

Abstract
The addition of cholesterol to the monoolein-based lipidic cubic phase (LCP) has been instrumental in obtaining high-resolution crystal structures of several G protein-coupled receptors. Here, we report the use of high resolution magic angle spinning NMR spectroscopy to record and assign the isotropic 13C chemical shifts of cholesterol in lipidic lamellar and cubic phases at different hydration levels with monoolein and chain deuterated DMPC as host lipids. The hydrogen bonding patterns of cholesterol in these phases were determined from the NMR data by quantum chemical calculations. The results are consistent with the normal orientation of cholesterol in lipid bilayers and with the cholesterol hydroxyl group located at the hydrophobic/hydrophilic interface. The 13C chemical shifts of cholesterol are mostly affected by the host lipid identity with little or no dependency on the hydration (20% vs. 40%) or the phase identity (lamellar vs. LCP). In chain deuterated DMPC bilayers, the hydroxyl group of cholesterol forms most of hydrogen bonds with water, while in monoolein bilayers it predominately interacts with monoolein. Such differences in the hydrogen-bonding network of cholesterol may have implications for the design of experiments in monoolein-based LCP.
Keywords: ab initio calculations, monoolein, NMR spectroscopy, sterol-membrane interactions, HRMAS
Introduction
Monoolein (MO) forms a range of lyotropic liquid crystal phases, depending on temperature and hydration.1, 2 The lipidic cubic phase (LCP) of MO has been used to facilitate the crystallization of various integral membrane proteins for X-ray structural studies.3 The success of this approach to membrane protein crystallization, often termed the in meso method, has been particularly notable for its use in obtaining the structures of several G protein-coupled receptors (GPCRs). The inclusion of cholesterol in the LCP was necessary to stabilize the receptors and to obtain high-resolution crystal structures for several GPCRs, including: the β2 adrenergic, the adenosine A2A, the dopamine D3, the chemokine CXCR4, the histamine H1, the muscarinic acetylcholine M2 and M3, the nociceptin, the μ-, κ- and δ-opioid, and the neurotensin 1 receptors. In certain cases with the β2 adrenergic and A2A adenosine receptors, apparent cholesterol binding sites were observed in the crystal structures, adding to rapidly growing data about the role of cholesterol in modulating the function and stability of several GPCRs (see a recent review for references and recent advances in the field).4
The success of using LCP for membrane protein crystallization can be rationalized by the ability of this phase to provide a stabilizing membrane-like environment for membrane proteins and to support type I crystal packing. Additionally, the MO-based LCP has a relatively high stability, and is tolerant of the inclusion of the detergents used to solubilize the membrane protein prior to incorporation in the LCP,5-7 and of other amphiphilic additives including cholesterol and certain phospholipids.8 The maximum solubility of cholesterol in LCPs is ~28 mol% in excess water at 20°C.8 However ~10 mol% cholesterol is more commonly used in crystallization studies. The LCP lattice parameter increases with increasing amounts of cholesterol,8 and the addition of cholesterol to LCP also results in a reduction in the self-diffusion coefficient of MO.9 Beyond these data describing the effect of cholesterol on the bulk structure, lipid diffusion and phase transitions of LCPs, relatively little is known regarding the properties of cholesterol-containing LCP at the molecular or atomic level. However, as interest in LCP, and its use in protein crystallization, continues to increase, there is a need for an improved knowledge of the effects of additives, such as cholesterol, on the structure and dynamics of this phase. Equally, it is important to understand how cholesterol interactions with membrane proteins in LCP are affected by solvation in monoolein, as compared with cholesterol protein interactions in natural lipid bilayers made predominately of phospholipids.
In contrast to the relatively limited knowledge of the physical-chemical properties of cholesterol containing LCP, there is a wealth of data describing the effect of cholesterol on the properties of lipidic lamellar phases, at least in part as a result of the ‘lipid raft’ hypothesis.10-12 Consequently, a wide variety of biophysical techniques have been developed and used to study the properties of cholesterol-containing lamellar phases in both synthetic and biological systems (see references11, 13 for recent reviews). Many of these methods can also be adapted to characterize LCP samples. For example, fluorescence-based techniques have been used to measure diffusion and clustering of membrane components within bilayers,14-16 and methods such as fluorescence recovery after photobleaching (FRAP) have been applied to LCPs.17, 18 Small- and wide-angle X-ray scattering has been used to measure bilayer thickness and inter-chain packing distances,19 and again, these methods have been applied to the study of LCPs.8, 20 Finally, a range of NMR spectroscopic tools have been used to analyze structural and dynamic properties of lipid bilayers. These methods include the use of 2H NMR of lipids with deuterated chains to measure acyl chain order21 and bilayer fluctuations,22 the use of 31P NMR to investigate the head-groups of phospholipids,23, 24 and the use of the 13C chemical shifts to investigate the hydrogen-bonding partners of cholesterol.25, 26
The application of methods that require the interpretation of anisotropic NMR parameters, such as 2H quadrupolar splitting and 31P chemical shift anisotropy, is not possible in LCP, because the rapid reorientation of the lipid molecules within the isotropic phase leads to an averaging of these values. However, various laboratories have begun to extend and apply NMR methods that rely on parameters accessible in isotropic systems, such as polarization transfer rates,27 relaxation times28 and line-shapes,29 to LCP. There are certain advantages to conducting NMR experiments in LCP, because the rapid isotropic reorientation of the lipid components leads to almost liquid-like spectra in terms of line-width and intensity, and the NMR spectra of LCP samples have even been recorded in a solution state probe30, although the use of an HR-MAS probe presents a definite advantage by removing inhomogeneous line broadening effects.
Here, we report the first quantification of H-bond partners of cholesterol in LCP, using cholesterol 13C isotropic chemical shifts. We use a methodology previously established to describe the hydrogen bonding of cholesterol in chain deuterated DMPC (DMPC-d54) bilayers.25 This approach uses the exquisite and quantitative sensitivity of cholesterol carbon chemical shifts to the local environment and hydrogen bonding partners of the hydroxyl group. The results for four different systems are presented: DMPC-d54 multi-lamellar vesicles (MLVs) with 10 mol% cholesterol at low (20 w/w%) and moderate (40 w/w%) hydration; and MO with 10 mol% cholesterol in a lamellar phase (20 w/w% hydration) and in an LCP (40 w/w% hydration). These hydration levels for MO were chosen because 40 w/w% is the hydration typically used for forming LCP in X-ray crystallography of GPCRs, while 20 w/w% hydration of MO gives lamellar phases and allows a direct comparison with DMPC-d54 and among the two MO major phases. These results are compared and discussed with reference to their implications for the in meso crystallization of membrane proteins.
Experimentals
Materials
Cholesterol and 3,4-13C2-cholesterol (used in parameter optimization) were purchased from Sigma-Aldrich Chemie Sarl, Saint-Quentin Fallavier, France and Euriso-Top-SA, Saint-Aubin, France, respectively. Monoolein (MO) was obtained from Nu-Chek Prep Inc, Elysian, MN, USA and 1,2-dimyristoyl(d54)-sn-glycero-3-phosphocholine (DMPC-d54) from Avanti Polar Lipids Inc, Alabaster, AL, USA. D2O (99.85 atom%) and 2H-depleted water were from Euriso-Top SA, Saint-Aubin, France, and chloroform (used in sample preparation) from VWR, Fontenay sous Bois, France. All materials were used without further purification.
Sample Preparation
MO or DMPC-d54 and cholesterol were dissolved in excess chloroform and mixed in the desired molar ratio. Chloroform was evaporated to dryness under a constant flow of N2 gas, and then under vacuum overnight. LCP samples containing MO and cholesterol at 40 w/w% hydration with D2O were prepared by mechanical mixing, according to the established protocol.3 40 w/w% hydration for the LCP sample was selected because this level of hydration is close to the full hydration capacity of LCP and frequently used in LCP crystallization studies, and therefore has practical relevance. In addition, in order to facilitate the preparation of homogeneous DMPC-d54 MLVs for direct comparison, the use of a sample with as much D2O as could be incorporated without significantly disrupting the LCP was appropriate. It was verified that the 13C chemical shifts and line-widths of cholesterol and MO in LCP are not significantly altered by changes in hydration between 30 and 40 w/w% (data not shown). Lamellar phase samples containing MO and cholesterol at 20 w/w% hydration were prepared by adding D2O to the dried lipids in the desired proportion and then mixing to homogeneity by centrifugation and freeze-thawing through at least five cycles. Lamellar phase samples containing DMPC-d54 and cholesterol were prepared by hydrating the dried lipid film in excess water, lyophilizing overnight, and then re-hydrating with the desired quantity of 2H-depleted water to obtain the 20 w/w% and 40 w/w% hydration levels. The samples were stored at 30 °C for 48 hours to reach the solvation equilibrium.
NMR Data Acquisition
Cubic phase experiments were performed on a Bruker DMX narrow bore spectrometer operating at a 1H Larmor frequency of 500 MHz. Spectra were acquired using a 4 mm Bruker double resonance HR-MAS gradient probe with a deuterium lock, at a magic angle spinning (MAS) frequency of 2.5 kHz. Various rotational frequencies were tested to determine the optimal frequency to obtain narrow line widths from LCP without causing disruption of the phase (monitored by broadening/inhomogeneity in the proton peak from residual water in the sample). Frequencies between 1 and 5 kHz were acceptable, and 2.5 kHz was chosen as a reasonable speed to obtain adequate line widths while minimizing the presence of spinning side bands in 1H and 13C spectra. Lamellar phase experiments were performed on a Bruker AVANCE standard bore operating at a 1H Larmor frequency of 700 MHz and equipped with a 4 mm double resonance 1H-[15N-13C] cross polarization (CP)-MAS probe. Lamellar phase samples containing MO and cholesterol were conducted at a MAS frequency of 5 kHz, and samples containing DMPC-d54 at 10 kHz. For all experiments, the probe temperature was calibrated using ethylene glycol in d6-DMSO. 2H NMR spectroscopy was used to check that the DMPC-d54 chain lipid quadrupolar splittings at 303K had standard values for hydrated MLVs. Static 2H spectra were recorded on a Bruker Avance narrow-bore spectrometer operating at a proton Larmor frequency of 500 MHz with a 5 mm single-resonance probe equipped with a solenoid coil oriented at 90° with respect to the magnetic field.
Cholesterol 13C chemical shifts for samples containing MO were obtained using a standard single-pulse experiment with power-gated 1H decoupling (waltz16 with a decoupling field of 2.5 kHz).31 Typical π/2 pulse lengths were 7 μs for 1H, and 7.65 μs for 13C. Cholesterol peaks were assigned with reference to published spectra of cholesterol in solution in CDCl3 and DMPC bilayers.25. For lamellar samples containing DMPC-d54, cholesterol 13C chemical shifts were obtained using a standard 1D CP-MAS sequence. A typical π/2 pulse length for 1H was 2.9 μs. The 1H-13C CP magnetisation transfer step employed a linear ramp (80 to 100% field strength) on the 1H channel with a contact time of 1.5 ms and a 13C radio frequency field of 55 kHz. High-power proton decoupling was obtained using the Spinal64 scheme32 and a decoupling field of 86 kHz.
Calculations of rotamers, hydrogen bonding, and theoretical chemical shifts
Theoretical NMR chemical shifts were calculated using a Hartree-Fock strategy, as described in a previous publication.26 The geometry of each of the supramolecular models, which combined the three major C3-O3 rotamer states of cholesterol (gauche (+), gauche (-), and anti, denoted as g+, g-, and anti, respectively) and the hydrogen-bonding partners of the hydroxyl group of cholesterol, was fully optimized at the HF/STO-3G level. Isotropic chemical shifts were determined for all atoms at the same theoretical level using a double-ζ-type basis set with a polarization function [i.e., 6-31G(d,p)]. Note that previous calculations using a DFT approach with the same basis set did not lead to any improvement in the calculated cholesterol chemical shifts compared with the Hartree-Fock strategy.26
The whole cholesterol molecule was used to perform calculations. To mimic the hydrogen-bonding environment of the sterol (i.e., solvent water and either MO or DMPC-d54), several hydrogen-bonding partners for the cholesterol hydroxyl group were considered: namely, the water molecules, the fatty acid ester bonds, and either the MO glycerol alcohol groups or the DMPC-d54 phosphodiester. Water molecules and glycerol alcohol groups were considered as both donors and acceptors of hydrogen bonds. The glycerol alcohol groups were modeled by ethanol (primary alcohol) and propan-2-ol (secondary alcohol). The fatty acid ester bonds and the DMPC-d54 head-group phosphate were modeled, as hydrogen-bond acceptors, by acetone and dimethyl phosphate, respectively.25, 26
Following the procedure described in,25 the theoretical carbon chemical shift (CS) differences between the CS obtained for the different models and the averaged CS over the three rotamers of cholesterol with no hydrogen-bonding interaction were calculated; i indicates the carbon number of the sterol, j the C3-O3 rotamer conformation, and k the type of hydrogen-bonding partner. This difference reflects the theoretical effect of a specific hydrogen-bonding interaction with one conformation of cholesterol that occurs in the lipid phase compared with cholesterol in CCl4 solution. For each carbon i (from 1 to 6), linear combinations of these CS variations were calculated in increments of 1% over each rotamer (%j, where j=g+,g- or anti, is the percentage of each rotamer), and over the different types of hydrogen-bond partners (%k, where k=a specific hydrogen-bond partner is the percentage each type of interaction). The values are provided in Supplementary Information Table S1.
| (eq. 1) |
Each is then compared to the experimental values
| (eq. 2) |
Two conditions must be fulfilled for each set of solutions: Σj=g+,g−,anti %j = 100% and Σk=H−bond partners %k ≤ 300%, which corresponds to a maximum of three simultaneous hydrogen-bonding interactions of the cholesterol hydroxyl group (one to the hydrogen of the hydroxyl group and two to the oxygen lone pairs).
The results were filtered to accept only values, for each carbon, for which the difference between the experimental and theoretical variations was less than 0.08ppm, to take into account both experimental accuracy and non-specific solvent effects on chemical shifts.25 The average value for the percentage of each rotamer and each H-bond partner was calculated over the 10% of solutions presenting the lowest value of root mean square deviation between theoretical and experimental CS variations.
Results and Discussion
Assignment of cholesterol 13C chemical shifts in lipid environment
The 13C chemical shifts for cholesterol (10 mol%) in DMPC-d54 and MO, each at 40 and 20 w/w% hydration, were recorded and assigned (see Table 1 and Figure 1). The use of cross polarization sequence, together with the perdeuterated chains of the DMPC-d54, enabled almost complete suppression of the lipid signal, facilitating the assignment of cholesterol carbon peaks. Only the two quaternary carbons were not assigned. The chemical shifts of cholesterol in DMPC-d54 at these two hydration levels were similar to each other, and to values reported previously for samples containing 30 mol% cholesterol with 50 w/w% hydration.25
Table 1.
13C chemical shifts of cholesterol in DMPC-d54 and in MO at different hydrations
| Sample | CCl4 | DMPC-d54 | DMPC-d54 | MO | MO |
|---|---|---|---|---|---|
| Hydration (w%) | 0 | 40 | 20 | 40 | 20 |
| Carbon | |||||
| 1 | 37.12 | 37.38 | 37.39 | 37.45 | 37.46 |
| 2 | 31.62 | 30.71 | 30.82 | 30.81 | 30.89 |
| 3 | 71.05 | 70.50 | 70.42 | 70.97 | 71.00 |
| 4 | 42.35 | 41.51 | 41.63 | 41.55 | 41.62 |
| 5 | 140.56 | 141.74 | 141.89 | 141.09 | 141.07 |
| 6 | 121.16 | 119.98 | 119.88 | 120.94 | 121.00 |
| 7 | 31.62 | 31.83 | 31.85 | – | – |
| 8 | 31.74 | 31.83 | 31.85 | – | – |
| 9 | 49.92 | 50.06 | 50.07 | 50.24 | 50.27 |
| 10 | 36.31 | – | – | 36.07 | 36.43 |
| 11 | 20.93 | 20.98 | 20.98 | 21.18 | 21.19 |
| 12 | 39.67 | 39.87 | 39.91 | 39.99 | 39.98 |
| 13 | 42.18 | – | – | 42.32 | 42.33 |
| 14 | 56.65 | 56.66 | 56.68 | 56.82/56.50 | 56.85/56.47 |
| 15 | 24.18 | 24.30 | 24.28 | 24.24 | 24.19 |
| 16 | 28.15 | 28.11 | 28.08 | 28.29 | 28.31 |
| 17 | 56.00 | 56.66 | 56.68 | 56.82/56.50 | 56.85/56.47 |
| 18 | 11.84 | 11.84 | 11.84 | 11.84 | 11.84 |
| 19 | 19.29 | 19.18 | 19.18 | 19.29 | 19.30 |
| 20 | 35.66 | 36.27 | 36.30 | 36.35 | 36.05 |
| 21 | 18.67 | 18.61 | 18.61 | 18.70 | 18.69 |
| 22 | 36.03 | 36.17 | 36.19 | 36.41 | 36.35 |
| 23 | 23.71 | 24.49 | 24.45 | 24.34 | 24.38 |
| 24 | 39.38 | 39.23 | 39.20 | 39.56 | 39.58 |
| 25 | 27.89 | 27.63 | 27.57 | 27.93 | 27.96 |
| 26 | 22.56 | 21.99 | 22.02 | 22.40/22.58 | 22.40 |
| 27 | 22.84 | 21.99 | 22.02 | 22.40/22.58 | 22.40 |
All DMPC-d54 and MO samples contained 10mol% cholesterol and data were recorded at 30°C. At 20w% hydration the DMPC-d54 and MO samples were both in a lamellar phase. At 40w% hydration the DMPC-d54 sample was in a lamellar phase whereas the MO sample was in a cubic phase. Chemical shifts were calibrated to C18 at 11.84ppm, and are accurate to ± 0.05ppm in DMPC-d54 and ± 0.03ppm in MO.
Abbreviations: DMPC-d54 1,2-dimyristoyl(d54)-sn-glycero-3-phosphocholine, MO monoolein
Figure 1.
The structure of cholesterol with carbon atom numbering
Despite the lack of perdeuterated acyl chains, and therefore the presence of large lipid signals in the spectra of the MO samples, the chemical shifts of all but two of the carbon atoms of cholesterol – C7 and C8 – could be resolved at both 40 and 20 w/w% hydration. These two unresolved carbons peaks were presumed to be situated underneath a strong lipid peak at ~32 ppm (based on their positions in the spectra in DMPC-d54 and in CCl4). A substantial difference between the intensity and line-width of the peaks in carbon spectra of the MO samples at 40 and 20 w/w% hydration was observed. At 40 w/w% hydration the sample is in an LCP, and the peaks are narrow (typical line widths of 6-12 Hz) with good signal-to-noise. Interestingly, the standard ‘solution’-type INEPT-based NMR experiments, including DEPT and HSQC, which were used to corroborate the assignment of the cholesterol chemical shifts, gave good signal-to-noise and line-widths, demonstrating a practical effect of the rapid reorientation of the lipid molecules in LCPs. In the lamellar phase, at 20 w/w% hydration, the peaks are much broader (typically 12-24 Hz) and have poorer signal to noise. However, the chemical shifts of cholesterol in the two MO phases do not differ substantially.
Note that it was necessary to use perdeuterated DMPC but not perdeuterated MO in order to obtain full assignments because the increased complexity of the carbon spectra of the DMPC samples (with DMPC having two non-equivalent acyl chains) in comparison of the single-chained MO. Furthermore, the cross polarization based excitation of cholesterol 13C carbons filters out the deuterated DMPC resonances while increasing sensitivity.
Cholesterol chemical shift variations in the different lipid phases from NMR data
The differences between the chemical shifts of cholesterol dissolved in CCl4 (isotropic environment) and of cholesterol incorporated in lipidic mesophases made of DMPC-d54 or of MO are shown in Figure 2. These chemical shift differences can be divided into three broad groups. First, C7 to C19 (except C17) – that is the carbons found in rings B, C and D of the rigid sterol structure – are relatively insensitive to changes in their environment, reflecting their lack of conformational flexibility, and their distance from any potential hydrogen bonding or other electrostatic interaction of the cholesterol hydroxyl group. Second, the more flexible carbons of the cholesterol tail region – C17 and C20-27 – are more sensitive to their environment and show distinct variations between the CCl4, DMPC-d54 and MO phases. Interestingly, the chemical shifts of these carbons in the MO lamellar phase (at 20 w/w% hydration) are more similar to the chemical shifts in MO in the LCP (at 40 w/w% hydration) than to those in the DMPC-d54 lamellar phases. The variations in the chemical shifts of C17 and C20-27 likely result predominantly from differences in side chain dynamics (i.e., the distribution of trans/gauche conformers along the side chain33), implying that the dynamics of the chains in the center of the MO bilayer are different from the dynamics of the chains in the center of the DMPC-d54 bilayer, and are more similar to the dynamics of the chains in the LCP. The middle part of the lipid bilayer is more ‘isotropic’ in both of the MO phases than in the DMPC-d54 bilayer, as illustrated by the carbon chemical shifts of positions 25-27, which are closer to the values in CCl4,(i.e., in an isotropic environment) than the corresponding chemical shifts in the DMPC-d54 bilayer. Because chain deuterated DMPC is used, it should be noted that the deuteration of DMPC or the hydration with D2O has been shown to produce negligible effect on its lipid chain order parameters.34, 35 Similarly, we have controlled that DMPC-cholesterol samples give the same 13C chemical shifts when hydrated in H2O or in D2O (supplementary information, Fig S2). Finally, the chemical shifts of C1-6, in the A ring of the rigid sterol structure and in close proximity to the hydroxyl group at position 3, are most sensitive to their environment, and specifically to the nature and quantity of potential hydrogen-bonding partners for the hydroxyl group, as was established previously in 25. There are small variations in the chemical shift differences for C1, C2 and C4 in the four different samples. However, there is a significant reduction in the chemical shift differences of C3, C5 and C6 in the MO phases compared with the differences in the DMPC-d54 phases. The fact that this reduction appears in the MO samples in both the cubic and lamellar phases implies that it is the result of some property of the MO itself, and not a function of the macromolecular organization (i.e., not an effect of curvature, lipid packing, lateral pressure, …).
Figure 2.
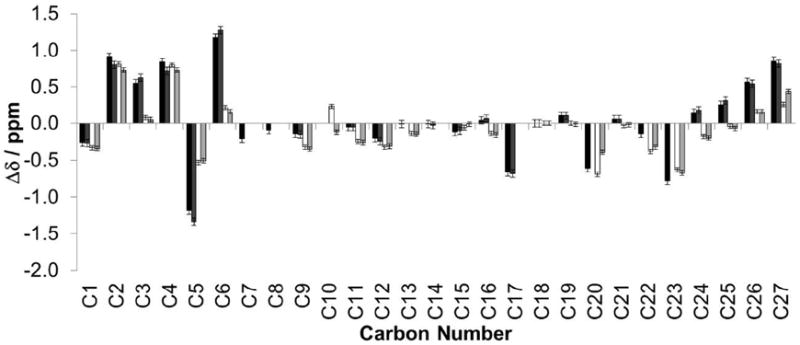
Plot of the differences between the 13C chemical shifts of assigned carbon positions in CCl4 and in various lipid phases
Comparison of chemical shift differences (Δδ, defined as the chemical shift in CCl4 minus the chemical shift in the conditions described) of assigned cholesterol carbons in DMPC-d54 at 40 w/w% (black) and 20 w/w% (dark grey) hydration, and MO at 40 w/w% (white) and 20 w/w% (light grey) hydration. All DMPC-d54 and MO samples contained 10 mol% cholesterol and data were recorded at 30°C. At 20 w/w% hydration the DMPC-d54 and MO samples were both in a lamellar phase. At 40 w/w% hydration the DMPC-d54 sample was in a lamellar phase whereas the MO sample was in a cubic phase. Chemical shifts were calibrated to C18 at 11.84ppm, and are accurate to ± 0.05ppm in DMPC-d54 and ± 0.03ppm in MO.
Determination of the distribution of hydrogen-bond partners: the chemical shift variations of cholesterol C1-6 were used to calculate the distribution of the rotameric states as described in the Materials and Methods section. The average values of the linear combination coefficients for the rotamers (%j) and hydrogen-bonding partners (%k) were calculated using only the 10% of solutions presenting the lowest rmsd values between the experimental and calculated CS variations (see below for discussion and full details in Table S2 in the Supplementary Information).
As a verification of the accuracy of the hydrogen-bonding calculations, these averaged values are combined with the variations (Table S2, in the Supplementary Information) to generate theoretical chemical shifts for carbons C1-6 in each sample using eq. 1. As seen in Supplementary Information Figure S1, there was excellent agreement between the experimental and theoretical chemical shifts for all six carbons in the two DMPC-d54 samples, and for C1, C2, C4 and C5 in the MO samples. The discrepancy between the experimental and theoretical chemical shifts for C3 and C6 in the MO samples was within the experimental error, but was greater than for the other calculated chemical shifts. In all of the samples, the hydroxyl group was calculated to be approximately evenly distributed between the three rotameric states (Supplementary Information, Table S2). This calculated distribution of rotamers provides a good indication of the accuracy of the calculations, because the sample is isotropic and dynamic and the hydroxyl group expected to be free to flip between the rotamers, and none of the rotamers is expected to be favored over the others. The same observation had been made in our earlier work for cholesterol in DMPC bilayers].25
Cholesterol/DMPC-d54 hydrogen-bond partner distribution
The hydrogen-bond partners of cholesterol (10 mol%) in DMPC-d54 at 40 and 20 w/w% hydration are similar to each other, and to those reported previously for cholesterol (30 mol%) in DMPC at 50 w/w% hydration.25 The average fraction of hydrogen bonding through the hydrogen of the cholesterol hydroxyl group to ester bond (0.28 ± 0.18, 0.27 ± 0.17 and 0.26 ± 0.19 for the samples with 40, 20 and 50 w/w% hydration, respectively), to head group phosphate (0.16 ± 0.02, 0.16 ± 0.02 and 0.18 ± 0.03, respectively) and to water (0.17 ± 0.10 for all three samples) were within one standard deviation of each other across the three samples (see Figure 3A and values in 25). There was more variation in the results for hydrogen bonding to water through the oxygen of the cholesterol hydroxyl group, with average values of 0.89 ± 0.09, 0.70 ± 0.08 and 0.76 ± 0.14 for the samples with 40, 20 and 50 w/w% hydration, respectively (Figure 3A). Overall, these data suggest that cholesterol resides predominantly ‘upright’ in the DMPC bilayer, with its hydroxyl group participating in hydrogen bonds with the ester and phosphate region of the phospholipid and with water at the lipid-water interface. This conclusion is generally consistent with small-angle neutron scattering data and with molecular modeling results, which suggest the perpendicular orientation of cholesterol in PC bilayers.36, 37
Figure 3.
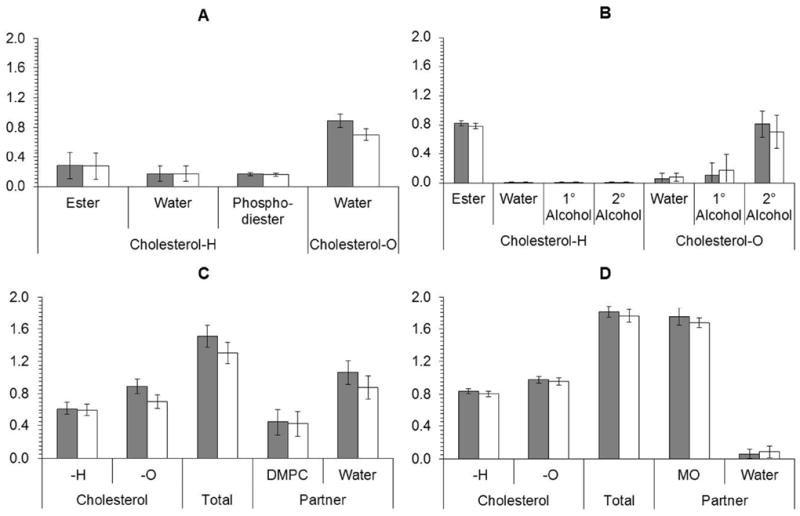
Calculated hydrogen-bonding partners of cholesterol
A break-down of the interactions of cholesterol with specific hydrogen-bonding partners (fragments of the respective lipid modeled as small molecules) is provided in A) for DMPC-d54 and B) for MO at 40 w/w% (black) and 20 w/w% (dark grey) hydration. A summary of the calculated hydrogen-bonding interactions of cholesterol at 40 w/w% (black) and 20 w/w% (dark grey) hydration is provided in C) DMPC-d54 and D) MO. In plots C and D, the data are expressed in two different ways: left, distinguished by whether the interaction is through the hydrogen of the cholesterol hydroxyl group – cholesterol-H – or through the oxygen of the cholesterol hydroxyl group – cholesterol-O; and right, distinguished by whether the interaction is with water or the lipid. The total probability of a hydrogen-bonding interaction is shown in the center. The vertical axes in these plots represent the probability of observing a hydrogen bond per cholesterol ± the standard deviation, where the standard deviation reflects the distribution of the results. The maximum number of hydrogen bonds per cholesterol hydroxyl group was normalized to 1 for bonds through the hydrogen of the hydroxyl (i.e., the hydrogen-bond partner acting as an H donor), and to 2 for bonds through the oxygen of the hydroxyl (i.e., the hydrogen-bond partner acting as an H acceptor). The three possible hydrogen bonds were considered independently in the calculations. The sum of the probability of a hydrogen bond occurring is always less than the maximum because the calculations did not constrain cholesterol to participate in a hydrogen-bonding interaction. The experimental data on which these calculations were based were obtained from DMPC-d54 and MO samples containing 10 mol% cholesterol and data were recorded at 30°C. At 20 w/w% hydration, the DMPC-d54 and MO samples were both in a lamellar phase. At 40 w/w% hydration, the DMPC-d54 sample was in a lamellar phase whereas the MO sample was in a cubic phase.
Cholesterol/MO hydrogen-bond partner distribution
The main differences between the hydrogen-bonding environment of cholesterol in the DMPC-d54 and in the MO phases are the absence of the phosphodiester group and the addition of the primary and secondary alcohol groups in the MO system as donor of hydrogen to cholesterol (summarized graphically in Figure 4). In the MO samples, which contained secondary and primary alcohols as potential partners instead of phosphate, the preference for donating a hydrogen to the ester bond rather than to water or an alcohol was much stronger (e.g., 0.82 ± 0.03, 0.01 ± 0.01, 0.01 ± 0.01, and 0.01 ± 0.01 of 1 for the ester bond, water, secondary alcohol and primary alcohol, respectively, in the 40 w/w% hydration sample – Figure 3B). The average total donation of hydrogen was greater for the MO samples (e.g., 0.84 ± 0.03, in the 40 w/w% hydration sample – Figure 3D) than for the DMPC-d54 samples (e.g., 0.62 ± 0.08, in the 40 w/w% hydration sample – Figure 3C).
Figure 4.
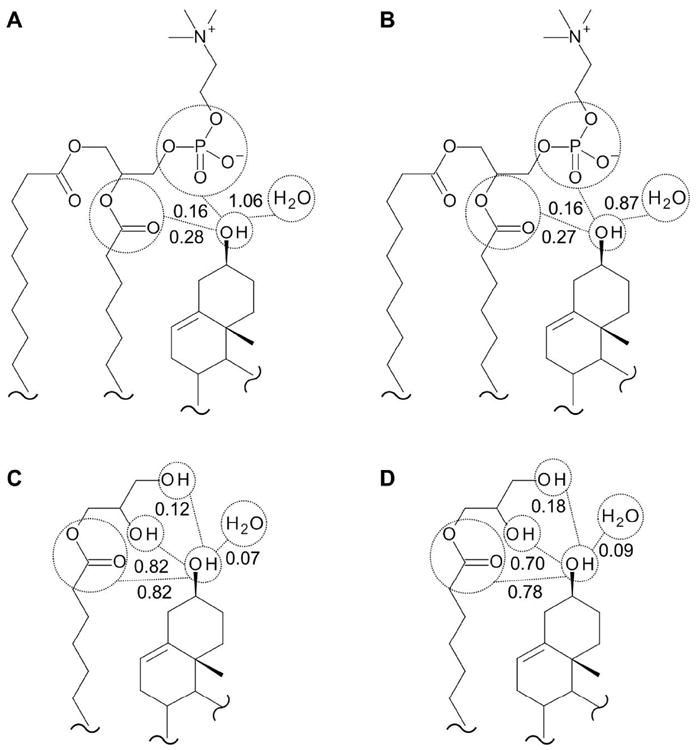
Summary of hydrogen-bonding interactions of cholesterol
Graphical summary of the hydrogen-bonding interactions of cholesterol in DMPC-d54 at A) 40 and B) 20 w/w% hydration, and in MO at C) 40 and D) 20 w/w% hydration. The values represent the percentage of interactions with the cholesterol of the different partners as either donor or acceptor of H-bond. The maximum number of hydrogen bonds per cholesterol hydroxyl group is three. Although all the figures indicate H atoms for clarity, the MO samples were hydrated with D2O.
Similar to the situation with donation of hydrogen, the overall propensity of the oxygen of the cholesterol hydroxyl group to accept a hydrogen from a partner is greater in the MO phases (0.98 ± 0.04 and 0.96 ± 0.05 of 2 in the 40 and 20 w/w% hydration samples, respectively – Figure 3B) than in the DMPC-d54 phase at the same hydration (0.89 ± 0.09, 0.70 ± 0.08 of 2, respectively – Figure 3A). In the same samples, the proportion of this interaction that is with water is less in the MO phase (0.06 ± 0.07 and 0.08 ± 0.05 in the 40 and 20 w/w% hydration samples, respectively – Figure 3B) compared with in the DMPC-d54 phase at the same hydration (0.89 ± 0.09 and 0.70 ± 0.08, respectively – Figure 3A). The additional hydrogen-bonding interactions with the oxygen of the cholesterol hydroxyl group in the MO phases (Figure 3B) are predominantly with the secondary alcohol (0.81 ± 0.18 and 0.70 ± 0.23 in the 40 and 20 w/w% hydration samples) and a little with the primary alcohol (0.11 ± 0.16 and 0.17 ± 0.21, respectively). These changes in hydrogen-bonding partners have a significant effect on the overall distribution of hydrogen bonding, including increasing the preference of cholesterol to form hydrogen bonds with a lipid partner rather than with water in the MO phases (1.75 ± 0.11 and 0.06 ± 0.05 for MO and water in 40% hydration sample, respectively – Figure 3D) compared with in the DMPC-d54 at the same hydration (0.45 ± 0.16 and 1.06 ± 0.15 for MO and water, respectively – Figure 3C).
These data cannot be interpreted to report on the relative penetration of water at the polarapolar interface in the two systems, but raise an interesting question about the penetration of water at the different interfaces, and the extent of lipid-lipid hydrogen-bond formation that could be addressed by additional experiments and/or computer simulations. However, the hydrogen-bonding data do indicate that the cholesterol hydroxyl group interacts with the polar-apolar interfacial region of the MO lamellar and cubic phases to a comparable or greater extent than in the DMPC-d54 lamellar phases, supporting the hypothesis that cholesterol maintains a conventional ‘upright’ position in these phases, as opposed to a ‘horizontal’ position at the center of the two lipid leaflets, as has been observed in certain systems with polyunsaturated phospholipids.36 A further demonstration of this predominant orientation of cholesterol in LCPs could be obtained using the elegant strategy developed by D. Huster and collaborators, which is based on 1H MAS NOESY and the analysis of intermolecular cross relaxation rates. 38, 39
Taken together, the results presented in this paper have potential implications for using LCP as a membrane mimetic matrix for biophysical studies and crystallization of membrane proteins. The role of cholesterol in facilitating crystallization of GPCRs is well recognized although not fully understood on a molecular level4. The exact nature of the lipid composition is known to play a role of the structure determination process,40 both via specific lipid protein contacts and via global physical properties such as the spontaneous curvature and lateral pressure profiles 41-43. The greater extent of cholesterol-lipid hydrogen-bond formation in the MO membranes compared with the DMPC-d54 membranes may affect the equilibrium between cholesterol-lipid and cholesterol-protein interactions in LCP in comparison with ‘native’ phospholipid membranes. Cholesterol’s side chain 13C chemical shifts in MO were found to be close to those in an isotropic organic solvent solution, in contrast with those in DMPC-d54 bilayers, suggesting a different lateral pressure profile at the acyl chain level. These considerations may be particularly important when studying membrane proteins for which significant conformational changes or flexibility in the transmembrane region of the protein are required for function. In such cases doping LCP with native lipids8 may help to restore the specific protein conformation and function in LCP.
Conclusions
In summary, the experimentally determined 13C chemical shifts of cholesterol C1-6 were used to calculate the hydrogen-bonding partners of cholesterol in four lipid phases: DMPC-d54 MLVs with 10 mol% cholesterol at low (20 w/w%) and moderate (40 w/w%) hydration, and MO with 10 mol% cholesterol in MLVs (20 w/w% hydration) and in an LCP (40 w/w% hydration). This original methodology based on the combination of 13C chemical shifts and quantum chemistry, initially developed in DMPC-d54 lipid bilayers, proves to be efficient in a completely different lipid phase and to afford a unique way of assessing the local environment around cholesterol. There were significant differences observed in the 13C chemical shifts, and hence in the hydrogen-bonding partners, of cholesterol in the MO phases in comparison with the DMPC-d54 phases, with a slight increase in overall hydrogen-bond formation, a decrease in cholesterol-water hydrogen bonding and an increase in lipid-cholesterol hydrogen bonding in the MO phases. The properties characteristic of MO bilayers may also have implications for conformational equilibrium and crystallization of membrane proteins in LCP. They should be kept in mind when discussing cholesterol – GPCR interactions (or any other lipid – protein interaction) based on X-ray structures in LCP.
Supplementary Material
Supplementary Table S1: theoretical chemical shift variations (in ppm) of cholesterol carbons 1 to 6 as a function of their C3-O rotameric states and hydrogen-bonding partners.
Supplementary Table S2: calculated rotameric states and hydrogen-bonding partners of cholesterol in various phases.
Supplementary Figure S1: comparison of experimental and theoretical chemical shifts
Supplementary Figure S2: comparison of cholesterol 13C chemical shifts in DMPC bilayers hydrated with either H2O or D2O.
Acknowledgments
Funding sources
These studies and the IPBS NMR equipment were financed by the French Research Ministry, CNRS, Université Paul Sabatier, the Région Midi-Pyrénées and European structural funds. The research leading to these results has received funding from the EU’s Seventh Framework Programme [FP7/2007-2013] under grant agreement # [211800], and by the NIH grant GM089857.
ABBREVIATIONS
- CP
cross polarisation
- CS
chemical shift
- DMPC-d54
1,2-dimyristoyl(d54)-sn-glycero-3-phosphocholine
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- FRAP
fluorescence recovery after photobleaching
- GPCR
G protein-coupled receptor
- LCP
lipidic cubic phase
- Lo
liquid ordered phase
- MAS
magic angle spinning
- MLV
multi-lamellar vesicle
- MO
monoolein
- rmsd
root mean squared deviation
Footnotes
Author contribution
The Manuscript was written through contribution of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information Available
This material is available free of charge via the internet at http://pubs.acs.org
Contributor Information
Franck Jolibois, Email: franck.jolibois@univ-tlse3.fr.
Prof. Vadim Cherezov, Email: vcherezo@scripps.edu.
References
- 1.Larsson K. Two cubic phases in monoolein-water system. Nature. 1983;304(5927):664–664. [Google Scholar]
- 2.Qiu H, Caffrey M. The phase diagram of the monoolein/water system: metastability and equilibrium aspects. Biomaterials. 2000;21(3):223–234. doi: 10.1016/s0142-9612(99)00126-x. [DOI] [PubMed] [Google Scholar]
- 3.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4(5):706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens RC, Cherezov V, Katritch V, Abagyan R, Kuhn P, Rosen H, Wuthrich K. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nature Rev Drug Discov. 2013;12(1):25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ai X, Caffrey M. Membrane protein crystallization in lipidic mesophases: Detergent effects. Biophys J. 2000;79(1):394–405. doi: 10.1016/S0006-3495(00)76301-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misquitta Y, Caffrey M. Detergents destabilize the cubic phase of monoolein: Implications for membrane protein crystallization. Biophys J. 2003;85(5):3084–3096. doi: 10.1016/S0006-3495(03)74727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Persson G, Edlund H, Amenitsch H, Laggner P, Lindblom G. The 1-monooleoyl-rac-glycerol/n-octyl-beta-D-glucoside/water system. Phase diagram and phase structures determined by NMR and X-ray diffraction. Langmuir. 2003;19(14):5813–5822. [Google Scholar]
- 8.Cherezov V, Clogston J, Misquitta Y, Abdel-Gawad W, Caffrey M. Membrane protein crystallization in meso: Lipid type-tailoring of the cubic phase. Biophys J. 2002;83(6):3393–3407. doi: 10.1016/S0006-3495(02)75339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eriksson PO, Lindblom G. Lipid and Water Diffusion in Bicontinuous Cubic Phases Measured by Nmr. Biophys J. 1993;64(1):129–136. doi: 10.1016/S0006-3495(93)81347-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouritsen OG. The liquid-ordered state comes of age. BBA-Biomembranes. 2010;1798(7):1286–1288. doi: 10.1016/j.bbamem.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 11.Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nature Rev Mol Cell Bio. 2010;11(10):688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- 12.Coskun U, Simons K. Membrane rafting: from apical sorting to phase segregation. FEBS lett. 2010;584(9):1685–93. doi: 10.1016/j.febslet.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 13.Mannock DA, Lewis RN, McMullen TP, McElhaney RN. The effect of variations in phospholipid and sterol structure on the nature of lipid-sterol interactions in lipid bilayer model membranes. Chem Phys Lipids. 2010;163(6):403–48. doi: 10.1016/j.chemphyslip.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 14.He HT, Marguet D. Detecting nanodomains in living cell membrane by fluorescence correlation spectroscopy. Annual rev phys chem. 2011;62:417–36. doi: 10.1146/annurev-physchem-032210-103402. [DOI] [PubMed] [Google Scholar]
- 15.Kenworthy AK. Fluorescence recovery after photobleaching studies of lipid rafts. Methods Mol Biol. 2007;398:179–92. doi: 10.1007/978-1-59745-513-8_13. [DOI] [PubMed] [Google Scholar]
- 16.Loura LM, Prieto M. Fluorescence resonance energy transfer to characterize cholesterol-induced domains. Methods Mol Biol. 2007;400:489–501. doi: 10.1007/978-1-59745-519-0_33. [DOI] [PubMed] [Google Scholar]
- 17.Cherezov V, Liu J, Griffith M, Hanson MA, Stevens RC. LCP-FRAP Assay for Pre-Screening Membrane Proteins for In Meso Crystallization. Cryst Growth Des. 2008;8(12):4307–4315. doi: 10.1021/cg800778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu F, Liu W, Hanson MA, Stevens RC, Cherezov V. Development of an Automated High Throughput LCP-FRAP Assay to Guide Membrane Protein Crystallization in Lipid Mesophases. Cryst Growth Des. 2011;11(4):1193–1201. doi: 10.1021/cg101385e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinn PJ, Wolf C. An X-ray diffraction study of model membrane raft structures. FEBS J. 2010;277(22):4685–98. doi: 10.1111/j.1742-4658.2010.07875.x. [DOI] [PubMed] [Google Scholar]
- 20.Conn CE, Darmanin C, Sagnella SM, Mulet X, Greaves TL, Varghese JN, Drummond CJ. Incorporation of the dopamine D2L receptor and bacteriorhodopsin within bicontinuous cubic lipid phases. 1. Relevance to in meso crystallization of integral membrane proteins in monoolein systems. Soft Matter. 2010;6(19):4828–4837. [Google Scholar]
- 21.Bartels T, Lankalapalli RS, Bittman R, Beyer K, Brown MF. Raftlike mixtures of sphingomyelin and cholesterol investigated by solid-state 2H NMR spectroscopy. J Am Chem Soc. 2008;130(44):14521–32. doi: 10.1021/ja801789t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrache HI, Brown MF. X-ray scattering and solid-state deuterium nuclear magnetic resonance probes of structural fluctuations in lipid membranes. Methods Mol Biol. 2007;400:341–53. doi: 10.1007/978-1-59745-519-0_23. [DOI] [PubMed] [Google Scholar]
- 23.Seelig J. 31P nuclear magnetic resonance and the head group structure of phospholipids in membranes. Biochim Biophys Acta. 1978;515(2):105–40. doi: 10.1016/0304-4157(78)90001-1. [DOI] [PubMed] [Google Scholar]
- 24.Holland GP, McIntyre SK, Alam TM. Distinguishing individual lipid headgroup mobility and phase transitions in raft-forming lipid mixtures with 31P MAS NMR. Biophys J. 2006;90(11):4248–60. doi: 10.1529/biophysj.105.077289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soubias O, Jolibois F, Reat V, Milon A. Understanding sterol-membrane interactions, Part II: Complete H-1 and C-13 assignments by solid-state NMR spectroscopy and determination of the hydrogen-bonding partners of cholesterol in a lipid bilayer. Chem Eur J. 2004;10(23):6005–6014. doi: 10.1002/chem.200400246. [DOI] [PubMed] [Google Scholar]
- 26.Jolibois F, Soubias O, Reat V, Milon A. Understanding sterol-membrane interactions Part I: Hartree-Fock versus C-13 and H-1 NMR isotropic chemical shifts of sterols in solution and analysis of hydrogen-bonding effects. Chem Eur J. 2004;10(23):5996–6004. doi: 10.1002/chem.200400245. [DOI] [PubMed] [Google Scholar]
- 27.Nowacka A, Mohr PC, Norrman J, Martin RW, Topgaard D. Polarization Transfer Solid-State NMR for Studying Surfactant Phase Behavior. Langmuir. 2010;26(22):16848–16856. doi: 10.1021/la102935t. [DOI] [PubMed] [Google Scholar]
- 28.Hakansson P, Westlund PO. Nuclear magnetic relaxation study of the microstructure of a bicontinuous cubic phase. Phys Chem Chem Phys. 2004;6(17):4321–4329. [Google Scholar]
- 29.Momot KI, Takegoshi K, Kuchel PW, Larkin TJ. Inhomogeneous NMR line shape as a probe of microscopic organization of bicontinuous cubic phases. J Phys Chem B. 2008;112(21):6636–6645. doi: 10.1021/jp8006415. [DOI] [PubMed] [Google Scholar]
- 30.Boyle-Roden E, Hoefer N, Dey KK, Grandinetti PJ, Caffrey M. High resolution HNMR of a lipid cubic phase using a solution NMR probe. J Magn Res. 2007;189(1):13–19. doi: 10.1016/j.jmr.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Shaka AJ, Keeler J, Freeman R. Evaluation of a New Broadband Decoupling Sequence: WALTZ-16. J Magn Res. 1983;53:313–340. [Google Scholar]
- 32.Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Res. 2000;142(1):97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 33.Quinn PJ, Wolf C. The liquid-ordered phase in membranes. BBA-Biomembranes. 2009;1788(1):33–46. doi: 10.1016/j.bbamem.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Gross JD, Warschawski DE, Griffin RG. Dipolar recoupling in MAS NMR: A probe for segmental order in lipid bilayers. J Am Chem Soc. 1997;119(4):796–802. [Google Scholar]
- 35.Beranova L, Humpolickova J, Sykora J, Benda A, Cwiklik L, Jurkiewicz P, Grobner G, Hof M. Effect of heavy water on phospholipid membranes: experimental confirmation of molecular dynamics simulations. Phys Chem Chem Phy s. 2012;14(42):14516–14522. doi: 10.1039/c2cp41275f. [DOI] [PubMed] [Google Scholar]
- 36.Harroun TA, Katsaras J, Wassall SR. Cholesterol is found to reside in the center of a polyunsaturated lipid membrane. Biochemistry. 2008;47(27):7090–7096. doi: 10.1021/bi800123b. [DOI] [PubMed] [Google Scholar]
- 37.Henin J, Chipot C. Hydrogen-bonding patterns of cholesterol in lipid membranes. Chem Phys Lett. 2006;425(4-6):329–335. [Google Scholar]
- 38.Huster D, Arnold K, Gawrisch K. Influence of docosahexaenoic acid and cholesterol on lateral lipid organization in phospholipid mixtures. Biochemistry. 1998;37(49):17299–17308. doi: 10.1021/bi980078g. [DOI] [PubMed] [Google Scholar]
- 39.Scheidt HA, Badeau RM, Huster D. Investigating the membrane orientation and transversal distribution of 17 beta-estradiol in lipid membranes by solid-state NMR. Chem Phys Lipids. 2010;163(4-5):356–361. doi: 10.1016/j.chemphyslip.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Li DF, Lee J, Caffrey M. Crystallizing Membrane Proteins in Lipidic Mesophases. A Host Lipid Screen. Cryst Growth Des. 2011;11(2):530–537. doi: 10.1021/cg101378s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Botelho AV, Gibson NJ, Thurmond RL, Wang Y, Brown MF. Conformational energetics of rhodopsin modulated by nonlamellar-forming lipids. Biochemistry. 2002;41(20):6354–6368. doi: 10.1021/bi011995g. [DOI] [PubMed] [Google Scholar]
- 42.Lindblom G, Larsson K, Johansson L, Fontell K, Forsen S. Cubic Phase of Monoglyceride-Water Systems - Arguments for a Structure Based Upon Lamellar Bilayer Units. J Am Chem Soc. 1979;101(19):5465–5470. [Google Scholar]
- 43.Kamo T, Nakano M, Kuroda Y, Handa T. Effects of an amphipathic alpha-helical peptide on lateral pressure and water penetration in phosphatidylcholine and monoolein mixed membranes. J Phys Chem B. 2006;110(49):24987–24992. doi: 10.1021/jp064988g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: theoretical chemical shift variations (in ppm) of cholesterol carbons 1 to 6 as a function of their C3-O rotameric states and hydrogen-bonding partners.
Supplementary Table S2: calculated rotameric states and hydrogen-bonding partners of cholesterol in various phases.
Supplementary Figure S1: comparison of experimental and theoretical chemical shifts
Supplementary Figure S2: comparison of cholesterol 13C chemical shifts in DMPC bilayers hydrated with either H2O or D2O.