

Abstract
Targeting therapeutic gene expression to skeletal muscle following intravenous administration is an attractive strategy for treating peripheral arterial disease (PAD), except that vector access to the ischemic limb could be a limiting factor. Since AAV serotype 9 transduces skeletal muscle at high efficiency following systemic delivery, we employed AAV-9 vectors bearing luciferase or enhanced green fluorescent protein (eGFP) reporter genes to test the hypothesis that increased desialylation of cell surface glycans secondary to hindlimb ischemia (HLI) might help offset the reduction in tissue perfusion that occurs in mouse models of PAD. The utility of the creatine kinase-based (CK6) promoter for restricting gene expression to skeletal muscle was also examined by comparing it to the cytomegalovirus (CMV) promoter after systemic administration following surgically-induced HLI. Despite reduced blood flow to ischemic limbs, CK6 promoter-driven luciferase activities in ischemic gastrocnemius (GA) muscles were ~34-, ~28-, and ~150-fold higher than in fully-perfused contralateral GA, heart, and liver, respectively, 10 days after intravenous administration. Furthermore, luciferase activity from the CK6 promoter in ischemic GA muscles was ~2-fold higher than with CMV, while in the liver CK6-driven activity was ~42-fold lower than with CMV, demonstrating that the specificity of ischemic skeletal muscle transduction can be further improved with muscle-specific promoters. Studies with Evans blue dye and fluorescently-labeled lectins revealed that vascular permeability and desialylation of cell surface glycans were increased in ischemic hindlimbs. Furthermore, AAV9/CK6/Luc vector genome copy numbers were ~6-fold higher in ischemic muscle compared to non-ischemic muscle in the HLI model, whereas this trend was reversed when the same genome was packaged in the AAV-1 capsid (which binds sialylated, as opposed to desialylated glycans), further underscoring the importance of desialylation in the ischemic enhancement of transduction displayed by AAV-9. Taken together, these findings suggest two complementary mechanisms contributing to the preferential transduction of ischemic muscle by AAV-9: increased vascular permeability and desialylation. In conclusion, ischemic muscle is preferentially targeted following systemic administration of AAV-9 in a mouse model of HLI. Unmasking of the primary AAV-9 receptor as a result of ischemia may contribute importantly to this effect.
Keywords: Adeno-associated virus, gene therapy, hindlimb ischemia, muscle-specific promoter
Introduction
Peripheral arterial disease (PAD) is mainly caused by atherosclerosis, which results in obstructions in arterial beds other than the coronary arteries, and the most common site is the lower extremity where occlusive disease leads to impaired perfusion. PAD affects about 3–10% of adults in the world and 15–20% in those over 70 years1, 2. Many patients are not candidates for surgical or catheter based revascularization and while patients with PAD should be treated with medications that target atherosclerosis, medications like statins and angiotensin converting enzyme inhibitors have yet to prove effective at increasing blood flow to ischemic limbs. Gene therapy protocols for PAD using genes that, for example, encode angiogenic growth factors to augment collateral blood flow to ischemic tissues have been pursued for more than a decade3. Results from clinical trials of gene therapy for PAD, which to date have primarily used plasmid and adenovirus-based vectors delivered intra-muscularly, or sometimes intravascularly, have been almost uniformly disappointing4, 5. Among the likely reasons for previous failures in human studies are the use of vectors that have short durations of expression and are inefficient at gene delivery when they are present in the target tissue, but perhaps no gap is greater than the fact that most of the ischemic muscle mass in a patient with PAD never receives gene therapy using the intramuscular injection methods employed in most clinical trials.
An ideal vector for skeletal muscle gene transfer would provide sustained gene expression, and could be administered with minimally invasive procedures without inducing significant vector-related inflammatory responses in the host. Over the past decade, adeno-associated viral (AAV) vectors have emerged as arguably the single most promising gene delivery system for human gene therapy. Recombinant AAV vectors transduce a wide variety of tissues in vivo and provide for long-term gene expression without provoking significant cell-mediated immune responses6–8. To date, over 100 AAV serotypes have been reported9. A comparison of the more recently-discovered serotypes showed that AAV-9 transduction to heart, lung and tibialis anterior muscle after intravenous (IV) injection is superior to all other serotypes and is age independent, whereas transduction to liver and kidney is age dependent10.
The natural tissue tropism of the various AAV serotypes can be exploited to favor gene delivery to one organ over another. This tropism is based on the viral capsids recognizing specific viral receptors expressed on specific cell types, thus allowing a degree of cell specific targeting within a given organ11. Cell-specific expression may be further aided by the use of tissue-specific promoters conferring gene expression restricted to a specific cell type11. This is desirable for gene therapy applications targeting organ specific diseases, as this will help avoid any possible harmful side effects due to gene expression in off target organs. Recently, several muscle specific promoter constructs based on the muscle creatine kinase (MCK) regulatory region were shown to provide striated muscle-restricted gene expression12–15. Of the several regulatory cassettes based on the MCK regulatory element, the CK6 promoter has been shown to provide skeletal muscle restricted gene expression with reduced expression in cardiac muscle12, 13. This is particularly desirable in the context of using AAV-9 for PAD gene therapy via systemic administration since AAV-9 has a known preference for cardiac over skeletal muscle. However, the use of skeletal muscle-specific promoters in combination with the more recent AAV serotypes in the context of PAD is largely unexplored and indeed the entire approach could, in theory, be limited by the fact that blood flow to the ischemic limb is reduced thus creating a barrier to intravascular gene delivery. Recently, cell surface N-linked glycans with terminal galactosyl residues were shown to serve as the primary receptor for AAV-916. Desialylation of these galactosylated glycans was shown to markedly increase cell surface binding and transduction by AAV-9 and significantly decrease that by other AAV serotypes, like AAV-116. We hypothesized that ischemia induces desialylation of the cell surface glycans, resulting in increased availability of AAV-9 receptors, and this together with the increase in vascular permeability characteristic of ischemic tissue might suffice to overcome the barrier of reduced blood flow in ischemic tissues. In the present study, we first sought to compare the magnitude and specificity of reporter gene expression driven by the human cytomegalovirus (CMV) immediate early and the minimal CK6 promoters packaged into AAV-9 and administered by IV injection in a mouse model of hindlimb ischemia (HLI). Using a novel approach that combines a muscle-specific promoter with an AAV serotype that preferentially transduces skeletal muscle, we show here that targeted expression of reporter genes in ischemic muscles following systemic delivery is not only possible, it is markedly enhanced relative to non-ischemic muscles and other tissues.
Results
Magnitude and specificity of gene expression from intravenous injection of AAV-9 harboring the CMV promoter
The perfusion ratio of ischemic to non-ischemic hindlimbs in C57Bl/6 mice (n = 5) immediately post-HLI was 0.34 ± 0.12 (mean ± SEM). Consistent with prior reports from our group17–19, the perfusion ratio recovered to 0.48±14 by post-operative (post-op) day 7, at which time the mice received IV injections of AAV9/CMV/Luc (4.15×1011 vector genomes (vg)/animal) via the right internal jugular vein. Luciferase expression was then monitored by non-invasive in vivo bioluminescence imaging. Age-matched C57Bl/6 male mice that did not undergo HLI and did not receive any vector served as negative controls (Fig. 1a). As indicated by the purple color-coding, luciferase expression from the CMV promoter was observed throughout the body on post-AAV days 7 (Fig. 1b) and 14 (Fig. 1c). However, luciferase expression appeared strongest in the upper abdominal region corresponding to liver. Interestingly with regard to hindlimb expression, bioluminescence signals appeared stronger in the ischemic hindlimbs (mouse's left side, rightmost hindlimb in Fig. 1) than in the non-ischemic, contralateral hindlimbs on both days 7 and 14 post-AAV injection. ROI analysis was then used to estimate relative luciferase signal intensity in each hindlimb and the upper abdomen (corresponding to liver). On post-AAV day 7, the mean bioluminescence signal from ischemic hindlimbs was 2.7-fold higher than the non-ischemic limbs but 17.7-fold lower than in the liver (Fig. 1d). On post-AAV day 14, bioluminescence in the ischemic limbs was 4.3-fold higher than in the non-ischemic limbs but 4.5-fold lower than in the liver.
Fig. 1.

Time course and tissue distribution of CMV- and CK6-mediated luciferase expression from AAV-9 following intravenous (IV) injection 7–8 days after hindlimb ischemia (HLI) surgery. (a) Negative control consisting of an age-matched C57Bl/6 male mouse that did not undergo HLI and did not receive any vector. In the CMV group, b and c, adult C57Bl/6 mice (n = 5 per group) were injected with 4.15×1011 vector genomes/mouse via jugular vein. In vivo bioluminescence (IVIS) images were obtained on the 7th (b) and 14th day (c) following vector administration. In the CK6 group, f and g, adult C57Bl/6 mice (n = 4 per group) were injected with 4.15×1011 vector genomes/mouse via jugular vein. In vivo bioluminescence (IVIS) images obtained on the 6th (f) and 10th day (g) following vector administration. (d, h) For each group of mice, the mean values of bioluminescence as average radiance (photons/s·cm2·sr) were obtained from the regions of interest in Panels b, c, f and g and plotted as ratios of Ischemic limb to Non-ischemic limb and Ischemic limb to upper abdomen (corresponding to liver). (e, i) Bar graph showing luciferase activities in tissue extracts in the CMV and CK6 groups, respectively. Protein extracts from various tissues were collected 10–14 days after vector administration for homogenization and in vitro luciferase assays. Luciferase activities are expressed as relative light units per mg protein (RLU/mg protein) (mean ± SEM, * p<0.05).
While bioluminescence imaging provides a non-invasive estimate of relative luciferase activities in serial studies, it is difficult to compare values between tissues due to differences in tissue depth and the differential absorption of photons by different tissues. For this reason, rigorous quantitative measurement of luciferase activity was performed in tissue extracts from the various organs as shown in Fig. 1e. Luciferase activity in the ischemic gastrocnemius (GA) muscles of mice treated with AAV9/CMV/Luc was 7-fold higher than in the contralateral GA, 2.0-fold higher than in the heart, and 1.8-fold higher than in the liver. These results demonstrate that AAV-9 is highly effective for delivering gene(s) to ischemic skeletal muscle following systemic delivery, even when using a non-tissue-specific promoter.
Magnitude and specificity of gene expression from intravenous injection of AAV-9 harboring the CK6 promoter
HLI was surgically induced in left hindlimbs of adult C57Bl/6 mice (n = 4). Immediately after surgery on post-op day 0, the ratio of perfusion as measured by laser Doppler between ischemic and non-ischemic hindlimbs was 0.34 ± 0.12 (mean ± SEM). On post-op day 7, the perfusion ratio had partially recovered to 0.48 ± 35. On post-op day 8, all mice received IV injections of AAV9/CK6/Luc (4.15 × 1011 vector genomes (vg)/animal) via the right internal jugular vein. Luciferase expression was again monitored by bioluminescence imaging. Bioluminescence signals appeared strongest in the ischemic hindlimbs on post-AAV days 6 (Fig. 1f) and 10 (Fig. 1g). Using ROI analysis, the mean bioluminescence signal in the ischemic limbs was 50.5-fold higher than in non-ischemic limbs and 17.2-fold higher than liver on day 6 post-AAV (Fig. 1h). On day 10 post-AAV, bioluminescence was 37.8-fold higher in ischemic versus non-ischemic limbs and 9.8-fold higher than liver (Fig. 1h). Similar results were obtained in parallel experiments performed in BALB/c mice (data not shown).
The more rigorous, quantitative measurement of luciferase activity in tissue extracts from selected organs harvested on day 10 post-AAV is presented in Fig. 1i. Again, luciferase activity was significantly higher in the ischemic hindlimb muscles compared to contralateral non-ischemic muscles or liver. Luciferase activity in the ischemic GA muscle of mice treated with AAV9/CK6/Luc was 34.1-fold higher than in the contralateral GA, 28.1-fold higher than in the heart, and 150-fold higher than in the liver (all comparisons p<0.05). Luciferase activity in the non-ischemic GA was 1.2-fold higher than in the heart and 6.6-fold higher than in the liver. Furthermore, luciferase activity in the ischemic GA was 1.9-fold higher while that in the liver was 41.7-fold lower in the CK6 group when compared to the CMV group. These results clearly demonstrate that the combination of AAV-9 and the CK6 promoter is highly efficient and selective for delivering genes to ischemic skeletal muscle following systemic delivery.
Distribution of eGFP expression in ischemic hindlimb muscle confirms the efficiency of AAV-9
Vectors carrying the enhanced green fluorescence protein (eGFP) gene driven by the CMV or CK6 promoters (AAV9/CMV/eGFP and AAV9/CK6/eGFP) were systemically administered to adult C57Bl/6 mice (n = 5 for CMV and n = 2 for CK6) via jugular vein at a dose of 4.15×1011 vg per mouse on the 7th day following HLI surgery. Two weeks following vector injection, eGFP expression in the mouse hindlimb skeletal muscles was assessed by fluorescence microscopy (Fig. 2). The results show that the AAV-9 capsid together with the CK6 promoter transduces ischemic skeletal myofibers (Fig. 2b) far more efficiently than the non-ischemic ones (Fig. 2a) after systemic delivery. Microscopic analysis of >3000 myofibers taken from two sections per tibialis anterior (TA) muscle from both the ischemic and non-ischemic hindlimbs of the two mice treated with AAV9/CK6/eGFP revealed that 50–55% of the ischemic myofibers exhibited variable intensities of eGFP fluorescence signal intensity above background (Fig. 2b), whereas non-ischemic myofibers from those same mice displayed only background autofluorescence (Fig. 2a). Furthermore, the expression of eGFP in the skeletal myofibers following systemic delivery was relatively low when gene expression was driven by the CMV promoter (Fig. 2c&d). With the CMV promoter, <0.5% of the myofibers in the ischemic TA exhibited fluorescent signal above background. Thus, the level of eGFP expression from AAV-9 in ischemic skeletal myofibers was significantly higher with the CK6 promoter than with the CMV promoter after IV injection in adult mice. These results using an independent reporter system confirm that the combination of AAV-9 capsid and CK6 promoter is highly efficient for the selective delivery of gene(s) to ischemic skeletal muscles following systemic delivery.
Fig. 2.

Fluorescence microscopy of muscle cryosections from mice injected IV with AAV-9 vectors carrying CMV or CK6 promoters. The AAV vectors AAV/CK6/eGFP and AAV/CMV/eGFP were packaged into AAV-9 capsids. Adult C57Bl/6 mice were injected with 4.15×1011 vector genomes/mouse (n = 2 for the AAV/CK6/eGFP group and 5 for the AAV/CMV/eGFP group) via jugular vein. Two weeks following vector administration, 15 μm cryosections of the tibialis anterior (TA) muscles from ischemic and non-ischemic hindlimbs were prepared for analysis by confocal microscopy. All images shown here were captured at 5× magnification with a constant 0.5 sec exposure. Panels a (non-ischemic) and b (ischemic) show TA muscles from mice injected with AAV/CK6/eGFP, while panels c (non-ischemic) and d (ischemic) represent those injected with AAV/CMV/eGFP. Bar = 200 μm.
Vascular permeability is markedly increased by HLI
HLI was surgically induced in left hindlimbs of C57Bl/6 mice (n = 6). On post-op day 7, the mice received intraperitoneal injections of Evans blue dye and the animals were euthanized 24h later. Ex-vivo images of hindlimb muscles (Fig. 3a) show that the ischemic TA muscles (I) were markedly blue compared to the non-ischemic (NI) ones. The quantitative analysis of the muscle extracts is summarized in Fig. 3b. The Evans blue dye content in ischemic TA muscles (12.7 ± 0.6% of the injected dose per gram tissue) was nearly 5-fold higher than in non-ischemic TA muscles (2.6 ± 0.3%, p<0.05).
Fig. 3.
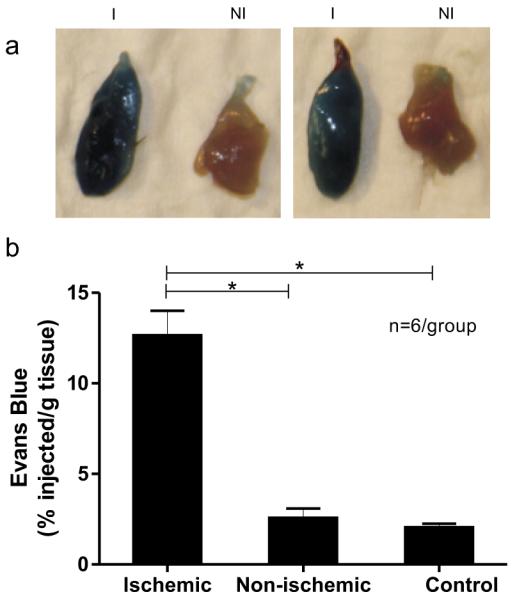
Vascular permeability is increased in the mouse model of hindlimb ischemia (HLI). (a) Ex vivo images of explanted ischemic (I) and non-ischemic (NI) TA muscle from adult C57Bl/6 mice that received Evans blue dye intraperitoneally on the 7th day after HLI surgery (n = 6). Tissues were harvested 24 hours after Evans blue administration. (b) Bar graph showing Evans blue quantification. Ischemic and non-ischemic TA muscles (shown in Panel a) were homogenized for Evans blue assay and results were expressed as percent of injected Evans blue dye per gram of tissue (% injected/g tissue). Tissues from mice that did not undergo HLI served as an additional negative control (n = 6). Mean ± SEM, * p<0.05.
HLI induces marked desialylation of cell surface N-linked glycans, thereby unmasking the primary receptor for AAV-9 binding
HLI was surgically induced in the left hindlimbs of adult male C57Bl/6 mice (n=3). Seven days following HLI, the distribution of sialylated versus desialylated cell surface glycans in mouse hindlimb skeletal muscles was assessed by fluorescence microscopy using lectin staining. Of the two lectins used, MAL-I binds to α2,3-sialylated glycans whereas ECL binds to the desialylated galactose residues of cell surface glycans. Myofibers from the ischemic TA showed abundant ECL staining along the cell surface compared to a weaker staining seen in the non-ischemic TA muscles (Fig. 4, top). Conversely, MAL-I staining was readily detected in non-ischemic TA muscles but was much weaker in ischemic TA muscles (Fig. 4, bottom). For each of the two lectins (MAL-I and ECL), four 40× fields of view were quantified for mean fluorescence intensity from each ischemic and non-ischemic muscle from each of the 3 mice entered into the study. The mean ratio of ECL fluorescence intensity in ischemic vs. non-ischemic muscle was 1.6, while the same fluorescence intensity ratio in MAL-1 stained tissue was 0.8. To incorporate an internal standard, the ratio of ECL to MAL-I fluorescence intensity in ischemic muscle (4.3 ± 0.6) was then compared to the same ratio in non-ischemic muscle (1.9 ± 0.1) and the difference was found to be significant (p<0.05). These results demonstrate that the induction of hindlimb ischemia causes marked desialylation of cell surface N-linked glycans, thus unmasking the primary cell surface attachment factor for AAV-9.
Fig. 4.
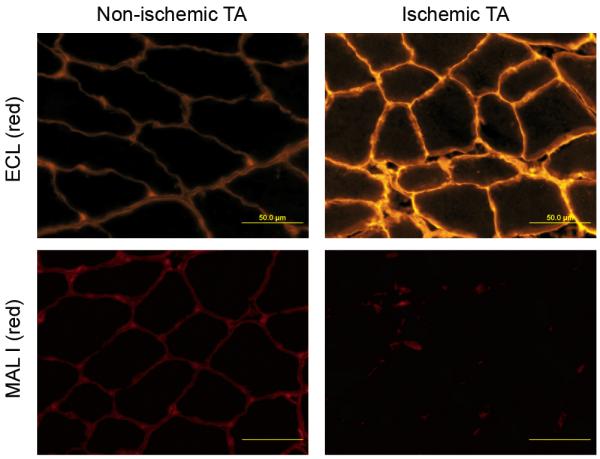
Differential distribution of sialylated and desialylated cell surface glycans in ischemic and non-ischemic muscle Fluorescence microscopy illustrating the distribution of sialylated and desialylated cell surface glycans in ischemic and non-ischemic TA muscles of adult C57Bl/6 mice on the 7th day after HLI surgery. Representative fluorescent photomicrographs are shown of the ischemic and non-ischemic TA sections stained with ECL (red, upper row), and MAL-I (red, lower row).
Magnitude of gene expression and tropism of tissue distribution following intravenous injection of AAV-1 and -9 harboring the CK6 promoter further implicates hindlimb ischemia in the unmasking of cell surface receptors, thereby facilitating selective transduction by AAV-9
HLI was surgically induced in left hindlimbs of adult C57Bl/6 mice (n = 5 per group) 7 days prior to the injection of AAV/CK6/Luc genomes packaged in either AAV-9 or AAV-1 capsids. On post-op day 7, the ratio of perfusion in ischemic vs. non-ischemic hindlimbs as measured by laser Doppler was 0.44 ± 0.13 (Mean ± SEM) for the AAV-9 group and 0.29 ± 11 for the AAV-1 group. After laser Doppler measurement on post-op day 7, five mice received IV injections of AAV9/CK6/Luc (4.15 × 1011 vector genomes (vg)/animal) via the right internal jugular vein, while the remaining 5 mice were similarly treated with AAV1/CK6/Luc. Luciferase expression was again monitored by bioluminescence imaging. In the AAV-9 group, bioluminescence signals again appeared strongest in the ischemic hindlimbs on post-AAV days 7 (Fig. 5a) and 14 (Fig. 5b) with markedly reduced expression in liver and little luciferase expression, if any, detected elsewhere. In the AAV-1 group, bioluminescence signal intensities in ischemic hindlimbs were visibly less intense than that seen in mice injected with the same vector genome packaged in AAV-9 capsids (Fig. 5c–d). Furthermore, the marginal bioluminescence signals from ischemic hindlimbs in the AAV-1 group were no stronger than those generated by liver.
Fig. 5.

Time course of CK6-mediated luciferase expression and tissue distribution of vector genomes from AAV-9 and AAV-1 following intravenous (IV) injection 7–8 days after hindlimb ischemia (HLI) surgery. Adult C57Bl/6 mice (n = 5 per group) were injected with 4.15×1011 vector genomes/mouse via jugular vein. In vivo bioluminescence (IVIS) images were obtained on the 7th day (a for AAV-9 and c for AAV-1 groups) and 14th day (b for AAV-9 and d for AAV-1 groups) following vector administration. Bar graphs showing luciferase activities (e) and vector genome copy numbers (f) in tissue extracts from the AAV-1 and AAV-9 groups. Protein extracts from various tissues were collected 14–16 days after vector administration for homogenization and in vitro luciferase assays. Luciferase activities are expressed as relative light units per mg protein (RLU/mg protein). Genomic DNA was isolated from each of the indicated tissues and used to determine vector genome copy number per μg host genomic DNA. RGA & LGA = non-ischemic right and ischemic left gastrocnemius anterior muscles, respectively. RTA & LTA = non-ischemic right and ischemic left tibialis anterior muscles, respectively. Both graphs depict mean ± SEM with asterisks (*) indicating p<0.05 vs. any other value by ANOVA.
The more rigorous, quantitative measurement of luciferase activity in tissue extracts from selected organs is presented in Fig. 5e. Again, luciferase activity in the AAV-9 group was significantly higher in ischemic hindlimb muscle compared to contralateral non-ischemic muscle, liver, or other organs. Luciferase activity in ischemic muscle from mice treated with AAV9/CK6/Luc reached a mean of 2.0×106 ± 7.8×105 RLU/mg protein (mean±SEM), whereas Luc activity in the same mice declined to only 4.4×104 ± 1.2×104 RLU/mg protein in non-ischemic muscle, 4.0×104 ± 1.8×104 RLU/mg protein in the liver, 1.4×104 ± 3.7×103 RLU/mg protein in the heart,1.5×103 ± 1.0×103 RLU/mg protein in the kidney and 7.9×102 ± 2.7×102 RLU/mg protein in the brain (see Fig. 5e). While luciferase activity in ischemic muscle was nearly 24-fold higher in the AAV-9 group as compared to the AAV-1 group (all comparisons vs. AAV-9 in ischemic muscle p<0.05 by ANOVA), luciferase activity from AAV-1 nevertheless trended higher in ischemic vs. non-ischemic muscle.
We next compared the vector genome (vg) copy numbers persisting in tissue samples at 14 days post-AAV injection using real-time PCR (Fig. 5f). Interestingly, AAV-9 vg copies were significantly higher in the liver (3.3×107 ± 1.8×107 vg copies/μg host genomic DNA) than in any other tissue examined (mean ±SEM, all comparisons p<0.05 by ANOVA). Nevertheless, the next highest concentration of vector genomes was found in ischemic muscle (1.7×105 ± 2.4×104 vg copies/μg host genomic DNA) followed by kidney, brain, non-ischemic muscle and heart. In contrast, AAV-1 did not exceed 3×105 vector genome (vg) copy numbers per μg host genomic DNA in any tissue examined; with the highest copy number found in the liver (2.6×105 ± 6.2×104 vg copies/μg host genomic DNA), followed by non-ischemic muscle, brain, ischemic muscle, kidney and heart. Finally, the vg/μg genomic DNA copy numbers for AAV-9 were 5.6-fold higher in ischemic vs. non-ischemic muscle; whereas this trend was reversed for AAV-1 which had 6.0-fold higher copy numbers in non-ischemic vs. ischemic muscle.
These results clearly demonstrate that AAV-9 selectively targets ischemic hindlimb muscle, and that the AAV serotype 9 capsid, in combination with the CK6 promoter, is highly efficient and selective for delivering genes to ischemic skeletal muscle following systemic delivery.
Discussion
PAD is a major health care problem and more than a decade of clinical trials of gene therapy for PAD has failed to bring this approach forward in any meaningful way3–5. Some of the plausible explanations for previous failures in human studies include: gene delivery vectors with inherently low magnitudes and durations of gene expression, and intra-muscular injection methods which are far more effective in pre-clinical studies of small animals with limited muscle mass where most of the muscle is accessible to the needle. In humans, studies have found no evidence of transgene expression20, 21 or when present was limited and heterogeneous in distribution22. Therefore, systemic delivery offers numerous theoretical advantages for treating patients with PAD, although two major concerns exist. First, blood flow to the ischemic limb is reduced in PAD and this may limit access of the vector to ischemic tissue. Second, it is desirable to restrict gene expression to the cell type of interest since the expression of therapeutic genes in off-target tissues could potentially lead to deleterious side effects. The results of the current study show, for the first time, that gene expression in ischemic hindlimb muscle can be achieved by systemic injection of an AAV-based vector system with a skeletal muscle-tropic capsid (AAV-9) and a tissue-specific promoter (a compact version of the muscle-specific MCK promoter/enhancer). In the present study, using an AAV serotype 9-based vector in an adult mouse model of hindlimb ischemia (HLI), we demonstrate that: 1) the CMV promoter is adequate to achieve ischemia-tropic gene expression in skeletal muscle following intravenous administration, 2) the CK6 promoter provides for more robust and highly specific gene expression in ischemic skeletal muscle, 3) vascular permeability and desialylation of cell surface glycans are both increased in post-ischemic hindlimbs, 4) luciferase protein expression from AAV-9 is significantly higher in ischemic tissues as compared to the same vector genome packaged in an AAV-1 capsid, and 5) vector genome copy numbers for AAV-9 (which binds desialylated glycans) are elevated in ischemic vs. non-ischemic muscle, whereas copy numbers for AAV-1 (which binds sialylated glycans) are reduced in ischemic vs. non-ischemic muscle. These last three findings are complementary, and together indicate that ischemic desialylation synergizes with increased vascular permeability to yield preferential transduction of ischemic muscle tissue following intravenous delivery of AAV-9.
Strong, non-selective, viral promoters such as CMV are typically used in animal studies as well as clinical trials of gene therapy for PAD21, 23–25. While tissue-specific promoters may be efficient at restricting gene expression to a particular cell or tissue type, their widespread use has not been realized because of a generally lower level of gene expression that is considered suboptimal for gene therapy applications. Furthermore, the “payload capacity” of the AAV capsid effectively limits the size of the recombinant AAV genome to approximately 4.9–5.3 kb26, 27. The choice of promoter for AAV-mediated, organ-specific gene expression should therefore be based on the size, specificity and strength of the promoter. Previous work in the field of gene therapy for muscular dystrophy led to the creation of hybrid promoter/enhancers in which various enhancers (including the MCK enhancer) have been introduced adjacent to the minimal MCK promoter12–15. In a recent comparison of five such hybrid constructs, Hauser et al. identified a compact (571 bp) combination of the MCK enhancer and promoter (CK6) that was 6-fold stronger than the full-length 3.3-kb MCK promoter/enhancer and almost 12% as strong as the CMV promoter in muscle cells12. Accordingly, we used the minimal CK6 promoter/enhancer in this study to achieve high-level, muscle-specific gene expression12–15, 28. Finally, in gene therapy protocols, the viral vector burden should be kept to a minimum to avoid vector-related side effects. While the specificity of gene expression needed for clinical efficacy will depend largely upon the nature of the therapeutic transgene, this study achieved efficient transduction of ischemic skeletal muscle without detectable adverse effects using a dose of 1.4 × 1013 vg/kg, which is comparable to intravenous doses of AAV vectors used in other small and large animal studies13, 29, 30.
One might anticipate lower expression levels in ischemic limbs compared to non-ischemic limbs based on the fact that ischemic limbs in this study had approximately one-half of the relative perfusion compared to non-ischemic limbs. Contrary to this expectation, the luciferase reporter gene and in vivo bioluminescence imaging (IVIS) clearly indicated that ischemic hindlimbs had higher luciferase activity than non-ischemic hindlimbs following intravenous delivery (Figs 1b,c,f,g). The ratios of in vitro luciferase activity in the ischemic skeletal muscle versus the other key organs such as liver and heart are summarized in Figs. 1e,i. Using the CK6 promoter, luciferase activity in the ischemic gastrocnemius (GA) muscle was found to be ~34-fold higher than in the contralateral GA and ~150-fold higher than in the liver. Luciferase activity in the ischemic GA was also ~2-fold higher while that in the liver was ~42-fold lower in the CK6 group when compared to the CMV group. Thus by combining the CK6 promoter with the AAV-9 capsid, we were able to harness the superior transduction efficiency of the AAV-9 capsid while retaining high selectivity for ischemic skeletal muscle over liver. Using in vivo bioluminescence imaging, similar data were obtained in a second inbred mouse strain (BALB/c), which has been previously documented to have extremely poor perfusion recovery after hindlimb ischemia17 (data not shown). These results confirm that the preferential transduction of ischemic skeletal muscle was not restricted to one mouse strain.
To the best of our knowledge, our results are the first to show robust and homogeneous gene expression in ischemic limbs compared to non-ischemic (contralateral) limbs following systemic delivery of an AAV vector. In further comparing the CMV and CK6 promoters, we found that the apparent tropism for ischemic skeletal muscle was much more pronounced with the CK6 promoter. One plausible explanation for this observation is that the increased vascular permeability and desialylation of cell surface glycans associated with ischemia may act in synergy with the natural muscle tropism of the AAV-9 capsid and the specificity of the CK6 promoter for skeletal muscle. The eGFP reporter gene was then used to characterize the distribution of gene expression and the rate of transduction in ischemic skeletal muscle after IV administration of AAV-9 vectors driven by the CMV and CK6 promoters. Using the CK6 promoter, the transduction rate in ischemic skeletal muscle was >50% at the dose used in this study. These results also demonstrated that AAV-9 achieves a relatively homogeneous distribution of gene expression in ischemic skeletal muscle after IV administration, particularly when deployed in combination with a muscle-specific promoter.
Studies with Evans blue dye were then conducted to test the hypothesis that increased vascular permeability may be one of the factors contributing to the preferential transduction of ischemic skeletal muscle. Consistent with previous studies31, the results indicated that vascular permeability was indeed increased in ischemic hindlimbs in our HLI model, suggesting a potential role in the increase in transduction efficiency observed under ischemic conditions. Recently, Shen et al. showed that N-linked glycans with terminal galactosyl residues serve as the primary receptor for AAV-9 in Chinese hamster ovary (CHO) cells16. While sialylated glycans serve as the cellular receptors for other AAV serotypes such as AAV-1, it was the desialylation of the N-terminal galactosylated glycans that increased cell surface binding and infectivity of AAV-916. Using two lectins, MAL-I (which binds to α2,3-sialylated glycans) and ECL (which binds to the desialylated galactose residues of cell surface glycans), we report here, for the first time, that ischemia markedly increases the desialylation of cell surface glycans in the mouse HLI model of PAD, suggesting a second contributing mechanism for the increase in transduction efficiency under ischemic conditions. Studies comparing the vector genome copy numbers and luciferase gene expression levels driven by the CK6 promoter between the AAV-9 and AAV-1 serotypes were then conducted to evaluate further the role of desialylated cell surface glycans in this system. The AAV-1 capsid was selected for this comparison because AAV-1 utilizes sialylated glycans for cell surface binding and transduction. We report here, for the first time, that the AAV-9 capsid is far more efficient at targeting gene expression to ischemic skeletal muscle, demonstrating a 24-fold advantage over AAV-1 in tissue luciferase activity after IV administration. These findings were consistent with results obtained from an analysis of vector genome (vg) copy number in various tissues, where the AAV-9 vg/μg genomic DNA copy numbers were 5.6-fold higher in ischemic vs. non-ischemic muscle; whereas this trend was reversed for AAV-1 which had 6.0-fold higher copy numbers in non-ischemic vs. ischemic muscle. These results provide convincing evidence in support of the hypothesis that ischemia-induced desialylation of galactosylated N-glycans unmasks the primary cellular receptor for AAV-9, thus promoting cell surface binding and transduction after IV injection, ultimately resulting in markedly increased and selective transgene expression in ischemic as compared to non-ischemic myofibers and other tissues, well above and beyond that afforded by increased vascular permeability alone. While these two mechanisms may complement one another in vivo, the effect of ischemia on AAV9-mediated gene expression is so pronounced that other mechanisms may also contribute. For example, the stress induced by fasting was recently shown to increase in vivo transduction efficiency by AAV vectors including AAV932, and the local stress induced by ischemia may function similarly to enhance the intracellular post-processing of AAV genomes.
Conclusions
This study shows for the first time that transgene expression is targeted to ischemic muscle following systemic administration of muscle-tropic AAV vectors. The specificity of ischemic skeletal muscle transduction can be further improved with the use of a muscle-specific promoter. Increased desialylation of the cell surface N-glycans is a mechanism that likely contributes to the ischemic enhancement of AAV-9 mediated gene transfer after systemic delivery. These findings will be of immediate utility in pre-clinical studies examining the role of various genes in the recovery from hindlimb ischemia, and may ultimately prove valuable in clinical gene therapy protocols targeting PAD.
Materials and Methods
Plasmids
The AAV vectors bearing the CMV promoter driving the expression of firefly luciferase (AAV/CMV/Luc) or eGFP (AAV/CMV/eGFP) have been described previously33. Construction of AAV vectors bearing the CK6 promoter driving the expression of firefly luciferase (AAV/CK6/Luc) or eGFP (AAV/CK6/eGFP) was accomplished in two steps. First, the CMV promoter was removed from AAV/CMV/Luc and AAV/CMV/eGFP by double digestion with XbaI and HindIII. Second, a PCR amplified 571 bp CK6 MCK enhancer/promoter was directionally inserted as an XbaI-HindIII fragment. The 571 bp muscle specific CK6 MCK enhancer/promoter construct was a kind gift of Dr. S. D. Hauschka12. The AAV-1 pseudotyped AAV/CK6/Luc vector used for the comparison of AAV-9 and AAV-1 was obtained from the Penn Vector Core in the School of Medicine Gene Therapy Program at the University of Pennsylvania.
AAV vector production
AAV vectors were packaged in AAV-293 cells by the triple transfection method, then purified by ammonium sulfate fractionation and iodixanol gradient centrifugation as described previously33, 34. Titers of the AAV vectors (vector genomes/ml) were determined by real-time PCR as described previously33, 35.
Animal procedures
Animal protocols used in this study were approved by the Institutional Animal Care and Use Committee and conformed to the “Guide for the Care and Use of Laboratory Animals” (NIH Publication 85–23, revised 1985). All mice (C57BL/6 and BALB/c) (15–21 weeks old) were purchased from The Jackson Laboratories (Bar Harbor, ME). Age-matched (15–21 week old) male mice were used for all the experiments to exclude estrogen as a potential confound in the HLI model described below.
Induction of hindlimb ischemia (HLI)
Mice underwent unilateral femoral artery ligation and excision on the left hindlimb as described previously17–19. Necrosis was visually assessed each day. Blood flow in the ischemic and contralateral non-ischemic limbs was measured as described previously with a laser Doppler perfusion imaging system (Perimed, Stockholm, Sweden)17–19.
AAV vector delivery
For intravenous (IV) injection, mice were anesthetized with isoflurane as described above and the AAV-1 or -9 solutions (50–100 μl containing 4.15 × 1011 vector genomes) were slowly injected via the right jugular vein on the 7–8th day following HLI surgery.
Bioluminescence imaging in vivo
Bioluminescence imaging was performed using an IVIS 100 system (Caliper Life Sciences, Hopkinton, MA). Luciferase expression in live mice was non-invasively detected after the IP injection of luciferin and images were processed as described previously35, 36. Equal-sized regions of interest (ROIs) were selected over each hindlimb and upper abdomen to obtain estimates of bioluminescence intensity.
Quantitative in vitro luciferase activity assays
Luciferase activity was measured using luciferase assay reagents from Promega Corp. (Madison, WI). After bioluminescence imaging and euthanasia at 10–14 days post vector injection; the heart, liver, skeletal muscle and other tissues were collected from experimental mice. Protein extracts were prepared and luciferase activities (Relative light units, RLU) were determined using a FLUOstar Optima micro-plate reader (BMG Labtech, Durham, NC).
Fluorescence imaging
eGFP expression and desialylation of cell surface glycans in mouse tissues were documented by fluorescence microscopy using a Zeiss LSM 700 confocal microscope (Gottingen, Germany). For eGFP expression fourteen days following vector administration, animals were euthanized for muscle collection and fixation in 3.7% paraformaldehyde at 4°C for 1 hour. After (3×) 5 min PBS washes, tissues were equilibrated with 30% sucrose in PBS overnight. Fifteen μm thick cryosections were then cut and used for quantifying eGFP expression by fluorescence signal intensity.
For assessing sialylated and desialylated cell surface glycans, animals were euthanized 7 days post-HLI. Ischemic and contralateral muscles were harvested and placed in OCT for snap freezing in liquid nitrogen. Seven μm cryostat sections were prepared to assess the differential distribution of sialylated or desialylated glycans in ischemic versus non-ischemic muscles. Staining was performed using the biotinlyated lectins, Maackia amurensis lectin (MAL-I) and Erythrina cristagalli lectin (ECL) (Vector Laboratories, Burlingame, CA). Lectins were visualized using Streptavidin-Alexa Fluor-555 (Invitrogen Carlsbad CA). Quantification of the fluorescence signal intensity of the lectin staining was done using Image J software.
Evans blue dye assay
Animals were anesthetized and maintained on 1–1.2% isoflurane in oxygen. Evans blue dye, 50 μl of a 20 mg/ml solution in saline, was injected intraperitoneally and tissues were harvested 24 h later. Evans blue dye was extracted from tissue following a protocol adapted from Bohmer et al.37. Briefly, the 28–70 mg samples were freeze-dried overnight. Formamide (300 μl) was added, and the samples were placed in a water bath at 60 °C for 2 h to extract Evans blue dye. After an additional 10 h at room temperature, the absorption of Evans blue dye was measured using a microplate reader (Biorad Model 3550) set at 595 nm. Sample concentrations were determined using a calibration curve generated in parallel from dilutions of Evans blue dye in formamide.
Determination of AAV vector genome copy number per μg genomic DNA
The AAV2 genomic backbone AAV/CK6/Luc was cross-packaged into capsids from AAV serotypes 9 and 1 for injection as described above. Two weeks after vector administration, total genomic DNA from a panel of tissues was prepared using a QIAamp DNA Mini Kit (Qiagen, Inc). Real-time PCR using the Bio-Rad iTaq Universal SYBR Green Supermix was performed on a Bio-Rad CFX Connect system (Hercules, CA, USA). The following primers were used for amplifying the firefly luciferase gene: 5′-AAGATTCAAAGTGCGCTGCTGGTG-3′ (forward) and 5′-TTGCCTGATACCTGGCAGATGGAA-3′ (reverse). Known copy numbers (103–109) of the plasmid AAV/CK6/Luc were used to construct the standard curve. The results were expressed as mean AAV vector genome copy numbers per μg of host genomic DNA.
Statistical analysis
Data were expressed as mean ± SEM. For statistical comparisons of luciferase activity and vector genome copy number, values from various tissues were compared using 1-way ANOVA and a Tukey's post-test was used to assess significance. P<0.05 was considered statistically significant in all of the comparisons.
Acknowledgements
The authors are grateful to Rebecca A. Maddux, Jane V. Carrick, Rachel E. Sullivan and John Burns for their valuable technical assistance and to Dr. S. D. Hauschka for the plasmid construct bearing the 571bp CK6 enhancer/promoter fragment. SH acknowledges support from 5 T32 HL007284-35 and ABK from T32 HL073555. This work was supported in part by the NHLBI Gene Therapy Resource Program, and by grants from the American Heart Association (12GRNT12040223 to BAF) and the National Institutes of Health (NIH R01 HL058582 to BAF and NIH R01 HL101200 to BHA).
Footnotes
Conflict of interest The authors declare that there are no conflicts of interest relevant to this manuscript.
References
- 1.Hirsch AT, Criqui MH, Treat-Jacobson D, Regensteiner JG, Creager MA, Olin JW, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. 2001;286(11):1317–24. doi: 10.1001/jama.286.11.1317. [DOI] [PubMed] [Google Scholar]
- 2.Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG, et al. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II) Eur J Vasc Endovasc Surg. 2007;33(Suppl 1):S1–75. doi: 10.1016/j.ejvs.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 3.Tongers J, Roncalli JG, Losordo DW. Therapeutic Angiogenesis for Critical Limb Ischemia: Microvascular Therapies Coming of Age. Circulation. 2008;118(1):9–16. doi: 10.1161/CIRCULATIONAHA.108.784371. [DOI] [PubMed] [Google Scholar]
- 4.Jones WS, Annex BH. Growth factors for therapeutic angiogenesis in peripheral arterial disease. Curr Opin Cardiol. 2007;22(5):458–63. doi: 10.1097/HCO.0b013e328236741b. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh R, Walsh SR, Tang TY, Noorani A, Hayes PD. Gene therapy as a novel therapeutic option in the treatment of peripheral vascular disease: systematic review and meta-analysis. International Journal of Clinical Practice. 2008;62(9):1383–90. doi: 10.1111/j.1742-1241.2008.01842.x. [DOI] [PubMed] [Google Scholar]
- 6.Snyder RO, Miao CH, Patijn GA, Spratt SK, Danos O, Nagy D, et al. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat Genet. 1997;16(3):270–276. doi: 10.1038/ng0797-270. [DOI] [PubMed] [Google Scholar]
- 7.Flotte TR, Afione SA, Conrad C, McGrath SA, Solow R, Oka H, et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(22):10613–7. doi: 10.1073/pnas.90.22.10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez YJ, Wang J, Kearns WG, Loiler S, Poirier A, Flotte TR. Latent Adeno-Associated Virus Infection Elicits Humoral but Not Cell-Mediated Immune Responses in a Nonhuman Primate Model. J. Virol. 1999;73(10):8549–8558. doi: 10.1128/jvi.73.10.8549-8558.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, et al. Robust Systemic Transduction with AAV9 Vectors in Mice: Efficient Global Cardiac Gene Transfer Superior to That of AAV8. Mol Ther. 2006;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bostick B, Ghosh A, Yue Y, Long C, Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Therapy. 2007;14(22):1605–9. doi: 10.1038/sj.gt.3303029. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds PN, Nicklin SA, Kaliberova L, Boatman BG, Grizzle WE, Balyasnikova IV, et al. Combined transductional and transcriptional targeting improves the specificity of transgene expression in vivo. Nat Biotech. 2001;19(9):838–842. doi: 10.1038/nbt0901-838. [DOI] [PubMed] [Google Scholar]
- 12.Hauser MA, Robinson A, Hartigan-O'Connor D, Williams-Gregory DA, Buskin JN, Apone S, et al. Analysis of muscle creatine kinase regulatory elements in recombinant adenoviral vectors. Molecular Therapy: the Journal of the American Society of Gene Therapy. 2000;2(1):16–25. doi: 10.1006/mthe.2000.0089. [DOI] [PubMed] [Google Scholar]
- 13.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10(8):828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salva MZ, Himeda CL, Tai PWL, Nishiuchi E, Gregorevic P, Allen JM, et al. Design of Tissue-specific Regulatory Cassettes for High-level rAAV-mediated Expression in Skeletal and Cardiac Muscle. Mol Ther. 2007;15(2):320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 15.Wang B, Li J, Fu FH, Chen C, Zhu X, Zhou L, et al. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Therapy. 2008;15(22):1489–99. doi: 10.1038/gt.2008.104. [DOI] [PubMed] [Google Scholar]
- 16.Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J Biol Chem. 2011;286(15):13532–40. doi: 10.1074/jbc.M110.210922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dokun AO, Keum S, Hazarika S, Li Y, Lamonte GM, Wheeler F, et al. A Quantitative Trait Locus (LSq-1) on Mouse Chromosome 7 Is Linked to the Absence of Tissue Loss After Surgical Hindlimb Ischemia. Circulation. 2008;117(9):1207–1215. doi: 10.1161/CIRCULATIONAHA.107.736447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, Annex BH. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. 2007;101(9):948–56. doi: 10.1161/CIRCRESAHA.107.160630. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Hazarika S, Xie D, Pippen AM, Kontos CD, Annex BH. In mice with type 2 diabetes, a vascular endothelial growth factor (VEGF)-activating transcription factor modulates VEGF signaling and induces therapeutic angiogenesis after hindlimb ischemia. Diabetes. 2007;56(3):656–65. doi: 10.2337/db06-0999. [DOI] [PubMed] [Google Scholar]
- 20.Creager MA, Olin JW, Belch JJF, Moneta GL, Henry TD, Rajagopalan S, et al. Effect of Hypoxia-Inducible Factor-1 Gene Therapy on Walking Performance in Patients With Intermittent Claudication. Circulation. 2011;124(16):1765–1773. doi: 10.1161/CIRCULATIONAHA.110.009407. [DOI] [PubMed] [Google Scholar]
- 21.Rajagopalan S, Mohler ER, 3rd, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, et al. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation. 2003;108(16):1933–8. doi: 10.1161/01.CIR.0000093398.16124.29. [DOI] [PubMed] [Google Scholar]
- 22.Baumgartner I, Chronos N, Comerota A, Henry T, Pasquet J-P, Finiels F, et al. Local Gene Transfer and Expression Following Intramuscular Administration of FGF-1 Plasmid DNA in Patients With Critical Limb Ischemia. Mol Ther. 2009;17(5):914–921. doi: 10.1038/mt.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byun J, Heard JM, Huh JE, Park SJ, Jung EA, Jeong JO, et al. Efficient expression of the vascular endothelial growth factor gene in vitro and in vivo, using an adeno-associated virus vector. J Mol Cell Cardiol. 2001;33(2):295–305. doi: 10.1006/jmcc.2000.1301. [DOI] [PubMed] [Google Scholar]
- 24.Pinkenburg O, Pfosser A, Hinkel R, Bottcher M, Dinges C, Lebherz C, et al. Recombinant adeno-associated virus-based gene transfer of cathelicidin induces therapeutic neovascularization preferentially via potent collateral growth. Hum Gene Ther. 2009;20(2):159–67. doi: 10.1089/hum.2007.178. [DOI] [PubMed] [Google Scholar]
- 25.Makinen K, Manninen H, Hedman M, Matsi P, Mussalo H, Alhava E, et al. Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: a randomized, placebo-controlled, double-blinded phase II study. Mol Ther. 2002;6(1):127–33. doi: 10.1006/mthe.2002.0638. [DOI] [PubMed] [Google Scholar]
- 26.Dong J-Y, Fan P-D, Frizzell RA. Quantitative Analysis of the Packaging Capacity of Recombinant Adeno-Associated Virus. Human Gene Therapy. 1996;7(17):2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- 27.Grieger JC, Samulski RJ. Packaging Capacity of Adeno-Associated Virus Serotypes: Impact of Larger Genomes on Infectivity and Postentry Steps. J. Virol. 2005;79(15):9933–9944. doi: 10.1128/JVI.79.15.9933-9944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodino-Klapac LR, Janssen PML, Montgomery CL, Coley BD, Chicoine LG, Clark KR, et al. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J Trans Med. 2007;5:45. doi: 10.1186/1479-5876-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arruda VR, Fields PA, Milner R, Wainwright L, De Miguel MP, Donovan PJ, et al. Lack of germline transmission of vector sequences following systemic administration of recombinant AAV-2 vector in males. Mol Ther. 2001;4(6):586–92. doi: 10.1006/mthe.2001.0491. [DOI] [PubMed] [Google Scholar]
- 30.Herzog RW, Fields PA, Arruda VR, Brubaker JO, Armstrong E, McClintock D, et al. Influence of vector dose on factor IX-specific T and B cell responses in muscle-directed gene therapy. Hum Gene Ther. 2002;13(11):1281–91. doi: 10.1089/104303402760128513. [DOI] [PubMed] [Google Scholar]
- 31.Ziv K, Nevo N, Dafni H, Israely T, Granot D, Brenner O, et al. Longitudinal MRI tracking of the angiogenic response to hind limb ischemic injury in the mouse. Magnetic Resonance in Medicine. 2004;51(2):304–311. doi: 10.1002/mrm.10687. [DOI] [PubMed] [Google Scholar]
- 32.Moulay G, Scherman D, Kichler A. Fasting Increases the In Vivo Gene Delivery of AAV Vectors. Clinical and Translational Science. 2010;3(6):333–336. doi: 10.1111/j.1752-8062.2010.00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prasad K-MR, Xu Y, Yang Z, Acton S, French BA. Robust Cardiomyocyte-Specific Gene Expression Following Systemic Injection of AAV: In Vivo Gene Delivery Follows a Poisson Distribution. Gene Therapy. 2010 doi: 10.1038/gt.2010.105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ried MU, Girod A, Leike K, Buning H, Hallek M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. Journal of Virology. 2002;76(9):4559–66. doi: 10.1128/JVI.76.9.4559-4566.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prasad K-MR, Xu Y, Yang Z, Toufektsian M-C, Berr SS, French BA. Topoisomerase inhibition accelerates gene expression after adeno-associated virus-mediated gene transfer to the mammalian heart. Molecular Therapy: the Journal of the American Society of Gene Therapy. 2007;15(4):764–71. doi: 10.1038/sj.mt.6300071. [DOI] [PubMed] [Google Scholar]
- 36.Wu JC, Inubushi M, Sundaresan G, Schelbert HR, Gambhir SS. Optical imaging of cardiac reporter gene expression in living rats. Circulation. 2002;105(14):1631–4. doi: 10.1161/01.cir.0000014984.95520.ad. [DOI] [PubMed] [Google Scholar]
- 37.Böhmer MR, Chlon CHT, Raju BI, Chin CT, Shevchenko T, Klibanov AL. Focused ultrasound and microbubbles for enhanced extravasation. Journal of Controlled Release. 148(1):18–24. doi: 10.1016/j.jconrel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]