

Abstract
Correlative microscopy techniques interrogate biological systems more thoroughly than is possible using a single modality. This is particularly true if disparate data types can be acquired from the same specimen. Recently, there has been significant progress towards combining the structural information obtained from soft x-ray tomography (SXT) with molecular localization data. Here we will compare methods for determining the position of molecules in a cell viewed by SXT, including direct visualization using electron dense labels, and by indirect methods, such as fluorescence microscopy and high numerical aperture cryo-light microscopy. We will also discuss available options for preserving the in vivo structure and organization of the specimen during multi-modal data collection, and how some simple specimen mounting concepts can ensure maximal data completeness in correlative imaging experiments.
Keywords: Cellular imaging, Cryo-Flourescence microscopy, Hybrid methods, Localization, Tomography
Introduction
Imaging is a key technique in cell biology, and the origin of much of our understanding of cell structure and organization (Leis et al., 2009). Cellular imaging is typically carried out to either visualize and quantify sub-cellular structures, or measure the cellular distribution of specific molecules (Sartori et al., 2005). Normally both types of information are needed to fully answer a biological question, irrespective of the question being asked or the type of cells being imaged (Leis et al., 2009). Clearly, the optimal situation is to collect both types of data from the same specimen because this imparts confidence in the validity of any conclusions drawn compared with the alternative of making assumptions based on data acquired from different specimens (Giepmans et al., 2005; Le Gros et al., 2009; Lucic et al., 2007; Martone et al., 2000; Sartori et al., 2007). As a result there has been an enormous upswing in the development and use of so-called ‘correlated microscopies’.
In correlated microscopies a specimen is imaged using two or more microscopes and the data is combined to form a composite view. Whilst this approach to imaging cells is highly desirable, the methodology required poses a number of technical and instrumental challenges, which until recently proved daunting and difficult to overcome (Leis et al., 2009; Leis et al., 2006; Sartori et al., 2005). Firstly, the specimen must remain loyal to the in vivo state for the duration of data collection, both in terms of the cell’s structure and organization. Secondly, data acquisition by one modality must not compromise either the fidelity of the specimen or the ability to carry out subsequent imaging methods. Thirdly, the data obtained from all modalities should be as complete as possible since missing data can mask or skew important features in the specimen, resulting in errors in assignment of location, quantification, or in determining the presence of absence of particular molecules. Here, we will discuss methods that have been developed for correlating soft x-ray tomography (SXT) with molecular localization methods, with a particular emphasis on fluorescence microscopy (FM).
Since SXT may not yet be familiar to all readers we will now briefly describe the characteristics and attributes of this modality as stand-alone techniques, prior to describing how it can be combined and correlated with molecular localization methods.
Soft X-ray Tomography
Soft x-ray microscopes currently used for studying biological material measure the transmission of “soft” x-ray photons through a specimen (Attwood, 1999). “Soft” x-ray photons have energies that fall within the so-called ‘water window’ region of the spectrum (Kirz et al., 1995). That is to say, between the K-absorption edges of oxygen at 280 eV and carbon at 530 eV (this equates to 2.34 and 4.4nm respectively) (McDermott et al., 2012b). At these energies, the illuminating light is attenuated an order of magnitude more strongly by biological materials than by water (Attwood, 1999; Kirz et al., 1995; Larabell and Le Gros, 2004a; Larabell and Nugent, 2010; Schneider, 1999; Schneider, 2003; Schneider et al., 2001; Schneider et al., 2003). This difference is linear, adheres to the Beer-Lambert Law and – because biological specimens are highly varied in terms of their internal composition - gives rise to excellent image contrast in most specimens, particularly biological cells (Attwood, 1999; Kirz et al., 1995; Larabell and Le Gros, 2004a; Larabell and Nugent, 2010; Schneider, 1999; Schneider, 2003; Schneider et al., 2001; Schneider et al., 2003).
Soft x-ray microscopes make use of Fresnel zone plate condenser and objective lenses that have low numerical aperture and relatively large depth of focus (Attwood, 1999; Kirz et al., 1995; Larabell and Le Gros, 2004a; Larabell and Nugent, 2010; Schneider, 1999; Schneider, 2003; Schneider et al., 2001; Schneider et al., 2003). Therefore, images taken using the x-ray microscope of specimens that are on the order of 10 μm in diameter are assumed to be 2-dimensional projections of the transmission through the specimen (Larabell and Le Gros, 2004a). Soft x-ray microscopy is combined with tomography, which involves simply imaging the specimen from a number of different angular viewpoints (Larabell and Le Gros, 2004a). If a sufficient number of 2-dimensional images are collected, a 3-dimensional reconstruction of the specimen can be calculated (Weiss et al., 2000).
The fluence of x-ray photons required for soft x-ray tomography could cause serious structural damage to a biological specimen. Damage is generally cumulative with dose, and therefore a serious concern in techniques when using tomography because the specimen is repeatedly illuminated (Fischer et al., 2006; Weiss et al., 2000). The long-standing solution to this problem has been to ‘preserve’ or ‘fix’ the specimen, either chemically using cross-linking aldehydes or by quickly cooling the specimen to low temperature (usually liquid nitrogen temperature, or lower) (Baumeister, 2002; Baumeister et al., 2008; Cyrklaff et al., 2007; Leis et al., 2009; Leis et al., 2006; Nicastro et al., 2000). There is an additional advantage to preserving the specimen; this process locks structures and molecules in place and prevents them from moving during data acquisition (Leis et al., 2006; Quintana, 1994; Ryan, 1992). Again this is particularly important in techniques where multiple images, or imaging modalities are required.
As with any process carried out prior to imaging, it is enormously important that the fixation procedure retains the in vivo structure and organization of the specimen (McDermott et al., 2009). Otherwise the resulting images will not represent reality, and be a source of confusion. Chemical fixation is a well-established technique that has been very commonly used in fluorescence, electron and other microscopies (Bell and SafiejkoMroczka, 1997). There is a substantial body of evidence – initially from electron microscopy and subsequently by soft x-ray microscopy - showing that chemical fixation can devastate the structural integrity of a specimen, even when carried out carefully by an expert (Leis et al., 2009). As a result, cryopreservation via vitrification is now considered the vastly preferred of the two approaches for any high resolution imaging study (Al-Amoudi et al., 2004; Baumeister et al., 2011; Dubochet et al., 1981; Dubochet et al., 1988; Leforestier et al., 1996; Leis et al., 2006). There are other significant benefits to cryopreservation for biological imaging. Specimens can be conveniently fixed at discrete points in time allowing time-dependent experiments to be carried out easily. For example, populations of cells can be frozen after a change in their environment, initiation of an event sequence, such as the cell cycle, or the introduction of a candidate drug molecule into the growth media. Moreover, large numbers of specimens can be collected and preserved, and then imaged when convenient or practical. This makes acquisition of statistically significant numbers of cellular imaging data set feasible. Consequently, all soft x-ray microscopes used for biological imaging incorporate instrumentation to maintain the specimen at cryogenic temperatures. Prior to being mounted in the microscope, the specimen can either be rapidly plunged into liquid cryogen, or fixed in a high-pressure freezer (Baumeister et al., 2011).
After selecting the type of preservation, the next consideration is how to mount the specimen for SXT imaging. There is a wide-range of possible options; for example, thin silicon nitride windows, or the grid system commonly used the electron microscopy community. Thin-walled glass capillaries have been developed and adopted as the specimen mount at the National Center for X-ray Tomography soft x-ray microscope (Larabell and Nugent, 2010; McDermott et al., 2012a; McDermott et al., 2012b). Two very important factors contribute to making the capillary a good decision. Firstly, the geometry of a cylinder – mounted along the axis of rotation –means the specimen can be imaged from any arbitrary angle around a full 360 degrees of rotation. This in turn allows projection series to be collected without the ‘missing wedge’ of data that is unavoidable if the specimen is mounted on a flat support (such as a silicon nitride window, or an electron microscopy grid) (Leis et al., 2009). Typically, such mounts can only be tilted a maximum of ±70 degrees, and as the flat specimen is tilted it appears significantly thicker, and therefore more strongly absorbing, to the point that there is insufficient transmission of the specimen illumination to the detector to provide adequate signal-to-noise. Missing data and the other factors associated with limited tilt tomography negatively impact the resultant tomographic reconstructions (Hagen et al., 2012; Leis et al., 2009). No such problems are encountered if the specimen is mounted in a capillary. In this geometry the sample can be rotated completely and therefore the tomographic reconstructions are significantly freer from artifact (Le Gros et al., 2005; McDermott et al., 2012a; McDermott et al., 2012b).
As mentioned above, soft x-ray microscopes produce two-dimensional projection images of the specimen (Falcone et al., 2011; Larabell and Nugent, 2010). Figure 1 shows a two-dimensional projection image of a human mammary epithelial cell taken using a soft x-ray microscope. The cell is obviously an internally complex three-dimensional object and as a consequence, such a projection image is virtually useless; all of the organelles and cellular structures appear confusingly superimposed on top of each other (Derosier and Klug 1968). Real information about cellular structure requires three-dimensional information. Fortunately, calculation of a three-dimensional tomographic reconstruction of the cell from the two-dimensional projection images is a straightforward, commonly used, and a very well established technique (Natterer, 1986; Natterer and Wübbeling, 2001). For example, this is the principle behind medical CT (computed tomography) that is ubiquitous in hospitals and clinics throughout the world. The basic concepts behind CT are used in SXT, but at significantly higher resolution (better than 50 nm). In SXT, the x-ray source and detector remain fixed and the specimen is rotated inside the microscope (Larabell and Nugent, 2010).
Figure 1.
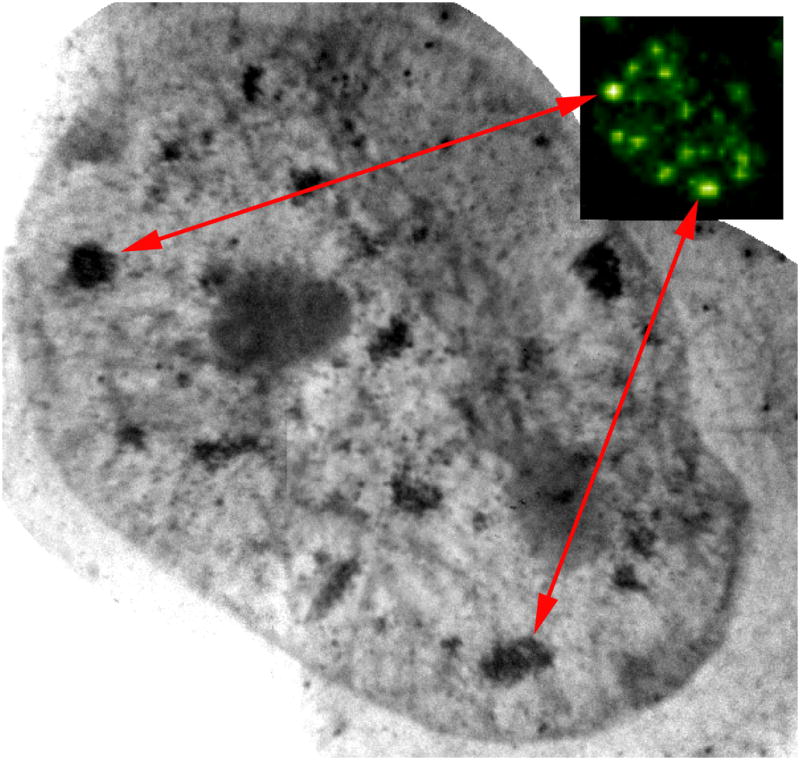
Localization of splicing factors in the nucleus of human mammary epithelial cell (T4) using correlated fluorescence and x-ray microscopy. Splicing factors tagged with FluoroNanogold® secondary antibodies can be seen using confocal microscopy (inset). The same splicing factors can be found in the x-ray microscope projection image after silver enhancement of the nanogold particles. Scale bar = 2 μm.
Figure 2F shows a computer-generated slice through the center of a soft x-ray reconstruction of a vitrified mouse lymphocyte. The spatial resolution of this soft x-ray reconstruction is ~50 nm, and the contrast reflects the distribution of water and organic matter inside the cell. It has been observed that in soft x-ray reconstructions of biological specimens the major organelle types display characteristic attenuation patterns (McDermott et al., 2012a). For example, lipid droplets appear dense (highly absorbing) due to the high density of biomolecules and low water content. Structures such as the vacuole and mitochondrion, are much less dense due to their relatively higher water content. The most common analysis/visualization technique is ‘segmentation’ (McDermott et al., 2012b). In this technique, regions of the reconstructed cell are isolated based on one or more particular characteristic(s), such as shape or x-ray Linear Absorption Coefficient (LAC), i.e. the measure of how strongly the material at each point in the tomographic reconstruction attenuates the x-ray illumination (McDermott et al., 2012b). The LAC values for a particular organelle type are characteristic, and highly conserved between the cells of the same type (McDermott et al., 2012b). Variations in LAC inside an organelle depict the internal structures, for example chromatin versus heterochromatin in the nucleus, or architectural features in the nucleolus.
Figure 2.

Localization of promyelocytic leukemia (PML) bodies in the nucleus of human lymphocytes using hybrid light and x-ray imaging. (A) Transmitted light microscope image of a lymphocyte showing localization of PML bodies in the nucleus, labeled using gold-enhanced FluoroNanogold® secondary antibodies. (B) Localization of PML bodies in the nucleus of a lymphocyte from the same population of cells seen using cryo x-ray tomography. This is a maximum intensity projection image of all orthoslices from the tomographic reconstruction showing only the segmented nucleus. (C) The same image seen in ‘B’ with distribution of PML bodies highlighted by gold dots. (D) One orthoslice from the tomographic reconstruction shown in ‘B, C’ showing two PML bodies (red arrows). (E) One orthoslice from control nucleus processed in parallel, omitting only the primary antibody labeling step. (F) One orthoslice from the tomographic reconstruction of a live, unlabeled, rapidly frozen lymphocyte for comparison. The nucleus is outlined in red and contains well-defined heterochromatin and a nucleolus (yellow arrow) that are not seen in ‘B – E’, presumably due to extensive processing for antibody labeling. (G) Labeled antibodies are easily identified using the Linear Absorption Coefficient (LAC), since only the gold enhanced particles they have a LAC > 0.6 μm-1, as seen in the plot of all voxels in the nucleus after aldehyde fixation (black dashed line), aldehyde fixation plus secondary antibodies (red solid line), and gold enhancement of antibodies (light blue solid line). Scale bars = 2 μm.
Localizing molecules in SXT reconstructions of a cell
There are two options for identifying the position of specific molecules in a soft x-ray tomogram; either directly or by using correlated imaging data. Consequently, there are two distinct methods for tagging molecules for identification, either by using:
Electron dense labels that can be identified directly in a SXT reconstruction by virtue of their x-ray absorption characteristics. Electron dense labels for this purpose are created by the conjugation of antibodies to an appropriate metal particle, such as nano-gold (Figures 1 and 2).
Fluorescent labels that can be located by correlating fluorescence microscopy/tomography data (Figures 1, 3 and 4). Fluorescent labels can either be exogenous (for example, vital dyes or antibodies conjugated to small organic dyes or nanoparticles) or genetically encoded, for example one of the engineered derivatives of Green Fluorescent Protein from the jellyfish Aequorea victoria (Tsien, 2005).
Figure 3.
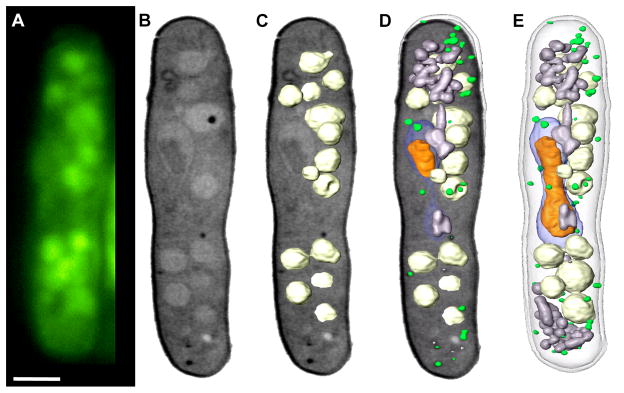
Correlated fluorescence and x-ray images of Schizosaccharomyces pombe. (A) Cryo-immobilized, high-aperture wide-field fluorescence image of S. pombe. The vacuoles were stained with CMFDA (5-chloromethyl fluorescein diacetate). (B) Soft X-ray tomography of the same cell shown in an orthoslice from the tomgraphic reconstruction. (C) Segmented vacuoles were overlaid on the orthoslice. (D) The completely segmented cell overlaid on the orthoslice. The segmented cell. Key: nucleus, blue; nucleolus, orange; mitochondria, grey; vacuoles, white; lipid-rich vesicles, green. Scale bar = 2 μm.
Figure 4.

Correlated lymphocyte Fluorescently Stained with LysoTracker. (A and B) Lymphocytes were fluorescently labeled with LysoTracker vital dye. A through-focus fluorescence dataset was collected using a cryo confocal light microscope. Weak non-specific Lysotracker staining was present throughout the cell, which was used to align the fluorescence and SXT reconstructions. Several round organelles were observed in the SXT reconstruction that correlated with the punctate Lysotracker fluorescence. Scale bar = 2 μm.
Each method has inherent advantages and disadvantages. Proteins expressed in tandem with fluorescent proteins have some likelihood that their biochemical function will be perturbed either due to steric hindrance or because the protein is present in the cell at a non-physiological concentration. Antibody-conjugated electron-dense labels bind, in principle, to molecules present in their native state in the cell at their physiological concentrations. However there are major disadvantages associated with immuno-labeling methods. First of all, the technique typically is limited to use on chemically fixed specimens. Secondly, antibodies may also have limited or differential access to the target epitope. Finally, antibodies do not readily pass through the plasma membrane of living cells. The first step during immuno-labeling is to find the best method of introducing the labeled antibodies. There are two basic approaches, one is to use detergents or other permeabilization agents to make holes large enough for passage of tagged immune-globulins and the other, which is harsher, is to use agents such as ethanol, methanol, or acetone.
For our early work we adopted whole-mount labeling approaches typically used for fluorescence microscopy, with FluoroNanogold as secondary antibodies. This enabled localization of the protein using confocal microscopy followed by detection in the x-ray microscope after silver-enhancement of the Nanogold particles. At 517 eV photon energy, the characteristic attenuation lengths of water, organic material, and silver are on the order of 10 μm, 0.5 μm, and 50 nm, respectively. The labels could therefore be identified by inspection as dark, highly absorbing foci. Correlation with the fluorescence is straightforward in this 2D projection image from the x-ray microscope, but precise spatial information is lacking. To obtain three-dimensional information of the distribution of proteins, we performed tomography after labeling, as seen in Figure 2. This approach made is possible to obtain more precise information about the location of PML bodies in the nucleus of whole, hydrated human lymphocytes. Figure 2B shows a maximum intensity projection of a SXT reconstruction of the immuno-labeled cell where the nucleus of the cell was segmented from the rest of the cell for clarity. A number of extremely dark, high-absorbing foci are observed throughout the nucleus, which are highlighted in gold in Fig. 2C. Orthoslices from the tomographic reconstruction show individual foci (Figure 2D) that correspond to the positions of the gold-enhanced PML antibodies, as unlabeled cells do not have voxels in the SXT reconstructions with similar LAC values (Figures 2E–G). The distribution is consistent with previous measurements (Spector, 1996; Spector, 2001), but it is clear that nuclear structures have been damaged due to processing. As discussed above, antibody labeling requires chemical fixation and membrane permeabilization, typically with high concentrations of detergent to localize proteins in the nucleus. Comparison of x-ray reconstructions of a treated and untreated cell shows the dramatic reorganizations that occur inside the nucleus (Figure 2E and F).
As a result of the potential for damage to cellular structures, plus the clear preference for cryogenic preservation and fluorescence labeling, correlated fluorescence microscopy of endogenous molecules is the molecular localization strategy of choice.
Fluorescence Correlative Microscopy
Fluorescence microscopy (FM) is ubiquitous in cell biology, to the point that it needs little introduction or description beyond stating that in general, it is now possible to fluorescently tag most proteins inside a cell (Giepmans et al., 2006; Shaner et al., 2005; Tsien, 2005). FM is without a doubt an enormously enabling technology, and has a huge presence in the literature. It is, therefore, natural that FM is the partner of choice in most efforts to develop correlative microscopy methods, and SXT is no different.
Initial efforts to correlate SXT and fluorescence used room-temperature confocal fluorescence microscopy of the same chemically fixed, dehydrated specimen as imaged using SXT. To correlate fluorescence in cryo-fixed cells with SXT demands a fluorescence microscope that can operate at cryogenic temperatures. There are currently commercially available low numerical aperture cryo-light microscopes available. However, these microscopes operate with air lenses and therefore FM images are of relatively low spatial resolution, and molecules could only be localized in a SXT reconstruction with poor precision. To overcome this shortfall, Le Gros and colleagues developed a new type of FM instrument that could operate at cryogenic temperatures (Le Gros et al., 2009; McDermott et al., 2009). This fulfilled the goals of allowing cryo-preserved specimens to be imaged in refractive index matched fluids, and therefore the use of high numerical-aperture lenses. We will now describe this breakthrough technology.
Cryogenic Fluorescence Microscopy
An original ‘proof of concept’ cryo-fluorescence microscope (CFM) designed by Le Gros and colleagues employed an objective lens that can operate at liquid nitrogen temperature (Le Gros et al., 2009). This feature permitted the use of refractive index-matched fluids, and, therefore, opened the door to cryogenic visible light microscopy with high numerical aperture collection (~1.3 NA). To date, liquid propane (refractive index =1.32) and iso-pentane (refractive index = 1.35) have been used successfully as the immersion fluid, with many more options still to be tested. This new microscope represented a major improvement over the use of ‘dry’ or air lenses, which are limited to a numerical aperture <1 (Le Gros et al., 2009). Imaging in such an equipped microscope suffers from signal degradation due to the fact that a smaller cone angle of light is collected when it crosses the boundary between two media of differing refractive indexes, for example, a biological specimen and air (or even a vacuum). The use of a cryogenic immersion fluid with a refractive index matched to the sample and objective lens greatly reduces such problems, allowing fluorescence imaging equal to that obtained with high NA lenses using conventional room temperature immersion media.
When fluorescence imaging is carried out at room temperature the concentration, brightness, and photo-stability of the labels determine the overall quality of the data (Thompson et al., 2002). Improving resistance to photon-induced damage is therefore a significantly important aim that can be achieved by imaging the specimen at low temperature (Moerner and Orrit, 1999). At liquid nitrogen temperature the working lifetime of a fluorescent protein can be extended by a factor of 30 or more (Le Gros et al., 2009). This allows collection of data with greatly improved signal-to-noise before ‘photo-bleaching’ occurs as compared with room temperature experiments (Le Gros et al., 2009). Moreover, this increase in working life makes it possible to consider new data collection strategies. For example, the specimen can be imaged using through-focus collection from many angles in a rotation series. Tomographic data collection opens the door to reconstructing fluorescence data with spherical symmetric point spread functions (i.e. each point is visualized as an isotropic sphere, rather than being anisotropic with poorer precision in one direction).
Adopting cryopreservation for fluorescence imaging offers several other advantages over room temperature microscopy. Firstly, the bandwidth of the emitted light may be narrower at cryogenic temperature compared with room temperature (Moerner and Orrit, 1999), allowing the use of narrow band emission filters, potentially increasing the fluorescence signal above the background. Secondly, cryo-immobilization significantly slows Brownian motion and dynamic cell processes inside the specimen, permitting long or repeated exposures. In conjunction with enhanced probe working life and improved spectral properties, this makes it possible to collect data for very high precision localization studies. Finally, in a vitrified specimen photo-bleaching is less sensitive to the chemical environment, therefore the local concentration of a labeled protein can be quantified more accurately than is possible at room temperature (Le Gros et al., 2005; Moerner and Orrit, 1999; Thompson et al., 2002). The increased collection efficiency of the microscope, combined with the increased working lifetime of the fluorescent probes and suppressed Brownian motion inside the cell, invites further improving the performance of the cryo light microscope to allow the use of ‘super resolution’ strategies. The exact form this will take remains an open question. Work is underway to determine how to best exploit the characteristics of fluorescent probes at low temperatures. For example, the increased stability of fluorophores at cryogenic temperatures is a significant advantage in methods such as ‘structured illumination’. Here the fluorescence signal must remain as constant as possible during the acquisition of large numbers of images. However, it remains unclear if probes can be found that ‘blink’ satisfactorily at low temperature, and thus allow the application of stochastic super resolution techniques, such as PALM and STORM. That said, CFM is very much unexplored territory, and as such opens up the opportunity for the development of completely novel approaches to the issue of imaging beyond the diffraction limit. This is an exciting new frontier, and one that holds much promise for the future.
Now that we have outlined the utilities of SXT and CFM we will discuss how these disparate data can be correlated.
Path to Correlative SXT and CFM
The basic problems involved in aligning two disparate types of data has been well studied, particularly in clinical imaging where this is routine in techniques such as correlated Positron Emission Tomography (PET) and Computer Tomography (CT) scans. In the medical community, algorithms have been developed to align two different diagnostic images, such as Positron Emission Tomography (PET) and Computer Tomography (CT) scans (Gilman et al., 2007; Krishnasetty et al., 2005). It may be possible to align some features in the soft x-ray and fluorescence reconstructions using similar algorithms, such as mutual information or normalized mutual information (Viola and Wells, 1997). Figure 3 shows correlated widefield cryo-fluorescence and soft x-ray images of Schizosaccharomyces pombe whose vacuoles were stained with CMFDA (5-chloromethyl fluorescein diacetate). Visual inspection of the fluorescence distribution (Fig. 3A) shows good correlation with large, low-absorbing vacuoles observed in the soft x-ray reconstruction (Fig. 3B). To emphasize the correlation between the two datasets, the vacuoles were segmented out of the soft x-ray reconstruction and overlaid with an orthoslice through the cell (Figure 3C), then put in context with additional organelles (Figures 3D, E). Figure 3 shows fluorescence data collected from a cryo-light microscope using standard widefield collection optics. To remove contributions from out of focus light and scatter, we added a spinning disk confocal unit to the cryo-light microscope. Through-focus fluorescence collected from a lymphocyte labeled with the vital dye LysoTracker is presented in Figures 4A and 4C. The fluorescence data was manually overlaid with the soft x-ray reconstruction of the same cell and presented in Figures 4B and 4D. Weak LysoTracker fluorescence was detected throughout the cell and used to roughly overlay the two datasets; round structures identified in the SXT reconstruction were used to align the punctate fluorescence dataset more precisely.
A more straightforward and objective correlation strategy is to use fiducial markers that are visible in both instruments. Fortunately, the technology to produce fluorescent nano-gold beads is becoming well established, and objects such as these can be readily incorporated onto the surface of the specimen-mounting capillary. Once the two types of data have been collected they can be aligned with respect to each other by simply maximizing the correlation between the signals in both reconstructions. This ongoing work will be the subject of future publications.
The alignment and correlation processes could be greatly aided if the data collected from both modalities were both isotropic and had similar resolution. This would minimize systematic uncertainties along a particular direction in cellular space, and maximize confidence in the assignment of a molecule to a particular cellular environment. Currently, along the optical axis (the z-axis) in CFM system, the spatial-resolution is approximately three times lower than the resolution obtained in the plane normal to the optical axis (the x,y plane). By rotating the sample around an axis normal to the optical axis the internal structures of the cell can be imaged sequentially at the higher (x,y) resolution. All of the data from each through-focus acquisition could then be recombined into a single reconstructed image with significantly improved, more isotropic spatial resolution (Heintzmann and Cremer, 2002; Satzler and Eils, 1997; Shaw et al., 1989; Swoger et al., 2003; Swoger et al., 2007)}.
Discussion
The ability to correlate SXT structural information with molecular localization data is an exciting development. From many perspectives, the future looks very bright and we anticipate significant growth in this area, in particular in the use of correlative CFM. In the case of CFM there is a huge range of work that can be carried out to improve this modality. For example, the cryogenic optical system can be optimized to minimize aberrations by using purpose-designed cryogenic immersion lenses. Different cryogenic immersion fluids can be tested to determine which offers the best combination of operating efficiency and refractive index matching. The software and algorithms for alignment of through-focus data can be made to run much faster by, for example, using code optimized to run on a Graphical Processing Unit (GPU).
For SXT, new generation zone plate optics have been developed that increase the maximum achievable spatial resolution to approaching 10 nm in test specimens (Chao et al., 2009). Such ultra-high resolution optics, such as these would decrease the depth of field (Chao et al., 2009). So, this enhancement would also require development of through-focus, deconvolution soft x-ray tomography. In practice, however, this problem is similar conceptually to that used in CFM, and therefore not considered a significant issue.
In terms of the development of correlative methods, there is much scope open for improving the CFM/SXT image registration. For example, landmark fiducials – visible in both modalities – can be further refined and optimized to reduce their physical size and enhance their quantum yield and brightness. Live cell imaging can also be incorporated into the workflow, allowing specific cells – containing rare structures or events – to be captured and mounted for imaging sequentially by CFM and SXT.
Conclusion
The addition of molecular localization capabilities has made SXT an even more valuable asset to cell imaging community. Of particular importance is the recent development of correlative high numerical aperture cryo-fluorescence microscopy. As has been established in high-resolution Electron Microscopy, cryogenic fixation results in specimens that are more closely representative of the native state (Dubochet et al., 1988). Retention of structural integrity is alone sufficient justification for considering adopting cryogenic light-based techniques for experiments where fixation is required. However, an even greater benefit to using cryo-light imaging is the observed increase in fluorophore working lifetimes. While low temperature microscopes have previously been used to view frozen cells, the introduction of a low temperature immersion lens approach has opened up the possibility of using the highest resolution fluorescence imaging methods on a vitrified sample prior to x-ray or electron microscopy. In our experience cryogenic imaging results in a significant increase in the useful working life of all fluorophores. This characteristic is highly desirable in general, but will be particularly useful in ‘super resolution’ imaging techniques such as 3-Dimensional Structured Illumination Microscopy (Carlton, 2008), or any other method that can benefit from the collection of comparatively large numbers of images.
Correlating CFM with SXT is an exciting new development in cellular imaging, and produces high-resolution, 3-dimensional structural views of a cell, including the location of specific molecules. This correlated microscopy technique can be applied to a wide range of cell types, from prokaryotes to eukaryotes, without the cells being sectioned, stained, or dehydrated. Therefore, the structural integrity of the cell is optimally retained during preparation, and then maintained during data collection by virtue of cryogenic preservation. The use of capillary mounting system permits cells to be imaged by multiple modalities, and more importantly, leads to highly completeness of data.
Materials and Methods
X-ray microscopy
Human mammary epithelial cells were imaged with a transmission X-ray microscope (XM-1) equipped with Fresnel zone plate condenser and objective (with 55nm and 45 nm outer zone widths respectively; the latter being the resolution-defining optical element). The cells were cultured on a thin (100 nm) low-absorption silicon nitride membrane and examined using photon energies just below the oxygen edge, i.e. 517 eV (corresponding to a wavelength of 2.4 nm). The data were collected using x-ray energies of 517 eV (2.4 nm), and 16-bit images were recorded using a Peltier-cooled, back-illuminated, 1340 X 1300 soft x-ray CCD camera (Roper Scientific Instruments Micromax system with SIT chip; Princeton Instruments, Trenton, NJ).
Soft X-ray Tomography
Projection images were collected using XM-2, the National Center for X-ray tomography soft x-ray microscope at the Advanced Light Source of Lawrence Berkeley National Laboratory. XM-2 was designed to investigate biological samples in their hydrated states. Rapidly frozen specimens were mounted in the cryogenic rotation stage (Le Gros et al., 2005) and maintained at low temperatures using a stream of helium gas that had been cooled to liquid nitrogen temperatures (Le Gros et al., 2005; McDermott et al., 2009). Cooling the specimen allows collection of projection images while mitigating the effects of exposure to radiation. Each dataset (i.e., 90 projection images spanning a range of 180°) was collected using Fresnel zone plate based objective lens with a resolution of 50 nm (Larabell and Le Gros, 2004b). Exposure times for each projection image ranged from 150 to 300 msec. Projection images were manually aligned using fiducial markers on adjacent images using the IMOD software package (Kremer et al., 1996). Tomographic reconstructions were calculated using the iterative reconstruction method (Mastronarde, 2005; Stayman and Fessler, 2004). The Amira software package (Version 5.3.1, Visage Imaging, San Diego, CA) was used to manually segment the reconstructed volumes.
Correlated cryo fluorescence and x-ray imaging
Schizosaccharomyces pombe were prepared and imaged as described previously (Le Gros et al., 1999). Lymphocytes were labeled with LysoTracker Green DND-26 vital dye (200 nM concentration, Molecular Probes, Grand Island, NY), following standard protocols. Labeled cells were pipetted into capillaries and vitrified by fast plunging in liquid propane. Vitrified specimens were cryo-transferred into a custom-built cryo light microscope (Le Gros et al., 1999) fitted with a Piezo-driven sample stage (Model E-501.00, PI, Auburn, MA), a spinning disk laser-based confocal unit (CSU-X1, Yokogawa, Tokyo, Japan), and an EMCCD camera (iXon DV887ECS-BV, Andor Technologies, Belfast, UK). A 491 nm solid-state laser (100 mW, Andor Technologies, Belfast, UK) excited the LysoTracker dye and a standard bandpass filter (525 +/− 50nm, Part number 41017, Chroma Technology Corp., Bellows Falls, VT) was used as the emission filter. After being imaged with the cryo-light microscope, the sample capillary was cryo-transferred to the x-ray microscope, where 92 projection images were collected with 2° of rotation between projections. Alignment and reconstruction of the soft x-ray data was performed as previously described (Parkinson et al., 2012). The through-focus fluorescence data was overlaid with the soft x-ray reconstruction using Amira (Version 5.3.1, Visage Imaging, San Diego, CA).
Immunocytochemistry
For localization of splicing factor, cells were permeabilized/extracted for 10 min at room temperature in a cytoskeletal stabilization buffer (CSB; 100 mm NaCl, 300 mm sucrose, 5 mm MgCl2, 10 mm PIPES, pH 6.8) with protease and phosphatase inhibitors (1 mm pefabloc, 10 mg mLP1 leupeptin, 10 mg mLP1 aprotinin, 10 mg mLP1 trypsin inhibitor II, 250 mm NaF, 0.5% Triton), washed in CSB with inhibitors (minus Triton), then fixed (2% paraformaldehyde, 0.1% glutaraldehyde in CSB). Cells were rinsed in PBS, blocked in CSB, then incubated in primary antibodies against splicing factor protein (SRm300), rinsed in blocking buffer, then incubated in secondary antibodies (FluoroNanogold, Nanoprobes Inc., Yaphank, NY, U.S.A.). After rinsing in PBS cells were imaged using confocal microscopy. Cells were then post fixed in 2% glutaraldehyde in PBS, rinsed in double distilled H2O, silver enhanced (Li Ag, Nanoprobes Inc.), rinsed in ddH2O, then imaged in the hydrated state with the X-ray microscope.
For localization of promyelocytic leukemia (PML) bodies, human lymphocytes were fixed (2% paraformaldehyde, 0.1% glutaraldehyde, and 0.1% Tween20 in 200mM HEPES, pH 7.2), rinsed, and incubated in blocking buffer (1% BSA, 0.05% Triton X100 in PBS). Cells were then incubated in primary antibodies (monoclonal mouse anti-PML, Santa Cruz sc-966), rinsed in blocking buffer, and incubated in secondary antibodies (FluoroNanogold described above). Nanogold particles were enhanced with gold for x-ray tomography.
Acknowledgments
This work was funded by the US Department of Energy, Office of Biological and Environmental Research (DE-AC02-05CH11231), the National Center for Research Resources of the National Institutes of Health (P41RR019664) and the National Institutes of General Medicine of the National Institutes of Health (GM63948).
Abbreviations
- CFM
cryogenic fluorescence microscopy
- FM
fluorescence microscopy
- LAC
linear absorption coefficient
- PSF
point spread function
- SXT
soft x-ray tomography
- YFP
yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Amoudi A, Chang JJ, Leforestier A, McDowall A, Salamin LM, et al. Cryo-electron microscopy of vitreous sections. Embo Journal. 2004;23:3583–3588. doi: 10.1038/sj.emboj.7600366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood DT. Soft x-rays and extreme ultraviolet radioation: principles and applications. Cambridge Uhiversity Press; Cambridge, New York: 1999. [Google Scholar]
- Baumeister W. Electron tomography: towards visualizing the molecular organization of the cytoplasm. Current Opinion in Structural Biology. 2002;12:679–684. doi: 10.1016/s0959-440x(02)00378-0. [DOI] [PubMed] [Google Scholar]
- Baumeister W, Gruska M, Medalia O, Leis A. Electron tomography of vitreous sections from cultured mammalian cells. Journal of Structural Biology. 2008;161:384–392. doi: 10.1016/j.jsb.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Baumeister W, Vanhecke D, Asano S, Kochovski Z, Fernandez-Busnadiego R, et al. Cryo-electron tomography: methodology, developments and biological applications. Journal of Microscopy. 2011;242:221–227. doi: 10.1111/j.1365-2818.2010.03478.x. [DOI] [PubMed] [Google Scholar]
- Bell PB, SafiejkoMroczka B. Preparing whole mounts of biological specimens for imaging macromolecular structures by light and electron microscopy. International Journal of Imaging Systems and Technology. 1997;8:225–239. [Google Scholar]
- Chao W, Kim J, Rekawa S, Fischer P, Anderson EH. Demonstration of 12 nm Resolution Fresnel Zone Plate Lens based Soft X-ray Microscopy. Optics Express. 2009;17:17669–17677. doi: 10.1364/OE.17.017669. [DOI] [PubMed] [Google Scholar]
- Cyrklaff M, Kudryashev M, Leis A, Leonard K, Baumeister W, et al. Cryoelectron tomography reveals periodic material at the inner side of subpellicular microtubules in apicomplexan parasites. Journal of Experimental Medicine. 2007;204:1281–1287. doi: 10.1084/jem.20062405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J, Knapek E, Dietrich I. Reduction of Beam Damage by Cryoprotection at 4k. Ultramicroscopy. 1981;6:77–80. [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, et al. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Falcone R, Jacobsen C, Kirz J, Marchesini S, Shapiro D, et al. New directions in X-ray microscopy. Contemporary Physics. 2011;52:293–318. [Google Scholar]
- Fischer P, Kim DH, Chao WL, Liddle JA, Anderson EH, et al. Soft X-ray microscopy of nanomagnetism. Materials Today. 2006;9:26–33. [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat Methods. 2005;2:743–749. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- Gilman MD, Fischman AJ, Krishnasetty V, Halpern EF, Aquino SL. Hybrid PET/CT of the thorax: when is computer registration necessary? J Comput Assist Tomogr. 2007;31:395–401. doi: 10.1097/01.rct.0000237817.18678.9c. [DOI] [PubMed] [Google Scholar]
- Hagen C, Guttmann P, Klupp B, Werner S, Rehbein S, et al. Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. Journal of Structural Biology. 2012;177:193–201. doi: 10.1016/j.jsb.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzmann R, Cremer C. Axial tomographic confocal fluorescence microscopy. Journal of Microscopy. 2002;206:7–23. doi: 10.1046/j.1365-2818.2002.01000.x. [DOI] [PubMed] [Google Scholar]
- Kirz J, Jacobsen C, Howells M. Soft x-ray microscopes and their biological applications. Quarterly reviews of biophysics. 1995;28:33–130. doi: 10.1017/s0033583500003139. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Krishnasetty V, Fischman AJ, Halpern EL, Aquino SL. Comparison of alignment of computer-registered data sets: combined PET/CT versus independent PET and CT of the thorax. Radiology. 2005;237:635–639. doi: 10.1148/radiol.2372041719. [DOI] [PubMed] [Google Scholar]
- Larabell C, Le Gros M. Whole cell cryo X-ray tomography and protein localization at 50 micron resolution. Biophysical Journal. 2004a;86:185A–185A. [Google Scholar]
- Larabell CA, Le Gros MA. X-ray tomography generates 3-D reconstructions of the yeast, saccharomyces cerevisiae, at 60-nm resolution. Molecular Biology of the Cell. 2004b;15:957–962. doi: 10.1091/mbc.E03-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larabell CA, Nugent KA. Imaging cellular architecture with X-rays. Current Opinion in Structural Biology. 2010;20:623–631. doi: 10.1016/j.sbi.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gros MA, McDermott G, Larabell CA. X-ray tomography of whole cells. Curr Opin Struct Biol. 2005;15:593–600. doi: 10.1016/j.sbi.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Le Gros MA, McDermott G, Uchida M, Knoechel CG, Larabell CA. High-aperture cryogenic light microscopy. J Microsc. 2009;235:1–8. doi: 10.1111/j.1365-2818.2009.03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leforestier A, Richter K, Livolant F, Dubochet J. Comparison of slam-freezing and high-pressure freezing effects on the DNA cholesteric liquid crystalline structure. Journal of Microscopy. 1996;184:4–13. doi: 10.1046/j.1365-2818.1996.1090666.x. [DOI] [PubMed] [Google Scholar]
- Leis A, Rockel B, Andrees L, Baumeister W. Visualizing cells at the nanoscale. Trends in Biochemical Sciences. 2009;34:60–70. doi: 10.1016/j.tibs.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Leis AP, Beck M, Gruska M, Best C, Hegerl R, et al. Cryo-electron tomography of biological specimens. Ieee Signal Processing Magazine. 2006;23:95–103. [Google Scholar]
- Lucic V, Kossel AH, Yang T, Bonhoeffer T, Baumeister W, et al. Multiscale imaging of neurons grown in culture: from light microscopy to cryo-electron tomography. J Struct Biol. 2007;160:146–156. doi: 10.1016/j.jsb.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Martone ME, Deerinck TJ, Yamada N, Bushong E, Ellisman MH. Correlated 3D light and electron microscopy: Use of high voltage electron microscopy and electron tomography for imaging large biological structures. Journal of Histotechnology. 2000;23:261–270. [Google Scholar]
- Mastronarde DN. Fiducial Marker and Hybrid Alignment Methods for Single-and Double-Axis Tomography. In: Frank J, editor. Electron tomography : methods for three-dimensional visualization of structures in the cell. Springer; New York, NY: 2005. pp. 163–185. [Google Scholar]
- McDermott G, Le Gros MA, Larabell CA. Visualizing Cell Architecture and Molecular Location Using Soft X-Ray Tomography and Correlated Cryo-Light Microscopy. Annual Review of Physical Chemistry. 2012a;63:225–239. doi: 10.1146/annurev-physchem-032511-143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott G, Le Gros MA, Knoechel CG, Uchida M, Larabell CA. Soft X-ray tomography and cryogenic light microscopy: the cool combination in cellular imaging. Trends Cell Biol. 2009;19:587–595. doi: 10.1016/j.tcb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott G, Fox DM, Epperly L, Wetzler M, Barron AE, et al. Visualizing and quantifying cell phenotype using soft X-ray tomography. Bioessays. 2012b;34:320–327. doi: 10.1002/bies.201100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner WE, Orrit M. Illuminating single molecules in condensed matter. Science. 1999;283:1670–1676. doi: 10.1126/science.283.5408.1670. [DOI] [PubMed] [Google Scholar]
- Natterer F. The Mathematics of Computerized Tomography. Wiley; New York, NY: 1986. [Google Scholar]
- Natterer F, Wübbeling F. Mathematical Methods in Image Reconstruction. Cambridge University Press; Cambridge, New York: 2001. [Google Scholar]
- Nicastro D, Frangakis AS, Typke D, Baumeister W. Cryo-electron tomography of Neurospora mitochondria. Journal of Structural Biology. 2000;129:48–56. doi: 10.1006/jsbi.1999.4204. [DOI] [PubMed] [Google Scholar]
- Parkinson DY, Knoechel C, Yang C, Larabell CA, Le Gros MA. Automatic alignment and reconstruction of images for soft X-ray tomography. Journal of Structural Biology. 2012;177:259–266. doi: 10.1016/j.jsb.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana C. Cryofixation, Cryosubstitution, Cryoembedding for Ultrastructural, Immunocytochemical and Microanalytical Studies. Micron. 1994;25:63–99. doi: 10.1016/0968-4328(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Ryan KP. Cryofixation of Tissues for Electron-Microscopy - a Review of Plunge Cooling Methods. Scanning Microscopy. 1992;6:715–743. [Google Scholar]
- Sartori A, Gatz R, Beck F, Rigort A, Baumeister W, et al. Correlative microscopy: Bridging the gap between fluorescence light microscopy and cryo-electron tomography. Journal of Structural Biology. 2007;160:135–145. doi: 10.1016/j.jsb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Sartori A, Gatz R, Beck F, Kossel A, Leis A, et al. Correlation microscopy: Bridging the gap between light- and cryo-electron microscopy. Microscopy and Microanalysis. 2005;11:16–17. [Google Scholar]
- Satzler K, Eils R. Resolution improvement by 3-D reconstructions from tilted views in axial tomography and confocal theta microscopy. Bioimaging. 1997;5:171–182182. [Google Scholar]
- Schneider G. High-resolution X-ray Microscopy of Radiation Sensitive Material. Cuvillier Verlag; Gottingen, Germany: 1999. [Google Scholar]
- Schneider G. X-ray microscopy: methods and perspectives. Analytical & Bioanalytical Chemistry. 2003;376:558–561. doi: 10.1007/s00216-003-2007-x. [DOI] [PubMed] [Google Scholar]
- Schneider G, Denbeaux G, Anderson E, Pearson A, Bates W, et al. High resolution X-ray tomography with applications in biology and materials science. Journal de Physique IV. 2003;104:607–613. [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Agard DA, Hiraoka Y, Sedat JW. Tilted view reconstruction in optical microscopy - 3-Dimensional reconstruction of Drosophila melanogaster embryo nuclei. Biophysical Journal. 1989;55:101–110. doi: 10.1016/S0006-3495(89)82783-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL. Nuclear organization and gene expression. Experimental Cell Research. 1996;229:189–197. doi: 10.1006/excr.1996.0358. [DOI] [PubMed] [Google Scholar]
- Spector DL. Nuclear domains. Journal of Cell Science. 2001;114:2891–2893. doi: 10.1242/jcs.114.16.2891. [DOI] [PubMed] [Google Scholar]
- Stayman JW, Fessler JA. Compensation for nonuniform resolution using penalized-likelihood reconstruction in space-variant imaging systems. IEEE Transactions on Medical Imaging. 2004;23:269–284. doi: 10.1109/TMI.2003.823063. [DOI] [PubMed] [Google Scholar]
- Swoger J, Huisken J, Stelzer EHK. Multiple imaging axis microscopy improves resolution for thick-sample applications. Optics Letters. 2003;28:1654–1656. doi: 10.1364/ol.28.001654. [DOI] [PubMed] [Google Scholar]
- Swoger J, Verveer P, Greger K, Huisken J, Stelzer EHK. Multi-view image fusion improves resolution in three-dimensional microscopy. Optics Express. 2007;15:8029–8042. doi: 10.1364/oe.15.008029. [DOI] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. Building and breeding molecules to spy on cells and tumors. FEBS Lett. 2005;579:927–932. doi: 10.1016/j.febslet.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Viola P, Wells WM. Alignment by maximization of mutual information. International Journal of Computer Vision. 1997;24:137–154. [Google Scholar]
- Weiss D, Schneider G, Niemann B, Guttmann P, Rudolph D, et al. Computed tomography of cryogenic biological specimens based on X-ray microscopic images. Ultramicroscopy. 2000;84:185–197. doi: 10.1016/s0304-3991(00)00034-6. [DOI] [PubMed] [Google Scholar]