

Abstract
Hypoxia-inducible factor-1α (HIF-1α) has been reported to regulate over 100 gene expressions in response to hypoxia and other stress conditions. In the present study, we found that arsenite could induce HIF-1α protein accumulation in both mouse epidermal Cl41 cells and mouse embryonic fibroblasts (MEFs). Knockout of p85α, a regulatory subunit of PI-3K, in MEFs (p85α−/−) dramatically decreased the arsenite-induced HIF-1α accumulation, indicating that p85α is crucial for arsenite effects on the stabilization of HIF-1α protein. Our further studies suggest that arsenite could induce inducible Hsp70 expression, and transfection of inducible Hsp70 into p85α−/− MEFs could restore HIF-1α protein accumulation. Moreover, the results using EMSA and Supershift assays indicate that p85α is crucial for arsenite-induced activation of the heat-shock transcription factor 1 (HSF-1), which is responsible for transcription of inducible Hsp70. Taken together, p85α-mediated HIF-1α stabilization upon arsenite exposure is specifically through HSF-1 activation and subsequent up-regulation of the inducible Hsp70 expression.
Keywords: Hsp70, HIF-1α, p85α, Akt, Arsenite
Introduction
Hypoxia-inducible factor-1 (HIF-1) is an important transcription factor that is responsible for regulating over 100 genes related to cellular growth and migration upon hypoxia and other stresses [1–3]. The hypoxic condition is reported to be involved in the pathogenesis of many diseases, including cancers [4, 5]. The increased HIF-1 expression in cancer cells is associated with resistance to radiotherapy and chemotherapy as well as poor prognosis [6, 7]. The involvement of HIF-1 in pathophysiological conditions and its value as a therapeutic target have attracted considerable attention to obtain the understanding of HIF-1 regulation.
The HIF-1 transcriptional unit is a heterodimer that is composed of the regulatory HIF-1α subunit and the constitutively expressed HIF-1β subunit. The biological function of HIF-1 is largely dependent on the expression of HIF-1α. HIF-1α protein is maintained at low levels under normoxia due to continuous degradation via the 26S proteasome pathway [8], whereas the protein levels increase rapidly in response to hypoxia and other stress conditions. The regulation of HIF-1α has mainly been addressed at translational and post-translational levels (reviewed in [9]).
Class IA phosphatidylinositol 3-kinase (PI-3K) is a central component for transducing signals essential for multiple cellular processes, including cell proliferation, differentiation, motility, and survival [10]. PI-3K is a heterodimer consisting of a 110-kDa catalytic subunit (p110) and a regulatory subunit, p85. Among several isoforms of PI-3K, p110/p85α is predominantly expressed in most tissues and is thought to be the major element responsible for cellular response to most stimuli [11, 12]. In addition to forming a complex with the p110 catalytic subunit, p85α also exists in a monomeric form due to the greater abundance of p85α than p110 in many cell types [12]. In our previous study, we have demonstrated that p85α plays an important role in cellular apoptotic response due to UV radiation in a PI-3K independent manner [13]. It has also been reported that monomeric p85α is involved in activation of several signal pathways [14, 15]. Although a few previous studies have reported that PI-3K/Akt pathway is involved in the regulation of HIF-1α in some experimental systems, there are contradictory results in some other experimental systems [16–19]. Currently, the detailed molecular mechanisms linking PI-3K and its major component p85α to HIF-1α expression remain unclear.
Arsenic is a well-recognized human carcinogen [20] that is distributed ubiquitously in soil and water [21]. Arsenic exposure is related to an increased risk for many human cancers, including lung, bladder, and skin cancers [22]. Unlike other carcinogens, arsenic itself does not induce significant DNA damage or gene mutation [23]. Instead, arsenic exposure promotes carcinogenesis by activating several signaling pathways, which lead to trans-activation of transcription factors and their downstream genes [23–25]. Although HIF-1α has been reported to be activated by arsenite in both in vivo and in vitro [26–28], the detailed molecular mechanisms leading to HIF-1α expression and activation due to arsenite exposure are not well understood at this time.
In the current study, we found that arsenite exposure induced HIF-1α protein accumulation in both mouse epidermal Cl41 cells and mouse embryonic fibroblasts (MEFs). Through the introduction of p85α−/− MEFs and the corresponding wild-type p85α+/+ MEFs into our experimental system, we found that p85α was essential in this process. Unlike its role in UV-induced cellular apoptotic response, p85α exerted its effect on HIF-1α protein accumulation by transcriptionally increasing inducible Hsp70 expression through a PI-3K/Akt/HSF-1-dependent pathway in cellular response to arsenite exposure.
Materials and methods
Plasmids, antibodies, and other reagents
pcDNA3-hsp70 plasmid was kindly provided by Dr. Hector Wong from the Children’s Hospital Medical Center, Cincinnati, OH, USA [29]. Hsp70-luciferase reporter was a gift from Dr. Alice Liu (Rutgers State University of New Jersey) [30]. Inducible hsp70 shRNAs were bought from Open Biosystems (Huntsville, AL, USA) with the hairpin sequence: (1) ccg ggc tga cga aga tga agg aga tct cga gat ctc ctt cat ctt cgt cag ctt ttt, and (2) ggg aac ccg cag aac acc gtg ttc tcg aga aca cgg tgt tct gcg ggt tct tttt. MG132 and CHX were purchased from Calbiochem (San Diego, CA, USA), and Wortmannin was bought from Sigma (St. Louis, MO, USA). Antibodies specific against Hsp70, inducible Hsp70, p-Akt473, p-Akt-308, and Akt were bought from Cell Signaling (Beverly, MA, USA), antibodies against HIF-1α were purchased from Bethyl (Montgomery, TX, USA), anti-HSF-1 and anti-Hsp90 were purchased from Stressgene (Ann Arbor, MI, USA), anti-β-actin and anti-myc were purchased from Sigma, anti-Lamin B was bought from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and anti-GAPDH was purchased from Abcam (Cambridge, MA, USA). The luciferase assay substrate was purchased from Promega (Madison, WI, USA).
Cell culture and transfection
p85α−/− mouse embryo fibroblasts (MEF cells) and its corresponding p85α+/+ MEFs were described in our previous study [13] and cultured in 37°C with 5% CO2 using DMEM containing 10% FBS, 1% penicillin/streptomycin, and 2 mM l-glutamine (Life Technologies). Mouse epidermal cell line Cl41 cells and their stable transfectants, Cl41 ∆p85α mass1 and Cl41 DN-Akt T308A/S473A [31], were cultured with Eagle’s MEM with 5% FBS, 2 mM l-glutamine, and 25 μg/ml gentamicin. HSF1−/− and HSF1+/+ MEF cells were kindly provided by Dr. Hector R. Wong (Children’s Hospital Medical Center, Cincinnati, OH, USA), and were cultured as previously described [32]. Cell transfections were conducted with FuGENE® HD (Roche Applied Science) following the manufacturer’s instructions. For stable transfection, cells were co-transfected with pSUPERIOR.puro (Invitrogen, Carlsbad, CA, USA), and subjected to selection with puromycin (Alexis, Plymouth, PA, USA) at a concentration of 2–4 μg/ml. After selection for 4–6 weeks, the surviving cells were cultured in normal medium for the ensuing experiments.
RT-PCR
Cells were treated with arsenite (20 μM) and MG132 (10 μM) for the indicated time. Total RNA was extracted using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. First-strand cDNA was synthesized with Oligo (dT)20 primers using SuperScript™ III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA), and the total RNA quantities used to perform reverse transcription were 1 μg. Specific primer pairs were designed for amplifying murine hsp70 (forward: 5′-cga cct gaa caa gag cat ca-3′, backward: 5′-atg acc tcc tgg cac ttg tc-3′), hsp90 (forward: 5′-gtg tgc aac agc tga agg aa-3′, backward: 5′-aca gca gca ctg gtg tca tc-3′), hif1-α (forward: 5′-tcc atg tga cca tga gga aa-3′, backward: 5′-tgg caa gca tcc tgt act gt-3′), β-Actin (forward: 5′-gac gat gat att gcc gca ct-3′, backward: 5′-gat acc acg ctt gct ctg ag-3′), respectively. The quantities of cDNA templates used for specific amplifications were 50 ng.
Quantitative RT-PCR
The same cDNAs that were used for the above RT-PCR were also analyzed for real-time PCR using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Specific primers pairs were designed for amplifying hif1-α (forward: 5′-gaa gac aac gcg ggc acc ga-3′, backward: 5′-tgc ttc gcc gag atc ttg ctg c-3′), β-Actin (forward: 5′-cct gtg gca tcc atg aaa ct-3′, backward: 5′-gtg cta gga gcc aga gca gt-3′), respectively. The real-time PCR was conducted following the protocol for Fast SYBR Green Master Mix kit (Applied Biosystems). In detail, an initial activation was performed at 95°C for 20 s, followed by 40 cycles of denaturation at 95°C for 1 s, and annealing and extension at 60°C for 20 s. The relative transcription levels were obtained by exponentially transforming ΔC
T values to 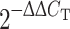 and the mean values were calculated with one standard deviation.
and the mean values were calculated with one standard deviation.
Western blotting
Whole-cell extracts were prepared with the cell lysis buffer (10 mM Tris–HCl, pH 7.4, 1% SDS, and 1 mM Na3VO4). Cytoplasmic and nuclear proteins were prepared with the Nuclear/Cytosol Fractionation Kit (BioVision, Mountain View, CA, USA) following the manufacturer’s protocols. Protein concentrations were determined either by the protein quantification assay kit (Bio-Rad Laboratories) or by Nano Drop® 1000 (Thermo Scientific). Proteins with the quantity 30–60 μg were resolved by SDS-PAGE, probed with the indicated primary antibodies, and incubated with the AP-conjugated second antibody (Cell Signaling). Signals were detected by the enhanced chemifluorescence Western blotting system as described in our previous reports [33]. The images were acquired by scanning with the phosphoimager (model Storm 860; Molecular Dynamics).
Immunoprecipitation
P85+/+ and P85−/− cells were treated by arsenite (20 μM) and/or MG132 (10 μM) for 6 h. The cells were then washed by PBS, and cell lysate was used for immunoprecipitation using an anti-HIF-1α antibody (Sigma, St. Louis, MO, USA) as in our previous report [34]. The immunoprecipitated samples were subjected to Western blotting assay with an anti-ubiquitin antibody (Cell Signaling).
EMSA and super gel shift
Cells were cultured in 10-cm dishes until they reached 80–90% confluence. After incubation with 0.1% FBS DMEM for 12 h, arsenite (20 μM) was added into the medium for 6 h. Nuclear extracts were isolated with the Nuclear/Cytosol Fractionation Kit (BioVision). The specific probe pair designed for activated HSF-1 included 5′-aga cgc gaa act gct gga aga ttc c-3′ and 5′-gga atc ttc cag cag ttt cgc gtc t-3′. The probe was conjugated with biotin with the Biotin 3′ End DNA Labeling Kit (Pierce) following the manufacturer’s instructions. Nuclear extracts were incubated with poly (dI·dC), 10× binding buffer, and specific probe for about 20 min, and then a PAGE gel was run for separating the activated transcription factor and the free probes. Luminescent signal was developed with the LightShift® Chemiluminescent EMSA Kit (Pierce, Rockford, IL, USA), and detected by an automatic developing machine (Kodak).
Immunofluorescence staining
p85α+/+ and p85α−/− MEF cells were seeded in the cell culture dishes. After reaching 90% confluence, the medium was removed and cells were washed in PBS, and fixed with 4% paraformaldehyde. PBS with 0.1% saponin was then used to facilitate cells permeable for antibody. The cells were then stained with FITC conjugated Hsp70 antibody and DAPI at room temperature for 30 min. After washing off the excess antibody, the cells were observed with a confocal microscope (Leica).
Luciferase assay
p85α+/+ and p85α−/− MEF cells stably transfected with Hsp70 promoter luciferase reporter were seeded in 96-well plates. After exposure to arsenite as indicated, the cells were lysed to detect firefly and internal TK signal with the Dual-Luciferase Reporter Assay System according to the manufacturer’s instructions (Promega, San Luis Obispo, CA, USA).
Results
p85α was critical for HIF-1α induction by arsenite in C141 and MEF cells independent of transcriptional alteration
Since skin is a major tissue that is affected by arsenic exposure, mouse epidermal cell line C141 cells were chosen as the model for this study. Cl41 cells were treated with arsenite at the dose of 20 μM for 0, 3, 6, and 12 h, respectively; the result of Western blotting indicated that HIF-1α accumulated upon arsenite exposure in a time-dependent manner (Fig. 1a). A similar manner of HIF-1α accumulation was also found in MEF cells upon arsenite exposure (Fig. 1b). Because the PI-3K/Akt pathway had been reported to be involved in the induction of HIF-1α under some stress conditions, we proposed that PI-3K/Akt and especially the predominant form of PI-3K, p85α/p110 heterodimer, could be at least partially involved in arsenite-induced accumulation of HIF-1α. To investigate our hypothesis, p85α−/− and the paired wild-type (WT) MEF cells were introduced into the experimental system, and the expression levels of p85α were checked in both cell lines with or without the condition of arsenite exposure (Fig. 1c). By treating both p85α−/− cells and WT counterparts with 20 μM of arsenite for different periods, we found that the absence of p85α abolished the induction of HIF-1α by arsenite, as is shown in Fig. 1d. To test whether p85α influenced the expression of HIF-1α protein at the transcriptional level, total RNA from both p85α−/− and WT MEF cells post-arsenite exposure was extracted, and the levels of hif-1α mRNA were checked by RT-PCR. As shown in Fig. 1e, knockout of p85α did not show any observable inhibition of hif-1α mRNA expression determined by RT-PCR assay in comparison to those in p85α+/+ MEFs (Fig. 1e). Although the result from real-time PCR showed that arsenite exposure slightly increased hif-1α mRNA level in p85α+/+ MEFs, the deficiency of p85α in p85α−/− cells did not lead to any reduction of hif-1α mRNA expression as compared to those in p85α+/+ MEFs (Fig. 1f). These results lead us to conclude that p85α-mediated HIF-1α up-regulation does not occur at the transcriptional level.
Fig. 1.

Arsenite induced HIF-1α accumulation in both Cl41 cells and p85α+/+ MEFs, but not in p85α−/− MEFs. Cl41 cells, p85α+/+ and p85α−/− MEFs were exposed to 20 μM of arsenite as indicated. HIF-1α protein levels in whole cellular extracts were determined by Western blotting in Cl41 cells (a) and p85α+/+ MEFs (b). Identification of p85α deficiency in p85α−/− MEFs in comparison to that in p85α+/+ MEFs (c). Comparison of HIF-1α protein induction due to arsenite (20 μM) exposure between p85α+/+ and p85α−/− MEFs (d). hif-1α mRNA levels were detected by RT-PCR (e) or quantitative PCR in both p85α+/+ and p85α−/− MEFs after 20 μM arsenite treatment (f)
p85α regulated HIF-1α protein expression through inhibiting HIF-1α degradation upon arsenite exposure
Since the transcriptional regulation of HIF-1α by p85α had been excluded, it was reasonable for us to attempt to determine whether p85α regulated HIF-1α at the level of protein modification and degradation. As shown in Fig. 2a, pre-incubation of cells with both MG132 and arsenite led to similar HIF-1α protein accumulation in both p85α+/+ and p85α−/− MEFs. When MG132 and arsenite were removed from the culture followed by the addition of cyclohexamide (CHX), a protein synthesis inhibitor used to block the de novo production of proteins, the pre-accumulated HIF-1α was able to undergo a gradual degradation (Fig. 2a). Although HIF-1α protein level in p85α−/− MEFs at the O time point is slightly higher than that in p85α+/+ MEFs, and HIF-1α protein degradation was observed in both p85α+/+ and p85α−/− MEFs, over 70% of HIF-1α protein was degraded at time point of 2 h in p85α−/− MEFs, while less than 25% of HIF-1α protein was degraded in p85α+/+ MEFs under the same experimental condition (Fig. 2a, b). Our results indicate that p85α provides an inhibitory effect on HIF-1α protein degradation. This notion was supported by our findings that similar levels of modification signal were observed in both p85α+/+ and p85α−/− MEFs when they were exposed to MG132 or MG132 plus arsenite (Fig. 2c), indicating that HIF-1α protein might degraded in p85α−/− MEFs after the protein modification. The detection of ubiquitin levels in immunoprecipitated protein by using an anti-HIF-1α antibody showed that HIF-1α protein modification is ubiquitination. Those results suggest that arsenite is able to protect ubiquitinized hif-1α protein undergoing to degradation, and such inhibition is dependent on p85α protein expression. Furthermore, treatment of cells with MG132 or MG132 plus arsenite did not show any effect on hif-1α mRNA level in either the p85α+/+ or p85α−/− MEFs (Fig. 2e). Our results demonstrate that p85α plays a crucial role in arsenite-induced up-regulation of HIF-1α protein expression through suppression of HIF-1α protein degradation.
Fig. 2.

p85α attenuated the degradation of HIF-1α protein in arsenite-treated MEFs. a, b p85α+/+ and p85α−/− MEFs were pretreated with MG132 (10 μM) and arsenite (20 μM) for 6 h and then exposed to CHX (10 μM) for the indicated time points after the removal of MG132 and arsenite. Levels of HIF-1α were determined by Western blotting (a), and normalized to the internal control of β-actin (b). c–e p85α+/+ and p85α−/− MEFs were treated with arsenite (20 μM), MG132 (10 μM), or a combination of these two reagents for 6 h. The whole-cell extracts were probed with anti-HIF-1α, Akt, and phosphorylation of Akt at Ser473 antibodies (c); the ubiquitin levels of HIF-1α protein in precipitants were subjected to Western blotting assay using an anti-ubiquitin antibody (d); and hif-1α mRNA was assessed by RT-PCR (e)
p85α inhibited HIF-1α protein degradation via a PI-3K/Akt-dependent pathway due to arsenite exposure
Although p85α is characterized as a regulatory subunit of PI-3K, monomeric p85α has been reported to function independently in some studies [13–15]. To evaluate whether p85α engages in the arsenite-induced accumulation of HIF-1α in a PI-3K-associated manner or independently, the activation levels of Akt, a major PI-3K downstream kinase, were measured in both p85α+/+ and p85α−/− MEFs upon the arsenite treatment. The knockout of p85α in p85α−/− MEFs led to an increase in the basal level of Akt phosphorylation at Ser473, in comparison to that in p85α+/+ MEFs (Fig. 3a). An explanation to this phenomenon would be the compensatory activation of Akt by other alternative signal pathways in response to the malfunction of PI-3K in p85α−/− MEFs. This notion can be supported by the finding of a similar situation of a high basal level of activated Akt in Cl41 DN-Akt transfectant in our previous study [35, 36]. Our results further showed that the phosphorylation of Akt at Ser473 was induced by arsenite in a time-dependent manner in p85α+/+ MEFs, while there was no inducible Akt phosphorylation at Ser473 in p85α−/− MEFs under the same experimental conditions (Fig. 3a). These results suggest that PI-3K/Akt pathway might be associated with p85α-mediated HIF-1α protein accumulation. Cl41 stable Δp85α transfectant (Cl41 AP-1 Δp85 mass1) and Cl41 stable domain-negative Akt (DN-Akt) transfectant (Cl41 AP-1 DN-Akt T308A/S473A) were used for further verification. As shown in Fig. 3b, c, phosphorylation levels at Serine 473 and Tyrosine 308 of Akt were suppressed to a notable extent in both stable transfectants. Consistently, inhibition of PI-3K activity by overexpression of Δp85 in C141 AP-1 Δp85 mass1 impaired HIF-1α protein accumulation in response to arsenite exposure (Fig. 3b). Similarly, HIF-1α protein accumulation was also blocked in the transfectant of Cl41 AP-1 DN-Akt T308A/S473A (Fig. 3c). This result indicates that without the normal function of Akt, p85α cannot mediate HIF-1α accumulation due to arsenite exposure. Therefore, we conclude that instead of functioning independently, p85α regulates arsenite-induced HIF-1α protein accumulation in the classical PI-3K/Akt-dependent manner.
Fig. 3.

The blockage of HIF-1α degradation by p85α depended on activation of PI-3K/Akt in both Cl41 and MEF cells. a The phosphorylation levels of Akt at Serine 473 were measured in p85α+/+ and p85α−/− MEFs after treatment of cells with arsenite (20 μM) for the indicated time points; b, c Cl41 cells stable transfected with dominant negative mutants of p85α (Δp85α) and Akt (DN-Akt) or vector control (AP-1 mass1) were treated with arsenite (20 μM) for the indicated time spans. The induction of HIF-1α and phosphorylation of Akt at Serine 473 and Tyrosine 308 were detected as indicated
p85α regulated Hsp70, but not Hsp90 expression, in cellular response to arsenite exposure
Previous studies have shown that Hsp90 is able to protect HIF-1α from degradation [37, 38], and that Hsp70 could bind to the ODD domain of HIF-1α, thus stabilize HIF-1α protein under hypoxia in a PI-3K/Akt-dependent manner [39]. Therefore, we were interested to learn whether Hsp70 and/or Hsp90 are/is associated with the cellular response to arsenite treatment. The expression levels of Hsp70 and Hsp90 in both Cl41 and MEFs were measured for this purpose. The results indicated that arsenite exposure led to the increase of Hsp70 in a similar pattern to HIF-1α protein accumulation in both Cl41cells and MEFs; but it did not show any effect on Hsp90 protein expression (Fig. 4a, b). Moreover, the induction of Hsp70 was largely blocked as a result of the lack of p85α in p85α−/− MEFs (Fig. 4b). Corresponding to the change of HIF-1α levels, Hsp70 could also be induced by the treatment of either MG132 solely or MG132 and arsenite co-treatment in both p85α+/+ and p85α−/− MEFs, while it can only be induced in p85α+/+ but not p85α−/− MEFs under arsenite treatment (Fig. 4c). The result suggests that p85α regulates the expression of Hsp70, but not Hsp90, in response to arsenite exposure.
Fig. 4.

Inducible Hsp70, but not constitutive Hsp70 or Hsp90, was up-regulated by arsenite in a PI-3K/Akt-dependent manner. a, b The induction of overall Hsp70 was evaluated in Cl41 cells (a) and p85α+/+ and p85α−/− MEFs (b) upon 20 μM of arsenite treatment. c The induction of overall Hsp70 was determined in p85α+/+ and p85α−/− MEFs upon treatment of cells for 6 h with arsenite (20 μM) or MG132 (10 μM), or combination of both; d p85α+/+ and p85α−/− MEFs were subject to pretreatment with 200 nM of Wortmannin for 30 min, and then exposed to 20 μM of arsenite for additional 6 h. Overall Hsp70 and inducible Hsp70 were detected by Western blotting. e, f The induction of overall Hsp70 and inducible Hsp70 was assessed in Cl41 cells stable transfected with Δp85α (e) or DN-Akt (f) in comparison to that in vector control transfectant
PI-3K/Akt pathway was required for inducible Hsp70 induction due to arsenite exposure
The human Hsp70 family has eight members. Hsp70-1α and Hsp70-1β are 99% identical in terms of amino acid sequence and were initially identified to be inducible during the process of cellular response to heat shock, and thus was named accordingly Hsp70-1 or inducible Hsp70 [40]. Previous studies have shown that inducible Hsp70 functions as an important chaperone in the maintenance for protein natural structure and maturation [40]. To identify whether the increased level of Hsp70 that we observed here is inducible Hsp70, antibodies specifically against overall Hsp70 or inducible Hsp70 were used to measure overall Hsp70 and inducible Hsp70 levels in paired MEFs or Cl41 cells. In the Western blotting result using overall-Hsp70 specific antibody in both Cl41 cells and MEFs, two major bands were detected, an upper one with an approximate molecular weight of 72 kDa, and a lower one with an approximate molecular weight 70 kDa (Fig. 4a, b). Arsenite exposure induced an increase of the lower band but not the upper one (Fig. 4a−f). Then the sample was incubated with the inducible Hsp70-specific antibody; only the lower band was detected this time (Fig. 4d–f), suggesting that the induced expression of the inducible Hsp70 counted for the overall increase of Hsp70. Furthermore, the results also indicated that inhibition of PI-3K or Akt by either pretreatment of cells with Wortmannin (Fig. 4d) or by transfection of Δp85 (Fig. 4e) or DN-Akt (Fig. 4f) led to a dramatic reduction of the levels of arsenite-induced inducible Hsp70 expression. Especially the expression of inducible Hsp70 and HIF-1α share a similar tendency in paired MEFs (Fig. 4d). Therefore, it would be interesting to further investigate whether inducible Hsp70, the chaperone whose expression level is dramatically affected by arsenite stimulation, is associated with the PI-3K/Akt mediated up-regulation of HIF-1α in response to arsenite exposure.
Hsp70 translocates into the nucleus along with its accumulation in response to arsenite exposure
Since inducible Hsp70 showed a similar pattern of induction to HIF-1α in response to arsenite, we were interested to know whether it also had a similar location pattern with HIF-1α. It is well known that HIF-1α is generally accumulated within the nucleus, thus the immunostaining method was adopted to detect the sub-cellular localization of Hsp70 under arsenite stimulation. It was found that in p85α+/+ MEFs, inducible Hsp70, once induced, localized mainly in the nucleus, while there was no increase of Hsp70 in p85α−/− MEFs exposed to arsenite (Fig. 5a). It has been noted that a certain level of Hsp70 expression was observed in the cytoplasm in both cell types with or without arsenite treatment, which should be the constitutively expressed sub-types of Hsp70. To further confirm the localization of inducible Hsp70, nuclear and cytoplasmic fractions of p85α+/+ MEFs were isolated, and the levels of Hsp70 and HIF-1α were measured via Western blotting. As shown in Fig. 5b, both Hsp70 and HIF-1α that were induced by arsenite mainly existed in the nucleus. These results suggest that induced Hsp70 shared a similar pattern of response to arsenite treatment with HIF-1α, not only in terms of temporal tendency but also in terms of localization.
Fig. 5.
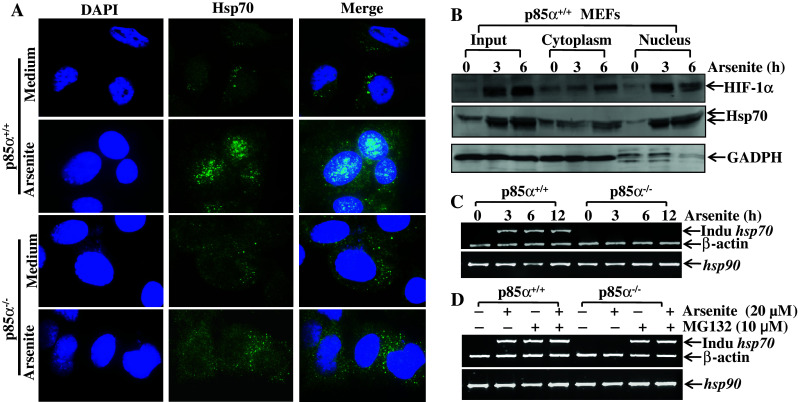
PI-3K/Akt pathway regulated both Hsp70 nuclear translocation and the level of inducible hsp70 mRNA. a Immunofluorescence staining was conducted as described in ‘Materials and methods’ on both p85α+/+ and p85α−/− MEFs with or without 20 μM of arsenite treatment. The representative DAPI, inducible Hsp70 staining and merged pictures are shown, respectively. b The cytoplasm and nucleus fractions were isolated from p85α+/+ MEFs with arsenite treatment for different time spans (0–6 h). HIF-1α and overall Hsp70 were detected with specific antibodies in each fraction as well as in whole cellular extract (input). GADPH was used here as an indicator of isolation efficacy. c, d RT-PCR was employed to detect the induction of inducible Hsp70 in both p85α+/+ and p85α−/− MEFs, under the conditions of 20 μM arsenite treatment for indicated time points (c), or under the treatment of cells with arsenite (20 μM), MG132 (10 μM) or combination of both for 6 h (d)
p85α regulated inducible Hsp70 transcription through the PI-3K/Akt/HSF-1 pathway upon arsenite exposure
To identify whether p85α was able to regulate inducible Hsp70 expression at the mRNA level in cellular response to arsenite, semi-quantitative reverse transcriptase (RT)-PCR was applied to compare inducible hsp70 mRNA levels between p85α+/+ and p85α−/− MEFs upon arsenite exposure. Consistent with the protein levels previously measured via Western blotting (Fig. 4b, c), the results showed that arsenite could notably induce increases in inducible hsp70 mRNA in p85α+/+ MEFs, but not in p85α−/− MEFs (Fig. 5c, d), indicating that p85α-mediated up-regulation of inducible Hsp70 protein expression occurs at the mRNA level. Moreover, it was observed that the hsp70 mRNA induction could be detected in p85α−/− MEFs with either MG132 treatment or MG132 plus arsenite co-treatment (Fig. 5d). The phenomenon that MG132 was able to induce inducible hsp70 mRNA expression (Fig. 5d) could be explained by the finding that MG132 itself serves as a heat-shock sponsor [41], and as an Akt activator [42]. This possibility could be supported by the result that MG132 could phosphorylate and thereby activate Akt in both p85α+/+ and p85α−/− MEFs (Fig. 2c). As a consequence, MG132 could possibly induce the expression of inducible Hsp70 via the activation of Akt.
Typically, the elevation of mRNA could result from two reasons: one is the transcriptional activation; the other is the increase in mRNA stability. To find whether p85α regulation of inducible hsp70 mRNA occurs via the transcriptional activation, the inducible Hsp70-promoter-luciferase reporter was transiently transfected into both p85α+/+ and p85α−/− MEFs. Exposure of the transfectants to arsenite led to a significant change in the level of hsp70 transcription in a dose-dependent manner in p85α+/+ MEFs, while there was no observable induction in p85α−/− MEFs (Fig. 6a). This result suggested that p85α elevated the level of hsp70 mRNA via transcriptional transactivation.
Fig. 6.
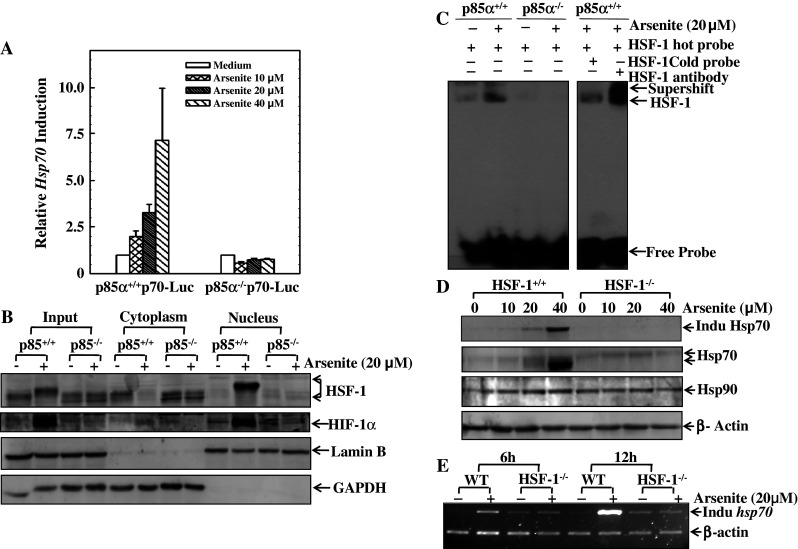
HSF-1 plays an indispensable role in transcriptional up-regulation of inducible Hsp70. a Hsp70 promoter-luciferase reporter was transiently transfected into p85α+/+ and p85α−/− MEFs, and were then subject to luciferase assay as described in the Materials and methods section. The relative intensities of fluorescence were shown proportionally in comparison between p85α+/+ and p85α−/− transfectants. b The cytoplasm and nucleus isolation assay was conducted in both p85α+/+ and p85α−/− MEFs with or without 20 μM of arsenite treatment for 6 h. The levels of HSF-1, HIF-1α, overall Hsp70, and inducible Hsp70 were evaluated, respectively. Lamin B and GAPDH were used as indicators of isolation efficacy. c The binding efficiency of HSF-1 on the promoter region of the hsp70 gene was determined by EMSA and super gel shift assay, as described in the Materials and methods section. Briefly, the sequences of HSF-1 binding sites on the Hsp70 promoter region were designed and incubated with nuclear extract from p85α+/+ or p85α−/− MEFs with or without 20 μM of arsenite treatment; each sample was subjected to PAGE gel. d, e HSF-1+/+ and HSF-1−/− MEFs were exposed to arsenite as indicated. The levels of overall and inducible Hsp70 were determined by Western blotting (d), and the levels of hsp70 mRNA were detected by RT-PCR (e)
Heat-shock inducible factor 1 (HSF-1) has been reported to be a transcriptional factor responsible for transcriptional regulation of hsp70 [43]. Therefore, the translocation of HSF-1 and its association with Hsp70 expression were compared between p85α+/+ MEFs and p85α−/− MEFs. As shown in Fig. 6b, arsenite exposure induced the nuclear translocation of HSF-1 and interestingly, a notable upwards shift of HSF-1 bands in p85α+/+ MEFs. Both the translocation of HSF-1 and the change of its molecular weight suggested arsenite-induced activation of HSF-1. In contrast, all changes were not observed in p85α−/− MEFs (Fig. 6b). Moreover, the differential activation of HSF-1 between p85α+/+ and p85α−/− MEFs is consistent with HIF-1α protein accumulation (Fig. 6b). Lamin B and GAPDH were used as the controls for nuclear and cytoplasmic proteins, respectively (Fig. 6b).
To address the function of the nuclear translocated HSF-1 as a transcriptional factor binding to the hsp70 promoter region, an oligo-nucleotide segment was synthesized based on the sequence of HSF-1 binding sites as reported in previous studies [43] and used as a probe in the gel shift assay. This oligo-nucleotide probe bound efficiently with the nuclear proteins extracted from the arsenite-treated p85α+/+ MEFs (lane 2 in Fig. 6c), but the formation of the retarded DNA–protein complexes did not appear in p85α−/− MEFs (lane 4 in Fig. 6c). At the same time, the competition experiments and the super gel shift assay were used to confirm the specific binding of HSF-1 protein with the probe (lanes 5 and 6 of Fig. 6c). The results suggest that HSF-1 was activated by arsenite exposure in a p85α-dependent manner.
To provide direct evidence that shows the role of HSF-1 in the transcriptional up-regulation of Hsp70, both HSF-1+/+ and HSF-1−/− MEFs were used for this purpose. As expected, arsenite exposure led to a high level of inducible Hsp70 protein expression in HSF-1+/+ MEFs, whereas the same treatment did not generate any detectable increase of inducible Hsp70 in HSF-1−/− MEFs (Fig. 6d). Consistently, arsenite-induced increase in inducible hsp70 mRNA was also blocked in HSF-1−/− MEFs, but not in HSF-1+/+ MEFs (Fig. 6e). These results demonstrate that HSF-1 is required for arsenite-induced inducible Hsp70 expression.
Inducible Hsp70 was a protective chaperone to stabilize HIF-1α due to arsenite exposure
Our result indicated that inducible Hsp70 and HIF-1α had a similar tendency in terms of both location and expression level during the arsenite treatment, and that the inhibition of the activity of PI-3K-Akt pathway would deter the induction of both of them. Therefore, we speculated that inducible Hsp70 might physically interact with (and thereby stabilize) HIF-1α. To address this notion, both inducible Hsp70 expressing construct (induc Hsp70) and constitutive Hsp70 expression construct (Hsc70) were transfected into p85α−/− MEFs separately. The expression efficiency of inducible Hsp70 in stable transfectant was identified by Western blotting (Fig. 7a); while the level of Hsc70 in the counterpart was determined indirectly by measuring the conjugated Myc-Hsp70 expression level with anti-Myc antibody (left panel of Fig. 7b). As expected, arsenite induced both inducible Hsp70 expression and HIF-1α protein accumulation in p85α+/+ MEFs, and there was no observable induction of either of those two protein expressions in p85α−/− (Vector) transfectant (Fig. 7a). Most importantly, overexpression of inducible Hsp70 in p85α−/− (induc Hsp70) transfectant led to an increase in basal HIF-1α protein accumulations (Fig. 7a). In contrast to p85α−/− (induc Hsp70) transfectant, over-expression of constitutive Hsp70 (Myc-Hsc70) in p85α−/− MEFs did not show any observable increase in HIF-1α protein expression (Fig. 7b), dramatically demonstrating that only inducible Hsp70, rather than constitutive form of Hsp70, is responsible for stabilization of the HIF-1α protein. It should be noted that in p85α−/− (induc Hsp70) MEFs, arsenite treatment appeared to induce HIF-1α protein expression in a dose-dependent manner, although inducible Hsp70 is constitutively expressed. This might be due to the involvement of other pathways in the regulation of HIF-1α protein expression through an inducible Hsp70-independent manner. This notion is under investigation in our laboratory at present. To further confirm the role of inducible Hsp70 in HIF-1α protein induction upon arsenite exposure, shRNAs specifical for inducible hsp70 were introduced into p85α+/+ MEFs. The stable transfectants with nonsense shRNA, and the inducible hsp70 shRNA 1 and shRNA 2, respectively, were exposed to different concentrations of arsenite for 6 h. The levels of inducible Hsp70 and HIF-1α measured by Western blotting indicated that the intervention of inducible Hsp70 expression in WT MEF cells decreased the accumulation of HIF-1α (Fig. 7c). Inducible Hsp70 was proven to function as a protective chaperone in stabilizing HIF-1α protein under arsenite exposure. Together with the results in Fig. 2c and d, we speculated that different from the hypoxia situation, arsenite stabilizes HIF-1α by producing inducible Hsp70, which chaperoned ubiquitinated HIF-1α and keep it from degradation in the proteasome-dependent pathway.
Fig. 7.

Inducible Hsp70-specific stabilized HIF-1α protein upon arsenite exposure. a The construct expressing inducible Hsp70 and vector construct (as control) were introduced separately into p85α−/− MEFs, and the corresponding stable transfectant was established. The induction of HIF-1α due to arsenite exposure was determined as indicated in p85α−/− (Vector), p85α−/− (Induc Hsp70), and p85α+/+ MEFs. The expression levels of inducible Hsp70 were verified by Western blotting. b p85α−/− MEFs were stably transfected with constitutive Hsp70 (Hsc70) expressing construct or the control vector. The success of transfection was confirmed by Western blotting (left panel). The arsenite-induced HIF-1α was detected in both transfectants (right panel). c WT MEFs were stably transfected with nonsense shRNA or two specific shRNAs targeting inducible Hsp70. The efficiency of knockdown was identified by Western blotting and the arsenite-induced HIF-1α protein expression was determined as indicated
Discussion
Arsenic-containing compounds play seemingly paradoxical roles as both ubiquitous environmental contaminants and tumor therapeutic agents [44, 45]. On one hand, exposure to inorganic arsenite can induce adverse health effects like hyperkeratosis, hyper-pigmentation, cardiovascular disease, reproductive defects, and cancers of several tissues [46–48]. On the other hand, arsenic trioxide is considered to be valuable because of its induction of apoptosis in leukemia and pancreatic cancer cells [49, 50]. Our previous studies have illustrated that the function of arsenite is possibly dose-dependent, with the ability to promote cell cycle via induction of cyclin D1 expression through the IKKβ–NFκB pathway at low doses (i.e., 1.25–5 μM) [51, 52] and also the ability to signal programmed cell apoptosis at high doses (i.e., >10 μM) [53]. Song et al. [53] found that the relatively high-dose-arsenite-induced apoptosis is linked to NFκB p50- and Gadd45α-dependent promotion of JNKs activation. In this study, we have proposed another arsenite-induced signaling pathway through PI-3K/Akt/HSF-1/Hsp70-regulated accumulation of HIF-1α protein.
HIF-1α is characterized by up-regulation of its expression level in response to stress. HIF-1α combines with its constitutively expressed partner HIF-1β to form a HIF-1 heterodimer that plays an important role in cellular response to various stresses [9]. The most well-studied regulation of HIF-1α is its post-translational accumulation under hypoxic condition [8, 9, 54]. Under normoxia, the oxygen-dependent degradation (ODD) domain of HIF-1α is hydroxylated by specific prolyl hydroxylases (PHD1, 2, and 3). Hydroxylated HIF-1α recruits von Hippel-Lindau tumor suppressor (pVHL) to the ODD domain, leading to the recognition by E3 ubiquitin ligase and subsequent ubiquitination and proteasome-dependent degradation of HIF-1α protein [8]. However, under hypoxia condition, this degradation route is suspended due to lack of oxygen, and HIF-1α thereby accumulates. Recent studies indicate that this hypoxia-induced accumulation of HIF-1α further involves sumoylation [55] and/or even de-sumoylation [54]. However, the arsenite-induced accumulation of HIF-1α seems irrelevant with this classical hydroxylation-dependent degradation model, because arsenic compounds are normally associated with the accumulation of reactive oxygen species within cells [56, 57], which is indicative of a relatively hyperoxic environment rather than hypoxia. Hence, it was of great interest to us to investigate the underlying mechanism by which arsenite leads to HIF-1α protein accumulation.
As is shown in our studies, p85α is indispensable in arsenite-induced HIF-1α protein accumulation. p85α, a regulatory subunit of PI-3K, was reported to implement its own function in a PI-3K-independent manner under certain circumstances [13]. However, we prove here that this is not the case in regulating arsenite-induced HIF-1α, but that arsenite-induced HIF-1α is via a PI-3K/Akt-dependent manner. The PI-3K/Akt signaling pathway is another important module reported to participate in the regulation of HIF-1α expression level under various circumstances, including insulin/interleukin-1β treatment [58], estrogen stimulation [59], UVB exposure [60], and even hypoxia [61]. According to the knowledge gathered so far, the PI-3K/Akt pathway involves HIF-1α regulation mainly in two post-transcriptional manners: either by enhancing the translation rate of HIF-1α mRNA or by protecting HIF-1α protein from degradation. Translational regulation of HIF-1α happens under heregulin [62] or some growth factor stimulation (e.g., insulin) [63]. To be specific, PI-3K/Akt increases the synthesis of HIF-1α by activating FRAP (FK506 binding protein-Rapamycin Associated Protein), a kinase that enhances the translation rate of hif-1α mRNA [62]. The prevention of HIF-1α degradation by PI-3K/Akt involves suppression of GSK3β activity, which in turn decreases the phosphorylation of HIF-1α and thereby enables HIF-1α to avoid being ubiquitinated [64]. Another preventive function of PI-3K/Akt on HIF-1α degradation is associated with the physical interaction between HIF-1α and two heat-shock proteins, Hsp70 and Hsp90 [39]. Provoked expression of both Hsp70 and Hsp90 contributes to HIF-1α stabilization under the circumstance of either hypoxia or heat shock [39]. Moreover, a study by Gao et al. [28] shows that reactive oxygen species (ROS) induced by a relatively high dose of arsenite (100 μM to 2 mM) might mediate phosphorylation of Akt and subsequent stabilization of HIF-1α in DU145 human prostate carcinoma cells. However, the linkage between phosphorylation of Akt and accumulation of HIF-1α under arsenite treatment (especially at a lower dose) still remained vague. In our present study, the consistency of induction of HIF-1α and inducible Hsp70 (but neither Hsp90 nor constitutive Hsp70) under arsenite treatment was observed in both Cl41 cells and MEF cells, which intrigued enough to cause us to investigate the new pathway by which PI-3K/Akt regulates the level of HIF-1α protein expression. Our data further demonstrated the indispensable role of inducible Hsp70 in stabilizing HIF-1α. The Hsp70 family had eight members, among which hsp70-1a and hsp70-1b are both inducible under various stress situations, and are 99% identical in terms of amino acid sequences [65]. They are collectively called inducible Hsp70 (or Hsp70-1), and function as important chaperones in the maintenance of the proteins’ natural structure and maturation, and to attenuate the degradation of their clients [40]. Zhou et al. [39] report that HIF-1α is one of the clients, with its ODD domain assigned to be the binding region of Hsp70. However, they do not distinguish inducible Hsp70 from the general term Hsp70, which includes other members in Hsp70 family, especially the constitutively expressed one named Hsp70-8 (or Hsc70) [65]. In the present studies, we show that the inducible Hsp70 was up-regulated to a relatively high level in comparison to constitutive Hsp70 (Hsc70), and that it played a critical role in stabilizing HIF-1α in cellular response to arsenite stimulation; while Hsc70 did not change its level during the whole process, and therefore played a much less important role in such HIF-1α protein induction. Similar to Hsp70, Hsp90 is another heat-shock protein reported to physically interact with HIF-1α [66, 67]. Hsp90 was previously reported to cooperate with Hsp70 to stabilize HIF-1α under hypoxia or heat-shock circumstance [39]. Moreover, it has been reported that MG132 could induce cellular apoptosis through the JNK1 pathway [68]. Our most recent studies have also demonstrated that JNK1 activation is crucial for HIF-1α protein accumulation due to hypoxia and nickel exposure through induction of both Hsp90/Hsp70 [34]. However, there is no change in either protein or mRNA level of Hsp90 upon arsenite exposure in both Cl41 and MEFs from our studies. Interestingly, Hsp90 has been reported by Somji et al. to be up-regulated by arsenite in the human kidney and in human proximal tubule (HPT) cells [69], indicative of the potential existence of diverse mechanisms by which different types of cells respond to arsenite exposure. Since skin is a major tissue subject to the toxicity of arsenite exposure, mouse epidermal Cl41 cells could be a more plausible model to demonstrate how arsenite influences the functions of skin tissue.
Our results show that the transcriptional up-regulation of hsp70 by activated PI-3K/Akt pathway is strictly mediated by heat-shock transcription factor-1 (HSF-1). This conclusion is supported by previous findings that PI-3K/Akt can activate HSF-1 [70]. Since the activation of HSF-1 needs full phosphorylation, and PI-3K downstream signal component PKCζ has been proven to phosphorylate HSF-1 [71], we anticipate that PKCζ might be a p85α/Akt downstream mediator responsible for HSF-1 transactivation in arsenite exposure.
HSF-1 activates transcription of a set of genes encoding heat-shock proteins, which share a common HSF binding sequence in their promoter regions called the heat-shock element (HSE) [72]. Because of their shared binding region of HSE, inducible Hsp70 and Hsp90 were reported to be activated simultaneously by HSF-1 under certain stress or growth factor stimulation (e.g., hypoxia [73], heat shock [74], and interleukin-6 [75]). However, inducible Hsp70 turns out to be the only one of the two that is enhanced by activated HSF-1 under relative high-dose (20 μM) arsenite exposure, and seemed to be sufficient in stabilizing HIF-1α under this scenario. Moreover, under the situation of arsenite exposure, a tendency of Hsp70 to translocate from cytoplasm into nucleus was observed both in immune-fluorescence assay and in immunoblot coupled with cytoplasm-nucleus isolation, chronologically roughly overlapped with the occurrence of HIF-1α accumulation in the nucleus. Since HIF-1α is known to possess a nuclear localization signal (NLS) in its C-terminal region, stabilized HIF-1α can rapidly bind to nuclear pore proteins and translocate into the nucleus [76]. It will be interesting to determine whether inducible Hsp70 physical binds with HIF-1α and thereby co-translocation into nucleus in the instance of arsenite stimulation and whether that could influence its other functions as a protein chaperone or stabilizer.
In summary, the present study demonstrates that the PI-3K/Akt pathway and inducible Hsp70 play a very important role in the stabilization of HIF-1α upon relatively high-dose arsenite exposure. Our study, together with other recent findings, suggests the existence of complicated mechanisms underlying the post-transcriptional regulation of HIF-1α under different types of stress. The identified PI-3K/Akt/HSF-1/inducible Hsp70 pathway in current studies will enrich our understanding of regulation of HIF-1α protein degradation.
Acknowledgments
This work was supported in part by grants from NIH/NCI CA112557, CA119028-05S110, and from NIH/NIEHS ES012451, ES010344. We thank Dr. Hector R. Wong (Children’s Hospital Medical Center, Cincinnati, OH, USA) for providing inducible Hsp70 constructs and HSF-1+/+ and HSF-1−/− cells, and Dr. Alice Liu (Rutgers State University of New Jersey, Piscataway, NJ, USA) for providing Hsp70 luciferase reporter plasmid. We also thank Ms. Nedda Tichi for her critical reading of the manuscript.
Conflict of interest
There is no conflict interest for all authors listed.
Abbreviations
- DMEM
Dulbecco’s modified Eagle’s medium
- EMSAs
Electrophoretic mobility shift assays
- FBS
Fetal bovine serum
- JNKs
c-Jun N-terminal kinases
- HIF-1
Hypoxia-inducible factor-1
- HSF
Heat-shock transcription factor
- Hsps
Heat shock or stress proteins
- MEFs
Mouse embryonic fibroblasts
- RT-PCR
Reverse transcription polymerase chain reaction
- PI-3K
Phosphatidylinositol-3-kinase
- shRNA
Short hairpin RNA
Footnotes
W. Guo and Z. Yang contributed equally to this work.
References
- 1.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsuzuki Y, Fukumura D, Oosthuyse B, Koike C, Carmeliet P, Jain RK. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha→hypoxia response element→VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 2000;60:6248–6252. [PubMed] [Google Scholar]
- 3.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 5.Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 6.Dachs GU, Patterson AV, Firth JD, Ratcliffe PJ, Townsend KM, Stratford IJ, Harris AL. Targeting gene expression to hypoxic tumor cells. Nat Med. 1997;3:515–520. doi: 10.1038/nm0597-515. [DOI] [PubMed] [Google Scholar]
- 7.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 8.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 9.Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. doi: 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 11.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 12.Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22:965–977. doi: 10.1128/MCB.22.3.965-977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song L, Li J, Ye J, Yu G, Ding J, Zhang D, Ouyang W, Dong Z, Kim SO, Huang C. p85alpha acts as a novel signal transducer for mediation of cellular apoptotic response to UV radiation. Mol Cell Biol. 2007;27:2713–2731. doi: 10.1128/MCB.00657-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol. 2005;170:455–464. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia Z, Silio V, Marques M, Cortes I, Kumar A, Hernandez C, Checa AI, Serrano A, Carrera AC. A PI3K activity-independent function of p85 regulatory subunit in control of mammalian cytokinesis. EMBO J. 2006;25:4740–4751. doi: 10.1038/sj.emboj.7601324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arsham AM, Plas DR, Thompson CB, Simon MC. Phosphatidylinositol 3-kinase/Akt signaling is neither required for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J Biol Chem. 2002;277:15162–15170. doi: 10.1074/jbc.M111162200. [DOI] [PubMed] [Google Scholar]
- 17.Alvarez-Tejado M, Alfranca A, Aragones J, Vara A, Landazuri MO, del Peso L. Lack of evidence for the involvement of the phosphoinositide 3-kinase/Akt pathway in the activation of hypoxia-inducible factors by low oxygen tension. J Biol Chem. 2002;277:13508–13517. doi: 10.1074/jbc.M200017200. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Davidson G, Huang Y, Jiang BH, Shi X, Costa M, Huang C. Nickel compounds act through phosphatidylinositol-3-kinase/Akt-dependent, p70(S6k)-independent pathway to induce hypoxia inducible factor transactivation and Cap43 expression in mouse epidermal Cl41 cells. Cancer Res. 2004;64:94–101. doi: 10.1158/0008-5472.CAN-03-0737. [DOI] [PubMed] [Google Scholar]
- 19.Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279:45643–45651. doi: 10.1074/jbc.M404097200. [DOI] [PubMed] [Google Scholar]
- 20.Lee TC, Tanaka N, Lamb PW, Gilmer TM, Barrett JC. Induction of gene amplification by arsenic. Science. 1988;241:79–81. doi: 10.1126/science.3388020. [DOI] [PubMed] [Google Scholar]
- 21.Gebel TW. Arsenic and drinking water contamination. Science. 1999;283:1458–1459. doi: 10.1126/science.283.5407.1455e. [DOI] [PubMed] [Google Scholar]
- 22.Simeonova PP, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, Luster MI. Arsenic mediates cell proliferation and gene expression in the bladder epithelium: association with activating protein-1 transactivation. Cancer Res. 2000;60:3445–3453. [PubMed] [Google Scholar]
- 23.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 24.Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci USA. 2001;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossman TG, Stone D, Molina M, Troll W. Absence of arsenite mutagenicity in E. coli and Chinese hamster cells. Environ Mutagen. 1980;2:371–379. doi: 10.1002/em.2860020307. [DOI] [PubMed] [Google Scholar]
- 26.Kamat CD, Green DE, Curilla S, Warnke L, Hamilton JW, Sturup S, Clark C, Ihnat MA. Role of HIF signaling on tumorigenesis in response to chronic low-dose arsenic administration. Toxicol Sci. 2005;86:248–257. doi: 10.1093/toxsci/kfi190. [DOI] [PubMed] [Google Scholar]
- 27.Duyndam MC, Hulscher TM, Fontijn D, Pinedo HM, Boven E. Induction of vascular endothelial growth factor expression and hypoxia-inducible factor 1alpha protein by the oxidative stressor arsenite. J Biol Chem. 2001;276:48066–48076. doi: 10.1074/jbc.M106282200. [DOI] [PubMed] [Google Scholar]
- 28.Gao N, Shen L, Zhang Z, Leonard SS, He H, Zhang XG, Shi X, Jiang BH. Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol Cell Biochem. 2004;255:33–45. doi: 10.1023/B:MCBI.0000007259.65742.16. [DOI] [PubMed] [Google Scholar]
- 29.Wong HR, Menendez IY, Ryan MA, Denenberg AG, Wispe JR. Increased expression of heat shock protein-70 protects A549 cells against hyperoxia. Am J Physiol. 1998;275:L836–L841. doi: 10.1152/ajplung.1998.275.4.L836. [DOI] [PubMed] [Google Scholar]
- 30.Khalil S, Luciano J, Chen W, Liu AY. Dynamic regulation and involvement of the heat shock transcriptional response in arsenic carcinogenesis. J Cell Physiol. 2006;207:562–569. doi: 10.1002/jcp.20599. [DOI] [PubMed] [Google Scholar]
- 31.Huang C, Ma WY, Dong Z. Requirement for phosphatidylinositol 3-kinase in epidermal growth factor-induced AP-1 transactivation and transformation in JB6 P+ cells. Mol Cell Biol. 1996;16:6427–6435. doi: 10.1128/mcb.16.11.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malhotra V, Kooy NW, Denenberg AG, Dunsmore KE, Wong HR. Ablation of the heat shock factor-1 increases susceptibility to hyperoxia-mediated cellular injury. Exp Lung Res. 2002;28:609–622. doi: 10.1080/01902140260426724. [DOI] [PubMed] [Google Scholar]
- 33.Huang C, Ma WY, Li J, Goranson A, Dong Z. Requirement of Erk, but not JNK, for arsenite-induced cell transformation. J Biol Chem. 1999;274:14595–14601. doi: 10.1074/jbc.274.21.14595. [DOI] [PubMed] [Google Scholar]
- 34.Zhang D, Li J, Costa M, Gao J, Huang C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res. 2010;70:813–823. doi: 10.1158/0008-5472.CAN-09-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Chen H, Tang M-S, Shi X, Amin S, Desai D, Costa M, Huang C. PI-3K and Akt are mediators of AP-1 induction by 5-MCDE in mouse epidermal Cl41 cells. J Cell Biol. 2004;165:77–86. doi: 10.1083/jcb.200401004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Tang M-s, Liu B, Shi X, Huang C. A critical role of PI-3K/Akt/JNKs pathway in benzo[lsqb]a[rsqb]pyrene diol-epoxide (B[lsqb]a[rsqb]PDE)-induced AP-1 transactivation in mouse epidermal Cl41 cells. Oncogene. 2004;23:3932–3944. doi: 10.1038/sj.onc.1207501. [DOI] [PubMed] [Google Scholar]
- 37.Kim WY, Oh SH, Woo JK, Hong WK, Lee HY. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1alpha. Cancer Res. 2009;69:1624–1632. doi: 10.1158/0008-5472.CAN-08-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu YV, Semenza GL. RACK1 vs. HSP90: competition for HIF-1 alpha degradation vs. stabilization. Cell Cycle. 2007;6:656–659. doi: 10.4161/cc.6.13.4435. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, Schmid T, Frank R, Brune B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation. J Biol Chem. 2004;279:13506–13513. doi: 10.1074/jbc.M310164200. [DOI] [PubMed] [Google Scholar]
- 40.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 41.Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem. 1997;272:9086–9092. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- 42.Kwak HB, Lee MS, Kim HS, Cho HJ, Kim JW, Lee ZH, Oh J. Proteasome inhibitors induce osteoclast survival by activating the Akt pathway. Biochem Biophys Res Commun. 2008;377:1–6. doi: 10.1016/j.bbrc.2008.05.048. [DOI] [PubMed] [Google Scholar]
- 43.Kim HP, Wang X, Zhang J, Suh GY, Benjamin IJ, Ryter SW, Choi AM. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38 beta MAPK and heat shock factor-1. J Immunol. 2005;175:2622–2629. doi: 10.4049/jimmunol.175.4.2622. [DOI] [PubMed] [Google Scholar]
- 44.Dong Z. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environ Health Perspect. 2002;110(Suppl 5):757–759. doi: 10.1289/ehp.02110s5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol Hematol. 2002;42:5–24. doi: 10.1016/S1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- 46.Stephanopoulos DE, Willman DA, Shevlin D, Pinter L, Gummin DD. Treatment and toxicokinetics of acute pediatric arsenic ingestion: danger of arsenic insecticides in children. Pediatr Crit Care Med. 2002;3:74–80. doi: 10.1097/00130478-200201000-00017. [DOI] [PubMed] [Google Scholar]
- 47.Haque R, Mazumder DN, Samanta S, Ghosh N, Kalman D, Smith MM, Mitra S, Santra A, Lahiri S, Das S, De BK, Smith AH. Arsenic in drinking water and skin lesions: dose-response data from West Bengal, India. Epidemiology. 2003;14:174–182. doi: 10.1097/00001648-200303000-00011. [DOI] [PubMed] [Google Scholar]
- 48.Cohen SM, Ohnishi T, Arnold LL, Le XC. Arsenic-induced bladder cancer in an animal model. Toxicol Appl Pharmacol. 2007;222:258–263. doi: 10.1016/j.taap.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Li X, Ding X, Adrian TE. Arsenic trioxide induces apoptosis in pancreatic cancer cells via changes in cell cycle, caspase activation, and GADD expression. Pancreas. 2003;27:174–179. doi: 10.1097/00006676-200308000-00011. [DOI] [PubMed] [Google Scholar]
- 50.Che XF, Zheng CL, Owatari S, Mutoh M, Gotanda T, Jeung HC, Furukawa T, Ikeda R, Yamamoto M, Haraguchi M, Arima N, Akiyama S. Overexpression of survivin in primary ATL cells and sodium arsenite induces apoptosis by down-regulating survivin expression in ATL cell lines. Blood. 2006;107:4880–4887. doi: 10.1182/blood-2005-08-3423. [DOI] [PubMed] [Google Scholar]
- 51.Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis. 2006;27:864–873. doi: 10.1093/carcin/bgi321. [DOI] [PubMed] [Google Scholar]
- 52.Ouyang W, Ma Q, Li J, Zhang D, Liu ZG, Rustgi AK, Huang C. Cyclin D1 induction through IkappaB kinase beta/nuclear factor-kappaB pathway is responsible for arsenite-induced increased cell cycle G1-S phase transition in human keratinocytes. Cancer Res. 2005;65:9287–9293. doi: 10.1158/0008-5472.CAN-05-0469. [DOI] [PubMed] [Google Scholar]
- 53.Song L, Li J, Zhang D, Liu ZG, Ye J, Zhan Q, Shen HM, Whiteman M, Huang C. IKKbeta programs to turn on the GADD45alpha-MKK4-JNK apoptotic cascade specifically via p50 NF-kappaB in arsenite response. J Cell Biol. 2006;175:607–617. doi: 10.1083/jcb.200602149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, Holsboer F, Arzt E. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131:309–323. doi: 10.1016/j.cell.2007.07.044. [DOI] [PubMed] [Google Scholar]
- 56.Shi H, Hudson LG, Ding W, Wang S, Cooper KL, Liu S, Chen Y, Shi X, Liu KJ. Arsenite causes DNA damage in keratinocytes via generation of hydroxyl radicals. Chem Res Toxicol. 2004;17:871–878. doi: 10.1021/tx049939e. [DOI] [PubMed] [Google Scholar]
- 57.Pourahmad J, O’Brien PJ, Jokar F, Daraei B. Carcinogenic metal induced sites of reactive oxygen species formation in hepatocytes. Toxicol In Vitro. 2003;17:803–810. doi: 10.1016/S0887-2333(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 58.Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Burgel T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002;512:157–162. doi: 10.1016/S0014-5793(02)02247-0. [DOI] [PubMed] [Google Scholar]
- 59.Kazi AA, Koos RD. Estrogen-induced activation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology. 2007;148:2363–2374. doi: 10.1210/en.2006-1394. [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Bi Z, Yan B, Wan Y. UVB radiation induces expression of HIF-1alpha and VEGF through the EGFR/PI3K/DEC1 pathway. Int J Mol Med. 2006;18:713–719. [PubMed] [Google Scholar]
- 61.Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell. 2007;18:4691–4697. doi: 10.1091/mbc.E07-04-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–27981. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- 64.Flugel D, Gorlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol. 2007;27:3253–3265. doi: 10.1128/MCB.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 66.Gradin K, McGuire J, Wenger RH, Kvietikova I, Fhitelaw ML, Toftgard R, Tora L, Gassmann M, Poellinger L. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the Arnt transcription factor. Mol Cell Biol. 1996;16:5221–5231. doi: 10.1128/mcb.16.10.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minet E, Ernest I, Michel G, Roland I, Remacle J, Raes M, Michiels C. HIF1A gene transcription is dependent on a core promoter sequence encompassing activating and inhibiting sequences located upstream from the transcription initiation site and cis elements located within the 5′UTR. Biochem Biophys Res Commun. 1999;261:534–540. doi: 10.1006/bbrc.1999.0995. [DOI] [PubMed] [Google Scholar]
- 68.Wang H-Q, Liu B-Q, Gao Y-Y, Meng X, Guan Y, Zhang H-Y, Du Z-X. Inhibition of the JNK signalling pathway enhances proteasome inhibitor-induced apoptosis of kidney cancer cells by suppression of BAG3 expression. Br J Pharmacol. 2009;158:1405–1412. doi: 10.1111/j.1476-5381.2009.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Somji S, Ann Sens M, Garrett SH, Gurel V, Todd JH, Sens DA. Expression of Hsp90 in the human kidney and in proximal tubule cells exposed to heat, sodium arsenite and cadmium chloride. Toxicol Lett. 2002;133:241–254. doi: 10.1016/S0378-4274(02)00205-9. [DOI] [PubMed] [Google Scholar]
- 70.Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3beta in the regulation of HSF-1 activity. J Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- 71.Baek SH, Lee UY, Park EM, Han MY, Lee YS, Park YM. Role of protein kinase Cdelta in transmitting hypoxia signal to HSF and HIF-1. J Cell Physiol. 2001;188:223–235. doi: 10.1002/jcp.1117. [DOI] [PubMed] [Google Scholar]
- 72.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 73.Mivechi NF, Koong AC, Giaccia AJ, Hahn GM. Analysis of HSF-1 phosphorylation in A549 cells treated with a variety of stresses. Int J Hyperthermia. 1994;10:371–379. doi: 10.3109/02656739409010281. [DOI] [PubMed] [Google Scholar]
- 74.Cotto J, Fox S, Morimoto R. HSF1 granules: a novel stress-induced nuclear compartment of human cells. J Cell Sci. 1997;110(Pt 23):2925–2934. doi: 10.1242/jcs.110.23.2925. [DOI] [PubMed] [Google Scholar]
- 75.Stephanou A, Isenberg DA, Nakajima K, Latchman DS. Signal transducer and activator of transcription-1 and heat shock factor-1 interact and activate the transcription of the Hsp-70 and Hsp-90beta gene promoters. J Biol Chem. 1999;274:1723–1728. doi: 10.1074/jbc.274.3.1723. [DOI] [PubMed] [Google Scholar]
- 76.Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]