

Summary
Stromal interaction molecules (STIM1 and STIM2) are critical components of store-operated calcium entry (SOCE). Sensing depletion of ER Ca2+ stores, STIM couples with plasma membrane Orai channels, resulting in the influx of Ca2+ across the PM into the cytosol. Although best recognized for their primary role as ER Ca2+ sensors, increasing evidence suggests STIM proteins have a broader variety of sensory capabilities than first envisaged, reacting to cell stressors such as oxidative stress, temperature and hypoxia. Further, the array of partners for STIM proteins is now understood to range far beyond the Orai channel family. Here we discuss the implications of STIM’s expanding role, both as a stress sensor and a general modulator of multiple physiological processes in the cell.
Introduction to Ca2+ signaling
Ca2+ is the most ubiquitous and abundant signaling ion in the body, mediating a vast array of physiological processes from fertilization to apoptosis [8]. Despite this ubiquity, specificity can be achieved through tight control of the location, duration, amplitude and frequency of Ca2+ release into the cell cytoplasm, the so called “Ca2+ signature.” Due to the relatively low resting cytosolic Ca2+ concentration (~100 nM), Ca2+-induced cell responses can be generated with even relatively small and discrete increases in the local Ca2+ concentration. However, Ca2+ extrusion by the plasma membrane Ca2+-ATPase (PMCA) [14] and reuptake into intracellular stores via the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [107] both contribute to the shape and nature of the Ca2+ signature (Figure 1). In addition, other proteins, including the Na+/Ca2+ exchanger (NCX) [74], the secretory pathway Ca2+ ATPase (SPCA) [95] pump and buffering by calretinin [82], calbindin-D28k [50] and parvalbumin [49] also modulate the Ca2+ signature.
Figure 1. Receptor-mediated control of Ca2+ Signaling.

The proteins and pathways pertinent to SOCE are shown, with relative localized Ca2+ concentration indicated by blue shading. Inositol 1,4,5-trisphosphate (IP3) is generated by the stimulation of G-protein coupled receptors (GPCRs) or tyrosine kinase receptors (TKR) on the plasma membrane (PM), both of which activate isoforms of phospholipase C (PLC). PLC cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and IP3, the later acting on endoplasmic reticulum (ER) localized IP3-receptors (IP3Rs) to release Ca2+, which can be subsequently be extruded from the cell by the plasma membrane Ca2+-ATPase (PMCA). Luminal Ca2+ concentration of the ER therefore decreases, leading to a dissociation of Ca2+ from the EF-hand motifs of the ER membrane protein, stromal interaction molecule 1 (STIM1). Ca2+ dissociation from STIM1 causes the protein to oligomerize from its resting dimer state and cluster at ER/PM junctions where STIM1 can interact with members of the Orai family of hexameric PM Ca2+ channels to initiate Ca2+ influx into the cytosol. Ca2+ is loaded from the cytosol back into the ER by the sarco/endoplasmic reticulum ATPase (SERCA) to restore luminal Ca2+ levels.
Increases in cytosolic Ca2+ concentration can be generated either by Ca2+ entry from the extracellular space or Ca2+ release from organelles that function as intracellular Ca2+ stores [8]. The primary store of intracellular Ca2+ in the cell is the endoplasmic reticulum (ER), with a luminal Ca2+ concentration of ~400 to 800 μM, although the Golgi [83] and the endo-lysosomal network [79] have also been shown to serve as intracellular Ca2+ stores. Stimulation of G-protein coupled receptors (GPCRs) or tyrosine kinase receptors (TKRs) can lead to the activation of phospholipase C (PLC) which in turn generates diacylglycerol (DAG) and the intracellular second messenger inositol 1,4,5-triphosphate (InsP3) via cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) (Figure 1) [61]. InsP3 then stimulates the release of Ca2+ from the ER and Golgi via the InsP3 receptor (InsP3R) [61]. In addition, cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP) can also elicit intracellular Ca2+ release [101]. While cADPR acts on another ER localized Ca2+ channel, the ryanodine receptor [52], the two-pore channel (TPC) family, localized to the endo-lysosomal system has been defined as the target of NAADP [11,15], although recent investigation have led to new questions regarding the true roles of these channels [7,42].
Early characterization of intracellular Ca2+ signaling recognized that Ca2+ release from intracellular ER stores elicited by InsP3 could lead to a subsequent Ca2+ influx into the cell [86]. It was later shown that a highly specific, non-voltage activated Ca2+ current across the plasma membrane was generated in response to ER Ca2+ depletion [55,42]. This current was proposed to replenish intracellular Ca2+ stores after receptor-mediated activation and was termed calcium-release activated Ca2+ current (ICRAC). It was speculated that ICRAC may be generated by direct coupling between proteins in the ER and plasma membrane [80,83], although it would be another 13 years before the molecular components of this pathway were identified. An alternative hypothesis for the activation for SOCE also arose based on the idea of a “Ca2+-influx factor” (CIF), a diffusible messenger that was proposed to be generated by cells upon store-depletion, to act on plasma membrane channels and initiate Ca2+ entry [10]. In this model CIF release relieves the inhibition of the membrane-associated phospholipase iPLA2β by CaM allowing it to generate undefined products which stimulate CRAC channels [23]. However, whereas CIF has not been identified to date, the identification and characterization of STIM1 and Orai1 has led to new support for the conformational coupling model as outlined below.
STIM1 and Orai mediate store-operated Ca2+ entry
Over the last 8 years, the roles of STIM1 and STIM2 as the ER Ca2+ sensors and activators of Orai1, the pore-forming unit of SOCE have been carefully defined. STIM1 and STIM2 were originally identified in 1996 [77] and 2001 [118], respectively, as single-pass ER membrane proteins with a variety of distinct domains (see Figure 2) but no clear functional roles. That one of those domains was a luminal Ca2+-binding EF hand [118] resulted in the inclusion of STIMs in selective siRNA screens of drosophila S2 [92] and Hela [57] cells and their discovery as required components of SOCE. Although it was initially believed that this EF hand was unpaired, it has since been revealed that the Ca2+-binding ‘canonical’ EF hand (cEF) is indeed the first of a pair with a second non-Ca2+-binding ‘hidden’ EF hand (hEF) [105]. Also within the ER lumen is a sterile α motif (SAM) thought to be required for EF hand-mediated induction of STIM oligomerization. The function of the cytosolic portion of the molecule is to bind with and modulate PM targets of STIM, particularly but not exclusively Orai1. A critical consideration is the need for this to occur only in the activated state; at rest, inactive STIM1 exists as a dimer maintained primarily by cytosolic interactions between the coiled-coil domain (CC1) and STIM-Orai activating region (SOAR) [22,120]. Upon store-depletion, conformational changes in the EF-SAM domains lead to the formation of large STIM oligomers followed by STIM clustering at tightly juxtaposed ER-PM junctions, driven in part by interactions between the positively charged poly-lysine rich domain of STIM and negatively charged lipids in the PM [19]. These lipid interactions are thought to serve a supporting role for the binding and activation of Orai1 by multimeric SOAR [123] (also known as CAD [75]). Interestingly, all vertebrate STIMs exhibit several hundred additional amino acids C-terminal to the SOAR domain which are not required for Orai1 activation. Nevertheless, important roles have been ascribed to a number of domains located in this region of STIM1 such as the inhibitory domain (ID) which participates in Ca2+-dependent Orai1 inactivation [27,53,69], a microtubule-interacting site (TRIP) [35,99] and the proline/serine-rich domain (PS) which we have shown to be required for inhibition of PMCA [89]. Future investigations will undoubtedly provide new insight into the functions of these C-terminal STIM domains as well as define roles for portions of the molecules that are not currently understood.
Figure 2. Domain architecture of STIM proteins.
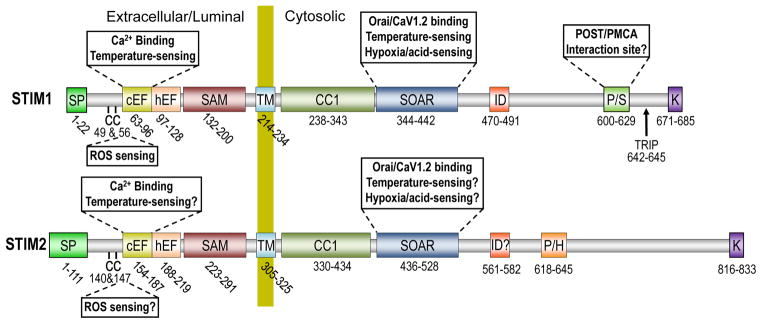
STIM1 (685 amino acids) and STIM2 (833 aa) are single membrane spanning proteins with luminal/extracellular N-termini (when localized to the ER or PM respectively) and cytosolic C-termini. The STIM N-terminus incorporates a signal peptide (SP), the canonical and hidden EF-hands (c/hEF) and the sterile α-motif (SAM) upstream of a single transmembrane domain (TM). The C-termini of both STIM1 and STIM2 contain a coiled-coil domain (CC1), the STIM-Orai activating region (SOAR), an inhibitory domain (ID) responsible for Ca2+-dependent inhibition of Orai1 function and a poly-lysine (K) region at the distal C-terminus responsible for association with the PM. Unique to STIM1 is a proline-serine (P/S) rich domain, while a non-homologous proline-histidine (P/H) rich region is found in STIM2. Many of these domains have been ascribed key roles in the multifaceted functions of STIM as outlined on the diagram.
While the role of STIM1 in SOCE has been well documented, our understanding of the functional role of STIM2 remains somewhat less clear. In most cell types, STIM2 knockdown has either a minor effect on SOCE [57,72] or no effect at all [102,92]. In contrast, neurons [6] and dendritic cells [5] both seem to be dependent predominantly on STIM2 for SOCE. The reasons for the dominance of STIM2 in these cell types is not currently clear, but may reflect potential advantages conferred by its distinct properties. Hence, despite the 66% sequence identity between the key domains (EF/SAM domains, CC1, SOAR) of STIM1 and STIM2, those differences that do exist confer distinct functional properties on these proteins. For example, the Ca2+-binding domains of STIM1 and STIM2 are identical in all but three amino acids, yet STIM2 exhibits a two-fold lower Ca2+ sensitivity than STIM1 [13], resulting in constitutive Ca2+ entry in some cell types [103,129,78], presumably due to lower resting ER Ca2+ levels. There is also evidence that STIM2 is a weaker activator of Orai1 [9], at least in part due to differences in the time course of STIM2 activation [78,129] that have been attributed to structural differences in the N-terminal random coil [129] and SAM [128] domains. This relative weakness in its ability to activate Orai1 can lead to apparent SOCE inhibition when comparing STIM1- vs. STIM2-mediated SOCE [102] and is likely a critical protective feature for a molecule prone to constitutive activation under resting conditions. Still unclear is the extent to which sequence differences in other portions of the protein might also contribute to their distinct characteristics. In addition, as discussed at length below, the extent to which STIM2 contributes to other types of stress sensing and targeting has not been carefully examined.
Orai channels are phylogenetically distinct from other characterized channel families, consisting of four transmembrane domains with cytosolic N and C termini. The first line of evidence implicating this family of transmembrane proteins as potential channels was that loss of Orai1 led to loss of SOCE [30,126,114]. This is something of a curiosity, as there are two additional members of the Orai family (Orai2 and Orai3) [30], all of which can mediate CRAC currents when overexpressed [65,58,26]. However, recent investigations suggest that the different Orai family members may dominate in a cell type-specific manner [67,68] and/or in alternative store-independent, but STIM1- and arachidonate/leukotriene-dependent modes [66,33]. The next key finding was that co-expression of STIM1 and Orai1 led to massive enhancement of Ca2+ influx following store-depletion [103,65,81,126]. Notably, very little elevation in Ca2+ influx was observed when STIM1 was expressed alone, while Orai1 expression actually inhibited endogenous SOCE, implying distinct roles for each in this process. While these findings had led many to speculate that Orai1 was the pore-forming unit of the CRAC channel, the matter was not considered settled until the critical role of glutamate 106 in Ca2+ selectivity was revealed near-simultaneously by three independent groups [84,112,121]. Hence, whereas mutation of this glutamate to either glutamine or alanine resulted in dominant-negative inhibition of CRAC, a conservative mutation to aspartate resulted in profound differences in ion selectivity. The identities of all of the pore-lining residues were since determined via cysteine scanning [64,130]; their identity confirmed with the recently solved structure of the drosophila Orai channel [12]. Based on this structure, it is now clear that Orai channels exhibit a hexameric structure at rest. However, precisely how the channel interacts with STIM proteins and the nature of the structural changes that occur as a result of this binding remain the subject of future investigation.
The processes described above have become the central dogma of SOCE, a mechanism by which receptor-mediated depletion of ER Ca2+ stores leads to the entry of Ca2+ across the PM. However, it has become increasingly apparent that the role of STIM proteins extends further than a simple sensor of ER Ca2+ levels, with an ever expanding number of physiological roles ascribed to the protein and proven interactions with a range of partners beyond Orai1.
Beyond Ca2+ sensing – STIM1 gets stressed
Although InsP3-mediated release of ER Ca2+ content is a fairly common signaling event, loss of ER Ca2+ is also a stress condition, as outlined in the next section. Recent investigations have revealed that this is only one of the stress conditions sensed by STIM proteins, as STIM function is also modulated by ROS overproduction, temperature variation, hypoxic stress and pH changes.
STIM and ER Ca2+ stress
The ability of STIMs to sense decreases in luminal ER Ca2+ concentration and activate Orai1 was the primary context in which STIM proteins were first defined [101]. This serves a dual purpose; in addition to the critical signaling role played by SOCE, Ca2+ entry may also fulfill a homeostatic role by supporting ER Ca2+ refilling. Hence, the primary mediator of ER Ca2+ content is the SERCA pump, which pumps Ca2+ from the cytoplasm to the ER. While SERCA does not require SOCE per se, the speed and efficiency of ER Ca2+ refilling are greatly improved when cytosolic Ca2+ levels are elevated in response to store-operated Ca2+ signals [63,1]. With that in mind, it is somewhat surprising that knockout STIM1, STIM2 or Orai1 does not reveal any differences in ER Ca2+ concentration [4,72,113]. One possible explanation for this is that compensation between STIM1 and STIM2 may mask the effects of knocking out single isoforms on ER Ca2+ content, particularly in the absence of any stressors that would increase the demand for efficient SERCA function.
Maintenance of ER Ca2+ content in its normal physiological range (400 – 800 μM) is critical for many ER functions such as protein synthesis, subsequent protein folding and post-translation modifications which are regulated by Ca2+-sensitive proteins such as the lectin chaperones calnexin (CNX) and calreticulin (CRT) [28]. Prolonged decreases in ER Ca2+ levels can induce the ER stress response in which protein synthesis is inhibited, unfolded proteins are catabolized by autophagy, and ultimately cell death can ensue. As with STIM1, calnexin and calreticulin fail to bind Ca2+ below a certain ER concentration and this dissociation primes them to retain unfolded proteins in the ER. Accumulation of unfolded proteins causes the chaperone protein Grp78 to dissociate from Ire1 and PERK, leading to PERK/Ire1 oligomerization, PERK phosphorylation of eIF2α and inhibition of protein translation [21], thereby limiting the potential damage of additional unfolded proteins. In addition, release of Ire1 leads to splicing of Xbp1 which, together with ATF6 stimulate expression of new ER chaperones and induction of autophagy for elimination of protein accumulation to reverse unfolded protein accumulation. Interestingly, STIM1 and STIM2 have been shown to directly bind calnexin independently of their glycosylation state [93]. However, this binding is likely to be lost when STIMs are activated by ER Ca2+ decreases, providing an additional mechanism to increase the availability of calnexin to help limit potential ER stress when ER Ca2+ is decreased.
STIM1 and Oxidative Stress
Reactive Oxygen Species (ROS) such as superoxide anion (O2·–) and hydrogen peroxide (H 2O2) play important roles as regulatory mediators in cellular physiological function through activation of various enzymatic cascades or transcription factors [94]. However, increased ROS levels arising from exposure to exogenous ROS producers, dysregulation of oxidative phosphorylation in mitochondria or excessive NAD(P)H oxidases (NOX) stimulation leads to oxidative stress, a condition in which the delicate dynamic balance between ROS levels and the antioxidants that counteract them is disrupted [94]. Oxidative stress, if not ameliorated, leads to cell death through apoptotic, necroptotic and necrotic pathways.
Recent investigations have implicated STIM1 and Orai1 as mediators of ROS-induced Ca2+ elevation. Hence, chemical antioxidants such as N-acetyl-l-cysteine or dibenziodolium chloride (DPI) decreased both Ca2+ entry and Ca2+ oscillation duration in response to agonists such as Receptor Activator of NF-κB Ligand (RANKL) [47] and lipopolysaccharide (LPS) [38] as well as under hypoxic conditions [37,70]. Further, removal of STIM1 through either RNA interference or genetic knockout ablated ROS-dependent modulation of Ca2+ signals [38]. It was further revealed in this study that cysteine-56 (C56) within the N terminus of STIM1 was crucial for ROS-dependent STIM1 activation [38]. Hence, ROS elevation through multiple mechanisms led to S-glutathionylation of C56 due to increased levels of oxidized glutathione which interacts with C56 on STIM1 producing a reversible post-translational modification. This modification results in constitutive activation of STIM1, independent of ER Ca2+ depletion. Because C56 is in close proximity to the Ca2+ binding EF hand, it is thought that S-glutathionylation of C56 disrupts the ability of the EF hand to bind to Ca2+, triggering oligomerization of STIM1 and Orai activation (Figure 3) [38]. It should be noted that, since this reactive cysteine is conserved in STIM2, ROS-mediated Ca2+ signaling may involve both STIM isoforms, although the sensitivity of STIM2 to ROS has not been tested. Irrespective, there is now clear evidence for ROS-dependent/store-independent STIM activation, highlighting new mechanisms behind ROS-dependent modulation of Ca2+ signals in both physiological and pathophysiological settings.
Figure 3. Multifaceted sensing strategies by STIM1.

RIGHT: Under normal physiological conditions, decreases in ER Ca2+ concentration lead to STIM oligomerization Orai channel activation and Ca2+ influx (Normoxia). TOP: Under hypoxic conditions, lack of ATP production prevents SERCA-mediated ER Ca2+ uptake, leading to decreased ER Ca2+ content. However, the concurrent accumulation of protons alters STIM/Orai coupling, preventing SOCE from occurring (Hypoxia/Acidity). BOTTOM: ROS overproduction promotes STIM activation of Orai by ROS-induced S-Glutathionylation of STIM, triggering store-independent Ca2+ influx (Oxidative Stress). LEFT: Increasing temperature causes a conformational change leading to oligomerization and clustering of STIM, without activation of Orai channels. Channels are activated upon cooling, due to the primed state of STIM and Ca2+ influx ensues (Heat-off Response).
In addition to direct regulation of STIM1 function, ROS can modulate multiple aspects of Ca2+ signaling. In a recent study investigating the impact of RANKL-mediated of production of ROS on osteoclastogenesis [47], it was shown that ROS elevation was required for long term Ca2+ oscillations (crucial for completion of osteoclast differentiation) whereas ablation of ROS resulted in inhibition of these long term Ca2+ oscillations. In this study, it was found that PLCγ activity was ROS-dependent, while prior studies have revealed that ROS production can increase the sensitivity of intracellular Ca2+ stores to InsP3 [43]. This indicates that in addition to store-independent STIM1 activation, ROS can also activate SOCE via store-dependent mechanisms. An additional consideration from the osteoclastogenesis study was the importance of ROS signaling for long term Ca2+ signals. Many of the cellular mechanisms regulating increased ROS production (eg. NOX5 and DUOX [106], mitochondrial ROS production [29]) can be stimulated by intracellular Ca2+ elevation while Ca2+ elevation is also stimulated by ROS. As such, ROS, PLC, InsP3R and STIM1 may mediate a complex positive feedback loop critical for sustained Ca2+ signaling with profound contributions to both physiological and pathophysiological implications.
Depending on the strength and duration of the signal, ROS-mediated Ca2+ elevation can be either protective or a major contributor to ROS-mediated cell damage [70,98]. An example of the former was demonstrated in osteosarcoma cells, where it was shown that, under mildly hypoxic conditions, ROS is elevated, leading to protective STIM1 activation [70]. This protection was due to compensation for the loss of energy production associated with hypoxic conditions, highly consistent with prior studies establishing that Ca2+ boosts mitochondrial energy output in an oxygen-independent manner [18]. On the other hand, Ca2+ overload due to excessive ROS elevation is associated with apoptosis, primarily due to mitochondrial damage [98]. Hence, due to their high membrane potential, mitochondria fill with Ca2+ as cytosolic Ca2+ levels rise. If too much Ca2+ enters the mitochondria, membrane potential lowers, triggering the mitochondrial permeability transition and cell death. Interestingly, STIM1/Orai1-mediated Ca2+ entry has been specifically shown to contribute to Ca2+ overload-induced cell death in both hippocampal neurons [39] and in endothelial cells during acute lung injury [31].
STIM and temperature sensing
In warm-blooded animals, core body temperature is stringently regulated with modest deviations leading to hypothermia or fever. Despite this, the temperature differential between body core and peripheral extremities can be over 5°C with implications for circulating cells such as those of the immune system. Typically a subset of thermosensitive TRP channels, primarily the TRPV family, although also TRPA1 and TRPM8, have been thought of as key sensors of temperature [111]. These channels have a sensory range of activation thresholds from around 15 °C (TRPA1) to greater than 52°C (TRPV2) [111]. The primary residues responsible for temperature activation of TRPV channels remain to be fully elucidated, however, variations in temperature likely cause a change in structural conformation of protein regions, such as the “turret” region of TRPV1, that shift the voltage-activation threshold to promote channel opening [40].
Like the TRP channels, STIM1 was recently shown to be temperature-sensitive. Increases in temperature to above 40°C caused STIM to cluster in ER-PM junctions of Jurkat T cells, independent of ER store depletion. Interestingly, Orai1 clustering was not observed at these temperatures, but did occur during subsequent cooling to around 37°C in a “heat-off” response (Figure 3) [119]. The heat-induced STIM1 clustering likely reflects the biophysical properties of the STIM EF-hand/SAM domain. Hence, these domains expressed in vitro have been shown to exhibit altered folding at temperatures of ~45°C [104]. This effectively denatures the EF hands and leads to Ca2+ dissociation; assuming that the full length molecule behaves similarly this would then lead to STIM1 activation. However, since activation was not observed until the temperature was lowered, this cannot be the only temperature-sensitive domain. Indeed, the authors also showed that C-terminal polybasic K domain of STIM1 (see Figure 2) was required for heat-induced STIM1 clustering. While the CAD/SOAR domain of STIM1 directly interacts with Orai to promote store-dependent clustering [75], the loss of the polybasic domain ablated heat-sensitive clustering of STIM1 [119]. This implies that SOAR-Orai1 interactions are also temperature-sensitive, a phenomenon that could reflect temperature sensitivity of SOAR, Orai1 or both. Future investigations revealing the mechanisms of SOAR-Orai1 interactions may shed new insight into which components of this interaction are likely to exhibit the greatest temperature sensitivity.
This temperature-dependent increase of basal Ca2+ influx through Orai activation moderately increased NFAT activation and downstream gene transcription, a key mediator of T cell activation. It is therefore speculated that the higher core body temperature may prime immune cells to function at cooler peripheral sites in the body but there may also be as yet undefined implications of blocking SOCE during the high body temperatures associated with fever. Fever has traditionally been thought of as a symptom of disease needing to be treated however, a recent study came to the conclusion that fever aids the immune defense by promoting T cell activation and differentiation [60]. Given the central role played by STIM/Orai-mediated Ca2+ entry in both T cell activation and differentiation, it is reasonable to speculate that this fever effect occurs by increasing the temperature-induced STIM/Orai activation that would be predicted to occur as cells move from the core to the periphery.
STIM1 is expressed throughout the animal kingdom, including in “cold-blooded” ectotherms. This raises the question as to the role of STIM temperature sensitivity in such animals during the process of raising and lowering body temperature, especially for example, in desert-dwelling ectotherms, which are exposed to large temperature fluctuations. It could be that these species have less temperature-sensitive STIM1 variants or as conservation of EF-hand/SAM domains may be critical for function, STIM1 could be specifically down-regulated in peripheral tissue in preference for STIM2. Peripheral down regulation of thermosensitive TRPV4 channels has been demonstrated in certain reptile species [56]. Alternatively temperature-sensitive STIM1 may be beneficial for ectotherm function. Trout experience water temperature fluctuations and varying energy output during upstream migration. A study in trout atrial myocytes suggested that cooler temperature promoted greater SR Ca2+ cycling due to decreased heart rate, leading to greater heart contraction with each beat [96]. While the role of STIM/Orai in cardiac function in mammals remains highly controversial, temperature-sensitive differences in STIM1 function may provide a crucial compensatory mechanism for cardiac and/or other cell types in ectotherms to help compensate for temperature-dependent differences in ion flow.
Interestingly, ability of STIM2 to activate Orai1 in a temperature-sensitive manner has not been assessed. However, the temperature sensitivity of the STIM2 EF-hand/SAM domains were measured in vitro [105], revealing a greater resistance to temperature-induced denaturation. Hence, if STIM2 also exhibits temperature sensitivity, the temperature range that it would respond to would likely be somewhat higher. On the other hand, since the SOAR and polybasic K domains of STIM1 and STIM2 exhibit high sequence homology, it seems reasonable to predict that STIM2 would activate Orai1 in a similar ‘heat-off’ manner.
Acidic and hypoxic stress
A recent study found that in human airway smooth muscle cells (hASMCs) hypoxia diminished SOCE by approximately 50% [62]. Preliminary efforts to assess this mechanism revealed no effect on STIM translocation to ER-PM junctions where it can activate Orai1; on the contrary, STIM1 accumulation into ER-PM junctions was enhanced under hypoxic conditions, likely due to decreased ER Ca2+ content reflecting loss of Ca2+ pumping due to reduced ATP levels. However, despite the presence of STIM1 within these junctions, a marked reduction in STIM/Orai FRET was observed. Since this difference could reflect either loss of STIM/Orai binding or binding in an altered conformation, the authors also examined the ability of the SOAR fragment to bind Orai1. Whereas SOAR and Orai1 associate constitutively at rest, this association was rapidly lost under hypoxic conditions, implicating an effect of hypoxia on STIM-Orai binding. Finally, since hypoxic cells exhibit marked acidification [74], the authors examined and confirmed the hypothesis that this decrease in intracellular pH was responsible for inhibition of STIM/Orai binding and SOCE (Figure 3). Prior studies have shown that electrostatic interactions serve a critical role between SOAR and Orai1 [16,17], and it is tempting to speculate that pH-mediated inhibition of SOAR-Orai interaction reflects electrostatic interference by H+ ions. Further, the fact that greater pH-mediated inhibition of STIM2-Orai1 FRET was observed may indicate a greater dependence for STIM2 than STIM1 on electrostatic interaction, although future studies are required to support this concept.
Acute oxygen deprivation is a relatively common event in the vascular system, skeletal muscle, cardiac tissue and the central nervous system, leading to short-term hypoxic stress managed by rapid cellular responses. For example, in exercising muscle, increased metabolic demands outstrip the supply of circulating oxygen and deplete oxygen stored in myoglobin. This leads to anaerobic respiration, whereby glucose is broken down to pyruvate which in turn is converted to lactate. Lactate quickly accumulates, decreasing cellular pH and inhibiting muscle contraction. While this is thought to result from competition between H+ and Ca2+ for troponin binding, this may also reflect decreased Ca2+ responses due to pH-mediated inhibition of store-operated Ca2+ entry.
In the vasculature, acute hypoxia-induced inhibition of contraction serves an important physiological role, as it causes vasodilation which then increases oxygen intake and blood flow [97]. Chronic hypoxia, however, can have severe consequences. One of the most severe examples is ischemia-reperfusion resulting from occlusion of the vasculature leading to extended loss of blood flow such as during a heart attack or stroke. There has been a longstanding interest in targeting Ca2+ entry as a treatment to this disease, although this has not been entirely successful, perhaps reflecting the complexity of the dysregulation of ion balance. During the ischemic period, protons accumulate in cells of the affected region, inhibiting STIM1 as discussed above and protecting cells from Ca2+ overload. However, upon reperfusion, protons are extruded by the H+/Na+ exchanger (HNE1) [34] and cytosolic Ca2+ concentration increases, leading to Ca2+ overload and extensive cell death via multiple mechanisms. Prior studies have revealed HNE1-mediated Na+ influx as one contributor [34], however, it is also tempting to speculate that relief of H+-mediated STIM1 inhibition could also contribute to Ca2+ overload during the reperfusion phase of ischemic injury. Interestingly, STIM1 has also been shown to inhibit Ca2+ extrusion by the plasma membrane Ca2+ ATPase (PMCA) [89] which could further contribute to runaway Ca2+ overload in the cytosol and cell death.
Hypoxia is also a relatively common event in cancer when solid tumors grow beyond the capacity of the vasculature to provide sufficient oxygen to the tumor cells [110]. The decreased availability of oxygen causes tumor cells to switch ATP production from oxidative phosphorylation (30–36 ATP molecules produced) to the less efficient process of glycolysis (2 ATP molecules), a phenomenon commonly referred to as the ‘Warburg effect’ [117]. The resultant decrease in ATP levels would be expected to lead to ER Ca2+ depletion, STIM activation and, potentially Ca2+ overload. In addition to pH-mediated inhibition of STIM activation there may also be mechanisms for decreasing the STIM1 expression during chronic hypoxia. Hence, Wilms Tumor Suppressor 1 (which inhibits STIM1 expression [90]) has been identified as a target of hypoxia-inducible factor 1 (HIF-1) [2]. Thus, inhibition of the expression and/or function of STIM may be a critical step for tolerance to hypoxic conditions.
Identification of new STIM targets
While Orai is the most established and best described STIM target, roles for STIM have been proposed in a number of processes other than store-operated Ca2+ entry. Perhaps the best described and most controversial of these targets are TRPC channels. Although STIM-dependent TRPC channel activation has been described in several high profile studies [44,73,124,125], conflicting evidence is offered by other groups [25,109]. However, in 2011, it was shown that the effect of store-depletion on TRPCs was somewhat indirect, in that it was dependent on Orai1-mediated Ca2+ entry which caused insertion of TRPCs into the PM [20]. Since measurement of store-operated Ca2+ currents requires buffering of cytosolic Ca2+, these findings may provide some insight into why different groups have drawn conflicting conclusions as to whether or not TRPCs are store- and/or STIM-dependent. Irrespective, the focus of this section is on the identification of new roles for STIM1 activation beyond store-operated Ca2+ entry. Perhaps the first demonstration of this concept was the finding that store depletion leads to STIM1-mediated activation of adenylate cyclase [54]. Although the precise characteristics of this interaction were not determined, subsequent investigations have to a wide variety of unexpected targets such as ER chaperones (Calnexin [93], ERp57 [85]), other channels (CaV1.2 [116,76]), Ca2+ pumps (SERCA [63], PMCA [89]) and numerous other classes of proteins. Here we will discuss the implications of these new findings on cell function.
STIM1-mediated PMCA inhibition; role of POST?
In a recent investigation, the Clapham group revealed Partner of STIM1 (POST) as an adaptor protein mediating interactions between STIM1 and SERCA, NCX, PMCA, the Na/K-ATPase and the nuclear transporters, importin-β and exportin [48]. Interestingly, we recently demonstrated inhibition of PMCA activity by STIM1 via close association at the immunological synapse (IS) of activated T cells (Figure 4) [89]. Hence, we were able to show that STIM1 colocalized with PMCA function at the IS where local Ca2+ clearance was inhibited. We further demonstrated that the proline/serine-rich domain of STIM1 was critical for PMCA inhibition, highly consistent with the fact that this phenomenon was isoform-specific given that STIM2 lacks this domain. Interestingly, local inhibition of Ca2+ clearance at the IS was also recently reported by another group, although in this case, inhibition of PMCA was proposed to be mediated by mitochondrial Ca2+ loading [87]. However, we found that the effect of STIM1 on PMCA function was independent of mitochondria function [89]; mitochondria-independent regulation of PMCA function was similarly established for POST itself [48]. Hence, irrespective of the impact that close apposition between mitochondria and PMCA has on Ca2+ clearance, this seems likely to be a distinct phenomenon from STIM1-mediated PMCA inhibition.
Figure 4. Localized Ca2+ dynamics in T-cell activation.

At rest, the various components of the Ca2+ homeostatic machinery are evenly distributed and mostly inactive. Upon activation, the T cell adopts distinct temporal and spatial Ca2+ signaling characteristics. At the immunological synapse (IS), STIM1 and POST associate with PMCA, resulting in inhibition of Ca2+ extrusion and local elevation of cytosolic Ca2+ levels. At the opposite side of the cell (the Cap), STIM1 activates Orai1, providing a source of Ca2+ which diffuses towards the IS. This model is based on findings reported in our recent paper showing that both the entry and extrusion of Ca2+ occurs predominantly at the Cap-side of activated T cells [89].
Sustained elevation of cytosolic Ca2+ serves a critical role in T-cell activation. Engagement of the T Cell Receptor leads to release of Ca2+ from the ER and initiation of Orai1-mediated Ca2+ entry via classical store-operated STIM1 activation. Under these conditions, Nuclear factor of activated T-cells (NFAT) is dephosphorylated by the Ca2+-dependent serine-threonine phosphatase calcineurin [71]. This leads to nuclear translocation of NFAT, where it promotes the transcription of cytokines and other genes necessary for the immune response [88]. However, within the nucleus, NFAT is rephosphorylated, at which point it is exported back into the cytoplasm. Hence, it is critical that cytosolic Ca2+ concentration remain elevated for an extended time period to continuously drive the nuclear localization of NFAT and achieve full T cell activation. While it is clear that Orai1-mediated Ca2+ influx is critical for this process, it is tempting to speculate that local inhibition of PMCA-mediated Ca2+ extrusion by STIM1 and POST are also crucial for this process to continue (Figure 4) [89,48]. Still many questions remain. Why is it important that cytosolic Ca2+ levels be elevated specifically at the IS? How are the disparate roles of STIM1 as an inducer of Ca2+ entry and inhibitor of Ca2+ efflux coordinated? What role is played by mitochondria in both processes? Future investigations will undoubtedly lead to the answers to these questions, thereby providing an improved understanding of the processes underlying T cell activation.
While the functional implications of interactions between POST and its other partners was not determined, it is intriguing that links between STIM1 function and each of the other POST interacting proteins with the exception of the Na/K-ATPase (thus far) have been reported. For example, STIM1 and SERCA have been proposed to be associated in microdomains, based on findings that refilling of ER Ca2+ content required the presence of STIM1 and occurred without increasing global cytosolic Ca2+ levels [45]. The potential impact of these microdomains on SERCA function were further demonstrated with the finding that Ca2+ levels directly impact SERCA-mediated Ca2+ pumping into the ER [63,1]. Whereas the relationship between STIM1 and NCX has been less extensively investigated, STIM1 has been shown to promote reverse mode NCX activity in airway smooth muscle [59]. Finally, STIM1 has previously been shown to immunoprecipitate with importin-β and exportin [93] which are involved in NFAT translocation to and from the nucleus [46], although the functional implications of this interaction was not determined. While the ability of POST to mediate interactions between STIM1 and these various binding partners was not assessed, there is nothing in the studies described above to exclude this possibility.
Inhibition of CaV1.2
The voltage-gated calcium channel, CaV1.2 has recently been revealed as a negatively regulated STIM target [116,76]. ER Ca2+ store depletion or intraluminal Ca2+ chelation were found to almost completely abolish CaV1.2 currents independent of cytosolic Ca2+ concentration [116]. Furthermore, over-expression of STIM1, and to a lesser extent STIM2, increased the inhibitory effect of store-depletion on CaV1.2 current. Conversely knock-down of STIM1 and Orai1 caused a reduction of store-depletion mediated CaV1.2 inhibition. STIM1-CaV1.2 interaction via the SOAR domain was demonstrated by co-immunoprecipitation of STIM1, while high-resolution imaging of ER-PM junctions revealed co-localization of STIM1/SOAR and CaV1.2 upon store depletion. Orai1 also appeared to be co-localized with CaV1.2 in STIM1 enriched ER-PM junctions, suggesting a complex of the three proteins exists. Indeed, Orai1 was found to assist in SOAR association with CaV1.2 independent of function, suggesting that it was serving a scaffolding role. Interestingly, investigations in hippocampal neurons revealed that STIM1 expression decreased surface expression of CaV1.2 by 73% [76]. Since the potential for internalization to mediate SOAR-mediated CaV1.2 inhibition is not clear, this may reflect fundamental differences between acute and chronic store depletion.
Cav1.2 represents the primary Ca2+ influx pathway in the smooth muscle of the arterial vasculature and is critical for modulation of vasculature tone [79]. As such L-type Ca2+ channel blockers are a widely used therapeutic agent for the treatment of hypertension [108]. Given the ability of STIM proteins to inhibit L-type channel activity, decreases in the expression or function STIM would be a potential mechanism for the increased vascular tone associated with hypertension. However, experiments performed in spontaneously hypertensive rats revealed that STIM1 levels were increased rather than decreased [32]. This is likely a reflection of ‘phenotypic switching’ from the contractile to the synthetic phenotype. Hence, in response to vascular damage, smooth muscle dedifferentiates into a non-contractile state capable of cell proliferation (ie ‘synthetic’) exhibiting increased STIM1 expression. Excessive smooth muscle growth leads to narrowing of vasculature and is a major cause of hypertension [28]. Interestingly, loss of either STIM1 or Orai1 expression has been shown to attenuate smooth muscle proliferation due to vascular damage in vivo [36,3,127]. This has primarily been attributed to a role for SOCE in phenotypic switching; however, STIM1-mediated inhibition of CaV1.2 may also contribute to this phenomenon since the synthetic celltype exhibits both enhanced SOCE and loss of L-type Ca2+ channel activity.
STIM1 microtubule/cytoskeletal interactions and mitosis
STIM1 has also been shown to interact with components of the microtubule network and cytoskeleton, regulating STIM localization with a wide variety of consequences. During interphase, STIM1 interacts with microtubule + TIP protein end binding 1 (EB1), promoting the association of the ER with microtubules to define ER spatial organization [99] via its TRIP domain (see figure 2). During mitosis however, STIM1 is phosphorylated near this site, leading to inhibition of the STIM1/EB1 interaction and dissociation from microtubules. This dissociation of STIM1 and EB1 prevents microtubules pulling the ER into the mitotic spindle and allows the correct partitioning of the organelles during cell division. Coincident to the inhibition of EB1 binding is a failure of STIM1 to activate Orai1 [99,100]. Unexpectedly, this phenomenon was independent of the loss of microtubule binding as a phospho-deficient mutant of STIM1 can restore SOCE independent of EB1 interaction, and loss of EB1 interaction does not block SOCE. Low sequence homology between STIM1 and STIM2 in the C-terminus around the EB1-binding domain [101] suggest that STIM2 may not bind microtubules directly, although it could need the close proximity of microtubules by the ER for STIM2 to also redistribute to ER/PM junctions and elicit SOCE. Inhibition of SOCE during cell division seems to be consistent between mitosis and meiosis [122], and it has been suggested that suppression of SOCE during cell division could be protective to prevent excessive Ca2+ influx [100]. Nevertheless, given the lack of demonstrated effect of phospho-deficient STIM1 mutants that signal SOCE throughout mitosis on cell cycle progression, the true impact of this finding remains somewhat unclear.
A splice variant of STIM1 with an extended C-terminus, STIM1L, was shown to interact with actin and increase the kinetics of SOCE activation in skeletal muscle [24]. Increased kinetics of SOCE are crucial for it to function in muscle where Ca2+ responses occur over a much more rapid time scale than non-excitable cells (for review of STIM/Orai function in skeletal muscle, see [51]). The interaction between the STIM1L-unique amino acids 589–599 with actin promotes the formation of STIM1L clusters in SR/PM junctions that colocalize with Orai, independently of store-content. Pre-clustering of STIM1L precludes the time taken for regular STIM1 to cluster and migrate to ER(SR)/PM junctions, seemingly the rate limiting step in SOCE. This enables the SR to rapidly refill upon emptying and sustain fast, repetitive Ca2+ release. Indeed silencing of endogenous STIM1L in myotubes lead to rapid rundown of Ca2+ and ablation of repetitive, high frequency Ca2+ signaling necessary for muscle activity. Thus STIM1L was most highly expressed in muscle and nervous system tissue where fast, regenerative signaling is required, and while it was detected in liver and spleen tissue it appeared to be expressed at much lower levels. Another group reported that STIM1L was expressed exclusively in skeletal muscle and bound with a higher capacity to Orai1 than the shorter STIM variant [41]. Furthermore SOCE was greater when STIM1L, opposed to standard STIM1, was over-expressed in HEK cells. It therefore seems STIM1L is expressed in tissues where high levels of regenerative SOCE are required to sustain frequent and robust Ca2+ signals.
Conclusions and Future Questions
STIM1 has been shown to be sensitive to a range of cellular stress conditions, to which it can mediate appropriate physiological responses. An increasing repertoire of STIM interaction partners has also been defined [48,91,89,115] and no doubt others will be elucidated. It therefore remains to be seen whether physiological stress conditions alter STIM interaction with other protein partners, and thus modulate the cell stress response in an Orai-independent manner. Furthermore, although primarily localized to the ER, a subset of STIM1 is found in the PM, with the N-terminal EF-hands exposed in the extracellular space. What role this PM STIM1 plays has yet to be defined, but it is conceivable that this STIM1 subpopulation could also be involved in sensing changes in extracellular Ca2+, pH, temperature and/or ROS in the external environment of the cell. Whether or not PM-STIM1 can associate with PM proteins in a manner similar to ER-STIM1 is not currently clear given its reverse orientation, however, the full interactome of STIM1 has yet to be identified. There could be any number of proteins in various intracellular compartments that could partner with PM-STIM1 in ways that cannot yet be conceptualized. As such, whereas precedent has led us to predominantly think of STIM1 as an ER Ca2+ sensor mediating Orai1 activation, its extensive capacity for stress sensing and relatively wide array of targets would seem to require a rethinking of this viewpoint.
Acknowledgments
This work was funded by NIH grant #GM097335 (JS).
References
- 1.Alonso MT, Manjarres IM, Garcia-Sancho J. Privileged coupling between Ca(2+) entry through plasma membrane store-operated Ca(2+) channels and the endoplasmic reticulum Ca(2+) pump. Molecular and cellular endocrinology. 2012;353 (1–2):37–44. doi: 10.1016/j.mce.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 2.Arif SH. A Ca(2+)-binding protein with numerous roles and uses: parvalbumin in molecular biology and physiology. Bioessays. 2009;31 (4):410–421. doi: 10.1002/bies.200800170. [DOI] [PubMed] [Google Scholar]
- 3.Aubart FC, Sassi Y, Coulombe A, Mougenot N, Vrignaud C, Leprince P, Lechat P, Lompre AM, Hulot JS. RNA interference targeting STIM1 suppresses vascular smooth muscle cell proliferation and neointima formation in the rat. Mol Ther. 2009;17 (3):455–462. doi: 10.1038/mt.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nature immunology. 2008;9 (1):81–88. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 5.Bandyopadhyay BC, Pingle SC, Ahern GP. Store-operated Ca2+ signaling in dendritic cells occurs independently of STIM1. Journal of leukocyte biology. 2010 doi: 10.1189/jlb.0610381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci Signal. 2009;2(93):ra67. doi: 10.1126/scisignal.2000522. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta. 2009;1793 (6):933–940. doi: 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1 (1):11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 9.Bird GS, Hwang SY, Smyth JT, Fukushima M, Boyles RR, Putney JW., Jr STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. 2009;19 (20):1724–1729. doi: 10.1016/j.cub.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolotina VM, Csutora P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends in biochemical sciences. 2005;30 (7):378–387. doi: 10.1016/j.tibs.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186 (2):201–209. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brailoiu E, Rahman T, Churamani D, Prole DL, Brailoiu GC, Hooper R, Taylor CW, Patel S. An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J Biol Chem. 2010;285 (49):38511–38516. doi: 10.1074/jbc.M110.162073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131 (7):1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89 (4):1341–1378. doi: 10.1152/physrev.00032.2008. [DOI] [PubMed] [Google Scholar]
- 15.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459 (7246):596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calloway N, Holowka D, Baird B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry. 2010;49 (6):1067–1071. doi: 10.1021/bi901936q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calloway N, Vig M, Kinet JP, Holowka D, Baird B. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Molecular biology of the cell. 2009;20 (1):389–399. doi: 10.1091/mbc.E07-11-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142 (2):270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annual review of biochemistry. 2011;80:973–1000. doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca(2)+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS biology. 2011;9 (3):e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Churamani D, Hooper R, Brailoiu E, Patel S. Domain assembly of NAADP-gated two-pore channels. Biochem J. 2012;441 (1):317–323. doi: 10.1042/BJ20111617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Molecular biology of the cell. 2010;21 (11):1897–1907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csutora P, Zarayskiy V, Peter K, Monje F, Smani T, Zakharov SI, Litvinov D, Bolotina VM. Activation mechanism for CRAC current and store-operated Ca2+ entry: calcium influx factor and Ca2+-independent phospholipase A2beta-mediated pathway. J Biol Chem. 2006;281 (46):34926–34935. doi: 10.1074/jbc.M606504200. [DOI] [PubMed] [Google Scholar]
- 24.Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol. 2011;194 (2):335–346. doi: 10.1083/jcb.201012157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, Putney JW., Jr TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587 (Pt 10):2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282 (24):17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 27.Derler I, Fahrner M, Muik M, Lackner B, Schindl R, Groschner K, Romanin C. A Ca2+ release-activated Ca2+ (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J Biol Chem. 2009;284 (37):24933–24938. doi: 10.1074/jbc.C109.024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellgaard L, Frickel EM. Calnexin, calreticulin, and ERp57: teammates in glycoprotein folding. Cell Biochem Biophys. 2003;39 (3):223–247. doi: 10.1385/CBB:39:3:223. [DOI] [PubMed] [Google Scholar]
- 29.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Molecular immunology. 2002;38 (10):713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 30.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441 (7090):179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 31.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, Koch WJ, Wang H, Soboloff J, Gill DL, Madesh M. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. The Journal of clinical investigation. 2013 doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giachini FR, Webb RC, Tostes RC. STIM and Orai proteins: players in sexual differences in hypertension-associated vascular dysfunction? Clin Sci (Lond) 2010;118 (6):391–396. doi: 10.1042/CS20090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Cobos JC, Zhang X, Zhang W, Ruhle BC, Motiani RK, Schindl R, Muik M, Spinelli AM, Bisaillon JM, Shinde AV, Fahrner M, Singer HA, Matrougui K, Barroso M, Romanin C, Trebak M. Store-Independent Orai1/3 Channels Activated by Intracrine LeukotrieneC4: Role in Neointimal Hyperplasia. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.111.300220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. Journal of cardiovascular pharmacology and therapeutics. 2011;16 (3–4):233–238. doi: 10.1177/1074248411409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW, Jr, Hoogenraad CC, Akhmanova A. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18 (3):177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo RW, Wang H, Gao P, Li MQ, Zeng CY, Yu Y, Chen JF, Song MB, Shi YK, Huang L. An essential role for stromal interaction molecule 1 in neointima formation following arterial injury. Cardiovascular research. 2009;81 (4):660–668. doi: 10.1093/cvr/cvn338. [DOI] [PubMed] [Google Scholar]
- 37.Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol. 2011;31 (17):3546–3556. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, Kurosaki T, Joseph SK, Gill DL, Madesh M. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190 (3):391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henke N, Albrecht P, Bouchachia I, Ryazantseva M, Knoll K, Lewerenz J, Kaznacheyeva E, Maher P, Methner A. The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis. 2013;4:e470. doi: 10.1038/cddis.2012.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hooper R, Patel S. NAADP on Target. Adv Exp Med Biol. 2012;740:325–347. doi: 10.1007/978-94-007-2888-2_14. [DOI] [PubMed] [Google Scholar]
- 41.Horinouchi T, Higashi T, Higa T, Terada K, Mai Y, Aoyagi H, Hatate C, Nepal P, Horiguchi M, Harada T, Miwa S. Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem Biophys Res Commun. 2012;428 (2):252–258. doi: 10.1016/j.bbrc.2012.10.034. [DOI] [PubMed] [Google Scholar]
- 42.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355 (6358):353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 43.Hu Q, Zheng G, Zweier JL, Deshpande S, Irani K, Ziegelstein RC. NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5-trisphosphate in human endothelial cells. J Biol Chem. 2000;275 (21):15749–15757. doi: 10.1074/jbc.M000381200. [DOI] [PubMed] [Google Scholar]
- 44.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol. 2006;8 (9):1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 45.Jousset H, Frieden M, Demaurex N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J Biol Chem. 2007;282 (15):11456–11464. doi: 10.1074/jbc.M609551200. [DOI] [PubMed] [Google Scholar]
- 46.Kehlenbach RH, Dickmanns A, Gerace L. Nucleocytoplasmic shuttling factors including Ran and CRM1 mediate nuclear export of NFAT In vitro. J Cell Biol. 1998;141 (4):863–874. doi: 10.1083/jcb.141.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim MS, Yang YM, Son A, Tian YS, Lee SI, Kang SW, Muallem S, Shin DM. RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J Biol Chem. 2010;285 (10):6913–6921. doi: 10.1074/jbc.M109.051557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krapivinsky G, Krapivinsky L, Stotz SC, Manasian Y, Clapham DE. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc Natl Acad Sci U S A. 2011;108 (48):19234–19239. doi: 10.1073/pnas.1117231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krishnaraju K, Hoffman B, Liebermann DA. The zinc finger transcription factor Egr-1 activates macrophage differentiation in M1 myeloblastic leukemia cells. Blood. 1998;92 (6):1957–1966. [PubMed] [Google Scholar]
- 50.Krishnaraju K, Hoffman B, Liebermann DA. Early growth response gene 1 stimulates development of hematopoietic progenitor cells along the macrophage lineage at the expense of the granulocyte and erythroid lineages. Blood. 2001;97 (5):1298–1305. doi: 10.1182/blood.v97.5.1298. [DOI] [PubMed] [Google Scholar]
- 51.Launikonis BS, Murphy RM, Edwards JN. Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflugers Arch. 2010;460 (5):813–823. doi: 10.1007/s00424-010-0856-7. [DOI] [PubMed] [Google Scholar]
- 52.Lee HC. Cyclic ADP-ribose and NAADP: fraternal twin messengers for calcium signaling. Sci China Life Sci. 2011;54 (8):699–711. doi: 10.1007/s11427-011-4197-3. [DOI] [PubMed] [Google Scholar]
- 53.Lee KP, Yuan JP, Zeng W, So I, Worley PF, Muallem S. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci U S A. 2009;106 (34):14687–14692. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11 (4):433–442. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 55.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell regulation. 1989;1 (1):99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, Marchant JS. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem. 2012;287 (4):2296–2307. doi: 10.1074/jbc.M111.305813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15 (13):1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, Penner R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17 (9):794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu B, Peel SE, Fox J, Hall IP. Reverse mode Na+/Ca2+ exchange mediated by STIM1 contributes to Ca2+ influx in airway smooth muscle following agonist stimulation. Respiratory research. 2010;11:168. doi: 10.1186/1465-9921-11-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mace TA, Zhong L, Kilpatrick C, Zynda E, Lee CT, Capitano M, Minderman H, Repasky EA. Differentiation of CD8+ T cells into effector cells is enhanced by physiological range hyperthermia. Journal of leukocyte biology. 2011;90 (5):951–962. doi: 10.1189/jlb.0511229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maifrede S, Liebermann DA, Hoffman B. Stress response gene EGR1 as tumor suppressor in BCR/ABL mediated leukemia. Blood; American Society of Hematology 54th Annual Meeting; Atlanta, GA. 2012; p. 33. [Google Scholar]
- 62.Mancarella S, Wang Y, Deng X, Landesberg G, Scalia R, Panettieri RA, Mallilankaraman K, Tang XD, Madesh M, Gill DL. Hypoxia-induced Acidosis Uncouples the STIM-Orai Calcium Signaling Complex. J Biol Chem. 2011;286 (52):44788–44798. doi: 10.1074/jbc.M111.303081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manjarres IM, Rodriguez-Garcia A, Alonso MT, Garcia-Sancho J. The sarco/endoplasmic reticulum Ca(2+) ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium. 2010;47 (5):412–418. doi: 10.1016/j.ceca.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 64.McNally BA, Yamashita M, Engh A, Prakriya M. Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci U S A. 2009;106 (52):22516–22521. doi: 10.1073/pnas.0909574106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281 (34):24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 2007;579 (Pt 3):703–715. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285 (25):19173–19183. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013;27 (1):63–75. doi: 10.1096/fj.12-213801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mullins FM, Park CY, Dolmetsch RE, Lewis RS. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31 (17):3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oh-hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol. 2008;20 (3):250–258. doi: 10.1016/j.coi.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nature immunology. 2008;9 (4):432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282 (12):9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ottolia M, John S, Xie Y, Ren X, Philipson KD. Shedding light on the Na+/Ca2+ exchanger. Annals of the New York Academy of Sciences. 2007;1099:78–85. doi: 10.1196/annals.1387.044. [DOI] [PubMed] [Google Scholar]
- 75.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136 (5):876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. 2010;330 (6000):101–105. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- 77.Parker NJ, Begley CG, Smith PJ, Fox RM. Molecular cloning of a novel human gene (D11S4896E) at chromosomal region 11p15.5. Genomics. 1996;37 (2):253–256. doi: 10.1006/geno.1996.0553. [DOI] [PubMed] [Google Scholar]
- 78.Parvez S, Beck A, Peinelt C, Soboloff J, Lis A, Monteilh-Zoller M, Gill DL, Fleig A, Penner R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. Faseb J. 2008;22 (3):752–761. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Patel S, Muallem S. Acidic Ca(2+) stores come to the fore. Cell Calcium. 2011;50 (2):109–112. doi: 10.1016/j.ceca.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 80.Patterson RL, van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98 (4):487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- 81.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8 (7):771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peng C, Chen Y, Yang Z, Zhang H, Osterby L, Rosmarin AG, Li S. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood. 2010;115 (3):626–635. doi: 10.1182/blood-2009-06-228130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pizzo P, Lissandron V, Capitanio P, Pozzan T. Ca(2+) signalling in the Golgi apparatus. Cell Calcium. 2011;50 (2):184–192. doi: 10.1016/j.ceca.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 84.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443 (7108):230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 85.Prins D, Groenendyk J, Touret N, Michalak M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO reports. 2011;12 (11):1182–1188. doi: 10.1038/embor.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7 (1):1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 87.Quintana A, Pasche M, Junker C, Al-Ansary D, Rieger H, Kummerow C, Nunez L, Villalobos C, Meraner P, Becherer U, Rettig J, Niemeyer BA, Hoth M. Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. Embo J. 2011;30 (19):3895–3912. doi: 10.1038/emboj.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annual review of immunology. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 89.Ritchie MF, Samakai E, Soboloff J. STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. Embo J. 2012;31 (5):1123–1133. doi: 10.1038/emboj.2011.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ritchie MF, Yue C, Zhou Y, Houghton PJ, Soboloff J. Wilms tumor suppressor 1 (WT1) and early growth response 1 (EGR1) are regulators of STIM1 expression. J Biol Chem. 2010;285 (14):10591–10596. doi: 10.1074/jbc.M109.083493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rogers JH. Calretinin: a gene for a novel calcium-binding protein expressed principally in neurons. J Cell Biol. 1987;105 (3):1343–1353. doi: 10.1083/jcb.105.3.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roos J, Digregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169 (3):435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saitoh N, Oritani K, Saito K, Yokota T, Ichii M, Sudo T, Fujita N, Nakajima K, Okada M, Kanakura Y. Identification of functional domains and novel binding partners of STIM proteins. Journal of cellular biochemistry. 2011;112 (1):147–156. doi: 10.1002/jcb.22910. [DOI] [PubMed] [Google Scholar]
- 94.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11 (4):173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 95.Shafarenko M, Liebermann DA, Hoffman B. Egr-1 abrogates the block imparted by c-Myc on terminal M1 myeloid differentiation. Blood. 2005;106 (3):871–878. doi: 10.1182/blood-2004-08-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shiels HA, Vornanen M, Farrell AP. Effects of temperature on intracellular Ca2+ in trout atrial myocytes. The Journal of experimental biology. 2002;205 (Pt 23):3641–3650. doi: 10.1242/jeb.205.23.3641. [DOI] [PubMed] [Google Scholar]
- 97.Shimoda LA, Polak J. Hypoxia. 4. Hypoxia and ion channel function. Am J Physiol Cell Physiol. 2011;300 (5):C951–967. doi: 10.1152/ajpcell.00512.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11 (4):473–485. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- 99.Smyth JT, Beg AM, Wu S, Putney JW, Jr, Rusan NM. Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr Biol. 2012;22 (16):1487–1493. doi: 10.1016/j.cub.2012.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smyth JT, Petranka JG, Boyles RR, DeHaven WI, Fukushima M, Johnson KL, Williams JG, Putney JW., Jr Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol. 2009;11 (12):1465–1472. doi: 10.1038/ncb1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13 (9):549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol. 2006;16 (14):1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 103.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281 (30):20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 104.Stathopulos PB, Zheng L, Ikura M. Stromal Interaction Molecule (STIM) 1 and STIM2 Calcium Sensing Regions Exhibit Distinct Unfolding and Oligomerization Kinetics. J Biol Chem. 2009;284 (2):728–732. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 105.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135 (1):110–122. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 106.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275 (13):3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 107.Sun S, Li W, Zhang H, Zha L, Xue Y, Wu X, Zou F. Requirement for store-operated calcium entry in sodium butyrate-induced apoptosis in human colon cancer cells. Bioscience reports. 2012;32 (1):83–90. doi: 10.1042/BSR20110062. [DOI] [PubMed] [Google Scholar]
- 108.Triggle DJ. L-type calcium channels. Curr Pharm Des. 2006;12 (4):443–457. doi: 10.2174/138161206775474503. [DOI] [PubMed] [Google Scholar]
- 109.Varga-Szabo D, Authi KS, Braun A, Bender M, Ambily A, Hassock SR, Gudermann T, Dietrich A, Nieswandt B. Store-operated Ca(2+) entry in platelets occurs independently of transient receptor potential (TRP) C1. Pflugers Arch. 2008;457 (2):377–387. doi: 10.1007/s00424-008-0531-4. [DOI] [PubMed] [Google Scholar]
- 110.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer metastasis reviews. 2007;26 (2):225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 111.Venkatachalam K, Montell C. TRP channels. Annual review of biochemistry. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16 (20):2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nature immunology. 2008;9 (1):89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312 (5777):1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Walsh CM, Doherty MK, Tepikin AV, Burgoyne RD. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem J. 2010;430 (3):453–460. doi: 10.1042/BJ20100650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330 (6000):105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124 (3215):269–270. [PubMed] [Google Scholar]
- 118.Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357 (Pt 3):673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xiao B, Coste B, Mathur J, Patapoutian A. Temperature-dependent STIM1 activation induces Ca(2+) influx and modulates gene expression. Nat Chem Biol. 2011;7 (6):351–358. doi: 10.1038/nchembio.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang X, Jin H, Cai X, Li S, Shen Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci U S A. 2012;109 (15):5657–5662. doi: 10.1073/pnas.1118947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443 (7108):226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu F, Sun L, Machaca K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc Natl Acad Sci U S A. 2009;106 (41):17401–17406. doi: 10.1073/pnas.0904651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11 (3):337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9 (6):636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32 (3):439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103 (24):9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, Singer HA, Matrougui K, Trebak M. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ Res. 2011;109 (5):534–542. doi: 10.1161/CIRCRESAHA.111.246777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zheng L, Stathopulos PB, Schindl R, Li GY, Romanin C, Ikura M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc Natl Acad Sci U S A. 2011;108 (4):1337–1342. doi: 10.1073/pnas.1015125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou Y, Mancarella S, Wang Y, Yue C, Ritchie M, Gill DL, Soboloff J. The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J Biol Chem. 2009;284 (29):19164–19168. doi: 10.1074/jbc.C109.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou Y, Ramachandran S, Oh-Hora M, Rao A, Hogan PG. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci U S A. 2010;107 (11):4896–4901. doi: 10.1073/pnas.1001169107. [DOI] [PMC free article] [PubMed] [Google Scholar]