [Google Scholar]
- 24.Van Hoonacker A, Englebienne P. 2006. Revisiting silver nanoparticle chemical synthesis and stability by optical spectroscopy. Curr. Nanosci. 2, 359–371. ( 10.2174/157341306778699310) [DOI] [Google Scholar]
- 25.Pastoriza-Santos I, Liz-Marzán LM. 2009. N,N-dimethylformamide as a reaction medium for metal nanoparticle synthesis. Adv. Funct. Mater. 19, 679–688. ( 10.1002/adfm.200801566) [DOI] [Google Scholar]
- 26.Shirtcliffe N, Nickel U, Schneider S. 1999. Reproducible preparation of silver sols with small particle size using borohydride reduction: for use as nuclei for preparation of larger particles. J. Colloid Interface Sci. 211, 122–129. ( 10.1006/jcis.1998.5980) [DOI] [PubMed] [Google Scholar]
- 27.Nickel U, Castell AZ, Poppl K, Schneider S. 2000. A silver colloid produced by reduction with hydrazine as support for highly sensitive surface-enhanced Raman. Langmuir 16, 9087–9091. ( 10.1021/la000536y) [DOI] [Google Scholar]
- 28.Fievet F, Francoise F-V, Lagier JP, Dumont B, Figlarz M. 1993. Controlled nucleation and growth of micrometre-size copper particles prepared by the polyol process. J. Mater. Chem. 3, 627–632. ( 10.1039/jm9930300627) [DOI] [Google Scholar]
- 29.Leopold N, Lendl B. 2003. A new method for fast preparation of highly surface-enhanced Raman scattering (SERS) active silver colloids at room temperature by reduction of silver nitrate with hydroxylamine hydrochloride. J. Phys. Chem. B 107, 5723–5727. ( 10.1021/jp027460u) [DOI] [Google Scholar]
- 30.Zhang Y, Peng H, Huang W, Zhou Y, Zhang X, Yan D. 2008. Hyperbranched poly (amidoamine) as the stabilizer and reductant to prepare colloid silver nanoparticles in situ and their antibacterial activity. J. Phys. Chem. C 112, 2330–2336. ( 10.1021/jp075436g) [DOI] [Google Scholar]
- 31.Yin Ye, Li Z-Y, Zhong Z, Gates B, Xia Y, Venkateswaran S. 2002. Synthesis and characterization of stable aqueous dispersions of silver nanoparticles through the Tollens process. J. Mater. Chem. 12, 522–527. ( 10.1039/b107469e) [DOI] [Google Scholar]
- 32.Liu Y-C, Lin L-H. 2004. New pathway for the synthesis of ultrafine silver nanoparticles from bulk silver substrates in aqueous solutions by sonoelectrochemical methods. Electrochem. Commun. 6, 1163–1168. ( 10.1016/j.elecom.2004.09.010) [DOI] [Google Scholar]
- 33.Raveendran P, Fu J, Wallen SL. 2003. Completely ‘green’ synthesis and stabilization of metal nanoparticles. J. Am. Chem. Soc. 125, 13 940–13 941. ( 10.1021/ja029267j) [DOI] [PubMed] [Google Scholar]
- 34.Pal S, Tak YK, Song JM. 2007. Does the antibacterial activity of silver nanoparticles depend on the shape of the nanoparticle? A study of the Gram-negative bacterium Escherichia coli. Appl. Environ. Microbiol. 73, 1712–1720. ( 10.1128/AEM.02218-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cobley CM, Rycenga M, Zhou F, Li Z-Y, Xia Y. 2009. Etching and growth: an intertwined pathway to silver nanocrystals with exotic shapes. Angew. Chem. Int. Ed. 48, 4824–4827. ( 10.1002/anie.200901447) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma VK, Yngard RA, Lin Y. 2009. Silver nanoparticles: green synthesis and their antimicrobial activities. Adv. Colloid Interface Sci. 145, 83–96. ( 10.1016/j.cis.2008.09.002) [DOI] [PubMed] [Google Scholar]
- 37.Abid J, Wark AW, Brevet PF, Girault HH. 2002. Preparation of silver nanoparticles in solution from a silver salt by laser irradiation. Chem. Commun. 2002, 792–793. ( 10.1039/b200272h) [DOI] [PubMed] [Google Scholar]
- 38.Troupis A, Hiskia A, Papaconstantinou E. 2002. Synthesis of metal nanoparticles by using polyoxometalates as photocatalysts and stabilizers. Angew. Chem. Int. Ed. 41, 1911–1914. () [DOI] [PubMed] [Google Scholar]
- 39.Xie J, Lee JY, Wang DIC, Ting YP. 2007. Silver nanoplates: from biological to biomimetic synthesis. ACS Nano 1, 429–439. ( 10.1021/nn7000883) [DOI] [PubMed] [Google Scholar]
- 40.Lee PC, Meisel D. 1982. Adsorption and surface-enhanced Raman of dyes on silver and gold sols. J. Phys. Chem. 86, 3391–3395. ( 10.1021/j100214a025) [DOI] [Google Scholar]
- 41.Pastoriza-Santos I, Liz-Marzán LM. 1999. Formation and stabilization of silver nanoparticles through reduction by N,N-dimethylformamide. Langmuir 15, 948–951. ( 10.1021/la980984u) [DOI] [Google Scholar]
- 42.Smetana AB, Klabunde KJ, Sorensen CM. 2005. Synthesis of spherical silver nanoparticles by digestive ripening, stabilization with various agents, and their 3-D and 2-D superlattice formation. J. Colloid Interface Sci. 284, 521–526. ( 10.1016/j.jcis.2004.10.038) [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Ong W, Kaifer AE, Peinador C. 2002. A ‘macrocyclic effect’ on the formation of capped silver nanoparticles in DMF. Langmuir 18, 5981–5983. ( 10.1021/la025956x) [DOI] [Google Scholar]
- 44.Hirai H, Nakao Y, Toshima N. 1979. Preparation of colloidal transition metals in polymers by reduction with alcohols or ethers. J. Macromol. Sci., Chem. 13, 727–750. [Google Scholar]
- 45.Luo Y, Sun X. 2007. Rapid, single-step preparation of dendrimer-protected silver nanoparticles through a microwave-based thermal process. Mater. Lett. 61, 1622–1624. ( 10.1016/j.matlet.2006.07.087) [DOI] [Google Scholar]
- 46.Navarro E, Piccapietra F, Wagner B, Marconi F, Kaegi R, Odzak N, Sigg L, Behra R. 2008. Toxicity of silver nanoparticles to Chlamydomonas reinhardtii. Environ. Sci. Technol. 42, 8959–8964. ( 10.1021/es801785m) [DOI] [PubMed] [Google Scholar]
- 47.Piccapietra F, Allue CG, Sigg L, Behra R. 2012. Intracellular silver accumulation in Chlamydomonas reinhardtii upon exposure to carbonate coated silver nanoparticles and silver nitrate. Environ. Sci. Technol. 46, 7390–7397. ( 10.1021/es300734m) [DOI] [PubMed] [Google Scholar]
- 48.Lowry GV, Gregory KB, Apte SC, Lead JR. 2012. Transformations of nanomaterials in the environment. Environ. Sci. Technol. 46, 6893–6899. ( 10.1021/es300839e) [DOI] [PubMed] [Google Scholar]
- 49.Tolaymat TM, El Badawy AM, Genaidy A, Scheckel KG, Luxton TP, Suidan M. 2010. An evidence-based environmental perspective of manufactured silver nanoparticle in syntheses and applications: a systematic review and critical appraisal of peer-reviewed scientific papers. Sci. Total Environ. 408, 999–1006. ( 10.1016/j.scitotenv.2009.11.003) [DOI] [PubMed] [Google Scholar]
- 50.Song JE, Phenrat T, Marinakos S, Xiao Y, Liu J, Wiesner MR, Tilton RD, Lowry GV. 2011. Hydrophobic interactions increase attachment of gum Arabic- and PVP-coated Ag nanoparticles to hydrophobic surfaces. Environ. Sci. Technol. 45, 5988–5995. ( 10.1021/es200547c) [DOI] [PubMed] [Google Scholar]
- 51.El Badawy AM, Luxton TP, Silva RG, Scheckel KG, Suidan MT, Tolaymat TM. 2010. Impact of environmental conditions (pH, ionic strength, and electrolyte type) on the surface charge and aggregation of silver nanoparticles suspensions. Environ. Sci. Technol. 44, 1260–1266. ( 10.1021/es902240k) [DOI] [PubMed] [Google Scholar]
- 52.Tejamaya M, Ramer I, Merrifield RC, Lead JR. 2012. Stability of citrate, PVP, and PEG coated silver nanoparticles in ecotoxicology media. Environ. Sci. Technol. 46, 7011–7017. ( 10.1021/es2038596) [DOI] [PubMed] [Google Scholar]
- 53.Huynh KA, Chen KL. 2011. Aggregation kinetics of citrate and polyvinylpyrrolidone coated silver nanoparticles in monovalent and divalent electrolyte solutions. Environ. Sci. Technol. 45, 5564–5571. ( 10.1021/es200157h) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Piccapietra F, Sigg L, Behra R. 2012. Colloidal stability of carbonate-coated silver nanoparticles in synthetic and natural freshwater. Environ. Sci. Technol. 46, 818–825. ( 10.1021/es202843h) [DOI] [PubMed] [Google Scholar]
- 55.OECD. 2006. OECD Guidelines for the testing of chemicals. Freshwater alga and cyanobacteria, growth inhibition test. Paris: OECD. [Google Scholar]
- 56.Burchardt AD, et al. 2012. Effects of silver nanoparticles in diatom Thalassiosira pseudonana and cyanobacterium Synechococcus sp. Environ. Sci. Technol. 46, 11 336–11 344. ( 10.1021/es300989e) [DOI] [PubMed] [Google Scholar]
- 57.Navarro E, et al. Submitted. Effects of different coatings of silver nanoparticles on silver bioavailability to algae. Environ. Sci. Technol. [Google Scholar]
- 58.Xiu ZM, Ma J, Alvarez PJJ. 2011. Differential effect of common ligands and molecular oxygen on antimicrobial activity of silver nanoparticles versus silver ions. Environ. Sci. Technol. 45, 9003–9008. ( 10.1021/es201918f) [DOI] [PubMed] [Google Scholar]
- 59.Liu J, Sonshine DA, Shervani S, Hurt RH. 2010. Controlled release of biologically active silver from nanosilver surfaces. ACS Nano 4, 6903–6913. ( 10.1021/nn102272n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gondikas AP, Morris A, Reinsch BC, Marinakos SM, Lowry GV, Hsu-Kim H. 2012. Cysteine-induced modifications of zero-valent silver nanomaterials: implications for particle surface chemistry, aggregation, dissolution, and silver speciation. Environ. Sci. Technol. 46, 7037–7045. ( 10.1021/es3001757) [DOI] [PubMed] [Google Scholar]
- 61.Bell RA, Kramer JR. 1999. Structural chemistry and geochemistry of silver-sulfur compounds: critical review. Environ. Toxicol. Chem. 18, 9–22. [Google Scholar]
- 62.Martinolich AJ, Park G, Nakamoto MY, Gate RE, Wheeler KE. 2012. Structural and functional effects of Cu metalloprotein-driven silver nanoparticle dissolution. Environ. Sci. Technol. 46, 6355–6362. ( 10.1021/es300901h) [DOI] [PubMed] [Google Scholar]
- 63.Liu J, et al. 2010. Controlled release of biologically active silver from nanosilver surfaces. ACS Nano 4, 6903–6913. ( 10.1021/nn102272n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marques MRC, Loebenberg R, Almukainzi M. 2011. Simulated biological fluids with possible application in dissolution testing. Dissolution Technol. 18, 15–28. [Google Scholar]
- 65.Bakke AM, Glover C, Krogdahl A. 2011. Feeding, digestion and absorption of nutrients In The multifunctional gut of fish (eds Grosell M, Farrell AP, Brauner CJ.), pp. 57–110. London, UK: Academic Press. [Google Scholar]
- 66.Herrin RT, Andren AW, Shafer MM, Armstrong DE. 2001. Determination of silver speciation in natural waters. II. Binding strength of silver ligands in surface freshwaters. Environ. Sci. Technol. 35, 1959–1966. ( 10.1021/es001510w) [DOI] [PubMed] [Google Scholar]
- 67.Reinfelder JR, Chang SI. 1999. Speciation and microalgal bioavailability of inorganic silver. Environ. Sci. Technol. 33, 1860–1863. ( 10.1021/es980896w) [DOI] [Google Scholar]
- 68.Lee DY, Fortin C, Campbell PGC. 2004. Influence of chloride on silver uptake by two green algae, Pseudokirchneriella subcapitata and Chlorella pyrenoidosa. Environ. Toxicol. Chem. 23, 1012–1018. ( 10.1897/03-145) [DOI] [PubMed] [Google Scholar]
- 69.Kaegi R, Voegelin A, Sinnet B, Zuleeg S, Hagendorfer H, Burkhardt M, Siegrist H. 2011. Behavior of metallic silver nanoparticles in a pilot wastewater treatment plant. Environ. Sci. Technol. 45, 3902–3908. ( 10.1021/es1041892) [DOI] [PubMed] [Google Scholar]
- 70.Ingvorsen K, Nielsen MY, Joulian C. 2003. Kinetics of bacterial sulfate reduction in an activated sludge plant FEMS. Microbiol. Ecol. 46, 129–137. ( 10.1016/S0168-6496(03)00209-5) [DOI] [PubMed] [Google Scholar]
- 71.Mylon SE, Benoit G. 2001. Subnanomolar detection of acid-labile sulfides by the classical methylene blue method coupled to HPLC. Environ. Sci. Technol. 35, 4544–4548. ( 10.1021/es010955x) [DOI] [PubMed] [Google Scholar]
- 72.Reinsch BC, Levard C, Li Z, Ma R, Wise A, Gregory KB, Brown GE, Lowry GV. 2012. Sulfidation of silver nanoparticles decreases Escherichia coli growth inhibition. Environ. Sci. Technol. 46, 6992–7000. ( 10.1021/es203732x) [DOI] [PubMed] [Google Scholar]
- 73.Hsu-Kim H. 2007. Stability of metal−glutathione complexes during oxidation by hydrogen peroxide and Cu(II)-catalysis. Environ. Sci. Technol. 41, 2338–2342. ( 10.1021/es062269+) [DOI] [PubMed] [Google Scholar]
- 74.Myshkin AE, Khromova VS. 2003. Peculiar features of the aggregation effect of silver(I) ion on hemoglobin. Biochim. Biophys. Acta-Proteins Proteomics 1651, 124–129. ( 10.1016/S1570-9639(03)00243-7) [DOI] [PubMed] [Google Scholar]
- 75.Kihlken MA, Singleton C, Le Brun NE. 2008. Distinct characteristics of Ag+ and Cd2+ binding to CopZ from Bacillus subtilis. J. Biol. Inorg. Chem. 13, 1011–1023. ( 10.1007/s00775-008-0388-1) [DOI] [PubMed] [Google Scholar]
- 76.Yang X, et al. 2012. Mechanism of silver nanoparticle toxicity is dependent on dissolved silver and surface coating in Caenorhabditis elegans. Environ. Sci. Technol. 46, 1119–1127. ( 10.1021/es202417t) [DOI] [PubMed] [Google Scholar]
- 77.Chen ZZ, Campbell PGC, Fortin C. 2012. Silver binding by humic acid as determined by equilibrium ion-exchange and dialysis. J. Phys. Chem. A 116, 6532–6539. ( 10.1021/jp212403r) [DOI] [PubMed] [Google Scholar]
- 78.Grosell M, Hogstrand C, Wood CM, Hansen HJM. 2000. A nose-to-nose comparison of the physiological effects of exposure to ionic silver versus silver chloride in the European eel (Anguilla anguilla) and the rainbow trout (Oncorhynchus mykiss). Aquat Toxicol 48, 327–342. ( 10.1016/S0166-445X(99)00029-6) [DOI] [PubMed] [Google Scholar]
- 79.Hogstrand C, Galvez F, Wood CM. 1996. Toxicity, silver accumulation and metallothionein induction in freshwater rainbow trout during exposure to different silver salts. Environ. Toxicol. Chem. 15, 1102–1108. ( 10.1002/etc.5620150713) [DOI] [Google Scholar]
- 80.McGeer JC, Wood CM. 1998. Protective effects of water Cl− on physiological responses to waterborne silver in rainbow trout. Can. J. Fish Aquat. Sci. 55, 2447–2454. ( 10.1139/f98-133) [DOI] [Google Scholar]
- 81.Monopoli MP, Aberg C, Salvati A, Dawson KA. 2012. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 7, 779–786. ( 10.1038/nnano.2012.207) [DOI] [PubMed] [Google Scholar]
- 82.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. 2009. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 8, 543–557. ( 10.1038/nmat2442) [DOI] [PubMed] [Google Scholar]
- 83.Smith AM. 2002. The structure and function of adhesive gels from invertebrates. Integr. Comp. Biol. 42, 1164–1171. ( 10.1093/icb/42.6.1164) [DOI] [PubMed] [Google Scholar]
- 84.Wotton RS. 2004. The ubiquity and many roles of exopolymers (EPS) in aquatic systems. Scientia Marina 68, 13–21. ( 10.3989/scimar.2004.68s113) [DOI] [Google Scholar]
- 85.Esteban MA. 2012. An overview of the immunological defenses in fish skin. ISRN Immunology. 2012, 853470 ( 10.5402/2012/853470) [DOI] [Google Scholar]
- 86.Stewart TJ, et al. 2012. Characterization of extracellular polymeric substances (EPS) from periphyton using liquid chromatography-organic carbon detection–organic nitrogen detection (LC-OCD-OND). Environ. Sci. Pollut. Res. Int. 20, 3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nagashima Y, Kikuchi N, Shimakura K, Shiomi K. 2003. Purification and characterization of an antibacterial protein in the skin secretion of rockfish Sebastes schlegeli. Comp. Biochem. Physiol. C-Toxicol. Pharmacol. 136, 63–71. ( 10.1016/S1532-0456(03)00174-1) [DOI] [PubMed] [Google Scholar]
- 88.Handy RD, Henry TB, Scown TM, Johnston BD, Tyler CR. 2008. Manufactured nanoparticles: their uptake and effects on fish—a mechanistic analysis. Ecotoxicology 17, 396–409. ( 10.1007/s10646-008-0205-1) [DOI] [PubMed] [Google Scholar]
- 89.Peulen TO, Wilkinson KJ. 2011. Diffusion of nanoparticles in a biofilm. Environ. Sci. Technol. 45, 3367–3373. ( 10.1021/es103450g) [DOI] [PubMed] [Google Scholar]
- 90.Federici G, Shaw BJ, Handy RD. 2007. Toxicity of titanium dioxide nanoparticles to rainbow trout, (Oncorhynchus mykiss): gill injury, oxidative stress, and other physiological effects. Aquat. Toxicol. 84, 415–430. ( 10.1016/j.aquatox.2007.07.009) [DOI] [PubMed] [Google Scholar]
- 91.Smith CJ, Shaw BJ, Handy RD. 2007. Toxicity of single walled carbon nanotubes to rainbow trout, (Oncorhynchus mykiss): respiratory toxicity, organ pathologies, and other physiological effects. Aquat. Toxicol. 82, 94–109. ( 10.1016/j.aquatox.2007.02.003) [DOI] [PubMed] [Google Scholar]
- 92.Bilberg K, Malte H, Wang T, Baatrup E. 2010. Silver nanoparticles and silver nitrate cause respiratory stress in Eurasian perch (Perca fluviatilis). Aquat. Toxicol. 96, 159–165. ( 10.1016/j.aquatox.2009.10.019) [DOI] [PubMed] [Google Scholar]
- 93.Jepson MA, Clark MA, Hirst BH. 2004. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv. Drug Delivery Rev. 56, 511–525. ( 10.1016/j.addr.2003.10.018) [DOI] [PubMed] [Google Scholar]
- 94.Yang T, Li Z, Wang L, Guo C, Sun Y. 2007. Synthesis, characterization, and self-assembly of protein lysozyme monolayer-stabilized gold nanoparticles. Langmuir 23, 10 533–10 538. ( 10.1021/la701649z) [DOI] [PubMed] [Google Scholar]
- 95.Walczak APEA. 2012. Behaviour of silver nanoparticles and silver ions in an in vitro human gastrointestinal digestion model. Nanotoxicology 1–13. ( 10.3109/17435390.2012.726382) [DOI] [PubMed] [Google Scholar]
- 96.Kirch J, Schneider A, Abou B, Hopf A, Schaefer UF, Schneider M, Schall C, Wagner C, Lehr C-M. 2012. Optical tweezers reveal relationship between microstructure and nanoparticle penetration of pulmonary mucus. Proc. Natl Acad. Sci. USA 109, 18 355–18 360. ( 10.1073/pnas.1214066109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu J, Wang Z, Liu FD, Kane AB, Hurt RH. 2012. Chemical transformations of nanosilver in biological environments. ACS Nano 6, 9887–9899. ( 10.1021/nn303449n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tennant J, Sharp PA. 2004. Mechanisms involved in copper uptake by human intestinal epithelial cells. Proc. Nutr. Soc. 63, 35A. [Google Scholar]
- 99.Muthusamy B, et al. 2005. Plasma Proteome Database as a resource for proteomics research. Proteomics 5, 3531–3536. ( 10.1002/pmic.200401335) [DOI] [PubMed] [Google Scholar]
- 100.Dziendzikowska K, et al. 2012. Time-dependent biodistribution and excretion of silver nanoparticles in male Wistar rats. J. Appl. Toxicol. 32, 920–928. ( 10.1002/jat.2758) [DOI] [PubMed] [Google Scholar]
- 101.Park K, Park E-J, Chun IK, Choi K, Lee SH, Yoon J, Lee BC. 2011. Bioavailability and toxicokinetics of citrate-coated silver nanoparticles in rats. Arch Pharm. Res. 34, 153–158. ( 10.1007/s12272-011-0118-z) [DOI] [PubMed] [Google Scholar]
- 102.Nevitt T, Ohrvik H, Thiele DJ. 2012. Charting the travels of copper in eukaryotes from yeast to mammals. Biochim. Biophys. Acta 1823, 1580–1593. ( 10.1016/j.bbamcr.2012.02.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee J, et al. 2002. Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 277, 4380–4387. ( 10.1074/jbc.M104728200) [DOI] [PubMed] [Google Scholar]
- 104.Bertinato J, Cheung L, Hoque R, Plouffe LJ. 2010. Ctr1 transports silver into mammalian cells. J. Trace Elem. Med. Biol. 24, 178–184. ( 10.1016/j.jtemb.2010.01.009) [DOI] [PubMed] [Google Scholar]
- 105.Solioz M, Odermatt A. 1995. Copper and silver transport by CopB–ATPase in membrane vesicles of Enterococcus hirae. J. Biol. Chem. 270, 9217–9221. ( 10.1074/jbc.270.16.9217) [DOI] [PubMed] [Google Scholar]
- 106.Andrés-Colás N, Perea-Garcia A, Puig S, Penarrubia L. 2010. Deregulated copper transport affects Arabidopsis development especially in the absence of environmental cycles. Plant Physiol. 153, 170–184. ( 10.1104/pp.110.153676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bury NR, Grosell M, Grover AK, Wood CM. 1999. ATP-dependent silver transport across the basolateral membrane of rainbow trout gills. Toxicol. Appl. Pharmacol. 159, 1–8. ( 10.1006/taap.1999.8706) [DOI] [PubMed] [Google Scholar]
- 108.Zimnicka AM, Maryon EB, Kaplan JH. 2007. Human copper transporter hCTR1 mediates basolateral uptake of copper into enterocytes: implications for copper homeostasis. J. Biol. Chem. 282, 26 471–26 480. ( 10.1074/jbc.M702653200) [DOI] [PubMed] [Google Scholar]
- 109.Kidane TZ, Farhad R, Lee KJ, Santos A, Russo E, Linder MC. 2012. Uptake of copper from plasma proteins in cells where expression of CTR1 has been modulated. Biometals 25, 697–709. ( 10.1007/s10534-012-9528-8) [DOI] [PubMed] [Google Scholar]
- 110.Hassett R, Kosman DJ. 1995. Evidence for Cu (II) reduction as a component of copper uptake by Saccharomyces cerevisiae. J. Biol. Chem. 270, 128–134. ( 10.1074/jbc.270.1.128) [DOI] [PubMed] [Google Scholar]
- 111.Wyman S, Simpson RJ, McKie AT, Sharp PA. 2008. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 582, 1901–1906. ( 10.1016/j.febslet.2008.05.010) [DOI] [PubMed] [Google Scholar]
- 112.Ohgami RS, et al. 2006. The Steap proteins are metalloreductases. Blood 108, 1388–1394. ( 10.1182/blood-2006-02-003681) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee J, Petris MJ, Thiele DJ. 2002. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter. identification of a Ctr1-indipendent copper transport system. J. Biol. Chem. 277, 40 253–40 259. ( 10.1074/jbc.M208002200) [DOI] [PubMed] [Google Scholar]
- 114.Tandy S, et al. 2000. Nramp2 expression is associated with pH-dependent iron uptake across the apical membrane of human intestinal Caco-2 cells. J. Biol. Chem. 275, 1023–1029. ( 10.1074/jbc.275.2.1023) [DOI] [PubMed] [Google Scholar]
- 115.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. 1997. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488. ( 10.1038/41343) [DOI] [PubMed] [Google Scholar]
- 116.Arredondo M, Munoz P, Mura CV, Nunez MT. 2003. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am. J. Physiol. Cell Physiol. 284, C1525–C1530. ( 10.1152/ajpcell.00480.2002) [DOI] [PubMed] [Google Scholar]
- 117.Handy RD, Eddy FB, Baines H. 1566. Sodium-dependent copper uptake across epithelia: a review of rationale with experimental evidence from gill and intestine. Biochim. Biophys. Acta 1566, 104–115. ( 10.1016/S0005-2736(02)00590-4) [DOI] [PubMed] [Google Scholar]
- 118.Bury NR, Wood CM. 1999. Mechanism of branchial apical silver uptake by rainbow trout is via the proton-coupled Na+ channel. Am. J. Physiol. Regul. Integr. Comp. Physiol. 277, R1385–R1391. [DOI] [PubMed] [Google Scholar]
- 119.Grosell M, Nielsen C, Bianchini A. 2002. Sodium turnover rate determines sensitivity to acute copper and silver exposure in freshwater animals. Comp. Biochem. Physiol Part C: Toxicol. Pharmacol. 133, 287–303. ( 10.1016/S1532-0456(02)00085-6) [DOI] [PubMed] [Google Scholar]
- 120.Schultz AG, Ong KJ, MacCormack T, Ma G, Veinot JGC, Goss GG. 2012. Silver nanoparticles inhibit sodium uptake in juvenile rainbow trout (Oncorhynchus mykiss). Environ. Sci. Technol. 46, 10 295–10 301. ( 10.1021/es3017717) [DOI] [PubMed] [Google Scholar]
- 121.Minghetti M, Leaver MJ, Carpen E, George SG. 2008. Copper transporter 1, metallothionein and glutathione reductase genes are differentially expressed in tissues of sea bream (Sparus aurata) after exposure to dietary or waterborne copper. Comp. Biochem. Physiol Part C: Toxicol. Pharmacol. 147, 450–459. ( 10.1016/j.cbpc.2008.01.014) [DOI] [PubMed] [Google Scholar]
- 122.Mackenzie NC, Brito M, Reyes AE, Allende ML. 2004. Cloning, expression pattern and essentiality of the high-affinity copper transporter 1 (ctr1) gene in zebrafish. Gene 328, 113–120. ( 10.1016/j.gene.2003.11.019) [DOI] [PubMed] [Google Scholar]
- 123.Minghetti M, Leaver MJ, George SG. 2010. Multiple Cu ATPase genes are differentially expressed and transcriptionally regulated by Cu exposure in Sea bream, Sparus aurata. Aquat. Toxicol. 97, 23–33. ( 10.1016/j.aquatox.2009.11.017) [DOI] [PubMed] [Google Scholar]
- 124.Handy RD, Musonda MM, Phillips C, Falla SJ. 2000. Mechanisms of gastrointestinal copper absorption in the African walking catfish: copper dose-effects and a novel anion-dependent pathway in the intestine. J. Exp. Biol. 203, 2365–2377. [DOI] [PubMed] [Google Scholar]
- 125.Aller SG, Unger VM. 2006. Projection structure of the human copper transporter CTR1 at 6-A resolution reveals a compact trimer with a novel channel-like architecture. Proc. Natl Acad. Sci. USA 103, 3627–3632. ( 10.1073/pnas.0509929103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Muhlfeld C, Gehr P, Rothen-Rutishauser B. 2008. Translocation and cellular entering mechanisms of nanoparticles in the respiratory tract. Swiss Med. Wkly 138, 387–391. [DOI] [PubMed] [Google Scholar]
- 127.Scown TM, et al. 2010. Effects of aqueous exposure to silver nanoparticles of different sizes in rainbow trout. Toxicol. Sci. Off. J. Soc. Toxicol. 115, 521–534. ( 10.1093/toxsci/kfq076) [DOI] [PubMed] [Google Scholar]
- 128.Farkas J, Christian P, Gallego-Urrea JA, Roos N, Hassellav M, Tollefsen KE, Thomas KV. 2011. Uptake and effects of manufactured silver nanoparticles in rainbow trout (Oncorhynchus mykiss) gill cells. Aquat. Toxicol. 101, 117–125. ( 10.1016/j.aquatox.2010.09.010) [DOI] [PubMed] [Google Scholar]
- 129.Wu Y, Zhou Q. 2013. Silver nanoparticles cause oxidative damage and histological changes in medaka (Oryzias latipes) after 14 days of exposure. Environ. Toxicol. Chem. 32, 165–173. ( 10.1002/etc.2038) [DOI] [PubMed] [Google Scholar]
- 130.Kwok KWH, Auffan M, Badireddy AR, Nelson CM, Wiesner MR, Chilkoti A, Liu J, Marinakos SM, Hinton DE. 2012. Uptake of silver nanoparticles and toxicity to early life stages of Japanese medaka (Oryzias latipes): effect of coating materials. Aquat. Toxicol. 120–121, 59–66. ( 10.1016/j.aquatox.2012.04.012) [DOI] [PubMed] [Google Scholar]
- 131.Xiu ZM, Zhang Q-B, Puppala HL, Colvin VL, Alvarez PJJ. 2012. Negligible particle-specific antibacterial activity of silver nanoparticles. Nano Lett. 12, 4271–4275. ( 10.1021/nl301934w) [DOI] [PubMed] [Google Scholar]
- 132.Griffitt RJ, Hyndman K, Denslow ND, Barber DS. 2009. Comparison of molecular and histological changes in zebrafish gills exposed to metallic nanoparticles. Toxicol. Sci. 107, 404–415. ( 10.1093/toxsci/kfn256) [DOI] [PubMed] [Google Scholar]
- 133.Gagné F, Andre C, Skirrow R, Gelinas M, Auclair J, van Aggelen G, Turcotte P, Gagnon C. 2012. Toxicity of silver nanoparticles to rainbow trout: a toxicogenomic approach. Chemosphere 89, 615–622. ( 10.1016/j.chemosphere.2012.05.063) [DOI] [PubMed] [Google Scholar]
- 134.Lee KJ, Nallathamby PD, Browning LM, Osgood CJ, Xu X-HN. 2007. In vivo imaging of transport and biocompatibility of single silver nanoparticles in early development of zebrafish embryos. ACS Nano 1, 133–143. ( 10.1021/nn700048y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Meyer JN, Lord CA, Yang XY, Turner EA, Badireddy AR, Marinakos SM, Chilkoti A, Wiesner MR, Auffan M. 2010. Intracellular uptake and associated toxicity of silver nanoparticles in Caenorhabditis elegans. Aquat. Toxicol. 100, 140–150. ( 10.1016/j.aquatox.2010.07.016) [DOI] [PubMed] [Google Scholar]
- 136.García-Alonso J, Khan FR, Misra SK, Turmaine M, Smith BD, Rainbow PS, Luoma SN, Valsami-Jones E. 2011. Cellular internalization of silver nanoparticles in gut epithelia of the estuarine polychaete Nereis diversicolor. Environ. Sci. Technol. 45, 4630–4636. ( 10.1021/es2005122) [DOI] [PubMed] [Google Scholar]
- 137.Miao AJ, Luo Z, Chen W-C, Santschi PH, Quigg A. 2010. Intracellular uptake: a possible mechanism for silver engineered nanoparticle toxicity to a freshwater alga Ochromonas danica. PLoS ONE 5, e15196 ( 10.1371/journal.pone.0015196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Simkiss K, Taylor M, Mason AZ. 1982. Metal detoxification and bioaccumulation in mollusks. Mar. Biol. Lett. 3, 187–201. [Google Scholar]
- 139.Brandenberger C, Clift MJD, Vanhecke D, Mahlfeld C, Stone V, Gehr P, Rothen-Rutishauser B. 2010. Intracellular imaging of nanoparticles: is it an elemental mistake to believe what you see? Part. Fibre Toxicol. 7 ( 10.1186/1743-8977-7-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Croteau M-N, Misra SK, Luoma SN, Valsami-Jones E. 2011. Silver bioaccumulation dynamics in a freshwater invertebrate after aqueous and dietary exposures to nanosized and ionic Ag. Environ. Sci. Technol. 45, 6600–6607. ( 10.1021/es200880c) [DOI] [PubMed] [Google Scholar]
- 141.Khan FR, Misra SK, Garcea-Alonso J, Smith BD, Strekopytov S, Rainbow PS, Luoma SN, Valsami-Jones E. 2012. Bioaccumulation dynamics and modeling in an estuarine invertebrate following aqueous exposure to nanosized and dissolved silver. Environ. Sci. Technol. 46, 7621–7628. ( 10.1021/es301253s) [DOI] [PubMed] [Google Scholar]
- 142.Handy R, et al. 2012. Practical considerations for conducting ecotoxicity test methods with manufactured nanomaterials: what have we learnt so far? Ecotoxicology 21, 933–972. ( 10.1007/s10646-012-0862-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wijnhoven SWP, et al. 2009. Nano-silver—a review of available data and knowledge gaps in human and environmental risk assessment. Nanotoxicology 3, 109–138. [Google Scholar]
- 144.Clift MJD, Gehr P, Rothen-Rutishauser B. 2011. Nanotoxicology: a perspective and discussion of whether or not in vitro testing is a valid alternative. Arch. Toxicol. 85, 723–731. ( 10.1007/s00204-010-0560-6) [DOI] [PubMed] [Google Scholar]
- 145.Chamakura K, Perez-Ballestero R, Luo Z, Bashir S, Liu J. 2011. Comparison of bactericidal activities of silver nanoparticles with common chemical disinfectants. Colloids Surf. B Biointerfaces 84, 88–96. ( 10.1016/j.colsurfb.2010.12.020) [DOI] [PubMed] [Google Scholar]
- 146.Median-Ramirez I, Bashir S, Luo Z, Liu JL. 2009. Green synthesis and characterization of polymer-stabilized silver nanoparticles. Colloids Surf. B Biointerfaces 73, 185–191. ( 10.1016/j.colsurfb.2009.05.015) [DOI] [PubMed] [Google Scholar]
- 147.Dror-Ehre A, Mamame H, Belenkova T, Markovich G, Adin A. 2009. Silver nanoparticle–E. coli colloidal interaction in water and effect on E. coli survival. J. Colloid Interface Sci. 339, 521–526. ( 10.1016/j.jcis.2009.07.052) [DOI] [PubMed] [Google Scholar]
- 148.Maysinger D, Lovric J, Eisenberg A, Savic R. 2007. Fate of micelles and quantum dots in cells. Eur. J. Pharm. Biopharm. 65, 270–281. ( 10.1016/j.ejpb.2006.08.011) [DOI] [PubMed] [Google Scholar]
- 149.Yuan Z, Li J, Cui L, Zu B, Zhang H, Yu C-P. 2013. Interaction of silver nanoparticles with pure nitrifying bacteria. Chemosphere 90, 1404–1411. ( 10.1016/j.chemosphere.2012.08.032) [DOI] [PubMed] [Google Scholar]
- 150.Mirzajani F, Aliahmadi A, Esmaeili MA. 2011. Antibacterial effect of silver nanoparticles of Staphylococcus aureus. Res. Microbiol. 162, 542–549. ( 10.1016/j.resmic.2011.04.009) [DOI] [PubMed] [Google Scholar]
- 151.McQuillan JS, Infante HG, Stokes E, Shaw AM. 2012. Silver nanoparticle enhanced silver ion stress response in Escherichia coli K12. Nanotoxicology 6, 857–866. ( 10.3109/17435390.2011.626532) [DOI] [PubMed] [Google Scholar]
- 152.Coutris C, Hertel-Aas T, Lapied E, Joner EJ, Oughton DH. 2012. Bioavailability of cobalt and silver nanoparticles to the earthworm Eisenia fetida. Nanotoxicology 6, 186–195. ( 10.3109/17435390.2011.569094) [DOI] [PubMed] [Google Scholar]
- 153.Kruszewski M, et al. 2011. Toxicity of silver nanomaterials in higher eukaryotes. In Advances in Molecular Toxicology (ed. Fishbein J.), vol. 5, pp. 179–218. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 154.van der Zande M, et al. 2012. Distribution, elimination, and toxicity of silver nanoparticles and silver ions in rats after 28-day oral exposure. ACS Nano 6, 7427–7442. ( 10.1021/nn302649p) [DOI] [PubMed] [Google Scholar]
- 155.Lee YEA. 2012. Serum kinetics, distribution and excretion of silver in rabbits following 28 days after a single intravenous injection of silver nanoparticles. Nanotoxicology 1–11. ( 10.3109/17435390.2012.710660) [DOI] [PubMed] [Google Scholar]
- 156.Herzog F, Clift MJD, Piccapietra F, Behra R, Schmid O, Petri-Fink A, Rothen-Rutishauser B. 2013. Exposure of silver-nanoparticles and silver-ions to lung cells in vitro at the air–liquid interface. Part Fibre Toxicol. 10, 11 ( 10.1186/1743-8977-10-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chernousova S, Epple M. 2013. Silver as antibacterial agent: ion, nanoparticle, and metal. Angew. Chem. Int. Ed. Engl. 52, 1636–1653. ( 10.1002/anie.201205923) [DOI] [PubMed] [Google Scholar]
- 158.Lansdown AB. 2006. Silver in health care: antimicrobial effects and safety in use. Curr. Probl. Dermatol. 33, 17–34. ( 10.1159/000093928) [DOI] [PubMed] [Google Scholar]