

Abstract
Studies of the structural organization and functions of the cell body of a neuron (soma) and its surrounding satellite glial cells (SGCs) in sensory ganglia have led to the realization that SGCs actively participate in the information processing of sensory signals from afferent terminals to the spinal cord. SGCs use a variety ways to communicate with each other and with their enwrapped soma. Changes in this communication under injurious conditions often lead to abnormal pain conditions. “What are the mechanisms underlying the neuronal soma and SGC communication in sensory ganglia” and “how do tissue or nerve injuries affect the communication?” are the main questions addressed in this review.
Keywords: purinergic receptor, pannexin, cytokine, gap junction, pain
Introduction
Dorsal root ganglion (DRG) and trigeminal ganglion (TG) neurons are the primary afferent neurons responsible for transmitting sensory information from the periphery to the central nervous system (CNS). DRGs are situated in the posterior root of spinal nerves; TGs are located on the cerebral surface of the temporal bone. Neurons in these ganglia have a unique pseudo-unipolar structure (Fig. 1). The cell body (soma) of each neuron in the ganglia gives out one axon that bifurcates into two branches. The peripheral branch of the neuron travels distally to various parts of the body, including skin and viscera (for DRG) and orofacial areas (for TG), to receive peripheral sensory stimuli through nerve endings. The central branch of the neuron travels proximally to the dorsal horns of the spinal cord or to the medullary dorsal horns in the trigeminal spinal tract nucleus to deliver incoming signals to the CNS through synaptic terminals. Even though afferent spikes initiating at the periphery can bypass the somata and reach spinal dorsal horns directly (Amir and Devor 2003a), spike invasion to somata provides the necessary information for protein synthesis and maintaining optimal levels of receptors and ion channels in both nerve terminals. In addition, the activity in somata actively contributes to the nerve signaling to dorsal horn cells (Amir and Devor 2003b). Following spinal nerve ligation injury, the neuronal somata in DRGs often develop persistent spontaneous firing (Liu et al. 1999). The ectopic discharges are thought to enhance peripheral signals reaching the spinal cord and trigger amplified responses, i.e., sensitization, in spinal neurons (Liu et al. 2001; Sukhotinsky et al. 2004). An increase in neuronal sensitivity to ATP (Zhou et al. 2001), sensitization of adjacent un-injured DRG neurons (Shim et al. 2005; Wu et al. 2001) and changes in transmitter expression (Devor 2009) have been proposed to be the mechanisms underlying the development of ectopic discharge and the production of chronic pain.
Fig. 1. Structural characteristics of dorsal root ganglia.
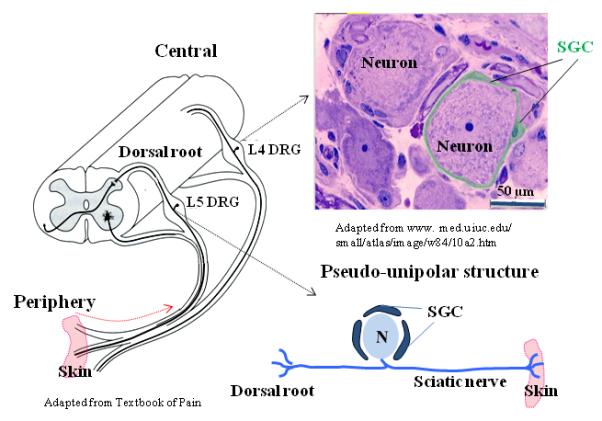
(Left) Locations of L4 and L5 DRGs in the spinal column. (Upper right) A micrograph showing a neuron being tightly wrapped by SGCs. (Lower right) An enlarged view of the pseudo-unipolar structure of a DRG neuron.
Recent studies of neuronal functions suggest that the neuron is not the only cell type contributing to neuronal signaling, non-neuronal cells, including astrocytes, oligodendrocytes and microglia play important roles in affecting neuronal activity through glia-neuron interactions (Chiang et al. 2011; Hanani 2005; Ji et al. 2006; Kettenmann et al. 2011; Tsuda et al. 2005; Watkins et al. 2007). An interesting structural feature of sensory ganglia is that somata of sensory neurons do not form synaptic contacts with one another in the ganglion (Pannese 1981). Instead, each soma is tightly enwrapped by a layer of satellite glial cells (SGCs) and enclosed by a connective tissue sheath to form a structural unit (Fig. 1). Multiple (2-3) soma-SGC unit clusters enclosed by one tissue sheath have been observed only in a small percentage (< 6%) of adult rat DRGs (Pannese 2010; Pannese et al. 1991). This structural arrangement between neuronal soma and SGCs suggests that their communication is a key determinant of somatic activity. Assessing changes in the communication after injury is essential for understanding the development of abnormal ectopic discharges in somata that influence afferent signaling.
Communication between SGCs through gap junctions
SGCs in sensory ganglia are similar to brain astrocytes with distinct characteristics (Franke et al. 2012; Verderio and Matteoli 2011). Like astrocytes in the CNS, SGCs express the intermediate filament protein, glial fibrillary acidic protein (GFAP). In response to injury, GFAP in SGCs is upregulated, i.e., reactive astrogliosis -- a defensive reaction of astroglia (Franke et al. 2012; Hanani 2005; Liu et al. 2012; Zhang et al. 2009). Astrocytes in the brain have complex branches of processes that make contacts with numerous neuronal somata, dendrites or sites of action potential generation, i.e., nodes of Ranvier, in myelinated axons (Franke et al. 2012; Freeman 2010). Unlike brain astrocytes, SGCs in peripheral ganglia form a glial sheath that envelops one or occasionally two neuronal somata (Pannese 2010; Pannese et al. 2003).
One of the ways that SGCs communicate with each other is through gap junctions. Under the electron microscope (EM), gap junctions were seen only between adjacent SGCs surrounding a single soma in control DRGs. Gap junctions have not been shown to exist between the SGC sheath and its enclosed neuronal soma or between somata (Huang et al. 2006; Pannese 2010). After nerve transection, SGCs were found to contact neighboring SGC sheaths surrounding another neuron through the formation of SGC bridges and new gap junctions (Hanani et al. 2002; Pannese 2010; Pannese et al. 2003). Gap-junction mediated coupling between SGCs has also been studied by injecting lucifer yellow fluorescent dye into individual SGCs and determining dye spread into other SGCs. In control ganglia, dye coupling is limited to SGCs that surround a single neuron, consistent with the EM observations. In ganglia isolated from rats with inflammation or nerve injury, the number of coupled SGCs surrounding a given neuron increased 2-3 fold (Dublin and Hanani 2007). In addition, a substantial number (up to 20%) of SGCs surrounding different neurons become dye-coupled in DRGs (Dublin and Hanani 2007; Hanani et al. 2002; Huang et al. 2010) and in TGs (Cherkas et al. 2004).
After inflammation or nerve injury, the threshold current for action potential firing in neuronal somata is significantly lowered, resulting in a large increase in the percentage of neuronal somata in DRGs exhibiting spontaneous firing (Huang et al. 2010). Gap junction blockers were found to reduce the spontaneous activity of neurons in injured DRGs, but have little effect on the electrical properties of neurons in control ganglia (Huang et al. 2010). This observation suggests that increased gap junctions mediate ectopic discharges of DRG neurons after injury. Since gap junction blockers also reduce injury-induced chronic pain behaviors, the results are consistent with the idea that gap junction-mediated ectopic discharges contribute to chronic pain conditions. The time course of changes in SGC-coupling was studied in whole ganglia following chronic compression of DRG (Zhang et al. 2009). The increase in the dye coupling among SGCs surrounding a single neuronal soma peaked at 7 days after injury and returned to normal 14 days later, even though the injury-induced pain behaviors lasted for at least 35 days (Song et al. 1999). The results suggest that an increase in SGC gap junction coupling contributes to the development but not the maintenance of the chronic pain state. The generality of this conclusion awaits studies of temporal changes in the SGC gap junction coupling in other injury models. It was found that knockdown of the expression of the gap junction, connexin 43 (Cx43), using RNA interference (RNAi) decreased orofacial pain behavior induced by chronic constriction injury of the infraorbital nerve (Jasmin et al. 2010; Ohara et al. 2008). This observation is consistent with the observations that non-selective pharmacological gap junction blockers inhibit DRG-mediated nociceptive pain behaviors (Dublin and Hanani 2007). Surprisingly, the same RNAi treatment in normal TGs was found to enhance the nocieptive pain behavior (Ohara et al. 2008). The mechanism underlying the opposite consequences of a block of SGC gap-junction coupling in uninjured and injured rats has yet to be determined.
SGC-mediated changes in the ionic environment
Changes in the expression of inwardly rectifying potassium channels
Voltage-dependent potassium (K+) channels, including the inwardly rectifying K+ channels (Kir) and voltage-dependent outward K+ channels and the small-conductance Ca2+-activated SK3 channel, are the major ion channels expressed in SGCs in DRGs and TGs (Cherkas et al. 2004; Takeda et al. 2011; Vit et al. 2006; Zhang et al. 2009). Since the inward K+ currents are sensitive to the Kir blockers, Ba2+ and Cs+ (Cherkas et al. 2004; Takeda et al. 2011; Vit et al. 2008; Zhang et al. 2009), much reduced by Kir4.1 RNAi treatment (Vit et al. 2008) and entirely absent in Kir4.1 -/- mutant mice (Tang et al. 2010), the inward K+ current in SGCs is mediated by Kir4.1 in DRGs and TGs and the Kir4.1 is the primary ion channel setting the resting membrane potential in SGCs (Tang et al. 2010). Furthermore, Kir4.1 was not found in neuronal somata (Vit et al. 2006). Following inflammation or nerve compression injury, Kir4.1-mediated currents in SGCs reduced significantly. One of the reasons for the reduction is a decrease in the expression of Kir4.1 (Takeda et al. 2011; Vit et al. 2008). Since reducing Kir4.1 expression in SGCs with RNAi treatment can induce pain-like behaviors in un-injured animals (Vit et al. 2008) and Kir4.1 is known to regulate extracellular K+ concentration in brain astrocytes (Neusch et al. 2006), K+ buffering hypothesis has been proposed. It is suggested that a decrease in Kir4.1 expression after injury reduces K+ buffering capacity of SGCs, raises extracellular K+ concentration in the vicinity of neurons and thus results in depolarization and hyperexcitability of neuronal somata to give rise to chronic pain (Tang et al. 2010; Vit et al. 2008; Zhang et al. 2009). On the other hand, the K+ buffering mechanism is not entirely consistent with the observation that the reversal potentials of K+ measured in in vivo SGCs of TGs did not change after inflammation (Takeda et al. 2011). Following chronic compression of DRG injury, many neuronal somata were found to be spontaneously active while the Kir-mediated currents in the SGCs associated with these neuronal somata were transiently reduced (Zhang et al. 2009). Seven days after the compression injury, the Kir currents in SGCs returned to the control level but the increased spontaneous activity in neurons persisted. The Kir is not likely to be responsible for the maintenance of the neuronal spontaneous activity in this pain model (Takeda et al. 2011; Zhang et al. 2009).
Changes in glutamate transporter expression
One of the major functions of astrocytes in the CNS is the rapid removal of glutamate from the perineuronal space to prevent the cytotoxic effects of a large accumulation of glutamate resulting from overstimulation of glutamate receptors (Anderson and Swanson 2000; Gadea and Lopez-Colome 2001). This is accomplished through the glutamate-glutamine cycle (Bak et al. 2006). The glutamate transporters, e.g., glutamate-aspartate transporter (GLAST) and glial glutamate transporter (GLT-1), in astrocytes are responsible for the uptake of glutamate released by neurons into synaptic clefts. Glutamate is then converted into glutamine by glutamine synthetase (GS) and released from astrocytes to the interstitial space. Glutamine is returned to presynaptic terminals of neurons through glutamine transporters and converted back to glutamate to be reused. In the spinal dorsal horns, the expression of glial GLAST and GLT-1 and the neuronal glutamate transporter, EAAC1, have been found to increase in the first 5 days after constriction sciatic nerve injury, followed by a decrease in their expression (Sung et al. 2003). Blocking the transient increase in the expression of these glutamate transporters was shown to exaggerate pain behaviors. These observations suggest that the initial increase in the transporters is to protect dorsal horn neurons from the damaging effect of glutamate accumulation induced by nerve injury. Enhancing transporter activity by a glutamate transporter activator, riluzole, during the late transporter reduction phase was found to reverse abnormal pain behaviors. Thus, glutamate uptake in spinal astrocytes has an important role in the development and maintenance of chronic pain (Sung et al. 2003). We have shown that vesicular release of glutamate occurs in the neuronal somata of DRGs (Gu et al. 2010). Immunocytochemical studies found that GLAST and GLT-1 are expressed in SGCs in DRGs (Berger and Hediger 2000; Carozzi et al. 2008; Hanani 2005) and in TGs (Ohara et al. 2009). Reducing the expression of GLAST and GLT-1 by RNAi in ganglia was found to enhance nociceptive behaviors induced by formalin or von Frey filament mechanical stimulation (Jasmin et al. 2010; Ohara et al. 2009). Therefore, SGCs surrounding neuronal somata in ganglia appear to have similar glutamate homeostasis functions as those observed in astrocytes in the spinal cord. It is of interest to determine if there is a similar time-dependent change in glutamate transporter expression in SGCs during different phases of development of chronic pain.
Neuronal-soma ---> SGC ---> soma communication through purinergic signaling
Transmitter release from neuronal somata
The common way that a neuron communicates with other neurons or cells is through the activation of receptors induced by transmitters. Being tightly wrapped by SGCs and lacking synaptic contact with each other, neuronal somata in DRGs are likely to communicate with other cells through somatic transmitter release. We showed that somata of DRG neurons undergo Ca2+-dependent exocytosis and substance P (SP) release in response to membrane depolarizations (Huang and Neher 1996). Capsaicin can evoke the release of SP, calcium gene-related peptide (CGRP) and adenosine-5′-triphosphate (ATP) from the somata of DRG and TG neurons (Matsuka et al. 2001; Ulrich-Lai et al. 2001). Using the sniffer patch technique, we found that electrical stimulation of the neuronal soma of DRG elicits Ca2+-dependent vesicular release of ATP and glutamate (Gu et al. 2010; Zhang et al. 2007). Since ATP release is much more abundant and robust than glutamate release, ATP is the major transmitter used by sensory ganglia for SGC-soma communication.
Involvement of P2XRs and P2YRs
ATP activates both ionotropic purinergic P2X receptor channels and G-protein coupled metabotropic P2YR receptors in the ganglia. Among the seven P2XR subtypes (P2X1-P2X7R) cloned, P2X7R is the only subtype that is not expressed in neurons of DRGs and TGs (Dunn et al. 2001; Kobayashi et al. 2005; Nakatsuka and Gu 2006). The P2X3R, which is often found in small-diameter sensory neurons and mediates nociceptive responses, is the most abundant (Dunn et al. 2001; North 2002; Xiang et al. 1998). Homomeric P2X3Rs and heteromeric P2X2/3Rs are the major receptors expressed in the neuronal somata (Burgard et al. 1999; North 2002). The data on the expression of P2XR subtypes in SGCs are less clear. P2X2, 4 and 5 receptors have been found in SGCs (Kobayashi et al. 2005; Kushnir et al. 2011). It is generally agreed that in SGCs, P2X1R and P2X3R are absent and P2X7R is the subtype most abundantly expressed (Chen et al. 2008; Kobayashi et al. 2005; Zhang et al. 2005). The exclusive expression of P2X3R in neurons and P2X7R in SGCs greatly facilitate our study of the communication between neuronal somata and SGCs.
Studies of the roles of metabotropic P2YRs in nociception began more recently. P2YRs have been divided in two groups based on their G protein coupling characteristics. The first group, consisting of P2Y1, 2, 4, 6 and 11 receptors, are coupled to Gq/G11 to activate phospholipase C (PLC)/IP3/Ca2+ signaling pathway. The second group P2YRs, including P2Y12, 13 and 14 receptors, are coupled to Gi/Go to inhibit adenyl cyclase and cAMP synthesis. The P2Y11R is dually coupled to both Gq/G11 and to Gs to activate adenyl cyclase (Malin and Molliver 2010). Among eight P2YR subtypes, i.e., P2Y1, 2, 4, 6, 11, 12, 13 and 14 receptors, all, except P2Y11R, were found in rodent DRG neurons (Magni and Ceruti 2013; Malin and Molliver 2010). P2Y1, 2, 4 and 6 receptors were expressed in TG neurons (Magni and Ceruti 2013). P2Y1R and P2Y2R are the two most abundant subtypes expressed in ganglion neurons. P2Y1, 12 and 14 receptors are found in DRG SGCs and P2Y1, 2, 4, 5, 13 and 14 receptors are expressed in TG SGCs (Magni and Ceruti 2013). Activation of Gi/Go coupled P2YRs was shown to reduce behavioral hyperalgesia, whereas activation of Gq/G11 coupled P2YRs promotes hyperalgesia (Malin and Molliver 2010). The effect of activation of Gq/G11 coupled P2YRs is not necessarily excitatory. P2Y1R activation has been found to reduce the P2X3R expression and activity in DRG neurons (Chen et al. 2008; Gerevich et al. 2005) and to inhibit N-type voltage-activated Ca2+ channels (Gerevich et al. 2004). P2Y2R activation inhibits P2X3R currents in DRG neurons (Mo et al. 2013). Unlike P2XRs, activation of P2YRs modulates the activity of other channels or receptors through second messengers. For example, P2Y1R activation inhibits Kv7 and P2Y2R activation increases TRPV1 currents (Yousuf et al. 2011). P2YR-mediated responses can be differentiated from those mediated by P2XRs through the analyses of intracellular Ca2+ ([Ca2+]i) responses. Activation of ionotropic P2XRs induces Ca2+ influx through open receptor channels, whereas activation of P2YRs gives rise to G-protein-mediated Ca2+ release from intracellular stores. Thus, P2X3R-mediated [Ca2+]i responses is sensitive to extracellular Ca2+ concentration. In contrast, an increase in [Ca2+]i due to P2YR activation persists in Ca2+ free external solution but is inhibited by cyclopiazonic acid, a Ca-ATPase inhibitor that depletes intracellular Ca2+ in the endoplasmic reticulum (Villa et al. 2010; Weick et al. 2003). P2YRs is indeed involved in SGCs-soma communication to produce chronic pain. For example, P2Y12 expression in SGCs is enhanced after lingual nerve crush and results in chronic pain production (Katagiri et al. 2012). Since CGRP is present only in TG neurons, stimulation of bradykinin receptors in neurons induced CGRP release from soma to activate P2YRs in SGCs resulting in an increase in cytokine release (Ceruti et al. 2011). The mechanism underlying P2YR-mediated neuropathic pain has not been extensively studied. To focus our discussion of the mechanisms underlying SGC-soma communication, P2YR receptor literature will not be extensively discussed here (see (Franke et al. 2012; Magni and Ceruti 2013) for additional references).
Neuronal soma ---> SGC communication
To determine how the neuronal soma in DRGs uses ATP somatic release to communicate with surrounding SGCs, i.e., soma ---> SGC communication, Ca2+ signaling between neuronal somata and SGCs in response to afferent fiber stimulation was studied (Gu et al. 2010; Zhang et al. 2007). Monitoring [Ca2+]i in the soma and surrounding SGCs simultaneously, we found that upon nerve stimulation, Ca2+ inflow through the activated voltage-dependent L-type Ca2+ channels increases [Ca2+]i in the neuronal soma first, followed by an [Ca2+]i increase in SCGs with a delay (Zhang et al. 2007). With increased nerve stimulus frequency, the [Ca2+]i increases in both soma and SGCs are larger and the delay between the two Ca2+ signals is shorter. Following the treatment with enzyme apyrase to degrade extracellular ATP and ADP, nerve stimulation still causes [Ca2+]i increase in the soma but can no longer produce any [Ca2+]i increase in SGCs. These observations strongly suggest that ATP is the transmitter mediating the soma ---> SGC communication (Zhang et al. 2007). Taking advantage of the observation that the P2X3R is expressed only in neuronal somata and the P2X7R is expressed only in SGCs in DRGs (Chen et al. 2008; Chessell et al. 2005; Kobayashi et al. 2005; Zhang et al. 2005), we tested the effects of P2X7R antagonists on [Ca2+]i signaling in DRGs following nerve stimulation and found that blocking P2X7R activity abolishes [Ca2+]i increase in SGCs without changing [Ca2+]i increase in the soma (Fig. 2, Left). These studies suggest that soma ---> SGC communication is accomplished through ATP release from the neuronal soma to activate P2X7Rs in SGCs (Gu et al. 2010; Zhang et al. 2007).
Fig. 2. Dependence of neuronal soma-SGC-soma communication on P2X7R activation.
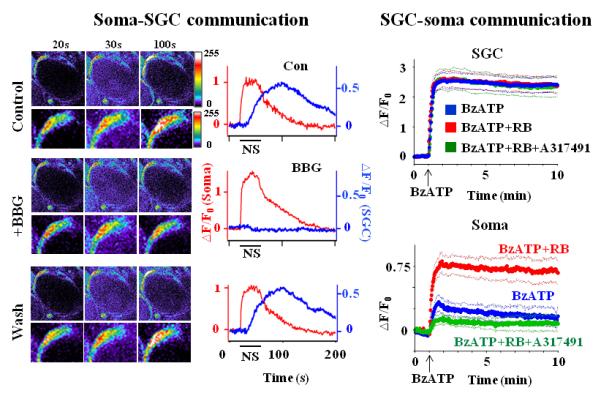
(Left) Soma-SGC communication. Images of Ca2+-dependent fluorescence changes in a neuronal soma and SGCs in response to a 20 Hz, 30 sec nerve stimulus are shown on the left. The number above each column is the time that the images were taken. Enlarged views of the SGC are shown directly below each panel. The time courses of the fluorescence change following nerve stimulation (NS) are given on the right. The NS caused an increase in the fluorescence in the neuronal soma ((F-F0)/F0 =△F/F0 (Soma)) and an increase in the SGC, i.e., (△F/F0 (SGC)) with a delay. F0 is the basal fluorescence in either the soma or in the SGC before NS. The △F/F0 in the soma was not affected by the application of the reversible P2X7R antagonist, BBG (1 μM) while △F/F0 in SGCs was inhibited by BBG. Thus, NS induced ATP release from the neuronal soma to activate P2X7Rs in SGCs. (Right) SGC-soma communication. Application of the P2X7R agonist, BzATP (100 μM) (arrows), evoked a large increase in △F/F0 in SGCs but a relatively small increase in somata. F0 is the basal fluorescence before the BzATP application. Preincubation of DRGs with the P2YR antagonist, RB (1 μM), had no effect on the BzATP-induced △F/F0 change in SGCs, but enhanced the △F/F0 change in somata. The enhancement was inhibited when P2X3R activity in somata was blocked by the P2X3R antagonist, A31749 (60 μM). Thus, stimulation of P2X7Rs in SGCs affects the activity of P2YRs and P2X3Rs in neuronal somata. The thick lines are the average fluorescences; the thin lines are the standard errors of the average values. Adapted from Zhang et al (2007) and Chen et al (2008)(copyright #2007 and #2008 by National Academy of Sciences, USA).
SGC --> soma communication
The mechanism used by SGCs to communicate with neuronal soma, i.e., SGC ---> soma communication, is less well understood. In the absence of voltage-dependent Na+ channels (Zhang et al. 2009), SGCs are not electrically excitable. Aside from inwardly rectifying and voltage-dependent K+ channels, SGCs express a variety of receptors, including P2XRs and P2YRs. Following the activation of P2Rs, the intracellular [Ca2+]i in SGCs cells increases as a result of Ca2+ inflow through open Ca2+-permeable ionotropic P2XRs (Ceruti et al. 2008; Gu et al. 2010; Suadicani et al. 2010; Zhang et al. 2007) or as a result of Ca2+ mobilization from intracellular Ca2+ stores induced by the activation of metabotropic P2YRs (Ceruti et al. 2008; Weick et al. 2003). We found that P2X7Rs in SGCs mediate a significant portion of the basal ATP release in DRGs. Furthermore, activation of P2Y1Rs in the somata inhibits the expression and activity of P2X3Rs in neurons (Chen et al. 2012; Chen et al. 2008). To determine the functional consequence of this modulation, the P2X7R-mediated [Ca2+]i changes in both SGCs and neurons in whole ganglia with and without the P2YR antagonist, reactive blue 2 (RB), were compared (Chen et al. 2008) (Fig. 2 Right). In the absence of RB, we found that the P2X7R agonist, BzATP, induced a large [Ca2+]i increase in SGCs and a rather small [Ca2+]i change in neuronal soma. Following RB treatment, BzATP produced a large [Ca2+]i increase in neurons while the P2X7R-mediated [Ca2+]i in SGCs remained unchanged. The RB-sensitive [Ca2+]i increase in neurons was blocked by the specific P2X3R antagonist, A317491 (Fig. 2, Right).Thus, the SGC--> soma communication is accomplished by P2X7R-mediated ATP release from SGCs to activate P2Y1Rs in neurons which in turn inhibit the activity of P2X3Rs in the soma, i.e., P2X7R--P2Y1R--P2X3R inhibitory control. Measuring the magnitude and propagation of intercellular Ca2+ waves allows one to monitor changes in the SGC-soma communication (Suadicani et al. 2010). Mechanical stimulation of SGCs has been shown to evoke Ca2+ waves that spread to the neighboring neuron and nearby SGCs in trigeminal neuron-SGC mixed cultures. The Ca2+ wave is largely inhibited by the P2R antagonist, suramin (Suadicani et al. 2010). Both of these observations suggest that SGCs use receptor-activated changes in the intracellular Ca2+ to induce ATP release to affect SGC---> neuronal soma communication. Aside from mediating ATP release, P2X7Rs have been found to mediate the release of glutamate, GABA, in astrocytes and microglia in the CNS (Franke et al. 2012). It is not known if those P2X7R-mediated transmitter releases also play a role in soma-SGC communication in sensory ganglia.
Soma and SGC interactions after injury
Since the transmission of neuronal signals from afferent-fibers to the spinal and medullary dorsal horns is strongly affected by purinergic receptor-mediated soma-SGC-soma communication in sensory ganglia (Chiang et al. 2011; Hanani 2005; Ohara et al. 2009), understanding changes in the communication in response to injury is essential for developing strategies to reduce transmission of chronic pain signals. An important characteristic of purinergic receptor-mediated nociceptive signaling in sensory neurons is its large amplification in response to injury. Studies of ATP-induced P2XR-mediated currents in DRG somata isolated from rats with inflammation or nerve injury have led us to conclude that a large increase in P2X3R expression and an enhancement in membrane trafficking of P2X3Rs are the two major mechanisms underlying the sensitization in DRG neurons (Chen et al. 2005; Xu and Huang 2002; Xu and Huang 2004). By producing much enhanced P2X3R-mediated responses after injury, ATP-induced depolarizations would be large enough to lead to action potential firing in nociceptive neurons (Burnstock et al. 2011; Xu and Huang 2002).
In addition to the intrinsic changes in P2Rs in neurons induced by injury, it is of interest to determine the participation of P2Rs in SGCs in changing neuronal activity. The observation that inflammation and nerve injury fail to produce hyperalgesia in the hindpaw of mice that are devoid of P2X7Rs, i.e., P2X7R-/- (Chessell et al. 2005) suggests that the P2X7R plays an important role in the development and maintenance of chronic pain states. The modulation of neuronal activity by the activation of P2X7R in SGCs is rather complex. Studying P2X7R-- P2Y1R--P2X3R inhibitory control in complete Freund adjuvant (CFA) rats, we found that even though the P2X7R-mediated inhibitory control persists under inflammation conditions, CFA treatment still produces hyperalgesia (Chen et al. 2008). The over-excitation of DRG neurons may result from CFA-induced upregulation of P2X3Rs and/or P2X7R-mediated cytokine release. Kushnir et al (2011) showed that the threshold concentration of ATP required to evoke the [Ca2+]i increase in SGCs in cultured and whole TGs is 100-fold lower following CFA-induced submandibular inflammation or infraorbital nerve axotomy. The [Ca2+]i response is mediated by P2YRs in the control cells but becomes P2X2, P2X5 and possibly P2X4 receptor-mediated after inflammation. The mechanism underlying the P2YR to P2XR switch has yet to be determined. Furthermore, P2X7R-mediated Ca2+ responses in TGs are not altered by the CFA treatment (Kushnir et al. 2011). This observation is in contrast with the observations that the P2X7R expression in SGCs, measured with immunostaining, is substantially upregulated in human DRGs after nerve injury (Chessell et al. 2005). Using Western analyses, we found that the P2X7R expression is significantly elevated in DRGs of CFA rats (Chen et al. 2008).
Under injurious conditions, the activation of P2X7Rs has been found to be closely associated with the maturation and release of cytokines from immune and glial cells (Colomar et al. 2003; Ferrari et al. 2006; Honore et al. 2009; Skaper et al. 2010; Solle et al. 2001). Cytokine released onto surrounding neurons enhances neuronal excitability and thus elicits pain and protective nocifensive behaviors (Heinzmann and McMahon 2011). The inflammatory cytokines, TNFα, IL-1β and IL-6, and their respective receptors TNFR1, IL-1R and IL-6R have been found in neurons and SGCs in sensory ganglia (Hanani 2005). In general, the expression of these cytokines in sensory ganglia is low under control conditions and increases by several-fold after inflammation (Takeda et al. 2007) and nerve injury (Dubovy et al. 2010; St-Jacques and Ma 2011). Injury-induced changes in cytokines and their receptors vary among pain models. After sciatic nerve crush injury, TNFα expression increases only in SGCs while the level of its receptor TNFR1 is elevated in both neurons and SGCs of DRGs (Ohtori et al. 2004). Following ventral root transection, TNFα expression is transiently elevated in both neurons and SGCs whereas TNFR1 expression increases only in DRG neurons (Xu et al. 2006). Since inhibition of TNFα synthesis blocks chronic pain only when it is applied right before the nerve transection not after hyperalgesia being established (Xu et al. 2006). The upregulation of TNFα and TNFR1 appears to be essential for the development, but not the maintenance of chronic pain.
Cytokines have been found to exert a variety of effects on the activity of sensory ganglia. IL-1β was found to reduce both sustained outward potassium current (IK) and transient potassium current (IA) in TG neurons. The inhibition is potentiated after inflammation (Takeda et al. 2008a). Studying the expression of IL-1β and its receptor IL-1RI in TGs, Takeda et al (2007) found that inflammation induces upregulation of IL-1β, which is expressed only in SGCs in TGs, and increases the expression of IL-1RI, which is found only in small diameter nociceptive neurons. Application of IL-1β onto acutely dissociated trigeminal neurons from inflamed rats significantly increases the depolarization-induced firing of these neurons and application of IL-1RI inhibitors reduces the firing activity of trigeminal neurons both in vitro (Takeda et al. 2007) and in vivo (Takeda et al. 2008b). Acute treatment of DRG neurons with another cytokine, TNFα, was found to evoke ryanodine- and thapsigargin-sensitive [Ca2+]i transients in DRG neurons (Pollock et al. 2002). Long-term exposure of TNFα was shown to enhance TRPV1R-mediated currents in DRG neurons through the production of PGE2 (Nicol et al. 1997) and to upregulate TRPV1 expression through ERK activation (Hensellek et al. 2007). In DRGs, we found that tetanic sciatic nerve stimulation, which mimics cellular responses to injury, evokes a large increase in P2X7R-mediated TNFα release from SGCs (Zhang et al. 2007). TNFα enhances ATP-induced depolarization and increases the firing of DRG neurons. These examples clearly show that SGCs use cytokine release, in addition to the release of small transmitter molecules, e.g., ATP, to modify neuronal somatic activity under injurious conditions.
Mechanisms of P2X7R-mediated ATP and cytokine release
Since P2X7R-mediated ATP and cytokine release have such an important role in SGC--> soma communication, it is of interest to understand the mechanisms underlying the P2X7R-dependent release from SGCs in order to identify a strategy for controlling the communication. Compared with other P2XRs, the P2X7R requires ~10-fold higher concentrations of ATP for its activation (North 2002). Furthermore, the ion permeability of the P2X7 receptor channel changes with ATP treatment. Responding to prolonged or repeated ATP stimulations, the P2X7R channel becomes less selective, i.e., pore dilation, allowing the passage of large molecules (up to 900 daltons) (Khakh et al. 1999; North 2002; Virginio et al. 1999). Can the large pore in the P2X7R channel provide a conduit for ATP release? The answer to this question has been controversial. Studying the permeation of N-methyl-D-glucamine (NMDG+, MW=195 daltons) and the uptake of a cationic propidium fluorescent dye, Yo-Pro-1 (MW=629 daltons) in P2X7R channels, Jiang et al (2005) found that NMDG+, but not Yo-Pro-1, depends on the external Na+ concentration and P2X7R composition for its permeation. The observation has led to the suggestion that NMDG+ permeates through the dilated pore in the P2X7R channel itself while Yo-Pro-1 enters cells through a different permeation pathway (Jiang et al. 2005). A hemichannel protein, pannexin (Panx), has been found to co-immunoprecipitate, thus physically associating with the P2X7R (Pelegrin and Surprenant 2006). Panx1 forms channels of large conductance (475 pS), is activated by the stimulation of P2X7Rs (Iglesias et al. 2009) and allows the passage of ATP (Bao et al. 2004; Locovei et al. 2006). Blocking Panx1 activity with an inhibitory peptide, 10Panx, or down-regulating Panx1 with Panx1-siRNA inhibits P2X7R-mediated Yo-Pro-1 uptake or the release of ATP without altering ATP-induced P2X7R-mediated currents in expressed HEK cells and astrocytes (Iglesias et al. 2009; Pelegrin and Surprenant 2006). These results are consistent with the idea that Panx1 is responsible for the P2X7R-mediated ATP release. The observation that ATP release and Yo-Pro-1 uptake are much reduced in astrocytes isolated from Panx1-/- mutant mice further supports the conclusion (Suadicani et al. 2012). On the other hand, a recent finding that the voltage-dependence of Yo-Pro-1 uptake is similar to that of ATP currents measured with an external saline solution containing normal concentration of Na+ suggests that the P2X7R channel allows direct passage of Yo-Pro-1 and possibly ATP. This observation, however, does not preclude the possibility of Panx1 participating in the P2X7R-mediated ATP release. Most of these studies were conducted in expressed cells. It is important to determine if P2X7R channels in SGCs have similar release properties. Studies of the involvement of Panx in SGC and soma interactions in sensory ganglia have just begun. We recently found that both Panx1 and Panx2 are expressed in DRG somata and SGCs, the Yo-Pro-1 uptake in SGCs is mediated by P2X7Rs and Panx1 (Huang and Gu 2012) and spared nerve injury-induced allodynia is partially blocked by 10Panx (preliminary observations). All suggest the involvement of Panx in SGC-soma communication. Hanstein et al (2012) (Hanstein et al. 2012) showed that Panx1 expression increased in TGs after inflammation and the hypersensitivity was blocked in TG cells isolated from Panx1-null, P2X7R-null and glia-specific deletion of Panx1 mice, supporting the participation of Panx1 in TG pain processing. We currently know very little about the mechanism underlying P2X7R-mediated cytokine release in SGCs. In macrophage, the activation of Panx1-P2X7R complex appears to be involved in the activation of inflammasome/caspase which leads to the release of cytokine IL -1β (Baroja-Mazo et al. 2013; MacVicar and Thompson 2010; Pelegrin et al. 2008; Schroder and Tschopp 2010).
Sorge et al (2012) (Sorge et al. 2012) studied the coding sequence of the P2X7R gene in various strains of mice that show tactile hypersensitivity after spinal nerve injury and genotyped human patients that display chronic pain following mastectomy or the development of osteoarthritis retrospectively. They found an association between the reduced pore dilation of the P2X7R channel and diminished pain responses under inflammatory or nerve injurious conditions. These results further highlight the essential role of the pore-dilation property of P2X7Rs in the production of chronic pain and the possible influence of P2X7R-mediated ATP and cytokine release from SGCs on the activity of sensory neurons and pain signaling.
Conclusion
SGCs use gap junctions to communicate with each other, use Kir and glutamate transporters to change the environment surrounding the neuronal soma and use ATP release and purinergic signaling to communicate with their enwrapped neuronal soma and other SGCs in sensory ganglia (Fig. 3). Following injury, there is a large increase in the expression of gap junction, a decrease in Kir expression, an enhancement in ATP release and an increase in P2XR and P2YR expression. The resultant changes, including an increase in gap-junction mediated SGC coupling, a reduction in Kir currents and glutamate recycling, a potentiation of P2X3R activity and an increase in P2X7R-mediated ATP and cytokine release, give rise to chronic pain conditions. A better understanding of the mechanisms by which SGCs interact with soma and with other SGCs under normal and injurious conditions will help us identify new therapeutic targets and design better approaches to treat chronic pain.
Fig. 3. Mechanisms involved in Neuronal soma - SGC- soma communication in the DRG.
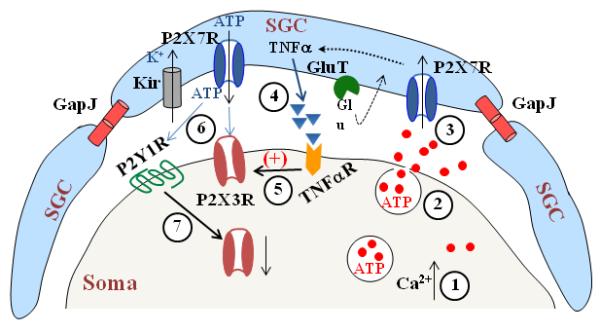
A schematic drawing to illustrate that gap junctions (GapJ) are involved in SGC--SGC communication, GluT (glutamate transporter) and Kir (inwardly rectifying K+ channels) regulate glutamate and K+ concentration surrounding the soma respectively and P2X7Rs in SGCs and P2Y1Rs and P2X3Rs in the soma participate in neuron---> SGC--> soma communication. Nerve stimulation evokes Ca2+-dependent ATP release from the soma (1, 2) to activate P2X7Rs in SGCs (3). P2X7R activation promotes the cytokine, e.g. TNFα, release from SGCs (4) to increase activity of P2X3Rs in the soma (5). ATP release resulting from P2X7R activation increases P2X3R activity (6). In addition, ATP also activates P2Y1Rs (6), which in turn down-regulate P2X3R expression (7).
Acknowledgement
Grant sponsor: National Institute of Dental and Craniofacial Research, Grant number: DE017813; National Institutes of Health National Institute of Neurological Disorders and Stroke, Grant numbers: NS030045.
References
- Amir R, Devor M. Electrical excitability of the soma of sensory neurons is required for spike invasion of the soma, but not for through-conduction. Biophys J. 2003a;84:2181–91. doi: 10.1016/S0006-3495(03)75024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R, Devor M. Extra spike formation in sensory neurons and the disruption of afferent spike patterning. Biophys J. 2003b;84:2700–8. doi: 10.1016/S0006-3495(03)75075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. Journal of neurochemistry. 2006;98:641–53. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- Bao L, Locovei S, Dahl G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004;572:65–8. doi: 10.1016/j.febslet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Baroja-Mazo A, Barbera-Cremades M, Pelegrin P. The participation of plasma membrane hemichannels to purinergic signaling. Biochimica et biophysica acta. 2013;1828:79–93. doi: 10.1016/j.bbamem.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. Distribution of the glutamate transporters GLAST and GLT-1 in rat circumventricular organs, meninges, and dorsal root ganglia. The Journal of comparative neurology. 2000;421:385–99. doi: 10.1002/(sici)1096-9861(20000605)421:3<385::aid-cne7>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Burgard EC, Niforatos W, van Biesen T, Lynch KJ, Touma E, Metzger RE, Kowaluk EA, Jarvis MF. P2X receptor-mediated ionic currents in dorsal root ganglion neurons. J Neurophysiol. 1999;82:1590–8. doi: 10.1152/jn.1999.82.3.1590. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Krugel U, Abbracchio MP, Illes P. Purinergic signalling: From normal behaviour to pathological brain function. Progress in neurobiology. 2011;95:229–74. doi: 10.1016/j.pneurobio.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Carozzi VA, Canta A, Oggioni N, Ceresa C, Marmiroli P, Konvalinka J, Zoia C, Bossi M, Ferrarese C, Tredici G. Expression and distribution of ’high affinity’ glutamate transporters GLT1, GLAST, EAAC1 and of GCPII in the rat peripheral nervous system. Journal of anatomy. 2008;213:539–46. doi: 10.1111/j.1469-7580.2008.00984.x. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti S, Fumagalli M, Villa G, Verderio C, Abbracchio MP. Purinoceptor-mediated calcium signaling in primary neuron-glia trigeminal cultures. Cell Calcium. 2008;43:576–90. doi: 10.1016/j.ceca.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Ceruti S, Villa G, Fumagalli M, Colombo L, Magni G, Zanardelli M, Fabbretti E, Verderio C, van den Maagdenberg AM, Nistri A. Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 Knock-in mice: implications for basic mechanisms of migraine pain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:3638–49. doi: 10.1523/JNEUROSCI.6440-10.2011. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li G, Huang LY. P2X7 receptors in satellite glial cells mediate high functional expression of P2X3 receptors in immature dorsal root ganglion neurons. Molecular pain. 2012;8:9. doi: 10.1186/1744-8069-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li GW, Wang C, Gu Y, Huang LY. Mechanisms underlying enhanced P2X receptor-mediated responses in the neuropathic pain state. Pain. 2005;119:38–48. doi: 10.1016/j.pain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang X, Wang C, Li G, Gu Y, Huang LY. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16773–8. doi: 10.1073/pnas.0801793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkas PS, Huang TY, Pannicke T, Tal M, Reichenbach A, Hanani M. The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain. 2004;110:290–8. doi: 10.1016/j.pain.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–96. doi: 10.1016/j.pain.2005.01.002. others. [DOI] [PubMed] [Google Scholar]
- Chiang CY, Dostrovsky JO, Iwata K, Sessle BJ. Role of glia in orofacial pain. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2011;17:303–20. doi: 10.1177/1073858410386801. [DOI] [PubMed] [Google Scholar]
- Colomar A, Marty V, Medina C, Combe C, Parnet P, Amedee T. Maturation and release of interleukin-1beta by lipopolysaccharide-primed mouse Schwann cells require the stimulation of P2X7 receptors. J Biol Chem. 2003;278:30732–40. doi: 10.1074/jbc.M304534200. [DOI] [PubMed] [Google Scholar]
- Devor M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res. 2009;196:115–28. doi: 10.1007/s00221-009-1724-6. [DOI] [PubMed] [Google Scholar]
- Dublin P, Hanani M. Satellite glial cells in sensory ganglia: Their possible contribution to inflammatory pain. Brain Behav Immun. 2007;21:592–8. doi: 10.1016/j.bbi.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Dubovy P, Klusakova I, Svizenska I, Brazda V. Satellite glial cells express IL-6 and corresponding signal-transducing receptors in the dorsal root ganglia of rat neuropathic pain model. Neuron glia biology. 2010;6:73–83. doi: 10.1017/S1740925X10000074. [DOI] [PubMed] [Google Scholar]
- Dunn PM, Zhong Y, Burnstock G. P2X receptors in peripheral neurons. Progress in neurobiology. 2001;65:107–34. doi: 10.1016/s0301-0082(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Franke H, Verkhratsky A, Burnstock G, Illes P. Pathophysiology of astroglial purinergic signalling. Purinergic signalling. 2012;8:629–57. doi: 10.1007/s11302-012-9300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR. Specification and morphogenesis of astrocytes. Science. 2010;330:774–8. doi: 10.1126/science.1190928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea A, Lopez-Colome AM. Glial transporters for glutamate, glycine and GABA I. Glutamate transporters. Journal of neuroscience research. 2001;63:453–60. doi: 10.1002/jnr.1039. [DOI] [PubMed] [Google Scholar]
- Gerevich Z, Borvendeg SJ, Schroder W, Franke H, Wirkner K, Norenberg W, Furst S, Gillen C, Illes P. Inhibition of N-type voltage-activated calcium channels in rat dorsal root ganglion neurons by P2Y receptors is a possible mechanism of ADP-induced analgesia. J Neurosci. 2004;24:797–807. doi: 10.1523/JNEUROSCI.4019-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerevich Z, Muller C, Illes P. Metabotropic P2Y1 receptors inhibit P2X3 receptor-channels in rat dorsal root ganglion neurons. Eur J Pharmacol. 2005;521:34–8. doi: 10.1016/j.ejphar.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Gu Y, Chen Y, Zhang X, Li GW, Wang C, Huang LY. Neuronal soma-satellite glial cell interactions in sensory ganglia and the participation of purinergic receptors. Neuron glia biology. 2010;6:53–62. doi: 10.1017/S1740925X10000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48:457–76. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Hanani M, Huang TY, Cherkas PS, Ledda M, Pannese E. Glial cell plasticity in sensory ganglia induced by nerve damage. Neuroscience. 2002;114:279–83. doi: 10.1016/s0306-4522(02)00279-8. [DOI] [PubMed] [Google Scholar]
- Hanstein R, Zhao JB, Gulinello M, Hanani M, Spray DC. Transient inflammation causes chronic tactile hypersensitivity: A new role for pannexin1 channels in pain. Society for Neuroscience; Neuroscience Meeting Planner New Orleans, LA. 2012. Program no:180.02. [Google Scholar]
- Heinzmann S, McMahon SB. New molecules for the treatment of pain. Current opinion in supportive and palliative care. 2011;5:111–5. doi: 10.1097/SPC.0b013e328345bb7e. [DOI] [PubMed] [Google Scholar]
- Hensellek S, Brell P, Schaible HG, Brauer R, Segond von Banchet G. The cytokine TNFalpha increases the proportion of DRG neurones expressing the TRPV1 receptor via the TNFR1 receptor and ERK activation. Molecular and cellular neurosciences. 2007;36:381–91. doi: 10.1016/j.mcn.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Honore P, Donnelly-Roberts D, Namovic M, Zhong C, Wade C, Chandran P, Zhu C, Carroll W, Perez-Medrano A, Iwakura Y. The antihyperalgesic activity of a selective P2X7 receptor antagonist, A-839977, is lost in IL-1alphabeta knockout mice. Behavioural brain research. 2009;204:77–81. doi: 10.1016/j.bbr.2009.05.018. others. [DOI] [PubMed] [Google Scholar]
- Huang LY, Gu Y. Purinergic P2X7 receptor-activated and pannexin-mediated responses in satellite glial cells in rat dorsal root ganglia. Society for Neuroscience; Neuroscience Meeting Planner New Orleans, LA. 2012. Program no:180.06. [Google Scholar]
- Huang LY, Neher E. Ca2+-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–45. doi: 10.1016/s0896-6273(00)80287-1. [DOI] [PubMed] [Google Scholar]
- Huang TY, Belzer V, Hanani M. Gap junctions in dorsal root ganglia: possible contribution to visceral pain. European journal of pain. 2010;14:49, e1–11. doi: 10.1016/j.ejpain.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Huang TY, Hanani M, Ledda M, De Palo S, Pannese E. Aging is associated with an increase in dye coupling and in gap junction number in satellite glial cells of murine dorsal root ganglia. Neuroscience. 2006;137:1185–92. doi: 10.1016/j.neuroscience.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J Neurosci. 2009;29:7092–7. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Vit JP, Bhargava A, Ohara PT. Can satellite glial cells be therapeutic targets for pain control? Neuron glia biology. 2010;6:63–71. doi: 10.1017/S1740925X10000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron glia biology. 2006;2:259–69. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LH, Rassendren F, Mackenzie A, Zhang YH, Surprenant A, North RA. N-methyl-D-glucamine and propidium dyes utilize different permeation pathways at rat P2X(7) receptors. Am J Physiol Cell Physiol. 2005;289:C1295–302. doi: 10.1152/ajpcell.00253.2005. [DOI] [PubMed] [Google Scholar]
- Katagiri A, Shinoda M, Honda K, Toyofuku A, Sessle BJ, Iwata K. Satellite glial cell P2Y12 receptor in the trigeminal ganglion is involved in lingual neuropathic pain mechanisms in rats. Molecular pain. 2012;8:23. doi: 10.1186/1744-8069-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological reviews. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Bao XR, Labarca C, Lester HA. Neuronal P2X transmitter-gated cation channels change their ion selectivity in seconds. Nature neuroscience. 1999;2:322–30. doi: 10.1038/7233. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Fukuoka T, Yamanaka H, Dai Y, Obata K, Tokunaga A, Noguchi K. Differential expression patterns of mRNAs for P2X receptor subunits in neurochemically characterized dorsal root ganglion neurons in the rat. J Comp Neurol. 2005;481:377–90. doi: 10.1002/cne.20393. [DOI] [PubMed] [Google Scholar]
- Kushnir R, Cherkas PS, Hanani M. Peripheral inflammation upregulates P2X receptor expression in satellite glial cells of mouse trigeminal ganglia: A calcium imaging study. Neuropharmacology. 2011;61:739–46. doi: 10.1016/j.neuropharm.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Liu FY, Sun YN, Wang FT, Li Q, Su L, Zhao ZF, Meng XL, Zhao H, Wu X, Sun Q. Activation of satellite glial cells in lumbar dorsal root ganglia contributes to neuropathic pain after spinal nerve ligation. Brain research. 2012;1427:65–77. doi: 10.1016/j.brainres.2011.10.016. others. [DOI] [PubMed] [Google Scholar]
- Liu X, Chung K, Chung JM. Ectopic discharges and adrenergic sensitivity of sensory neurons after spinal nerve injury. Brain Res. 1999;849:244–7. doi: 10.1016/s0006-8993(99)02165-4. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhou JL, Chung K, Chung JM. Ion channels associated with the ectopic discharges generated after segmental spinal nerve injury in the rat. Brain Res. 2001;900:119–27. doi: 10.1016/s0006-8993(01)02274-0. [DOI] [PubMed] [Google Scholar]
- Locovei S, Bao L, Dahl G. Pannexin 1 in erythrocytes: function without a gap. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7655–9. doi: 10.1073/pnas.0601037103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Thompson RJ. Non-junction functions of pannexin-1 channels. Trends in neurosciences. 2010;33:93–102. doi: 10.1016/j.tins.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Magni G, Ceruti S. P2Y purinergic receptors: New targets for analgesic and antimigraine drugs. Biochemical pharmacology. 2013;85:466–77. doi: 10.1016/j.bcp.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Malin SA, Molliver DC. Gi- and Gq-coupled ADP (P2Y) receptors act in opposition to modulate nociceptive signaling and inflammatory pain behavior. Molecular pain. 2010;6:21. doi: 10.1186/1744-8069-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuka Y, Neubert JK, Maidment NT, Spigelman I. Concurrent release of ATP and substance P within guinea pig trigeminal ganglia in vivo. Brain Res. 2001;915:248–55. doi: 10.1016/s0006-8993(01)02888-8. [DOI] [PubMed] [Google Scholar]
- Mo G, Peleshok JC, Cao CQ, Ribeiro-da-Silva A, Seguela P. Control of P2X3 channel function by metabotropic P2Y2 utp receptors in primary sensory neurons. Molecular pharmacology. 2013;83:640–7. doi: 10.1124/mol.112.082099. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Gu JG. P2X purinoceptors and sensory transmission. Pflugers Arch. 2006;452:598–607. doi: 10.1007/s00424-006-0057-6. [DOI] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. Journal of neurophysiology. 2006;95:1843–52. doi: 10.1152/jn.00996.2005. others. [DOI] [PubMed] [Google Scholar]
- Nicol GD, Lopshire JC, Pafford CM. Tumor necrosis factor enhances the capsaicin sensitivity of rat sensory neurons. J Neurosci. 1997;17:975–82. doi: 10.1523/JNEUROSCI.17-03-00975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Ohara PT, Vit JP, Bhargava A, Jasmin L. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J Neurophysiol. 2008;100:3064–73. doi: 10.1152/jn.90722.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara PT, Vit JP, Bhargava A, Romero M, Sundberg C, Charles AC, Jasmin L. Gliopathic pain: when satellite glial cells go bad. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2009;15:450–63. doi: 10.1177/1073858409336094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine. 2004;29:1082–8. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- Pannese E. The satellite cells of the sensory ganglia. Advances in anatomy, embryology and cell Biology. 1981;65:1–111. doi: 10.1007/978-3-642-67750-2. [DOI] [PubMed] [Google Scholar]
- Pannese E. The structure of the perineuronal sheath of satellite glial cells (SGCs) in sensory ganglia. Neuron glia biology. 2010;6:3–10. doi: 10.1017/S1740925X10000037. [DOI] [PubMed] [Google Scholar]
- Pannese E, Ledda M, Arcidiacono G, Rigamonti L. Clusters of nerve cell bodies enclosed within a common connective tissue envelope in the spinal ganglia of the lizard and rat. Cell and tissue research. 1991;264:209–14. doi: 10.1007/BF00313957. [DOI] [PubMed] [Google Scholar]
- Pannese E, Ledda M, Cherkas PS, Huang TY, Hanani M. Satellite cell reactions to axon injury of sensory ganglion neurons: increase in number of gap junctions and formation of bridges connecting previously separate perineuronal sheaths. Anat Embryol (Berl) 2003;206:337–47. doi: 10.1007/s00429-002-0301-6. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol. 2008;180:7147–57. doi: 10.4049/jimmunol.180.11.7147. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. Embo J. 2006;25:5071–82. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH. TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology. 2002;42:93–106. doi: 10.1016/s0028-3908(01)00163-0. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Shim B, Kim DW, Kim BH, Nam TS, Leem JW, Chung JM. Mechanical and heat sensitization of cutaneous nociceptors in rats with experimental peripheral neuropathy. Neuroscience. 2005;132:193–201. doi: 10.1016/j.neuroscience.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Debetto P, Giusti P. The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:337–45. doi: 10.1096/fj.09-138883. [DOI] [PubMed] [Google Scholar]
- Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–32. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- Song XJ, Hu SJ, Greenquist KW, Zhang JM, LaMotte RH. Mechanical and thermal hyperalgesia and ectopic neuronal discharge after chronic compression of dorsal root ganglia. J Neurophysiol. 1999;82:3347–58. doi: 10.1152/jn.1999.82.6.3347. [DOI] [PubMed] [Google Scholar]
- Sorge RE, Trang T, Dorfman R, Smith SB, Beggs S, Ritchie J, Austin JS, Zaykin DV, Vander Meulen H, Costigan M. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nature medicine. 2012;18:595–9. doi: 10.1038/nm.2710. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Ma W. Role of prostaglandin E2 in the synthesis of the pro-inflammatory cytokine interleukin-6 in primary sensory neurons: an in vivo and in vitro study. Journal of neurochemistry. 2011;118:841–54. doi: 10.1111/j.1471-4159.2011.07230.x. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Cherkas PS, Zuckerman J, Smith DN, Spray DC, Hanani M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron glia biology. 2010;6:43–51. doi: 10.1017/S1740925X09990408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suadicani SO, Iglesias R, Wang J, Dahl G, Spray DC, Scemes E. ATP signaling is deficient in cultured Pannexin1-null mouse astrocytes. Glia. 2012;60:1106–16. doi: 10.1002/glia.22338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhotinsky I, Ben-Dor E, Raber P, Devor M. Key role of the dorsal root ganglion in neuropathic tactile hypersensibility. Eur J Pain. 2004;8:135–43. doi: 10.1016/S1090-3801(03)00086-7. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:2899–910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Kitagawa J, Takahashi M, Matsumoto S. Activation of interleukin-1beta receptor suppresses the voltage-gated potassium currents in the small-diameter trigeminal ganglion neurons following peripheral inflammation. Pain. 2008a;139:594–602. doi: 10.1016/j.pain.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Takeda M, Takahashi M, Matsumoto S. Contribution of activated interleukin receptors in trigeminal ganglion neurons to hyperalgesia via satellite glial interleukin-1beta paracrine mechanism. Brain, behavior, and immunity. 2008b;22:1016–23. doi: 10.1016/j.bbi.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Takeda M, Takahashi M, Nasu M, Matsumoto S. Peripheral inflammation suppresses inward rectifying potassium currents of satellite glial cells in the trigeminal ganglia. Pain. 2011;152:2147–56. doi: 10.1016/j.pain.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Kadoi J, Nasu M, Takahashi M, Kitagawa J, Matsumoto S. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain. 2007;129:155–66. doi: 10.1016/j.pain.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Tang X, Schmidt TM, Perez-Leighton CE, Kofuji P. Inwardly rectifying potassium channel Kir4.1 is responsible for the native inward potassium conductance of satellite glial cells in sensory ganglia. Neuroscience. 2010;166:397–407. doi: 10.1016/j.neuroscience.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–7. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Flores CM, Harding-Rose CA, Goodis HE, Hargreaves KM. Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from rat trigeminal ganglion: evidence for intraganglionic neurotransmission. Pain. 2001;91:219–26. doi: 10.1016/S0304-3959(00)00439-5. [DOI] [PubMed] [Google Scholar]
- Verderio C, Matteoli M. ATP in neuron-glia bidirectional signalling. Brain research reviews. 2011;66:106–14. doi: 10.1016/j.brainresrev.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Villa G, Fumagalli M, Verderio C, Abbracchio MP, Ceruti S. Expression and contribution of satellite glial cells purinoceptors to pain transmission in sensory ganglia: an update. Neuron glia biology. 2010;6:31–42. doi: 10.1017/S1740925X10000086. [DOI] [PubMed] [Google Scholar]
- Virginio C, MacKenzie A, Rassendren FA, North RA, Surprenant A. Pore dilation of neuronal P2X receptor channels. Nat Neurosci. 1999;2:315–21. doi: 10.1038/7225. [DOI] [PubMed] [Google Scholar]
- Vit JP, Jasmin L, Bhargava A, Ohara PT. Satellite glial cells in the trigeminal ganglion as a determinant of orofacial neuropathic pain. Neuron Glia Biol. 2006;2:247–257. doi: 10.1017/s1740925x07000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vit JP, Ohara PT, Bhargava A, Kelley K, Jasmin L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J Neurosci. 2008;28:4161–71. doi: 10.1523/JNEUROSCI.5053-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56:148–69. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick M, Cherkas PS, Hartig W, Pannicke T, Uckermann O, Bringmann A, Tal M, Reichenbach A, Hanani M. P2 receptors in satellite glial cells in trigeminal ganglia of mice. Neuroscience. 2003;120:969–77. doi: 10.1016/s0306-4522(03)00388-9. [DOI] [PubMed] [Google Scholar]
- Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, Meyer RA. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:RC140. doi: 10.1523/JNEUROSCI.21-08-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Bo X, Burnstock G. Localization of ATP-gated P2X receptor immunoreactivity in rat sensory and sympathetic ganglia. Neurosci Lett. 1998;256:105–8. doi: 10.1016/s0304-3940(98)00774-5. [DOI] [PubMed] [Google Scholar]
- Xu GY, Huang LY. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci. 2002;22:93–102. doi: 10.1523/JNEUROSCI.22-01-00093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Huang LY. Ca2+/calmodulin-dependent protein kinase II potentiates ATP responses by promoting trafficking of P2X receptors. Proc Natl Acad Sci U S A. 2004;101:11868–73. doi: 10.1073/pnas.0401490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006;123:306–21. doi: 10.1016/j.pain.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Yousuf A, Klinger F, Schicker K, Boehm S. Nucleotides control the excitability of sensory neurons via two P2Y receptors and a bifurcated signaling cascade. Pain. 2011;152:1899–908. doi: 10.1016/j.pain.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Mei X, Zhang P, Ma C, White FA, Donnelly DF, Lamotte RH. Altered functional properties of satellite glial cells in compressed spinal ganglia. Glia. 2009;57:1588–99. doi: 10.1002/glia.20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen Y, Wang C, Huang LY. Neuronal somatic ATP release triggers neuron-satellite glial cell communication in dorsal root ganglia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9864–9. doi: 10.1073/pnas.0611048104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Han P, Faltynek CR, Jarvis MF, Shieh CC. Functional expression of P2X7 receptors in non-neuronal cells of rat dorsal root ganglia. Brain Res. 2005;1052:63–70. doi: 10.1016/j.brainres.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Zhou J, Chung K, Chung JM. Development of purinergic sensitivity in sensory neurons after peripheral nerve injury in the rat. Brain Res. 2001;915:161–9. doi: 10.1016/s0006-8993(01)02845-1. [DOI] [PubMed] [Google Scholar]