

Abstract
Background
Treatment of metastatic malignant melanoma patients harboring BRAF(V600E) has improved drastically after the discovery of the BRAF inhibitor, vemurafenib. However, drug resistance is a recurring problem, and prognoses are still very bad for patients harboring BRAF wild-type. Better markers for targeted therapy are therefore urgently needed.
Methodology
In this study, we assessed the individual kinase activity profiles in 26 tumor samples obtained from patients with metastatic malignant melanoma using peptide arrays with 144 kinase substrates. In addition, we studied the overall ex-vivo inhibitory effects of vemurafenib and sunitinib on kinase activity status.
Results
Overall kinase activity was significantly higher in lysates from melanoma tumors compared to normal skin tissue. Furthermore, ex-vivo incubation with both vemurafenib and sunitinib caused significant decrease in phosphorylation of kinase substrates, i.e kinase activity. While basal phosphorylation profiles were similar in BRAF wild-type and BRAF(V600E) tumors, analysis with ex-vivo vemurafenib treatment identified a subset of 40 kinase substrates showing stronger inhibition in BRAF(V600E) tumor lysates, distinguishing the BRAF wild-type and BRAF(V600E) tumors. Interestingly, a few BRAF wild-type tumors showed inhibition profiles similar to BRAF(V600E) tumors. The kinase inhibitory effect of vemurafenib was subsequently analyzed in cell lines harboring different BRAF mutational status with various vemurafenib sensitivity in-vitro.
Conclusions
Our findings suggest that multiplex kinase substrate array analysis give valuable information about overall tumor kinase activity. Furthermore, intra-assay exposure to kinase inhibiting drugs may provide a useful tool to study mechanisms of resistance, as well as to identify predictive markers.
Introduction
Metastatic malignant melanoma is associated with a poor prognosis. For decades, patients have been treated with palliative chemotherapy like dacarbazine (DTIC) monotherapy. However, only 10–15% of patients respond to this type of treatment, and for the majority, responses are of short duration only [1].
Recent advances in melanoma research have unraveled some of the complexity of the molecular mechanisms of this disease. The oncogene BRAF (v-raf murine sarcoma viral oncogene homolog B1) is frequently mutated in melanoma (40–50% of cases) and has resulted in the development of BRAF-targeting kinase inhibitors, like vemurafenib (PLX4032) and dabrafenib [2]–[4]. Following the recent approvals by both the Food and Drug Administration and the European Medicines Agency, vemurafenib is now increasingly used for treatment of patients with late-stage melanoma harboring BRAF(V600E/K) mutations. Initial clinical studies with vemurafenib showed remarkably positive results, with response rates approaching 80% [2]. However, the duration of response was recognized to last for a few months only [2], and occurrence of drug resistance was a major drawback [5], [6]. Thus, better understanding of the molecular mechanisms involved in resistance to vemurafenib therapy may identify interacting tumor signaling pathways that further can be exploited as alternative actionable therapy targets.
Kinases have become attractive targets for novel anticancer drugs [7]–[10]. Tumor kinase signaling comprising composite activities of effector proteins, both directly down-stream in the BRAF-signaling pathway, but also more indirectly participating within this particular signaling pathway, might be implicated in cancer progression and drug resistance, and could act as functional biomarkers. In this study, we assayed the kinase activity in protein lysates from tumor samples provided by metastatic melanoma patients using a multiplex kinase substrate array technology. Our primary aim was to identify specific kinase activity profiles of metastatic melanoma and normal skin tissue. Secondly, we aimed to study the ex-vivo inhibitory effects of vemurafenib in order to describe kinases and signaling pathways involved in vemurafenib response, and to compare the findings to the inhibitory effects of in-vitro vemurafenib treatment in metastatic melanoma cell lines. The experiments were repeated with sunitinib, a multi-targeted kinase inhibitor, for comparison of results obtained with vemurafenib, using the same methodological approach.
Materials and Methods
Ethics Statement
The Regional Committee for Medical and Health Research Ethics approved the study, and each patient provided written informed consent.
Tissue Specimens
In total, 26 fresh-frozen tumor samples from patients suffering from stage IV melanoma were collected prior to DTIC treatment at Haukeland University Hospital (Table 1). The patient material was collected from October 1999 to November 2007, and follow-up was terminated in May 2009. The tumor biopsies were collected from distant metastases or from locoregional relapse by incisional or tru-cut (liver) biopsies, and were immediately snap-frozen in liquid nitrogen (individual patient characteristics are summarized in Table S1). All tissue specimens have been histologically confirmed by a pathologist and have previously been described and screened for mutations in BRAF, NRAS (neuroblastoma RAS viral (v-ras) oncogene homolog), CDKN2A (cyclin-dependent kinase inhibitor 2A), and TP53 (Tumor protein p53) [11]–[13]. Additionally, four normal skin tissue samples were collected at Akershus University Hospital in 2010 from individuals not affected by melanoma. No clinical data was obtained from these patients.
Table 1. Patient Characteristics.
| All patients (n) | BRAF wild-type (n) | BRAF(V600E) (n) | |
| Patient demographic | |||
| Number of samples | 26 | 15 | 10 |
| Age at diagnosis, median (yrs) | 60 | 59 | 61 |
| Age at metastasis, median (yrs) | 65 | 65 | 65 |
| Sex | |||
| Male | 14 | 7 | 4 |
| Female | 11 | 8 | 6 |
| Localization of primary tumor | |||
| Lower extremity | 6 | 4 | 2 |
| Upper extremity | 3 | 3 | - |
| Head | 3 | 3 | - |
| Trunk | 9 | 2 | 7 |
| No primary detected | 4 | 3 | 1 |
| Pathological types of melanoma | |||
| Nodular melanoma | 8 | 7 | 1 |
| Superficial spreading | 11 | 5 | 6 |
| Unknown | 7 | 4 | 3 |
| Type of metastasis | |||
| Lymph node | 6 | - | 6 |
| Subcutaneous | 18 | 14 | 4 |
| Clinical stage at inclusion | |||
| Stage III | 1 | - | 1 |
| Stage IV | 25 | 15 | 9 |
| Response to DTIC | |||
| Responder | 13 | 9 | 4 |
| Non-responder | 13 | 7 | 6 |
| NRAS status | |||
| NRAS wild-type | 20 | 10 | 10 |
| NRAS(Q61) | 6 | 6 | - |
Tissue Preparation
The tissue specimens were sectioned with a microtome into 10 µm thick coupes, to a total volume of ∼3 mm3. The number of coupes needed was calculated based on the surface area of the tissue specimen. The tissue samples were kept frozen at all times during the procedure, and stored at −80°C until further use. To avoid contamination, the tumor and normal tissue specimens were prepared separately. The sectioned tissue was lysed with the mammalian protein extraction reagent (M-PER) buffer (Pierce Biotechnology, Inc., Rockford, IL), supplemented with phosphatase and protease inhibitors (Pierce Biotechnology, Inc), for the determination of kinase activity profiles in the presence and absence of two different inhibitors; vemurafenib (PLX4032; Axon Medchem B.V., Groningen, The Netherlands) and sunitinib (SU11248; Sigma Aldrich, Oslo, Norway). The protein concentration of lysates was determined using the BCA assay (Pierce Biotechnology, Inc.). For each experiment, 15 µg of protein lysate from melanoma tissue or 20 µg of protein lysate from normal skin tissue was added to the reaction mixture, in addition to 400 µM ATP and 12.5 mg/mL of monoclonal fluorescein isothiocyanate-conjugated anti- phosphotyrosine antibody (Exalpha Biologicals, Inc., Maynard, MA).
Kinase Activity Profiling of Metastatic Malignant Melanoma Tumors
Kinase activity profiling was performed using the Tyrosine Kinase PamChip® Array for Pamstation®12 (PamGene International B.V., ‘s-Hertogenbosch, The Netherlands) at Akershus University Hospital. Each array consists of 144 peptide substrates, primarily with known tyrosine residues, representing ∼100 different proteins. Three chips can be run simultaneously, and each chip consists of four arrays. The lysates are repeatedly pumped up and down through the porous array, allowing repeat substrate phosphorylation. Based on pilot experiments of increasing concentrations of the individual kinase inhibitors added to melanoma tissue lysates that were incubated on the arrays, concentrations that resulted in ∼50% inhibition of most kinase substrates were chosen for the main experiments. Hence, concentrations of 40 µM vemurafenib and 7.5 µM sunitinib were spiked into the assay mixtures prior to incubation, whereas 1.5% dimethyl sulfoxide was added to mixtures not containing the inhibitors. The samples were run in three technical replicates in the presence or absence of vemurafenib, as paired measurements with and without inhibitor on the same chip. The experimental procedure was repeated with sunitinib. Incubations were commenced for 60 cycles, followed by washing and fluorescence measurement of all peptide spots every fifth cycle. The experiments were run blinded, and the tumor and normal skin tissue lysates were run separately. The microarray data are submitted to ArrayExpress (http://www.ebi.ac.uk/arrayexpress/); accession number E-MTAB-1245.
Data Adaptation and Statistical Analysis of Malignant Melanoma Tumors
End-level signal intensities for each peptide spot were quantified and analyzed using BioNavigator version 5.1 (Pamgene International B.V.). Signals obtained after subtraction of local array background were used for further analysis. Negative numbers were set to 0.01 and log2-transformed.
For analysis of the ‘basal kinase activity profiles’ (measurements obtained without inhibitor), the technical replicates were averaged. The overall difference between the measurements in the first series, relative to that in the second series was corrected by subtracting the mean of each peptide in the corresponding experimental series (centering), and by averaging the centered results of both experimental series. For analysis of ‘inhibition profiles’ (measurement obtained with inhibitor), values were obtained by calculating the log-fold change (LFC) of each peptide without any further normalization of the data. LFC was calculated by subtracting the log2-transformed signal values with inhibitor from the corresponding values without inhibitor added. The pairing of measurements with and without inhibitor was taken into account by first calculating the LFC of each chip, and subsequently averaging the LFCs of each chip to obtain the value used in further analysis.
Per-peptide differences between conditions were evaluated using two-tailed t-tests, and unsupervised multivariate clustering of samples was evaluated with principal component analysis (PCA), both using BioNavigator interfaced to R (The R-project). Supervised multivariate analysis of conditions was performed by applying partial least squares discriminant analysis (PLS-DA), using BioNavigator interfaced to a custom PLS-DA implementation written in Matlab (MathWorks, Natick, MA). PLS-DA was performed without any pre-selection of kinase substrates. Prediction performance was evaluated using leave-one-out cross-validation (LOOCV), making sure that the model was optimized completely independent of the test sample [14]. Pathway connectivity of kinase substrates was determined by using the KEGG pathway database [15], [16] and literature search.
Kinase Activity Profiling and Statistical Analysis of Melanoma Cell Lines
The MelJD, patient-3-post and MM200 metastatic melanoma cell lines were obtained from Professor P. Hersey, University of Sydney, Sydney, NSW, Australia [17], [18]. The MelJD cell line is BRAF wild-type, whereas the patient-3-post and MM200 cell lines harbor the BRAF(V600E) mutation. In this manuscript we entitle the patient-3-post cell line as “vemurafenib-resistant” and the MM200 cell line as “vemurafenib-sensitive”, due to their difference in sensitivity to vemurafenib treatment, as also shown previously [17], [18]. All cell lines were maintained in RPMI 1640 medium (Sigma-Aldrich, Oslo, Norway) supplemented with 10% fetal calf serum and 1% Glutamax (Invitrogen, Oslo, Norway). The cells were routinely grown as a monolayer in 75 cm2 flasks at 37°C in 95% air/5% CO2, and subcultured twice a week to maintain exponential growth. The cell lines were confirmed to be mycoplasma-free prior to the experiments.
Cells were exposed to in-vitro treatment with vemurafenib (5 µM) or dimethyl sulfoxide (vehicle) for 1 hour. The cells were harvested by washing the cells twice with 10 ml ice-cold PBS, before adding 4 ml ice-cold PBS and loosening the cells by scraping. To obtain the pellet, the samples were centrifuged (10 minutes, 2500 rpm, 4°C) and supernatant was removed. Lysis buffer was added and the samples were vortexed and lysed for 15 minutes on ice. After centrifugation (15 minutes, 15000 rpm, 4°C), supernatants were aliqouted and immediately frozen at −80°C. Protein concentrations were measured using a BCA protein assay kit (Pierce Biotechnology, Inc).
Kinase activity profiling was assessed by using 10 µg of total protein from all samples. Lysates from each cell line were run in triplicates. The raw data was log2-transformed by identical procedures as the data from the patient specimens, before per-peptide differences between conditions (vemurafenib-treated versus untreated samples, and pair-wise comparison of cell lines) were evaluated using the two-tailed t-tests.
Results
Basal Kinase Activity in Metastatic Malignant Melanoma
The majority of the array kinase substrates (80–90%) showed higher phosphorylation levels upon incubation with metastatic melanoma lysates compared with normal skin tissue lysates. The difference ranged up to 5-fold between the two tissue types (Figure 1A and Table S2). Supervised and unsupervised clustering analysis of the samples showed no correlations between phosphorylation patterns of kinase substrates and known molecular (BRAF-, NRAS-, CDKN2A-, or TP53 mutational status) or clinical parameters (age, gender, stage, or anatomical location of tumor), including response to DTIC (Figure 1B).
Figure 1. Kinase activity profiles of metastatic malignant melanoma and normal skin tissue.

A) The heat map shows phosphorylation levels for all 144 kinase substrates (vertical axis) in response to incubation with lysates from metastatic malignant melanoma samples and normal skin tissue samples (horizontal axis). Color bar represents phosphorylation intensities; blue indicates low phosphorylation levels, whereas yellow indicates higher phosphorylation levels. B) Unsupervised hierarchical clustering including all samples and 144 kinase substrates did not reveal any correlation between phosphorylation profiles and different molecular and clinical parameters. Different variables are indicated by colors, including BRAF-, NRAS-, CDKN2A-, TP53- mutational status, and DTIC response.
Ex-vivo Kinase Inhibitory Effects of Vemurafenib
The inhibition profiles obtained with ex-vivo exposure of melanoma tumor lysates to vemurafenib showed reduced kinase substrate phosphorylation levels. Whilst phosphorylation levels of the majority of the kinase substrates were decreased by approximately 50% (Table S3), the inhibitory effect was weaker on kinase substrates with low basal phosphorylation levels.
BRAF(V600E) mutation was present in 10 out of 26 tumors (38.5%), whereas NRAS(Q61) mutation was present in 6 out of 26 tumors (23%). Unsupervised PCA showed a tendency of separation between BRAF(V600E) and BRAF wild-type tumors (Figure 2A). Prediction performance with PLS-DA was evaluated using LOOCV [14]. This type of supervised analysis classified BRAF wild-type and BRAF(V600E) samples based on the inhibition profiles, with an accuracy of 20/26 samples (77%) (Figure 2B). Classification of tumors harboring BRAF(V600E) was correct for 90% of the samples, whereas for BRAF wild-type tumors, only 75% of samples were correctly classified with PLS-DA, reflecting the interesting observation that a few BRAF wild-type tumors consistently grouped with BRAF(V600E) tumors.
Figure 2. Classification of melanoma samples based on BRAF mutational status.
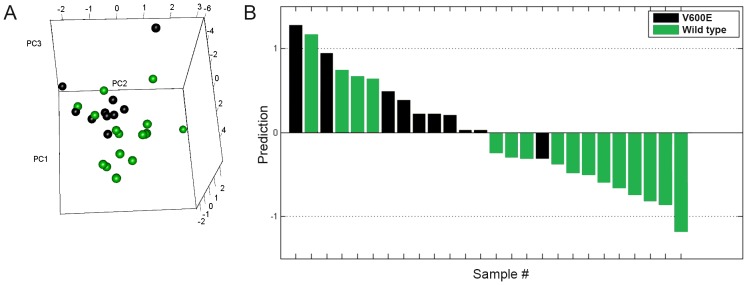
A) Unsupervised principal component analysis (PC1–3) including all 144 kinase substrates separated BRAF wild-type (green) and BRAF(V600E) (black) melanoma tumors in two groups based on the inhibition profiles obtained with ex-vivo vemurafenib. B) BRAF wild-type (green) and BRAF(V600E) (black) melanoma tumors were classified with partial least squares discriminant analysis. The prediction scores shown were obtained by testing the corresponding sample during leave-one-out cross-validation. Samples with prediction score lower than 0 were classified as BRAF wild-type, whereas samples with prediction score higher than 0 were classified as BRAF(V600E).
Furthermore, applying two-tailed t-tests identified 40 kinase substrates that were significantly affected by ex-vivo vemurafenib (P<0.05), and distinguished between BRAF(V600E) and BRAF wild-type tumors (Table S4). Supervised clustering analysis comprising these 40 kinase substrates showed a significantly stronger inhibitory effect of vemurafenib in BRAF(V600E) tumors than in BRAF wild-type tumors (Figure 3A). Again, a few BRAF wild-type tumors invariably grouped together with BRAF(V600E) tumors, exhibiting stronger inhibition in response to vemurafenib than the other BRAF wild-type tumors. No statistically significant differences in phosphorylation profiles were observed between BRAF(V600E) and BRAF wild-type tumors in the absence of ex-vivo vemurafenib incubation (Figure 3B). The kinase substrates that distinguished between BRAF wild-type and BRAF(V600E) tumors represented kinases mainly involved in the phosphatidylinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling network, including processes such as angiogenesis, proliferation, cell cycle progression and apoptosis (Figure 4 and Table S5). However, pathway exploration including all 144 peptides revealed that both these pathways were overrepresented on the array (Table S5).
Figure 3. Supervised clustering of BRAF(V600E) and BRAF wild-type melanoma tumors.

A) Supervised clustering of melanoma samples based on 40 kinase substrates (vertical axis) identified as significantly differentially affected by ex-vivo exposure to vemurafenib in BRAF wild-type (green) and BRAF(V600E) (black) samples (horizontal axis). Clustering using the inhibition profiles separated the samples in two groups according to BRAF mutational status. Color bar represents the level of inhibition; red indicates strong inhibition, whereas blue indicates weak inhibition. B) Clustering using the basal kinase activity data did not separate the melanoma samples according to BRAF mutational status. Color bar represents the level of phosphorylation; yellow indicates high phosphorylation, whereas blue indicates low phosphorylation of kinase substrates. Samples marked with asterisks (*) harbor NRAS mutations.
Figure 4. Kinases and pathways affected by ex-vivo vemurafenib in BRAF(V600E) melanoma tumors.

In dark blue color are array substrates representing kinases distinguishing between BRAF wild-type and BRAF(V600E) tumors (P<0.05) in response to vemurafenib. Marked with light blue color are kinase substrates showing reduced levels of phosphorylation in response to vemurafenib, but which are not identified as differentially inhibited according to BRAF mutational status. In yellow color are the main cellular processes (angiogenesis, apoptosis, proliferation, and cell cycle progression) affected in response to ex-vivo vemurafenib. Some kinase substrates may be represented in more than one cellular process. Note that RAF in this case is CRAF, not BRAF. Abbreviations: v-akt murine thymoma viral oncogene (AKT), cyclin-dependent kinase (CDK), epidermal growth factor receptor (EGFR), v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2), extracellular-signal-regulated kinases (ERK), fibroblast growth factor receptor (FGFR), growth factor receptor-bound protein 2 (Grb2), janus kinase (JAK), mitogen-activated protein kinase kinase (MEK), platelet-derived growth factor receptor (PDGFR), 3-phosphoinositide dependent protein kinase-1(PDK1), phosphatidylinositide 3-kinase (PI3K), protein kinase C (PKC), phospholipase C- gamma (PLCg), v-Raf murine sarcoma viral oncogene (RAF), rat sarcoma viral oncogene (RAS), ret proto-oncogene (RET), son of sevenless (SOS), signal transducer and activator of transcription (STAT), neurotrophic tyrosine kinase receptor (TRK).
Ex-vivo Kinase Inhibitory Effects of Sunitinib
Sunitinib, a multi-targeted receptor tyrosine kinase inhibitor, affected as expected a range of array kinase substrates, revealing inhibition profiles resembling those obtained with vemurafenib (Figure 5A and Table S3). However, in contrast to the results obtained with vemurafenib inhibition, attempts to correlate sunitinib inhibition profiles to BRAF mutational status or other molecular or clinical parameters, including supervised analysis with the panel of 40 kinase substrates, showed no significant findings. Unsupervised hierarchical clustering with inhibition profiles including all samples and kinase substrates is shown in Figure 5B.
Figure 5. Kinase inhibition profiles in response to ex-vivo exposure to vemurafenib or sunitinib.

A) Inhibition (y-axis) of all 144 kinase substrates (x-axis) in response to ex-vivo incubation with vemurafenib and sunitinib in metastatic malignant melanoma tumors. B) Heat map with sunitinib inhibition profiles of all 144 kinase substrates (vertical) and twenty-six metastatic malignant melanoma tumors (horizontal). Unsupervised hierarchical clustering did not show any correlation with BRAF- (BRAF wild-type (green), BRAF(V600E) (black)) or NRAS(Q61) (marked with *) mutations. Color bar represents inhibition intensities; red indicates strong inhibition, whereas blue indicates weak inhibition.
In-vitro Inhibitory Effects of Vemurafenib on Kinase Activity in BRAF(V600E) and BRAF Wild-type Melanoma Cell Lines
To further examine the role of BRAF mutational status on kinase activity, and as a mean to validate results obtained with patient specimens, we profiled the kinase activity of lysates from the three melanoma cell lines MelJD (BRAF wild-type), patient-3-post (BRAF(V600E)/“vemurafenib-resistant”) and MM200 (BRAF(V600E)/“vemurafenib-sensitive”).
The results showed a great variability in the reduction of kinase substrate phosphorylation levels in response to in-vitro vemurafenib treatment. Reduced phosphorylation levels were seen in all cell lysates, with the largest effect seen in lysates from the BRAF wild-type MelJD cells (Table S6). Inhibition profiles revealed that the phosphorylation levels of only 12 kinase substrates were significantly (P<0.05) reduced by vemurafenib in all three cell lines (Figure 6). Stronger kinase inhibition after vemurafenib treatment was seen in MelJD and MM200 cells, with 41 of the same kinase substrates affected. MelJD and vemurafenib-sensitive MM200 cells showed similar inhibition profiles compared to vemurafenib-resistant cells. In total, the phosphorylation level of 59 kinase substrates were significantly differentially affected by vemurafenib (P<0.05) in patient-3-post and MelJD cells, with 50% (20/40) of them being identical to the kinase substrates identified as differentially affected between BRAF wild-type and BRAF(V600E) in patient specimens (Table 2). A smaller number of kinase substrates were differentially affected by vemurafenib in MM200 and MelJD cells, respectively, with only 20% (8/40) of the kinase substrates being the same as the ones identified in patient specimens.
Figure 6. Venn diagram of kinase substrates that are significantly affected by vemurafenib in BRAF(V600E) and BRAF wild-type melanoma cell lines.

The MelJD cells harbor BRAF wild-type, whereas both patient-3-post and MM200 cells harbor BRAF(V600E) mutations. The patient-3-post cells are vemurafenib-resistant, whereas MM200 cells are sensitive to vemurafenib. The numbers given denote the number of kinase substrates that are significantly affected in each pair-wise comparison of the three different cell lines, as well as the number of kinase substrates that are commonly affected among the cell lines.
Table 2. Significantly differentially affected kinase substrates (P<0.05) between BRAF(V600E) and BRAF wild-type melanoma in lysates from cell lines and tumor tissue.
| Kinase substrate ID | Encoding protein | MelJD vs MM200 | MelJD vs patient-3-post | Melanoma tissue |
| 41_654_666 | Erythrocyte membrane protein band 4.1 | X | X | |
| ANXA1_14_26 | Annexin A1 | X | X | X |
| ANXA2_17_29 | Annexin A2 pseudogene 3; annexin A2; annexin A2 pseudogene 1 | X | ||
| C1R_199_211 | Complement component 1, r subcomponent | X | X | |
| CALM_93_105 | Calmodulin 3; calmodulin 2; calmodulin 1 | X | ||
| CD3Z_116_128 | CD247 molecule | X | ||
| CD79A_181_193 | CD79a molecule, immunoglobulin-associated alpha | X | X | X |
| CDK2_8_20 | Cyclin-dependent kinase 2 | X | ||
| CDK7_157_169 | Cyclin-dependent kinase 7 | X | ||
| CRK_214_226 | v-crk sarcoma virus CT10 oncogene homolog | X | X | |
| CTNB1_79_91 | Catenin (cadherin-associated protein), beta | X | ||
| DCX_109_121 | Doublecortin | X | ||
| DYR1A_312_324 | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A | X | X | X |
| EFS_246_258 | Embryonal Fyn-associated substrate | X | ||
| EGFR_1062_1074 | Epidermal growth factor receptor | X | ||
| EGFR_1103_1115 | Epidermal growth factor receptor | X | X | |
| EGFR_1165_1177 | Epidermal growth factor receptor | X | ||
| EGFR_1190_1202 | Epidermal growth factor receptor | X | X | |
| EGFR_862_874 | Epidermal growth factor receptor | X | X | |
| ENOG_37_49 | Enolase 2 (gamma, neuronal) | X | ||
| EPHA1_774_786 | EPH receptor A1 | X | X | X |
| EPHA2_581_593 | EPH receptor A2 | X | ||
| EPHA2_765_777 | EPH receptor A2 | X | ||
| EPHA7_607_619 | EPH receptor A7 | X | X | |
| EPHB1_771_783 | EPH receptor B1 | X | X | |
| EPOR_361_373 | Erythropoietin receptor | X | ||
| EPOR_419_431 | Erythropoietin receptor | X | ||
| ERBB2_1241_1253 | v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 | X | ||
| ERBB2_870_882 | v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 | X | ||
| FAK1_569_581 | PTK2 protein tyrosine kinase 2 | X | X | |
| FAK2_572_584 | PTK2B protein tyrosine kinase 2 beta | X | X | |
| FER_707_719 | Fer (fps/fes related) tyrosine kinase | X | X | |
| FES_706_718 | Feline sarcoma oncogene | X | ||
| FGFR1_761_773 | Fibroblast growth factor receptor 1 | X | ||
| FGFR2_762_774 | Fibroblast growth factor receptor 2 | X | ||
| FGFR3_753_765 | Fibroblast growth factor receptor 3 | X | X | X |
| FRK_380_392 | Fyn-related kinase | X | X | X |
| JAK1_1015_1027 | Janus kinase 1 | X | X | |
| JAK2_563_577 | Janus kinase 2 | X | ||
| K2C6B_53_65 | Keratin 6B | X | X | |
| K2C8_425_437 | Keratin 8 pseudogene 9 | X | X | |
| LAT_194_206 | Linker for activation of T cells | X | X | |
| LAT_249_261 | Linker for activation of T cells | X | X | |
| LCK_387_399 | Lymphocyte-specific protein tyrosine kinase | X | X | |
| MET_1227_1239 | Met proto-oncogene (hepatocyte growth factor receptor) | X | X | |
| MK01_180_192 | Mitogen-activated protein kinase 1 | X | X | |
| MK07_211_223 | Mitogen-activated protein kinase 7 | X | X | |
| MK14_173_185 | Mitogen-activated protein kinase 14 | X | X | |
| NPT2A_501_513 | Solute carrier family 34 (sodium phosphate), member 1 | X | X | |
| NTRK1_489_501 | Neurotrophic tyrosine kinase, receptor, type 1 | X | ||
| NTRK2_696_708 | Neurotrophic tyrosine kinase, receptor, type 2 | X | X | |
| P85A_600_612 | Phosphoinositide-3-kinase, regulatory subunit 1 (alpha) | X | X | |
| PAXI_111_123 | Paxillin | X | X | |
| PAXI_24_36 | Paxillin | X | ||
| PDPK1_2_14 | 3-phosphoinositide dependent protein kinase-1 | X | ||
| PDPK1_369_381 | 3-phosphoinositide dependent protein kinase-1 | X | X | |
| PECA1_706_718 | Platelet/endothelial cell adhesion molecule | X | ||
| PGFRB_1002_1014 | Platelet-derived growth factor receptor, beta polypeptide | X | ||
| PGFRB_1014_1028 | Platelet-derived growth factor receptor, beta polypeptide | X | X | |
| PGFRB_572_584 | Platelet-derived growth factor receptor, beta polypeptide | X | ||
| PGFRB_768_780 | Platelet-derived growth factor receptor, beta polypeptide | X | ||
| PGFRB_771_783 | Platelet-derived growth factor receptor, beta polypeptide | X | ||
| PLCG1_764_776 | Phospholipase C, gamma 1 | X | X | |
| PRRX2_202_214 | Paired related homeoboX 2 | X | X | |
| RAF1_332_344 | v-raf-1 murine leukemia viral oncogene homolog 1 | X | ||
| RASA1_453_465 | RAS p21 protein activator (GTPase activating protein) 1 | X | ||
| RET_1022_1034 | Ret proto-oncogene | X | X | |
| RON_1346_1358 | Macrophage stimulating 1 receptor (c-met-related tyrosine kinase) | X | ||
| SRC8_CHICK_476_488 | Cortactin | X | X | |
| SRC8_CHICK_492_504 | Cortactin | X | X | |
| STAT1_694_706 | Signal transducer and activator of transcription 1 | X | ||
| STAT4_714_726 | Signal transducer and activator of transcription 4 | X | ||
| TEC_512_524 | Tec protein tyrosine kinase | X | ||
| TYRO3_679_691 | TYRO3 protein tyrosine kinase | X | X | |
| VGFR1_1040_1052 | Fms-related tyrosine kinase 1 (vascular endothelial growth factor) | X | ||
| VGFR1_1049_1061 | Fms-related tyrosine kinase 1 (vascular endothelial growth factor) | X | X | |
| VGFR1_1235_1247 | Fms-related tyrosine kinase 1 (vascular endothelial growth factor) | X | X | |
| VGFR2_1046_1058 | Kinase insert domain receptor (a type III receptor tyrosine kinase) | X | ||
| VGFR2_1052_1064 | Kinase insert domain receptor (a type III receptor tyrosine kinase) | X | ||
| ZAP70_485_497 | Zeta-chain (TCR) associated protein kinase | X | X |
X denotes the kinase substrates that are significantly affected between BRAF(V600E) and BRAF wild-type.
X highlighted in bold denotes kinase substrates that were also identified as significant in melanoma tissue.
Discussion
Following several decades of nearly complete stagnation in the clinical treatment of patients with metastatic malignant melanoma, we are currently witnessing dramatic improvements regarding therapy. Activating mutations in BRAF (mainly V600E/K) have been identified in half of all melanoma cases [4], and the development of novel compounds targeting mutated BRAF [6], [19], or compounds boosting the immunological responses directed towards cancer cells [20], has given new hope to this group of patients. However, these studies have also revealed that early drug resistance occurs in the majority of patients, causing a considerable clinical challenge [2], [5], [6]. As kinases are part of key cellular process, and mutations herein are often implicated in both cancer progression and/or drug resistance [21], we examined the overall kinase activity profiles in metastatic malignant melanoma tumor samples, using a multiplex microarray technology previously proven to be robust and reliable [22]–[25].
We show that phosphorylation levels of kinase substrates were generally increased in lysates from metastatic melanoma compared to normal skin tissue, indicating high kinase activity. The phosphorylation patterns in melanoma did however not correlate to any clinical or molecular parameters, like BRAF- and NRAS mutational status, or response to DTIC therapy. The increased kinase activity observed in our melanoma samples is in agreement with previous studies which shows that kinases are hyperactive in many cancers, acting as a driving force towards tumor proliferation and other growth processes [7].
We further analyzed the ex-vivo inhibitory effects of the BRAF inhibitor, vemurafenib, and the multi-targeted tyrosine kinase inhibitor, sunitinib, in the same set of melanoma samples. Inhibition experiments with sunitinib were performed as an indication that the method worked as expected i.e. multiple kinase substrates were inhibited. Hence, no correlation in inhibition pattern was observed with regard to various clinical and biological parameters.
The ex-vivo inhibition profiles upon exposure to vemurafenib showed that a wide range of kinase substrates were affected, indicated by decreased levels of phosphorylation. This was also observed in cell lines treated with vemurafenib in-vitro, regardless of BRAF mutational status. Possible explanations for this comprehensive inhibitory pattern might be due to off-target effects of vemurafenib, as has been suggested by others [26]. Nevertheless, the majority of the affected substrates represented effector proteins participating within the signaling network mediating kinase activity through the BRAF-encoded pathway.
Interestingly, inhibition profiles obtained with ex-vivo vemurafenib revealed a panel of 40 kinase substrates distinguishing the BRAF wild-type and BRAF(V600E) melanoma tumors. Kinases involved in the PI3K and MAPK pathway were among the kinase substrates discriminating the two groups. However, bearing in mind that the 40 discriminating substrates in this analysis appeared from a total number of 144 peptides constituting the kinase activity profiles, the false discovery rate among peptides with a statistical significance level of P<0.05 can be estimated to be about 16%. The 40 kinase substrates were more strongly inhibited when incubated with lysates from tumors harboring BRAF(V600E) compared to BRAF wild-type tumors, which is consistent with previous studies showing that BRAF(V600E) tumors are more responsive to vemurafenib than BRAF wild-type tumors [4], [27], [28]. The unresponsiveness of BRAF wild-type tumors is thought to occur through a complex interplay between RAS and RAF dimers, leading to compensatory activation of the MAPK pathway [29]. Dimerization is promoted through RAS activation [30], and in the presence of activated NRAS, CRAF is preferred over BRAF, leading to loss of the inhibitory effect of vemurafenib [31]. Notably, BRAF and NRAS mutations are mutually exclusive in melanoma [32], which is also observed in our study. Thus, in BRAF(V600E) tumors, RAS activity is low, and the drug is able to bind to the BRAF monomer, blocking its activity completely.
In our study, both supervised and unsupervised analyses revealed that some wild-type BRAF samples also exhibited decreased levels of kinase substrate phosphorylation upon exposure to vemurafenib. These samples were classified together with the BRAF(V600E) tumors based on inhibition profiles. In the absence of clinical vemurafenib response data, we speculate whether these patients might benefit from vemurafenib treatment despite the lack of the V600E mutation. When profiling the kinase substrates in BRAF wild-type melanoma cell line (MelJD), we observed that kinase inhibition upon in-vitro vemurafenib treatment occurred to a similar degree as in the vemurafenib-sensitive cell line (MM200) harboring BRAF(V600E). This supports our findings from the patient specimens; that patients with wild-type BRAF may respond to vemurafenib treatment. Increased proliferation in BRAF wild-type cells in response to vemurafenib has, however, been reported as a possibly hazardous event [27], [29]. It would therefore be of interest to study these tumors for other activating mutations, either in the BRAF gene (e.g. BRAF(L597)) [33], or elsewhere, that has been shown to confer sensitivity to kinase inhibitors targeting the MAPK pathway.
Furthermore, some samples within the BRAF(V600E) group showed a lower degree of inhibition in ex-vivo response to vemurafenib. Lower degree of inhibition upon vemurafenib treatment in-vitro was also observed in our vemurafenib-resistant cell line (patient-3-post). This variability in sensitivity towards vemurafenib has previously been observed in several melanoma cell lines, where the presence of BRAF mutations did not guarantee a response [34], [35]. Further studies will be necessary to explore the potential of this differential degree of kinase inhibition in identifying patients that might respond poorly to vemurafenib, despite the presence of the indicative BRAF(V600E) mutation.
The 40 kinase substrate signature obtained between BRAF wild-type and BRAF(V600E) melanoma tumor samples after ex-vivo treatment with vemurafenib was similar to the signature obtained with in-vitro vemurafenib treatment between BRAF wild-type and vemurafenib-resistant BRAF(V600E) cells (Table 2). These results suggest that this signature may be useful in predicting patients benefiting from vemurafenib treatment.
Vemurafenib resistance is common in melanoma, and several mechanisms to how this occurs have been proposed. These include (a) BRAF splicing variants (p61BRAF(V600E)) lacking the RAS-binding domain [36]; (b) phosphatase and tensin homolog (PTEN) loss leading to elevated PI3K/AKT signaling [37]; (c) increased PDGFRβ expression leading to activation of survival pathways, or (d) NRAS(Q61K) mutations leading to activated MAPK pathway signaling [38]. In addition, increased EGFR expression in BRAF(V600E) colorectal tumors has been shown to correlate with vemurafenib resistance [39]. Expression of EGFR is generally low in melanoma compared to colorectal cancer [39]; however, EGFR overexpression in some melanoma tumors could explain the clinical resistance towards vemurafenib. Both EGFR and PDGFRβ are upstream of BRAF and affect both the MAPK and PI3K pathway (Figure 4). In our study, kinase substrates encoding for EGFR and PDGFRβ were found to be significantly differentially affected by ex-vivo vemurafenib in melanoma tumors harboring BRAF(V600E) and BRAF wild-type. This was also observed in-vitro, specifically in BRAF wild-type and vemurafenib-sensitive BRAF(V600E) cells, whereas no significant inhibition of EGFR and PDGFRβ was observed in the vemurafenib-resistant cell line (Table 3). Additionally, the kinase substrate encoding for RAF (C-RAF) was only significantly affected in the vemurafenib-sensitive cell line. Hence, our results support at least the notion that EGFR and PDGFRβ may be involved in the development of resistance to vemurafenib. Although resistance to BRAF inhibition is a challenge, recent evidence suggests that combinational therapy with inhibitors of the MAPK and PI3K pathway may be efficacious in melanoma patients with (V600E) mutations [5], [19], [40]. These findings make the signature of kinase substrates identified in this study as potential biomarker for such targeted therapy.
Table 3. EGFR, PDGFRβ and RAF kinase inhibitory effects of vemurafenib on lysates from cell lines harboring BRAF(V600E) mutations being resistant (patient-3-post) and sensitive (MM200) to vemurafenib, and BRAF wild-type (MelJD).
| BRAF(V600E) patient-3-post | BRAF(V600E) MM200 | BRAF wild-type MelJD | |
| Kinase substrate ID | P value | P value | P value |
| EGFR_1062_1074 | 3,71E-01 | 3,64E-01 | 1,84E-01 |
| EGFR_1103_1115 | 2,10E-01 | 1,32E-01 | 2,32E-02 |
| EGFR_1118_1130 | 1,14E-01 | 9,80E-01 | 6,97E-02 |
| EGFR_1165_1177 | 6,90E-01 | 2,06E-02 | 4,82E-02 |
| EGFR_1190_1202 | 5,20E-02 | 8,97E-01 | 1,69E-02 |
| EGFR_862_874 | 3,99E-01 | 1,01E-03 | 4,49E-01 |
| EGFR_908_920 | 8,03E-02 | 6,06E-01 | 2,96E-01 |
| PGFRB_1002_1014 | 1,33E-01 | 2,45E-02 | 1,21E-02 |
| PGFRB_1014_1028 | 1,72E-01 | 2,15E-02 | 1,40E-03 |
| PGFRB_572_584 | 1,83E-01 | 8,84E-01 | 9,70E-02 |
| PGFRB_709_721 | 3,28E-02 | 1,51E-02 | 1,77E-02 |
| PGFRB_768_780 | 1,54E-01 | 2,96E-02 | 2,18E-05 |
| PGFRB_771_783 | 2,12E-01 | 2,84E-02 | 5,52E-03 |
| RAF1_332_344 | 8,41E-02 | 2,46E-02 | 5,11E-02 |
Highlighted in bold are kinase substrates with P<0.05.
In conclusion, our findings show that metastatic malignant melanoma is characterized by high activity of a range of kinases. The multiplex kinase substrate array technology used in the present study proved to be robust and reliable, and provided valuable information. This method may therefore become an important tool for screening of disease-specific functional biomarkers, and thereby pave the way for individualized cancer treatment. Furthermore, ex-vivo exposure to drugs may identify kinase substrate signatures that correlate to clinical response.
Supporting Information
Individual patient characteristics of all metastatic malignant melanoma cases.
(XLS)
Mean phosphorylation intensity values of all kinase substrates across twenty-six metastatic malignant melanoma samples and four normal skin tissue samples in the basal data set.
(XLS)
Mean inhibition of 144 kinase substrates in metastatic malignant melanoma samples in response to ex-vivo vemurafenib and sunitinib.
(XLS)
Kinase substrates identified as significantly differentially affected (P<0.05) by ex-vivo vemurafenib incubation in BRAF wild-type and BRAF(V600E) melanoma samples.
(XLS)
Results from KEGG pathway analysis of kinase substrates involved in the PI3K and MAPK signalling pathways including all 144 kinase substrates, and 40 kinase substrates differentiating between BRAF(V600E) and BRAF wild-type tumors.
(XLS)
Kinase inhibitory effects of vemurafenib in melanoma cell lines harboring BRAF(V600E) mutations (MM200 and patient-3-post ) and BRAF wild type (MelJD).
(XLSX)
Acknowledgments
We would like to thank Torben Lüders for technical support and discussions during the preparation of this paper.
Funding Statement
This work was supported by Grant No. 2011030 from the South-Eastern Norway Regional Health Authority to VNK, and grants received from Division of Medicine, Akershus University Hospital. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Serrone L, Zeuli M, Sega FM, Cognetti F (2000) Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res 19: 21–34. [PubMed] [Google Scholar]
- 2. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, et al. (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363: 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380: 358–365. [DOI] [PubMed] [Google Scholar]
- 4. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, et al. (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467: 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Villanueva J, Vultur A, Herlyn M (2011) Resistance to BRAF inhibitors: unraveling mechanisms and future treatment options. Cancer Res 71: 7137–7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alcala AM, Flaherty KT (2012) BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res 18: 33–39. [DOI] [PubMed] [Google Scholar]
- 7. Sharma SV, Settleman J (2007) Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 21: 3214–3231. [DOI] [PubMed] [Google Scholar]
- 8. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, et al. (2012) Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet 379: 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, et al. (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncology 13: 239–246. [DOI] [PubMed] [Google Scholar]
- 10. Joensuu G, Joensuu T, Nupponen N, Ruutu M, Collan J, et al. (2012) A phase II trial of gefitinib in patients with rising PSA following radical prostatectomy or radiotherapy. Acta Oncologica 51: 130–133. [DOI] [PubMed] [Google Scholar]
- 11. Jonsson G, Busch C, Knappskog S, Geisler J, Miletic H, et al. (2010) Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res 16: 3356–3367. [DOI] [PubMed] [Google Scholar]
- 12. Busch C, Geisler J, Knappskog S, Lillehaug JR, Lonning PE (2010) Alterations in the p53 pathway and p16INK4a expression predict overall survival in metastatic melanoma patients treated with dacarbazine. J Invest Dermatol 130: 2514–2516. [DOI] [PubMed] [Google Scholar]
- 13. Busch C, Geisler J, Lillehaug JR, Lonning PE (2010) MGMT expression levels predict disease stabilisation, progression-free and overall survival in patients with advanced melanomas treated with DTIC. Eur J Cancer 46: 2127–2133. [DOI] [PubMed] [Google Scholar]
- 14. Westerhuis JA, Hoefsloot HCJ, Smit S, Vis DJ, Smilde AK, et al. (2008) Assessment of PLSDA cross validation. Metabolomics 4: 81–89. [Google Scholar]
- 15. Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research 40: 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lai F, Jiang CC, Farrelly ML, Zhang XD, Hersey P (2012) Evidence for upregulation of Bim and the splicing factor SRp55 in melanoma cells from patients treated with selective BRAF inhibitors. Melanoma Res 22: 244–251. [DOI] [PubMed] [Google Scholar]
- 18. Lai F, Jin L, Gallagher S, Mijatov B, Zhang XD, et al. (2012) Histone deacetylases (HDACs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective BRAF inhibitors. Adv Pharmacol 65: 27–43. [DOI] [PubMed] [Google Scholar]
- 19. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, et al. (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. New England Journal of Medicine 367: 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jeter JM, Cranmer LD, Hersh EM (2012) Ipilimumab pharmacotherapy in patients with metastatic melanoma. Clinical Medicine Insights Oncology 6: 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barouch-Bentov R, Sauer K (2011) Mechanisms of drug resistance in kinases. Expert Opin Investig Drugs 20: 153–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Folkvord S, Flatmark K, Dueland S, de Wijn R, Groholt KK, et al. (2010) Prediction of response to preoperative chemoradiotherapy in rectal cancer by multiplex kinase activity profiling. Int J Radiat Oncol Biol Phys 78: 555–562. [DOI] [PubMed] [Google Scholar]
- 23. Versele M, Talloen W, Rockx C, Geerts T, Janssen B, et al. (2009) Response prediction to a multitargeted kinase inhibitor in cancer cell lines and xenograft tumors using high-content tyrosine peptide arrays with a kinetic readout. Molecular Cancer Therapeutics 8: 1846–1855. [DOI] [PubMed] [Google Scholar]
- 24. Sikkema AH, Diks SH, den Dunnen WFA, ter Elst A, Scherpen FJG, et al. (2009) Kinome Profiling in Pediatric Brain Tumors as a New Approach for Target Discovery. Cancer Res 69: 5987–5995. [DOI] [PubMed] [Google Scholar]
- 25. Lindholm EM, Kristian A, Nalwoga H, Kruger K, Nygard S, et al. (2012) Effect of antiangiogenic therapy on tumor growth, vasculature and kinase activity in basal- and luminal-like breast cancer xenografts. Mol Oncol 6: 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai KY, Vin H, Leung M, Chitsazzadeh V, Ojeda S, et al.. (2012) Suppression of apoptosis by BRAF inhibitors through off-target inhibition of JNK signaling. 2012 ASCO Annual Meeting. Chicago, Illinois.
- 27. Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, et al. (2010) The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A 107: 14903–14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, et al. (2010) PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res 23: 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464: 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weber CK, Slupsky JR, Kalmes HA, Rapp UR (2001) Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res 61: 3595–3598. [PubMed] [Google Scholar]
- 31. Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, et al. (2006) In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res 66: 9483–9491. [DOI] [PubMed] [Google Scholar]
- 32. Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, et al. (2006) Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 25: 3357–3364. [DOI] [PubMed] [Google Scholar]
- 33. Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, et al. (2012) BRAFL597 mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer discovery 2: 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tap WD, Gong KW, Dering J, Tseng Y, Ginther C, et al. (2010) Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia 12: 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sondergaard JN, Nazarian R, Wang Q, Guo DL, Hsueh T, et al.. (2010) Differential sensitivity of melanoma cell lines with BRAF(V600E) mutation to the specific Raf inhibitor PLX4032. Journal of Translational Medicine 8. [DOI] [PMC free article] [PubMed]
- 36. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, et al. (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480: 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, et al. (2011) PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 71: 2750–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, et al. (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, et al. (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483: 100–103. [DOI] [PubMed] [Google Scholar]
- 40. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, et al. (2010) Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18: 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Individual patient characteristics of all metastatic malignant melanoma cases.
(XLS)
Mean phosphorylation intensity values of all kinase substrates across twenty-six metastatic malignant melanoma samples and four normal skin tissue samples in the basal data set.
(XLS)
Mean inhibition of 144 kinase substrates in metastatic malignant melanoma samples in response to ex-vivo vemurafenib and sunitinib.
(XLS)
Kinase substrates identified as significantly differentially affected (P<0.05) by ex-vivo vemurafenib incubation in BRAF wild-type and BRAF(V600E) melanoma samples.
(XLS)
Results from KEGG pathway analysis of kinase substrates involved in the PI3K and MAPK signalling pathways including all 144 kinase substrates, and 40 kinase substrates differentiating between BRAF(V600E) and BRAF wild-type tumors.
(XLS)
Kinase inhibitory effects of vemurafenib in melanoma cell lines harboring BRAF(V600E) mutations (MM200 and patient-3-post ) and BRAF wild type (MelJD).
(XLSX)