

Abstract
The Polydnaviridae (PDV), including the Bracovirus (BV) and Ichnovirus genera, originated from the integration of unrelated viruses in the genomes of two parasitoid wasp lineages, in a remarkable example of convergent evolution. Functionally active PDVs represent the most compelling evolutionary success among endogenous viral elements (EVEs). BV evolved from the domestication by braconid wasps of a nudivirus 100 Ma. The nudivirus genome has become an EVE involved in BV particle production but is not encapsidated. Instead, BV genomes have co-opted virulence genes, used by the wasps to control the immunity and development of their hosts. Gene transfers and duplications have shaped BV genomes, now encoding hundreds of genes. Phylogenomic studies suggest that BVs contribute largely to wasp diversification and adaptation to their hosts. A genome evolution model explains how multidirectional wasp adaptation to different host species could have fostered PDV genome extension. Integrative studies linking ecological data on the wasp to genomic analyses should provide new insights into the adaptive role of particular BV genes. Forthcoming genomic advances should also indicate if the associations between endoparasitoid wasps and symbiotic viruses evolved because of their particularly intimate interactions with their hosts, or if similar domesticated EVEs could be uncovered in other parasites.
Keywords: polydnavirus, Cotesia, genome evolution, parasitoid wasp, virus adaptation, obligatory mutualism
1. Introduction
Paleovirology and the study of endogenous viral elements (EVEs), corresponding to ancient viral sequence insertions in eukaryotic genomes, are unveiling the long and rich interactions viruses have entertained with their hosts [1–4]. Although polydnaviruses (PDVs) might still be considered atypical, they represent the most compelling evolutionary success among EVEs. The virus ancestors of the Polydnaviridae family were integrated into the genomes of parasitoid wasps (Hymenoptera, Ichneumonoidea, Braconidae), where they were subjected to complex genomic rearrangements. Now, wasps produce and use for their own ends functional infective virus particles that enclose fragmented dsDNA genomes solely encoding virulence genes [5]. The benefit to the virus genome resides in its vertical transmission free of the mutation load generally incurred by non-functional EVEs [6]. Extraordinarily, EVE domestication (i.e. implicating genetic changes and regulated viral particle production) has occurred at least twice during wasp evolution from independent virus families (figure 1) [10,11], and we are probably just beginning to unravel the diversity of parasitoid wasp–virus associations.
Figure 1.

Multiple origins of virus symbioses in the Ichneumonoidea. Phylogenies and molecular dating are modified from [7–9] for ichneumonoid wasp and [2] for the free insect DNA virus.
Within the insect order Hymenoptera, ichneumonoid wasps encompass the Braconidae and Ichneumonidae, two highly diverse parasitoid families, both in terms of species richness and parasitic strategies [12]. Their larvae develop to the detriment of arthropod hosts, principally of the order Lepidoptera, Coleoptera, Hymenoptera and Hemiptera [13]. Both families have seen the evolution of numerous koinobiont endoparasitoids, in which wasp larvae grow inside developing hosts [12]. This particular lifestyle imposes relatively long and intimate relationships between hosts and parasites, giving rise to complex immune and physiological interactions [14]. To overcome host defences, wasps have evolved an arsenal of virulence factors present in their venoms and/or produced in their parasitized hosts by symbiotic PDV genes (figure 1).
PDVs are essentially chimeric viruses composed of viral particles enclosing DNA circles encoding virulence genes supposedly of wasp origin. PDVs have original infection cycles split between two hosts. PDV particles are produced only in wasps, but infect cells of the caterpillar host (figure 2). PDV genomes are stably integrated into the genomes of parasitoid wasps [15]. They are composed of (i) proviral segments used to produce the multiple dsDNA circles that encode virulence genes and that are packaged in infectious particles, and of (ii) genes, encoding the so-called viral machinery that produce the particles (figure 2). Expression of the structural genes as well as excision and packaging of PDV dsDNA circles occurs in specialized cells of the calyx, a particular region of the wasp ovaries located at the bases of the oviducts. During oviposition of parasitoid eggs, PDV particles are injected into the lepidopteran host and infect many lepidopteran cell types but do not replicate. Virulence gene expression leads to modifications in lepidopteran host physiology, such as inhibition of wasp egg encapsulation and developmental manipulations, allowing wasp development and emergence (figure 2) [16–19].
Figure 2.

Bracovirus life cycle and genome organization. (a) The BV genome is integrated in the wasp genome (yellow). It is composed of (i) proviral segments (blue) used to produce the multiple dsDNA circles that encode virulence genes (coloured rectangles) and that are packaged in the particles, and of (ii) BV structural genes (nudiviral genes; black or grey rectangles) that are involved in particle production. (b) Nudiviral gene expression as well as amplification and excision of BV circles occurs in the calyx cells of the wasp ovaries. Direct repeat junctions (DRJs; red triangles) are involved in circularization. (c) DNA circles are packaged into BV particles. (d) BV particles are injected in the lepidopteran host during oviposition of parasitoid eggs and infect many lepidopteran cell types but do not replicate. (e) BV virulence gene expression leads to modifications in lepidopteran host physiology, such as inhibition of wasp egg encapsulation, allowing wasp development. (f) Emergence of adults carrying bracovirus genomes from wasp pupae. This figure is based on the life cycle of CcBV associated with C. congregata parasitoid wasp of M. sexta. (Photographs A. Bézier and A. Wild.)
2. Polydnavirus origins
(a). Diversity of wasp–virus association
The Polydnaviridae family encompasses two genera: Bracovirus (BV) and Ichnovirus (IV), both associated with thousands of wasp species from six Braconidae subfamilies and the Ichneumonidae subfamily Campopleginae, respectively. All these wasps are koinobiont parasitoids of lepidopteran larvae [20]. Both BV and IV are symbiotically associated with wasps, and are produced in large amount as large fragmented dsDNA viruses in wasp ovaries, but their particles have different morphological features. Within Ichneumonidae, wasps from the Banchinae subfamily are also associated with viruses. Based on virus particle morphology and wasp phylogenetic relationships (figure 1), banchine PDV were proposed to form a third PDV group [21]. Only the characterization of the viral machinery producing banchine PDV would indicate whether they have a different or common origin with IVs. Furthermore, virus-like particles (VLPs; particles resembling viruses but devoid of nucleic acids) are produced by several wasps from the Figitidae (Hymenoptera, Cynipoidea) and Euphorinae (Braconidae) [22,23], and most notably by the campoplegine Venturia canescens (Ichneumonidae) [24] (figure 1). The origins of these diverse VLPs remain to be elucidated. Venturia canescens VLPs were the first immunosuppressive particles described in the ovaries of a parasitoid wasp, in a seminal paper [25]. Venturia canescens belongs to the Campopleginae, and thus is expected to harbour a regular ichnovirus, but it produces VLPs, in which no DNA is incorporated. One hypothesis for the origin of V. canescens VLPs is that they might correspond to dysfunctional IVs having lost the ability to incorporate DNA, in which case it might be possible to identify the remnants of IVs in the wasp genome. Alternatively, they could be produced by as yet completely unknown cellular processes.
(b). Bracoviruses originate from an ancestral nudivirus
(i). Nudivirus genes are involved in Bracovirus production
The ovary transcriptomes of three braconid wasps, Chelonus inanitus (Cheloninae), Cotesia congregata (Microgastrinae) [10] and Microplitis demolitor [26], were analysed to identify genes involved in bracovirus production. A series of 29 nudivirus genes (nudiviruses are large dsDNA insect viruses related to baculoviruses) were expressed in the ovaries (table 1). Furthermore, one-third of these genes encoded BV particle components [10,27,28]. Last, 18 of these nudiviral genes, corresponding to baculovirus core genes [29], should perform essential functions of the virus cycle, based on functional characterization in baculoviruses [30] (table 1). Most viral functions [10,27] such as transcription, particle assembly and packaging and entry into host cells could be identified (table 1): (i) all RNA polymerase subunits involved in baculovirus transcription; (ii) genes encoding the equivalent of the major baculovirus capsid (VP39) [31,32] and a protein involved in nucleocapsid assembly (38K) [33]; (iii) all components of the baculovirus PIF complex involved in cell entry [34]. No transcripts involved in viral DNA replication could be identified, except for a nudiviral helicase gene in Microplitis demolitor [26].
Table 1.
Genes of nudiviral origin identified in braconid wasps. MdBV, Microplitis demolitor bracovirus; CcBV, Cotesia congregata bracovirus, CiBV, Chelonus inanitus bracovirus; HzNV1, Heliothis zea nudivirus-1; GbNV, Gryllus bimaculatus nudivirus; OrNV, Oryctes rhinoceros nudivirus; AcMNPV, Autographa californica multiple nucleopolyhedrovirus. +, gene present; −, gene absent; n.d. gene not isolated to date; n.a. not applicable.
| protein functiona | gene name | presence in bravoviruses |
ORF number in nudiviruses and baculoviruses |
variation in selectionb | |||||
|---|---|---|---|---|---|---|---|---|---|
| MdBV | CcBV | CiBV | HzNV1 | GbNV | OrNV | AcMNPV | (p-value) | ||
| nudivirus/baculovirus core genes | |||||||||
| replication | helicase | + | n.d. | n.d. | 104 | 88 | 34 | 95 | 0.45 |
| transcription | RNA polymerase p47 | + | + | n.d. | 75 | 69 | 20 | 40 | 0.04 |
| RNA polymerase lef-8 | + | + | + | 90 | 49 | 64 | 50 | <0.01 | |
| RNA polymerase lef-4 | + | n.d. | + | 98 | 96 | 42 | 90 | 0.03 | |
| RNA polymerase lef-9 | + | n.d. | n.d. | 75 | 24 | 96 | 62 | 0.07 | |
| initiation factor lef-5 | + | + | n.d. | 101 | 85 | 52 | 99 | 0.17 | |
| packaging and assembly | very late factor vlf-1 | + | n.d. | + | 121 | 80 | 30 | 77 | 0.01 |
| capsid protein vp91 | + | n.d. | + | 46 | 2 | 106 | 83 | <0.01 | |
| capsid protein vp39c | + | + | + | 89 | 64 | 15 | 89 | 0.31 | |
| viral phosphatase 38Kc | + | + | + | 10 | 1 | 87 | 98 | 0.17 | |
| sulfhydryloxidase p33 | + | n.d. | n.d. | 13 | 7 | 113 | 92 | 0.61 | |
| ODV envelope component (per os infectivity factor) | pif-0 (p74) | + | + | + | 11 | 45 | 126 | 138 | <0.01 |
| pif-1 | + | n.d. | + | 55 | 52 | 60 | 119 | <0.01 | |
| pif-2 | + | n.d. | + | 123 | 66 | 17 | 22 | <0.01 | |
| pif-3c | + | + | n.d. | 88 | 3 | 107 | 115 | <0.01 | |
| pif-4 (19 kDa) | + | + | + | 103 | 87 | 33 | 96 | 0.20 | |
| pif-5 (odv-e56) | + | + | + | 76 | 5 | 115 | 148 | 0.02 | |
| pif-6 (ac68)c | + | + | n.d. | 74 | 55 | 72 | 68 | 0.44 | |
| nudivirus/baculovirus genes | |||||||||
| envelope | odv-e66 | + | + | + | − | − | 12 | 46 | 0.10 |
| nudivirus-specific genes | |||||||||
| DNA processing | integrase | + | n.d. | + | 144 | 57 | 75 | − | n.a. |
| flap endonucleased | + | n.d. | n.d. | 68 | 65 | 16 | − | n.a. | |
| particle component | HzNVorf9-likec,e | + | + | + | 9 | − | − | − | n.a. |
| HzNVorf106-likee | + | + | + | 106 | − | − | − | n.a. | |
| HzNVorf118-likec,e,f | + | + | + | 118 | − | − | − | n.a. | |
| HzNVorf124-likeg | − | + | + | 124 | 95 | 41 | − | n.a. | |
| unknown | HzNVorf64-likef | + | + | n.d | 64 | − | − | − | n.a. |
| HzNVorf94-likec | + | + | nd | 94 | − | − | − | n.a. | |
| HzNVorf128-like | + | + | + | 128 | − | − | − | n.a. | |
| HzNVorf140-like | + | + | + | 140 | − | − | − | n.a. | |
| bracovirus-specific genes | |||||||||
| particle component | 17ae | + | + | + | − | − | − | − | n.a. |
| 30be | + | + | + | − | − | − | − | n.a. | |
| 35ae | + | n.d. | + | − | − | − | − | n.a. | |
| 97ae | + | n.d. | + | − | − | − | − | n.a. | |
| 97be | + | n.d. | + | − | − | − | − | n.a. | |
| 27bc,e | + | + | + | − | − | − | − | n.a. | |
| unknown | Cc50C22.5c | + | + | n.d. | − | − | − | − | n.a. |
| Cc50C22.6c | + | + | n.d. | − | − | − | − | n.a. | |
aProtein function assigned based on gene homology.
bBranch-site REL method was used to test whether the BV lineage was subjected to episodic diversifying selection compared with free-living viruses (significant p < 0.01).
cGene belonging to the nudiviral cluster identified within C. congregata wasp genome.
dProtein sequence similarity identified using probabilistic methods (HMMER program; http://hmmer.janelia.org/search/phmmer) and confirmed by orthologous sequence alignment.
eProtein identified as particle component in CcBV and/or CiBV.
fHzNVorf118-like is also referred to as PmV within bracovirus genomes and HzNVorf64-like as p51 within Heliothis zea nudivirus-1 genome.
gProtein identified as virion structural protein in HzNV-2 (11k).
Evolutionary rate analyses (GAbranch model [35]) performed on the whole nudiviral gene dataset showed they globally evolved under strong evolutionary constraints. Interestingly, the genetic algorithm showed the BV lineage had significantly higher evolutionary rates (0.06 < dN/dS < 0.2) than the free virus lineages (0.004 < dN/dS < 0.008). In particular, significant dN/dS increase (p < 0.01, table 1) was observed for a number of genes mostly involved in cell entry (pif genes). There are two hypotheses to explain these relaxed selection pressures in domesticated versus free viruses: (i) domestication could have lowered functional constraints on viral genes, to globally evolve at the same rate as the host genome; or (ii) the genes producing BV particles could have been subject to diversifying selection episodes leading, for example, to changes in the number of cell types the virus could enter.
(ii). Genomic organization of nudiviral genes and proviral segments
The integrated form of most nudiviral genes has been identified by analyses of a C. congregata genomic bacterial artificial chromosome clone library (see [15] in this issue). Consistent with nudivirus genome integration in ancestral wasps, half of the nudiviral genes are organized in a cluster within the wasp genome. The region is characterized by high gene density and contains intronless genes. This cluster encodes VP39 and 38K, the most abundant proteins in CiBV particles [28] and is amplified during CcBV virus particle production [36]. Other nudiviral genes are now dispersed in the C. congregata genome [10]. This scattering could be expected 100 myr after the ancestral nudivirus integration if there is no particular selective pressure to maintain these genes together [36].
The other part of the BV genome located within the wasp chromosomes is composed of proviral segments used to produce the multiple dsDNA circles packaged in the particles, which encodes virulence genes ensuring wasp larval development (figure 2). No nudiviral genes are contained in this packaged genome. In C. congregata, the proviral segment organization consists of a macrolocus comprising two-third of proviral sequences and seven dispersed loci each with one to three segments (see [15]). Comparisons between Cotesia and Glyptapanteles species [37], which diverged approximately 17 Ma [7], highlighted the homology and the remarkable genomic stability of the proviral integration sites, as orthologous hymenopteran genes were found in the flanking regions (see [15] in this issue).
A still unresolved question is the relative organization of nudiviral genes involved in particle production and the proviral segments in the wasp genome. Only one nudiviral gene, the odv-e66-like1 gene, which encodes a particle component in C. inanitus [28], has been found within the conserved proviral macrolocus [15]. This localization of odv-e66-like1 is unlikely to be random and thus sustain the hypothesis that the nudiviral machinery and proviral segments have a common origin [15]. The most important modification in BVs compared with pathogenic viruses resides in the fact that the nudiviral genes, including odv-e66-like1, have lost the ability to be incorporated in viral particles [10]. They have been totally replaced in the particles by genes originating from the wasp genome [15,37–39] or from mobile elements [40–42].
(iii). Bracovirus replication
So far, transcriptomic analyses have given no clues to elucidating BV replication. In BVs, viral DNA replication leads to the production of packaged circles from proviral segments (figure 2) [43]. First viral DNA sequences have to be amplified within wasp cells in nuclear virus factories. Contrary to initial hypotheses [44,45], it is not the circles themselves that are amplified from proviral segment excision but larger molecules [46] that are replicated linearly [36]. In C. congregata, two segments were found to amplify together within the same molecule as well as sequences not packaged in the particles [46]. Recent results showed that most proviral segments are similarly amplified as large DNA molecules comprising the sequences of several segments localized in tandem within the wasp genome [36]. This provirus amplification does not appear to involve nudiviral genes, because no DNA replication genes have been identified, apart from a helicase (table 1) [10,26]. However, BV DNA processing and encapsidation probably involves a number of nudiviral genes such as vlf-1, integrase and fen-like flap endonuclease (table 1) based on baculovirus functional homology and HMMER results [47].
(c). Ichnoviruses originate from an uncharacterized virus family
Transcriptomic analyses from the ovaries of the ichneumonid wasp Hyposoter didymator allowed the identification of the genes involved in IV particle production [11,48]. The genes expressed within ichneumonid wasp ovaries were not related to nudiviruses or to any other known viruses except for the p12 and p53 viral genes, which had previously been identified as structural proteins of Campoletis sonorensis IV (CsIV) [49]. Similarly as in BV, the genes encoding IV particle components do not possess introns and are organized within gene-rich regions of the wasp chromosomes. These regions are referred to as ichnovirus structural protein encoding regions (IVSPERs). In H. didymator, three IVSPERs sharing related genes belonging to seven gene families were identified. They are thought to correspond to the remnants of a duplicated virus genome [11,48]. IVSPER genes are conserved among phylogenetically distant IV-associated parasitoid wasps such as Tranosema rostrale [11] and Campoletis sonorensis [48]. Altogether, the data suggest IVSPERs derive from a common viral ancestor. However, the lack of similarity between IVSPER genes and any known pathogenic viral genes implies the IV ancestor belonged to a virus family for which no present-day members are described [11].
(d). Endogenous viral element domestication convergence
At present, there is no doubt that IV and BV particles originated from distinct viruses [10,11]. Virus domestication occurred at least twice as two parasitoid wasp lineages independently integrated viruses into their genomes. In a remarkable example of convergent evolution, this resulted in the wasps delivering into their hosts pathogenic genes contained in virus particles.
Why have several mutualist associations with large DNA viruses been described in parasitoid wasps and not elsewhere in the tree of life? Altogether, there are a number of arguments to explain why parasitoid wasps might have had enhanced probability of association with viruses resulting in long-term domestication. Parasitoid wasps use diverse strategies for controlling host physiology. They generally consist of the injection of virulence proteins produced in the venom gland or the ovaries [50]. The association with viruses might allow the production of a larger set of virulence factors at a lower physiological cost for the wasp. This evolutionary benefit may explain why PDVs have been selected repeatedly. Another explanation might be linked with the life-history traits of the wasps themselves: endoparasitoid wasps insert their ovipositors in several individuals of a lepidopteran population either to probe the host quality before oviposition or to feed on the host. This behaviour may have favoured encounters with viruses, which can themselves use the wasps as vectors for horizontal transmission, as is the case for ascoviruses [51,52]. It should be noted that pathogenic virus infection induced during oviposition may benefit the development of parasitoid larvae by inhibiting host defences [53]. Moreover, nudiviruses might be particularly prone to domestication because they are able to infect gonads [54] and/or to integrate their genomes in host cells [55].
3. Evolution of bracovirus genomes since integration
(a). Bracovirus genome evolution since integration within wasp genomes
Recent advances in wasp genomics combined with our knowledge of phylogenetics shed partial light on BV genome evolution. We can focus on two periods in the history of the microgastroid complex [15]. The first period concerns the events around the original viral genome integration over 100 Ma [7], and the second around 17 Ma relates to provirus divergence in Cotesia and Glyptapanteles (figures 1 and 3).
Figure 3.
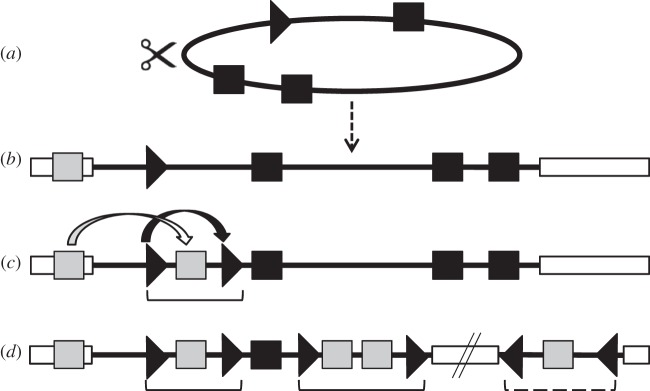
Bracovirus evolutionary model from an ancestral nudivirus. (a) Genome of the ancestral nudivirus; (b) initial nudivirus genome integration into a wasp genome; (c) formation of the first proviral segment and (d) simplified scheme of a present-day BV. Black squares are for nudiviral genes, black arrowhead for nudivirus derived circularization site, white rectangles for wasp genomes, grey squares are for wasp genes. Brackets are for BV proviral segments located in the macrolocus, hashed bracket is for an isolated proviral segment.
Bracoviruses evolved from integrated nudiviruses by several dramatic genomic transformations (figure 3). After integration of the ancestral nudivirus genome (figure 3a) into the wasp genome (figure 3b), the duplication of sequences, termed direct repeat junctions (DRJs; allowing virus genome excision and packaging), could have resulted in the packaging of wasp DNA instead of nudivirus genes in the particles (figure 3c,d). DRJs are specific DNA circularization sequence motifs flanking each segment and common to all bracoviruses [15]. This sequence motif could derive from the sequence of the ancestral nudivirus allowing the encapsidation of single genomes from concatemers produced during DNA replication [56]. Moreover, this period probably saw the first virulence gene translocations from the wasp genome towards proviral segments, resulting in the increase of viral particle capacity to counteract the immune system of the parasitized host (figure 3c). The acquisition of wasp genes by proviral segments and their subsequent diversification have produced a new entity that can be considered as an extended genome conferring to the wasp the ability to develop in their lepidopteran hosts [57], a notion akin to the extended phenotype [58]. It probably provided the braconid wasps with improved fitness, which favoured the diversification of both the microgastroid wasp complex and their symbiotic bracoviruses. Since genome integration 100 Ma, many events have occurred leading to diverse proviral genome organizations in different wasp lineages. However, comparative genomics on the Glyptapanteles and Cotesia genera revealed a surprising conservation in the localization of most proviral segments, which have remained stably integrated in braconid wasp genomes [15] since the separation of the wasps 17 Ma [7]. Proviral sequences were also subjected to specific gene transfers [37] and duplications [15] leading to unique PDV genome organizations in each wasp species.
(b). Gene transfers into bracoviruses
(i). Ancient wasp gene transfers into the provirus
Among the genes transferred into BVs, two families of virulence genes are particularly remarkable, the ankyrin and the ptp genes [59–62]. Apart from CiBV (Cheloninae), these genes are found in all BVs studied, including Toxoneuron nigriceps BV (Cardiochilinae) and MdBV, Glyptapanteles BV and Cotesia BV (Microgastrinae). Therefore, the insertion of the first ank and ptp genes must have happened before the separation of these different lineages 86 Ma (figure 1).
(ii). Recent gene transfers into the provirus
Although many PDV genes are similar to cellular genes, it has so far been difficult to formally demonstrate that they derived from gene transfer events between insect (wasp or lepidopteran) genomes and PDVs. High PDV gene divergence compared with insect homologues generally leads to loss of phylogenetic signal [63]. This divergence could be explained by the time (100 Myr) elapsed since integration in BV genomes but could also reflect high selection pressures imposed by the interaction with the lepidopteran host [60,63,64] (see §4). However, phylogenetic analyses of the sugar transporter gene family recently acquired by Glyptapanteles BVs showed the BV genes were more closely related to hymenopteran than lepidopteran genes [37]. To date, this represents the only robust example of gene transfer from wasps to PDVs. In addition, few packaged genes, such as a baculovirus p94 and an ascovirus gene, have most probably been acquired from other viruses by lateral transfer [41].
(c). Mechanisms involved in bracovirus gene evolution
Since integration of the nudivirus genome, different mechanisms, including duplications, gene mutations followed by selection and insertion of transposable elements (TEs), have been involved in shaping bracovirus genomes and droving the gene content diversity of contemporary BVs.
(i). Gene expansion is a hallmark of polydnaviridae genomes
A striking feature of PDV genomes is that over half of their genes belong to multigenic families. For example, the CcBV genome comprises 222 genes, 183 of which belong to 37 gene families. Combining phylogenetic analyses and provirus comparative genomics gave insights into the molecular evolution of the largest multigene family, the ptp gene family, with 13, 27, 32 and 42 members in MdBV, CcBV, GfBV and GiBV, respectively [64]. The ptp gene family expansion is linked to four major mechanisms: (i) large chromosomal segmental duplication of the provirus, (ii) tandem duplications of genes within segments, (iii) potential dispersed insertion of reverse transcribed RNA, and finally (iv) an original, bracovirus-specific novel duplication mechanism, which involves viral circle reintegration in the wasp genome [64] (see also §3d).
Comparisons of C. congregata BV proviral loci with those of related species gave further insights into the evolutionary dynamics of BV genomes. It appears that within the macrolocus, comprising two-third of the proviral segments, large tandem duplications encompassing several segments have played a major role in BV genome expansion [15]. Because duplication boundaries do not correspond to those of the segments [15], it is more likely that a chromosomal mechanism, such as those involved in duplications of insecticide resistance genes [65], rather than a specific viral process, is the basis of these duplications.
(ii). Transposable element and bracoviruses
A wide array of TEs of both class 1 (retrotransposons that mobilize via RNA intermediates) and class 2 (DNA transposons that mobilize via DNA intermediates) have been identified both in sequences flanking proviral segments and in circular encapsidated PDV genomes [66]. Like any eukaryotic genome, during over 100 Myr of evolution, proviral BVs have been largely exposed to mobile elements, many of which are now rearranged and no longer active. The recently described Maverick TEs were identified in parasitoid wasp genomes, and both in flanking wasp sequences next to GfBV proviral locus and within encapsidated CcBV sequences [37,42]. Functional Mavericks encode a retroviral-like integrase, and a number of proteins with homology to replication and packaging proteins of dsDNA viruses [67]. Phylogenetic analyses indicated the CcBV element derives from an endogenous wasp Maverick insertion within the provirus [42]. Mobile elements could therefore represent a means by which genes are transferred from parasitoid wasp genomes to proviral sequences. To date, however, few arguments support this hypothesis, with the exception of gene acquisition from cystatin and ptp-r transcripts that might have involved a retrotranscriptase activity [64,68], as described for retrogene production involving Line1 elements in vertebrates [69]. Some TE genes could also be used by PDVs in functional interactions with the lepidopteran host. For example, a BV gene showing sequence homology to a retroviral aspartyl protease is highly expressed in host haemocytes of the tobacco budworm, suggesting its implication in parasitism-induced host modifications [40].
(d). Bracovirus segment reintegration within wasp and lepidopteran host genomes
A re-emerging theme in the study of PDV evolution is the ability of viral circles to secondarily integrate into insect (lepidopteran or wasp) host genomic DNA [70]. Despite not replicating in lepidopteran host cells, chromosomal integration has been demonstrated both in vitro, in host and non-host derived cell cultures, and in vivo, in parasitized lepidopteran hosts for some PDV circles [70,71]. Viral circle reintegration (of ptp containing circles) in two different genomic locations of Cotesia sesamiae wasps have also been described [64]. Bracovirus DNA integration into lepidopteran host or secondary reintegration in wasp genomes might involve specific unknown mechanisms. Sequence comparison of circular and reintegrated viral sequences suggested that circle reintegration did not involve DRJs, but was mediated by specific sites in the circles, named left and right junction (figures 3 and 4 and table 2) [64,70]. Furthermore, reintegration also resulted in the deletion of a 40–53 bp viral region (indicated by Δ in figure 4). Similar viral reintegration boundaries have been identified in three BVs (CcBV, GiBV and MdBV), and a particular stretch of viral sequence was lost during the reintegration process (table 2). BV integration is likely more widespread than initially assumed, as for MdBV alone, all 15 circles can persist in cell cultures, and integration motifs could be identified in 12 segments [70]. Functionally, PDV genome integration into lepidopteran host DNA could be important for wasp parasitism success, if the integration process was required to maintain PDV expression in the late stages of parasitism [74,75], or during prolonged interactions of wasps with their hosts (during diapause [76]).
Figure 4.
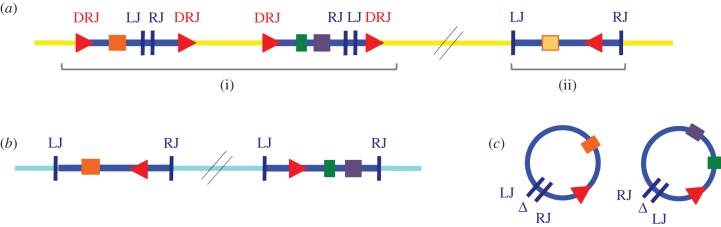
Bracovirus sequences integrated into insect genomic DNA and encapsidated circles. (a) BV segments integrated in wasp genomic DNA. Within the C. sesamiae wasp genome (in yellow), (i) classical proviral BV segments containing virulence genes (coloured rectangles) delimited by DRJ sequences (red triangles) and (ii) a reintegrated segment can be identified. The reintegrated segment is not delimited by DRJ, but is bordered by left junction (LJ) and right junction (RJ) sequences. (b) BV segments reintegrated in lepidopteran host. Within lepidopteran genomes (in light blue), reintegrated BV segments bordered by LJ and RJ sequences can also be identified. (c) Encapsidated BV circles injected in lepidopteran host. Sequence comparison between circular and reintegrated viral sequences (in wasp or lepidopteran genomes) show that circle reintegration in both cases involves loss of a stretch of viral sequence (indicated by Δ), and is mediated by similar reintegration boundaries (LJ, RJ), suggesting that BV use a specific but unknown mechanism to reintegrate into genomic DNA. See table 2 for sequences involved in BV circle circularization and reintegration.
Table 2.
Bracovirus circle motifs involved in genomic DNA integration. LJ, left junction, RJ, right junction.
| bracovirus | bracovirus encapsidated genome |
bracovirus circles reintegrated in genomic DNA |
||||||
|---|---|---|---|---|---|---|---|---|
| wasp host | DRJ | no. of segmentsa | host DNA | LJ | RJ | deletion (bp) | no. of segmentsa | |
| GiBV | G. indiensis | AGCTT | 24 [72] | Lepidopteran Lymantria dispar | CATGGT | n.d. | n.d. | 1 [71] |
| MdBV | M. demolitor | AGCTT | 13 [72] | Lepidopteran Pseudoplusia includens | ACCA | TAGT | 50–51 | 12 [70] |
| ACTA | TAGT | |||||||
| ACTT | TAGT | |||||||
| CsBV | C. sesamiae | AGCTT | 16 [73] | Hymenopteran | ACCA | TGGT | 40–53 | 3 [64] |
| C. sesamiae | TCCA | TGGT | ||||||
| ACCT | TGGA | |||||||
aNumber of segments on which the studies are based.
The fact that BV circles can reintegrate into the genomes of both wasp and lepidopteran hosts suggests BVs could mediate horizontal gene transfer between these insects. Initial experimental data indicate reintegrations do happen during natural interactions and could be more frequent than initially expected [64–70]. For PDV reintegration into wasp DNA, PDV circles would need to enter the wasp germline and to stably integrate into the wasp genome during the phase when parasitoid wasp eggs and larvae are exposed to virus circles in the haemolymph of the lepidopteran host. Concerning integration and transmission of PDV sequences in Lepidoptera, this implies wasp oviposition in semipermissive or non-permissive lepidopteran hosts that survive parasitism, and therefore ‘live to tell the tale’ of PDV integration [77,78].
4. Bracoviruses and wasp adaptation
Because all BVs originated from the capture of a single ancestral nudivirus by the common ancestor of microgastroid braconid parasitoid wasps, wasp and virus genomes are co-diversifying in a co-cladogenetic pattern [2,8,79]. Microgastrinae biodiversity analyses suggest that 94% of these wasps attack only one or two host species within a given geographical locality [80]. It is hypothesized that BVs, which have large genomes of highly diverse gene content, could drive the adaptation or specialization of parasitoid wasps to particular caterpillar hosts [8,73]. To date, the extent to which adaptation to hosts is determined or influenced by variation in BV gene content and sequence is rather fragmentary. Laboratory cross-protection experiments performed with three Microplitis BVs in two lepidopteran hosts suggested that BV-mediated immunosuppression is indeed one determinant of host range, along with other factors derived from the wasp larvae and the caterpillar [81]. Integrated approaches combining wasp ecological and life-history traits, phylogenetic analyses, comparative BV genomic data and controlled parasitism experiments should help determine whether BV gene sequences are genuinely linked to given host ranges.
We shall discuss the mechanisms that could be implicated in shaping BV genomic features involved in determining wasp host range and describe an ecological and phylogenomic framework where this work has been initiated.
(a). Mechanisms involved in shaping bracovirus genomes that could be involved in wasp adaptation
(i). Bracovirus gene content links with wasp life-history traits
Each packaged BV genome has a unique gene content, with most genes organized into gene families [15,82]. Among the 37 gene families so far identified, some are present in practically all BVs (i.e. ank, ptp), whereas others are specific to particular wasp lineages [5]. Specific genes or gene families could reflect how physiological interactions with different hosts have modelled these genomes. For example, despite originating from the same ancestral nudivirus, no common virulence genes have been identified to date between CiBV and other BV genomes [83]. Like all Cheloninae, C. inanitus oviposits into the eggs of lepidopteran hosts, whereas the other BV-associated wasps inject their eggs inside immune competent larvae (figure 1) [14]. Parasitism strategies involving the oviposition into host eggs versus larvae, exposing the wasp larvae to different physiological contexts, could explain the different panel of virulence proteins found in chelonines [83,84] compared with other wasps. Other differences in life-history strategies of the wasps could impact BV gene sets, for example, solitary versus gregarious development [5].
(ii). Bracovirus gene evolution: duplications and diversification
The remarkable expansion of many PDV genes into multigene families is a feature that could also be associated with wasp adaptive capacities. Gene duplications and their subsequent divergence play an important role in the evolution of novel gene functions [85,86]. In host–parasitoid interactions where endoparasitoid wasps develop within caterpillars and have to face the arsenal of a functional immune system, BV virulence genes are expected to be under strong evolutionary constraints. Selection pressures are imposed on BV genes to overcome new host resistances either in the process of a coevolutionary arms race with the host species in which the wasp is established, or in the process of host switch or host range expansion. Duplications and subsequent divergence of PDV genes could therefore be involved in wasp adaptation to new or evolving hosts.
In BVs, molecular evolutionary approaches gave insights into the evolutionary processes involved in the expansion and diversification of particular BV multigene families [64,73,87]. To assess whether BV genes are coevolving with their lepidopteran hosts, rates of non-synonymous versus synonymous substitutions were initially measured at the interspecific level. Higher rates of amino acid changes, leading to innovation of protein function [88,89], are expected in BV genes involved in wasp adaptation to hosts. High positive selection was identified in cystatins, which are inhibitors of C1 cysteine proteases, and the positively selected residues were located in the vicinity of active sites assumed to directly interact with host proteases [87]. In the case of different ptp genes, the largest BV gene family, some gene copies evolved under relaxed selection pressure, whereas others underwent positive selection episodes [64,73]. For example, we detected selected amino acids within PTPE and PTPX in regions predicted to be involved in PTP substrate specificity suggesting recent protein target shifts in the host [64]. The evolution of the ptp gene family shows (i) evidence for classical gene duplication models assuming fixation of the duplicated copy is a neutral process, as well as (ii) evidence for alternative models proposing the gene duplication process is itself under positive natural selection [86]. Evidence for gene loss implying a ‘birth and death’ model (iii) could also be observed [90]. According to the ‘birth and death’ model, genes arise continuously by duplication and are lost by deletion or by mutational events. An ongoing process of pseudogenization was also observed for copies corresponding to different ptp genes in different species [60]. As shown in primates, Drosophila or large DNA viruses such as poxviruses, gene expansion and contraction could explain important adaptive traits allowing physiological adaptations of their host species [91–93]. The expansion of the ptp gene family could therefore be an important source of evolutionary innovations conferring new adaptive traits to the wasps. In accordance, different ptp expression patterns and different functions have been described in the context of host–parasite interactions [16].
The challenge is now to link mutations, diversifying selection and gene gain and loss observed in BV genomes with potential wasp host shifts, or wasp counter-adaptations to resistant hosts. In order to do this, it is now important to be able to perform similar studies on wasps for which reliable ecological data is available. Only a complete dataset, combining wasp phylogeny, wasp ecology (i.e. host range) and selection analyses, could enable us to understand the consequences of BV gene content and evolution on wasp specialization.
(b). Case study: Cotesia sesamiae
To understand the potential adaptive role of bracoviruses, one needs to conduct detailed population studies of the wasps in an ecological context that includes accurate knowledge of their host range. However, there are currently few models linking BV genes to the ecological adaptation of their carrier wasps. One of the best examples, the wasp C. sesamiae, parasitizes over 20 species of African stem borers belonging to the Noctuidae and Crambidae families [94]. As its hosts include major cereal pests, such as Busseola fusca and Sesamia calamistis, detailed host preference studies have been done for biological control improvement. Busseola fusca was found to be either susceptible or resistant to parasitization by different C. sesamiae populations [95]. These wasp populations were found to carry different alleles of the CrV1 bracovirus gene. These alleles, labelled virulent and avirulent, are involved in the success or failure to parasitize B. fusca [96,97]. Only the wasps of overlapping geographical range with B. fusca were found to carry the virulent CrV1 BV allele, suggesting wasp adaptation is linked with this BV gene [96]. Comprehensive population studies further showed that host range was the main factor explaining C. sesamiae population structure based on eight CrV1 alleles [94]. This suggested that, even though most wasp populations were generalists parasitizing several host species, cryptic specialization could occur [94]. However, based on partial genomic data, C. sesamiae BVs encode over 130 genes, mostly involved in physiological and immune interactions [73]. Genome-wide molecular evolution studies revealed at least 17 genes, including CrV1, histone H4, ep1, ep2 and lectin, were under positive selection within the Cotesia genus and likely implicated in wasp ecological adaptation [73]. A comparative study of three positively selected genes, namely CrV1, histone-H4 and ep2, in 21 wild C. sesamiae populations associated with different host species, identified several haplotypes for the three BV genes. Different allelic combinations of these genes, implicated in several immune pathways, were found in different populations. In addition, the signature of positive selection was detected in the three genes, but in branches leading to different wasp groups. This suggested the three genes carry different adaptive potential, depending on local host–wasp interactions [73]. As they can parasitize many lepidopteran species, C. sesamiae wasps are involved in a complex landscape comprising multiple coevolutionary interactions, which can take them through different directions to different adaptive peaks. Cotesia sesamiae relies on several BV mediated molecular pathways to overcome multiple host resistance. There is a need for further integrative studies to detail the role BVs play in the ecological adaptation of the wasps.
(c). Evolutionary model for polydnavirus genome extension
In the context of parasitoid evolution, the multiple EVE domestications appear to have been particularly beneficial to the diversification of the wasps, based on species richness [12,13,98,99]. However, there might be a cost to the wasp for the replication of PDVs. Some wasps do not use PDVs at all, and the case of V. canescens [25,100] suggests reversal from PDV use could be possible. In this light, why are PDV genomes so large? Serial knockout experiments could help determine whether all PDV genes are absolutely required to foster wasp development. However, working in non-model systems has so far been problematic for this kind of approach. Furthermore, they would be unlikely to reflect the complete picture of the conditions a wasp could encounter in the wild. The maintenance of large PDV genomes, at least in the case of BVs, might be explained by the way viral DNA circles are packaged in the particles. There is no real constraint on the size of the DNA circles encapsidated in the virus particles, and the segmented nature of the genome allows the presence of many genes. If the main cost encountered by the wasp is on the production of the viral particles, the encapsidation of many versus few virulence genes might not be as costly as it seems at first sight. Most of the physiological cost is endorsed by the parasitized host, which express the viral genes using its own cellular machinery.
We would like to propose a model, based on the gene for gene coevolution model [101], to explain how ecological constraints could foster PDV genome extension (figure 5). The model assumes that the only issue to a parasitism challenge is the death of one of the partners, either the wasp or the caterpillar, as is the case in host–parasitoid interactions. In the gene for gene model, a single host mutation can counteract the virulence gene, but is in turn overcome by a single parasite gene mutation (SLR in figure 5a). Taking on a new locus or even a new resistance metabolic pathway to overcome virulence gives an immediate advantage to the host (MLR in figure 5a). In the case of PDVs, we can postulate the transfer of new virulence genes or duplication and diversification of existing virulence genes in PDV circles could overturn this kind of multilocus resistance (figure 5a). If the cost of encapsidation is low, several virulence genes could be assayed at once, but only the gene conferring a genuine selective advantage should be retained through time; non-adaptive genes could be lost through pseudogenization [15]. So far, we have placed this arms race in the context where the wasps parasitize a single host species. However, we know from C. sesamiae that the situation is far more complex in the field where the wasps can develop on several hosts within a locality [94]. We therefore extended our model to two hosts, both harbouring multilocus resistance (figure 5b). In this situation, the PDV has to produce twice as many virulence genes to overcome distinct resistances. Even if both hosts can rely on the same pathways (and even on orthologous genes) to overcome parasitism (MLS in figure 5b), point mutations in each host gene could be sufficient to confer resistance to a particular virulence protein (MLR in figure 5b). The acquisition by the PDV of new virulence genes targeting each of these resistance mutations would be necessary to overturn resistance in both host species (MLR in figure 5b). The model therefore proposes an explanation for the large gene family extension observed in PDV genomes in the ecological adaptive landscape in which parasitoid wasps evolve. Only detailed population genomics studies, in multiple host species, could validate these hypotheses for global genome extension.
Figure 5.

Polydnavirus mediated host parasite coevolution. (a) PDV adaptation to a single host species harbouring either a single locus susceptibility (SLS), a single locus resistance (SLR) or multilocus resistance (MLR); (b) PDV adaptation to two hosts species harbouring either a multilocus susceptibility (MLS), or multilocus resistance (MLR). Pie and semicircle shapes indicate PDV genes; squares and circles host resistance genes. Corresponding shapes and colours indicate an efficient PDV effector targeting of host factor allowing the wasp larvae to develop and the transmission of the PDV gene in their chromosomes. Lightning shapes depict mutations and virulence or resistance gene acquisitions.
5. Conclusion
Nudiviral genes in braconid wasp genomes can be considered as EVEs because they originated from a nudivirus integrated into the genome of wasp ancestor species [10]. The same holds true for ichneumonid IVSPER genes, even if the corresponding pathogenic virus has yet to be characterized [11]. Contrary to most EVEs studied [4], bracoviruses are not fossil genomic remnants but active viruses. Indeed, most viral functions described for pathogenic viruses have been conserved and BVs undergo all the steps of a classical virus cycle. The main difference between PDVs and conventional viruses resides in the separation of the virus cycle between two cell types and hosts: calyx cells in the wasp ovaries produce infectious particles, and parasitized lepidopteran host cells are infected. This conforms to the definition of viruses as infectious agents with nucleic acid genomes, which replicate inside living host cells to produce particles transferring their genome to other cells [102].
In viruses, however, the genome packaged in the particles is usually supposed to contain all the information required for replication, which is not the case for PDVs. As obligatory symbionts, PDVs contain a genome involved in wasp adaptation to its host, and the information required for particle production now resides permanently in the wasp genome. PDVs are not the only viruses transferring genetic material other than their own. In prokaryotes, several phages of independent origins have been identified in a wide range of species. They were called gene transfer agents (GTAs) [103]. GTAs mediate genetic exchanges between bacteria of the same species in particular environmental conditions by transmitting random bacterial DNA instead of their own. To account for this functional diversity in the viral world, Stoltz & Krell [104] recently proposed establishing three categories: classical viruses, GTAs and endogenous viruses.
Genomic advances in all branches of the tree of life should reveal the diversity of viruses and EVEs associated with cellular organisms. This would give clues as to whether symbiotic viruses are a particular feature of parasitoid wasps or are fairly common, such as GTA in prokaryotes, and have been ignored in less studied taxa. Within parasitoid wasps, deep sequencing of closely related wasps with contrasting life-histories and host ranges would give new insights as to whether particular PDV genes or gene sets are required for wasp adaptation. The PDV genome extension model predicts generalist wasps should harbour more PDV genes than specialists.
Acknowledgements
We thank G. Rohrmann for discussion on gene homology and P. Gayral, J. Gauthier and F. Dedeine for discussion.
Funding statement
This work was supported by the ERC project no. 205206 ‘GENOVIR’ and the ANR project ‘Paratoxose’.
References
- 1.Emerman M, Malik HS. 2010. Paleovirology: modern consequences of ancient viruses. PLoS Biol. 8, e1000301 (doi:10.1371/journal.pbio.1000301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thézé J, Bézier A, Periquet G, Drezen J-M, Herniou EA. 2011. Paleozoic origin of insect large dsDNA viruses. Proc. Natl Acad. Sci. USA 108, 15 931–15 935 (doi:10.1073/pnas.1105580108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katzourakis A, Gifford RJ, Tristem M, Gilbert MT, Pybus OG. 2009. Macroevolution of complex retroviruses. Science 325, 1512 (doi:10.1126/science.1174149) [DOI] [PubMed] [Google Scholar]
- 4.Katzourakis A, Gifford RJ. 2010. Endogenous viral elements in animal genomes. PLoS Genet. 6, e1001191 (doi:10.1371/journal.pgen.1001191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strand MR, Burke GR. 2012. Polydnaviruses as symbionts and gene delivery systems. PLoS Pathog. 8, e1002757 (doi:10.1371/journal.ppat.1002757) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmes EC. 2011. The evolution of endogenous viral elements. Cell Host Microbe 10, 368–377 (doi:10.1016/j.chom.2011.09.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy N, Banks JC, Whitfield JB, Austin AD. 2008. Phylogeny of the parasitic microgastroid subfamilies (Hymenoptera: Braconidae) based on sequence data from seven genes, with an improved time estimate of the origin of the lineage. Mol. Phylogenet. Evol. 47, 378–395 (doi:10.1016/j.ympev.2008.01.022) [DOI] [PubMed] [Google Scholar]
- 8.Whitfield JB, O'Connor JM. 2012. Molecular systematics of wasps and polydnavirus genomes and their coevolution. In Parasitoid viruses: symbionts and pathogens (eds Beckage NE, Drezen J-M.), pp. 89–97 Oxford, UK: Elsevier [Google Scholar]
- 9.Quicke DLJ, Laurenne NM, Fitton MG, Broad GR. 2009. A thousand and one wasps: a 28S rDNA and morphological phylogeny of the Ichneumonidae (Insecta: Hymenoptera) with an investigation into alignment parameter space and elision. J. Nat. Hist. 43, 1305–1421 (doi:10.1080/00222930902807783) [Google Scholar]
- 10.Bézier A, et al. 2009. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science 323, 926–930 (doi:10.1126/science.1166788) [DOI] [PubMed] [Google Scholar]
- 11.Volkoff AN, et al. 2010. Analysis of virion structural components reveals vestiges of the ancestral ichnovirus genome. PLoS Pathog. 6, e1000923 (doi:10.1371/journal.ppat.1000923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauld ID. 1988. Evolutionary patterns of host utilization by ichneumonoid parasitoids (Hymenoptera: Ichneumonidae and Braconidae). Biol. J. Linn. Soc. 35, 351–377 (doi:10.1111/j.1095-8312.1988.tb00476.x) [Google Scholar]
- 13.Quicke DLJ. 1997. Parasitic wasps. London, UK: Chapman and Hall [Google Scholar]
- 14.Godfray HCJ. 1994. Parasitoids: behavorial and evolutionary ecology. Princeton, NJ: Princeton University Press [Google Scholar]
- 15.Bézier A, et al. 2013. Functional endogenous viral elements in the genome of the parasitoid wasp Cotesia congregata: insights into the evolutionary dynamics of bracoviruses. Phil. Trans. R. Soc. B 368, 20130047 (doi:10.1098/rstb.2013.0047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreau S, Huguet E, Drezen J-M. 2009. Polydnaviruses as tools to deliver wasp virulence factors to impair lepidopteran host immunity. In Insect infection and immunity: evolution, ecology and mechanisms (eds Reynolds S, Rolff J.), pp. 137–158 Oxford, UK: Oxford University Press [Google Scholar]
- 17.Moreau SJM. 2003. Relationships between parasitoid Hymenoptera and the immune system of their hosts: the “active” and “passive” mechanisms redefined. Ann. Soc. Entomol. Fr. 39, 305–314 (doi:10.1080/00379271.2003.10697389) [Google Scholar]
- 18.Beckage NE, Gelman DB. 2004. Wasp parasitoid disruption of host development: Implications for new biologically based strategies for insect control. Annu. Rev. Entomol. 49, 299–330 (doi:10.1146/annurev.ento.49.061802.123324) [DOI] [PubMed] [Google Scholar]
- 19.Beckage NE. 2012. Polydnaviruses as endocrine regulators. In Parasitoid viruses: symbionts and pathogens (eds Beckage NE, Drezen J-M.), pp. 163–168 Oxford, UK: Elsevier [Google Scholar]
- 20.Strand MR, Drezen J-M. 2012. Polydnaviridae. In Virus taxonomy classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses (eds Adams LK, Carstens), pp. 267–278 Amsterdam, The Netherlands, Elsevier [Google Scholar]
- 21.Lapointe R, Tanaka K, Barney WE, Whitfield JB, Banks JC, Beliveau C, Stoltz D, Webb BA, Cusson M. 2007. Genomic and morphological features of a banchine polydnavirus: a comparison with bracoviruses and ichnoviruses. J. Virol. 81, 6491–6501 (doi:10.1128/JVI.02702-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizki RM, Rizki TM. 1990. Parasitoid virus-like particles destroy Drosophila cellular immunity. Proc. Natl Acad. Sci. USA 87, 8388–8392 (doi:10.1073/pnas.87.21.8388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barratt BI, Evans AA, Stoltz DB, Vinson SB, Easingwood R. 1999. Virus-like particles in the ovaries of Microctonus aethiopoides Loan (Hymenoptera: Braconidae), a parasitoid of adult weevils (Coleoptera: Curculionidae). J. Invertebr. Pathol. 73, 182–188 (doi:10.1006/jipa.1998.4826) [DOI] [PubMed] [Google Scholar]
- 24.Schmidt O, Schuchmann-Feddersen I. 1989. Role of virus-like particles in parasitoid-host interaction of insects. Subcell Biochem. 15, 91–119 [DOI] [PubMed] [Google Scholar]
- 25.Rotheram S. 1967. Immune surface of eggs of a parasitic insect. Nature 214, 700 (doi:10.1038/214700a0) [DOI] [PubMed] [Google Scholar]
- 26.Burke GR, Strand MR. 2012. Deep sequencing identifies viral and wasp genes with potential roles in replication of Microplitis demolitor bracovirus. J. Virol. 86, 3293–3306 (doi:10.1128/JVI.06434-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bézier A, Herbinière J, Lanzrein B, Drezen J-M. 2009. Polydnavirus hidden face: the genes producing virus particles of parasitic wasps. J. Invertebr. Pathol. 101, 194–203 (doi:10.1016/j.jip.2009.04.006) [DOI] [PubMed] [Google Scholar]
- 28.Wetterwald C, et al. 2010. Identification of bracovirus particle proteins and analysis of their transcript levels at the stage of virion formation. J. Gen. Virol. 91, 2610–2619 (doi:10.1099/vir.0.022699-0) [DOI] [PubMed] [Google Scholar]
- 29.Herniou EA, Olszewski JA, Cory JS, O'Reilly DR. 2003. The genome sequence and evolution of baculoviruses. Annu. Rev. Entomol. 48, 211–234 (doi:10.1146/annurev.ento.48.091801.112756) [DOI] [PubMed] [Google Scholar]
- 30.Rohrman GF. Bethesda, MD:: National Center for Biotechnology Information; 2011. Baculovirus molecular biology, 2nd edn (Internet) [Google Scholar]
- 31.Pearson MN, Russell RLQ, Rohrmann GF, Beaudreau GS. 1988. P39, A major baculovirus structural protein: immunocytochemical characterization and genetic location. Virology 167, 407–413 [PubMed] [Google Scholar]
- 32.Thiem SM, Miller LK. 1989. Identification, sequence, and transcriptional mapping of the major capsid protein gene of the baculovirus Autographa californica nuclear polyhedrosis virus. J. Virol. 63, 2008–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu W, Lin T, Pan L, Yu M, Li Z, Pang Y, Yang K. 2006. Autographa californica multiple nucleopolyhedrovirus nucleocapsid assembly is interrupted upon deletion of the 38K gene. J. Virol. 80, 11 475–11 485 (doi:10.1128/JVI.01155-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng K, van Oers M, Hu Z, van Lent J, Vlak J. 2010. Baculovirus per os infectivity factors form a complex on the surface of occlusion-derived virus. J. Virol. 84, 9497–9504 (doi:10.1128/JVI.00812-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pond SLK. 2004. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679 (doi:10.1093/bioinformatics/bti079) [DOI] [PubMed] [Google Scholar]
- 36.Louis F, Bézier A, Periquet G, Ferras C, Drezen J-M, Dupuy C. In press. Bracovirus DNAs and the nudiviral cluster involved in particle production are amplified in several replication units. J. Virol. (doi:10.1128/JVI.00886-13) [Google Scholar]
- 37.Desjardins CA, et al. 2008. Comparative genomics of mutualistic viruses of Glyptapanteles parasitic wasps. Genome Biol. 9, R183 (doi:10.1186/gb-2008-9-12-r183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Espagne E, Dupuy C, Huguet E, Cattolico L, Provost B, Martins N, Poirié M, Periquet G, Drezen J-M. 2004. Genome sequence of a polydnavirus: insights into symbiotic virus evolution. Science 306, 286–289 (doi:10.1126/science.1103066) [DOI] [PubMed] [Google Scholar]
- 39.Webb BA, et al. 2006. Polydnavirus genomes reflect their dual roles as mutualists and pathogens. Virology 347, 160–174 (doi:10.1016/j.virol.2005.11.010) [DOI] [PubMed] [Google Scholar]
- 40.Falabella P, Varricchio P, Gigliotti S, Tranfaglia A, Pennacchio F, Malva C. 2003. Toxoneuron nigriceps polydnavirus encodes a putative aspartyl protease highly expressed in parasitized host larvae. Insect Mol. Biol. 12, 9–17 (doi:10.1046/j.1365-2583.2003.00382.x) [DOI] [PubMed] [Google Scholar]
- 41.Drezen J-M, Bézier A, Lesobre J, Huguet E, Cattolico L, Periquet G, Dupuy C. 2006. The few virus-like genes of Cotesia congregata bracovirus. Arch. Insect Biochem. Physiol. 61, 110–122 (doi:10.1002/arch.20108) [DOI] [PubMed] [Google Scholar]
- 42.Dupuy C, Periquet G, Serbielle C, Bézier A, Louis F, Drezen J-M. 2011. Transfer of a chromosomal Maverick to endogenous bracovirus in a parasitoid wasp. Genetica 139, 489–496 (doi:10.1007/s10709-011-9569-x) [DOI] [PubMed] [Google Scholar]
- 43.Dupuy C, Gundersen-Rindal D, Cusson M. 2012. Genomics and replication of polydnaviruses. In Parasitoid viruses: symbionts and pathogens (eds Beckage NE, Drezen J-M.), pp. 47–58 Oxford, UK: Elsevier [Google Scholar]
- 44.Savary S, Beckage N, Tan F, Periquet G, Drezen J-M. 1997. Excision of the polydnavirus chromosomal integrated EP1 sequence of the parasitoid wasp Cotesia congregata (Braconidae, Microgastinae) at potential recombinase binding sites. J. Gen. Virol. 78, 3125–3134 (doi:10.1046/j.1365-2583.1999.83130.x) [DOI] [PubMed] [Google Scholar]
- 45.Gruber A, Stettler P, Heiniger P, Schümperli D, Lanzrein B. 1996. Polydnavirus DNA of the braconid wasp Chelonus inanitus is integrated in the wasp's genome and excised only in later pupal and adult stages of the female. J. Gen. Virol. 77, 2873–2879 (doi:10.1099/0022-1317-77-11-2873) [DOI] [PubMed] [Google Scholar]
- 46.Pasquier-Barre F, Dupuy C, Huguet E, Monteiro F, Moreau A, Poirie M, Drezen J-M. 2002. Polydnavirus replication: the EP1 segment of the parasitoid wasp Cotesia congregata is amplified within a larger precursor molecule. J. Gen. Virol. 83, 2035–2045 [DOI] [PubMed] [Google Scholar]
- 47.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (doi:10.1093/nar/gkr367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volkoff AN, Drezen J-M, Cusson M, Webb BA. 2012. The organisation of genes encoding icnovirus structural proteins. In Parasitoid viruses: symbionts and pathogens (eds Beckage NE, Drezen J-M.), pp. 33–45 Oxford, UK: Elsevier [Google Scholar]
- 49.Deng L, Stoltz DB, Webb BA. 2000. A gene encoding a polydnavirus structural polypeptide is not encapsidated. Virology 269, 440–450 (doi:10.1006/viro.2000.0248) [DOI] [PubMed] [Google Scholar]
- 50.Poirié M, Carton Y, Dubuffet A. 2009. Virulence strategies in parasitoid Hymenoptera as an example of adaptive diversity. C. R. Biol. 332, 311–320 (doi:10.1016/j.crvi.2008.09.004) [DOI] [PubMed] [Google Scholar]
- 51.Govindarajan R, Federici BA. 1990. Ascovirus infectivity and effects of infection on the growth and development of noctuid larvae. J. Invertbr. Pathol. 56, 291–299 (doi:10.1016/0022-2011(90)90115-M) [DOI] [PubMed] [Google Scholar]
- 52.Tilmann P, Styer E, Hamm J. 2004. Transmission of ascovirus from Heliothis virescens (Lepidoptera: Noctuidae) by three parasitoids and effects of virus on survival of parasitoid Cardiochiles nigriceps (Hymenoptera: Braconidae). Environ. Entomol. 33, 633–643 (doi:10.1603/0046-225X-33.3.633) [Google Scholar]
- 53.Renault S, Petit A, Benedet F, Bigot S, Bigot Y. 2002. Effects of the Diadromus pulchellus ascovirus, DpAV-4, on the hemocytic encapsulation response and capsule melanization of the leek-moth pupa, Acrolepiopsis assectella. J. Insect Physiol. 48, 297–302 (doi:10.1016/S0022-1910(01)00174-3) [DOI] [PubMed] [Google Scholar]
- 54.Raina AK, Adams JR. 1995. Gonad-specific virus of corn earworm. Nature 374, 770 (doi:10.1038/374770a0) [DOI] [PubMed] [Google Scholar]
- 55.Lin C-L, Lee J-C, Chen S-S, Wood HA, Li M-L, Li C-F, Chao Y-C. 1999. Persistent Hz-1 virus infection in insect cells: evidence for insertion of viral DNA into host chromosomes and viral infection in a latent status. J. Virol. 73, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanarsdall AL, Mikhailov VS, Rohrmann GF. 2007. Baculovirus DNA replication and processing. Curr. Drug Targets 8, 1096–1102 (doi:10.2174/138945007782151397) [DOI] [PubMed] [Google Scholar]
- 57.Periquet G. 2009. Transferts d'ADN, domestication et genomes étendus: le cas des virus d'hyménoptères parasitoïdes. C. R. Acad. Agric. Fr. 95, 99–102 [Google Scholar]
- 58.Dawkins R. 1999. The extended phenotype: the long reach of the gene. Oxford, UK: Oxford University Press [Google Scholar]
- 59.Thoetkiattikul H, Beck MH, Strand MR. 2005. Inhibitor kappa B-like proteins from a polydnavirus inhibit NF-kappa B activation and suppress the insect immune response. Proc. Natl Acad. Sci. USA 102, 11 426–11 431 (doi:10.1073/pnas.0505240102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Provost B, et al. 2004. Bracoviruses contain a large multigene family coding for protein tyrosine phosphatases. J. Virol. 78, 13 090–13 103 (doi:10.1128/jvi.78.23.13090-13103.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Falabella P, et al. 2007. Characterization of the IkappaB-like gene family in polydnaviruses associated with wasps belonging to different Braconid subfamilies. J. Gen. Virol. 88, 92–104 (doi:10.1099/vir.0.82306-0) [DOI] [PubMed] [Google Scholar]
- 62.Chen YP, Taylor PB, Shapiro M, Gundersen-Rindal DE. 2003. Quantitative expression analysis of a Glyptapanteles indiensis polydnavirus protein tyrosine phosphatase gene in its natural lepidopteran host, Lymantria dispar. Insect Mol. Biol. 12, 271–280 (doi:10.1046/j.1365-2583.2003.00411.x) [DOI] [PubMed] [Google Scholar]
- 63.Bézier A, Herbinière J, Serbielle C, Lesobre J, Wincker P, Huguet E, Drezen J-M. 2008. Bracovirus gene products are highly divergent from insect proteins. Arch. Insect Biochem. Physiol. 67, 172–187 (doi:10.1002/arch.20219) [DOI] [PubMed] [Google Scholar]
- 64.Serbielle C, Dupas S, Perdereau E, Héricourt F, Dupuy C, Huguet E, Drezen J-M. 2012. Evolutionary mechanisms driving the evolution of a large polydnavirus gene family coding for protein tyrosine phosphatases. BMC Evol. Biol. 12, 253 (doi:10.1186/1471-2148-12-253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weill M, et al. 2003. Comparative genomics: insecticide resistance in mosquito vectors. Nature 423, 136–137 (doi:10.1038/423136b) [DOI] [PubMed] [Google Scholar]
- 66.Gundersen-Rindal D, Dupuy C, Huguet E, Drezen J-M. 2013. Parasitoid polydnaviruses: evolution, pathology and applications. Biocontrol Sci. Technol. 23, 1–61 (doi:10.1080/09583157.2012.731497) [Google Scholar]
- 67.Pritham EJ, Putliwala T, Feschotte C. 2007. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 390, 3–17 (doi:10.1016/j.gene.2006.08.008) [DOI] [PubMed] [Google Scholar]
- 68.Espagne E, et al. 2005. A virus essential for insect host–parasite interactions encodes cystatins. J. Virol. 79, 9765–9776 (doi:10.1128/JVI.79.15.9765-9776.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Esnault C, Maestre J, Heidmann T. 2000. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 24, 363–367 (doi:10.1038/74184) [DOI] [PubMed] [Google Scholar]
- 70.Beck MH, Zhang S, Bitra K, Burke GR, Strand MR. 2011. The encapsidated genome of Microplitis demolitor bracovirus integrates into the host Pseudoplusia includens. J. Virol. 85, 11 685–11 696 (doi:10.1128/JVI.05726-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gundersen-Rindal D. 2003. Polydnavirus integration in lepidopteran host cells in vitro. J. Insect Physiol. 49, 453–462 (doi:10.1016/s0022-1910(03)00062-3) [DOI] [PubMed] [Google Scholar]
- 72.Desjardins CA, et al. 2007. Structure and evolution of a proviral locus of Glyptapanteles indiensis bracovirus. BMC Microbiol. 7, 61 (doi:10.1186/1471-2180-7-61) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jancek S, et al. 2013. Adaptive selection on bracovirus genomes drives the specialization of Cotesia parasitoid wasps. PLoS ONE 8, e64432 (doi:10.1371/journal.pone.0064432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bonvin M, Kojic D, Blank F, Annaheim M, Wehrle I, Wyder S, Kaeslin M, Lanzrein B. 2004. Stage-dependent expression of Chelonus inanitus polydnavirus genes in the host and the parasitoid. J. Insect Physiol. 50, 1015–1026 (doi:10.1016/j.jinsphys.2004.09.002) [DOI] [PubMed] [Google Scholar]
- 75.Weber B, Annaheim M, Lanzrein B. 2007. Transcriptional analysis of polydnaviral genes in the course of parasitization reveals segment-specific patterns. Arch. Insect Biochem. Physiol. 66, 9–22 (doi:10.1002/arch.20190) [DOI] [PubMed] [Google Scholar]
- 76.Cusson M, Stolz D, Lapointe R, Béliveau C, Nisole A, Volkoff AN, Drezen J-M, Maaroufi H, Levesque RC. 2012. Genomics of banchine ichnoviruses: insights into their relationship to bracoviruses and campoplegine ichnoviruses. In Parasitoid viruses: symbionts and pathogens (eds Beckage N, Drezen J-M.), pp. 79–87 Oxford, UK: Elsevier [Google Scholar]
- 77.Harwood SH, McElfresh JS, Nguyen A, Conlan CA, Beckage NE. 1998. Production of early expressed parasitism-specific proteins in alternate sphingid hosts of the braconid wasp Cotesia congregata. J. Invertebr. Pathol. 71, 271–279 (doi:10.1006/jipa.1997.4745) [DOI] [PubMed] [Google Scholar]
- 78.Beckage NE, Tan FF. 2002. Development of the braconid wasp Cotesia congregata in a semi-permissive noctuid host, Trichoplusia ni. J. Invertebr. Pathol. 81, 49–52 (doi:10.1016/S0022-2011(02)00112-X) [DOI] [PubMed] [Google Scholar]
- 79.Whitfield JB. 2002. Estimating the age of the polydnavirus/braconid wasp symbiosis. Proc. Natl Acad. Sci. USA 99, 7508–7513 (doi:10.1073/pnas.112067199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith MA, Rodriguez JJ, Whitfield JB, Deans AR, Janzen DH, Hallwachs W, Hebert PDN. 2008. Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proc. Natl Acad. Sci. USA 105, 12 359–12 364 (doi:10.1073/pnas.0805319105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kadash K. 2003. Cross-protection experiments with parasitoids in the genus Microplitis (Hymenoptera: Braconidae) suggest a high level of specificity in their associated bracoviruses. J. Insect Physiol. 49, 473–482 (doi:10.1016/s0022-1910(03)00064-7) [DOI] [PubMed] [Google Scholar]
- 82.Huguet E, Serbielle C, Moreau SJM. 2012. Evolution and origin of polydnavirus virulence genes. In Parasitoid viruses: symbionts and pathogens (eds Beckage NE, Drezen J-M.), pp. 63–78 London, UK: Elsevier [Google Scholar]
- 83.Annaheim M, Lanzrein B. 2007. Genome organization of the Chelonus inanitus polydnavirus: excision sites, spacers and abundance of proviral and excised segments. J. Gen. Virol. 88, 450–457 (doi:10.1099/vir.0.82396-0) [DOI] [PubMed] [Google Scholar]
- 84.Bonvin M, Marti D, Wyder S, Kojic D, Annaheim M, Lanzrein B. 2005. Cloning, characterization and analysis by RNA interference of various genes of the Chelonus inanitus polydnavirus. J. Gen. Virol. 86, 973–983 (doi:10.1099/vir.0.80833-0) [DOI] [PubMed] [Google Scholar]
- 85.Ohno S. 1993. Patterns in genome evolution. Curr. Opin. Genet. Dev. 3, 911–914 (doi:10.1016/0959-437X(93)90013-F) [DOI] [PubMed] [Google Scholar]
- 86.Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108 (doi:10.1038/nrg2689) [DOI] [PubMed] [Google Scholar]
- 87.Serbielle C, Chowdhury S, Pichon S, Dupas S, Lesobre J, Purisima EO, Drezen J-M, Huguet E. 2008. Viral cystatin evolution and three-dimensional structure modelling: a case of directional selection acting on a viral protein involved in a host–parasitoid interaction. BMC Biol. 6, 38 (doi:10.1186/1741-7007-6-38) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Z, Bielawski B. 2000. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503 (doi:10.1016/S0169-5347(00)01994-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (doi:10.1093/molbev/msm088) [DOI] [PubMed] [Google Scholar]
- 90.Nei M, Rooney AP. 2005. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 39, 121–152 (doi:10.1146/annurev.genet.39.073003.112240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Babushok D, Ostertag E, Kazazian H. 2007. Current topics in genome evolution: molecular mechanisms of new gene formation. Cell Mol. Life Sci. 64, 542–554 (doi:10.1007/s00018-006-6453-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McBride CS. 2007. Rapid evolution of smell and taste receptor genes during host specialization in Drosophila sechellia. Proc. Natl Acad. Sci. USA 104, 4996–5001 (doi:10.1073/pnas.0608424104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150, 831–841 (doi:10.1016/j.cell.2012.05.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Branca A, Le Ru BP, Vavre F, Silvain J-F, Dupas S. 2011. Intraspecific specialization of the generalist parasitoid Cotesia sesamiae revealed by polyDNAvirus polymorphism and associated with different Wolbachia infection. Mol. Ecol. 20, 959–971 (doi:10.1111/j.1365-294X.2010.04977.x) [DOI] [PubMed] [Google Scholar]
- 95.Ngi-Song AJ, Overholt WA, Stouthamer R. 1998. Suitability of Busseola fusca and Sesamia calamistis (Lepidoptera: Noctuidae) for the development of two populations of Cotesia sesamiae (Hymenoptera: Braconidae) in Kenya. Biol. Control 12, 208–214 (doi:10.1006/bcon.1998.0628) [Google Scholar]
- 96.Dupas S, Gitau CW, Branca A, Le Ru BP, Silvain JF. 2008. Evolution of a polydnavirus gene in relation to parasitoid–host species immune resistance. J. Hered. 99, 491–499 (doi:10.1093/jhered/esn047) [DOI] [PubMed] [Google Scholar]
- 97.Gitau CW, Gundersen-Rindal D, Pedroni M, Mbugi PJ, Dupas S. 2007. Differential expression of the CrV1 haemocyte inactivation-associated polydnavirus gene in the African maize stem borer Busseola fusca (Fuller) parasitized by two biotypes of the endoparasitoid Cotesia sesamiae (Cameron). J. Insect Physiol. 53, 676–684 (doi:10.1016/j.jinsphys.2007.04.008) [DOI] [PubMed] [Google Scholar]
- 98.Pennacchio F, Strand MR. 2006. Evolution of developmental strategies in parasitic Hymenoptera. Annu. Rev. Entomol. 51, 233–258 (doi:10.1146/annurev.ento.51.110104.151029) [DOI] [PubMed] [Google Scholar]
- 99.Santos AMC, Quicke DLJ. 2011. Large-scale diversity patterns of parasitoid insects. Entomol. Sci. 14, 371–382 (doi:10.1111/j.1479-8298.2011.00481.x) [Google Scholar]
- 100.Reineke A, Asgari S, Schmidt O. 2006. Evolutionary origin of Venturia canescens virus-like particles. Arch. Insect Biochem. Physiol. 61, 123–133 (doi:10.1002/arch.20113) [DOI] [PubMed] [Google Scholar]
- 101.Ehrlich PR, Raven PH. 1964. Butterflies and plants: a study in coevolution. Evolution 18, 586–608 (doi:10.2307/2406212) [Google Scholar]
- 102.Cann AJ. 2001. Principles of molecular virology, 3rd edn San Diego, CA: Academic Press [Google Scholar]
- 103.Lang AS, Zhaxybayeva O, Beatty JT. 2012. Gene transfer agents: phage-like elements of genetic exchange. Nat. Rev. Microbiol. 10, 472–482 (doi:10.1038/nrmicro2802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stoltz D, Krell P. 2012. The origins and early history of polydnavirus research. In Parasitoid viruses: symbionts and pathogens (eds Beckage NE, Drezen J-M.), pp. 5–13 Oxford, UK: Elsevier [Google Scholar]