

Abstract
The 99 residue transmembrane C-terminal domain (C99, also known as β-CTF) of the amyloid precursor protein (APP) is the product of β-secretase cleavage of full length APP and the substrate for γ-secretase cleavage. The latter cleavage releases the amyloid-β polypeptides that are closely associated with Alzheimer’s disease. C99 is thought to form homodimers; however, the free energy in favor of dimerization has not previously been quantitated. It was also recently documented that cholesterol forms a 1:1 complex with monomeric C99 in bicelles. Here, the affinities for both homodimerization and cholesterol binding to C99 were measured in bilayered lipid vesicles using both electron paramagnetic resonance (EPR) and Förster resonance energy transfer (FRET) methods. Homodimerization and cholesterol binding were seen to be competitive processes, which center on the transmembrane G700XXXG704XXXG709 glycine zipper motif and the adjacent Gly709. The observed Kd for cholesterol binding (Kd = 2.7 ± 0.3 mol%) is on the low end of the physiological cholesterol concentration range in mammalian cell membranes. On the other hand, the observed Kd for homodimerization (Kd = 0.47 ± 0.15 mol%) likely exceeds the physiological concentration range for C99. These results suggest that the 1:1 cholesterol:C99 complex will be more highly populated than C99 homodimers under most physiological conditions, observations that are of relevance to understanding γ-secretase cleavage of C99.
It is generally believed that the etiology of Alzheimer’s disease (AD) is closely related to the production of the amyloid-β (Aβ) polypeptides, with factors that result in higher overall Aβ concentrations or that favor an increased production of the long forms of Aβ relative to the short forms being regarded as pro-AD factors.(1–4) Accordingly, there is great interest in elucidating the molecular mechanisms of amyloidogenesis. This study focuses on the biochemical and biophysical properties of a key protein intermediate in the amyloidogenic pathway, C99 (Figure 1). C99 is the transmembrane 99 residue C-terminal domain of the amyloid precursor protein (APP) that is generated upon cleavage of APP by β-secretase. C99 is also the immediate precursor of the Aβ polypeptides, which are released when C99 is cleaved by γ-secretase. The mechanisms responsible for regulation of substrate recognition and subsequent cleavage of C99 by γ-secretase are not well understood. However, there is considerable evidence that elevated cholesterol levels enhance the amyloidogenic pathway of APP processing and inhibit a competing non-amyloidogenic cleavage pathway(5–14), observations that may be related to the recent discovery that C99 (and possibly full length APP) is a cholesterol binding protein.(15)
Figure 1.

Topology map of C99 protein. Not depicted is the hinge-like kink located in the transmembrane helix that occurs at G708G709(15).
Much attention has also been paid to the possible homodimerization of C99 and its impact on cleavage by γ-secretase. The transmembrane domain (TMD) of C99 contains both a glycine zipper sequence (G700XXXG704XXXG708) and an adjacent G709XXXA713 motif. These common motifs sometime contribute to homo- or hetero-dimerization of membrane proteins by promoting intimate and adhesive helix-to-helix contacts.(16–19) There is now considerable evidence that C99 can form homodimers(20–35), with most data pointing to the surface provided by the Gly sites in the G700XXXG704XXXG708 zipper as the central interface.(20, 26, 28, 29, 31, 35) This notion has prompted studies in which the effect of Gly mutations on γ-secretase cleavage in model mammalian cell lines have been tested.(26–29) The results of these studies were originally interpreted mainly in terms of the potential impact of alterations in homodimerization on γ-secretase cleavage, but may not need to be revisited in light of data that suggests these same sites are involved in cholesterol binding.(15)
The actual avidity of homodimerization (as reflected by Kd) has not been rigorously quantitated. Moreover, the fact that both dimerization and cholesterol binding seem to involve the same glycine zipper motif in the transmembrane domain raises questions about the possible relationship of cholesterol binding to homodimerization and γ-secretase cleavage. Most importantly, are these competitive processes? Given that the cholesterol concentration present in the plasma membranes of mammalian cells is well above the 5 mol% Kd measured for 1:1 complex formation with C99 in bicelles,(15, 36) an understanding of the relationship between homodimerization and cholesterol binding is needed to illuminate the final biochemical step of the amyloidogenic pathway in living cells.
Here we quantitate homodimerization of C99 in bilayered lipid vesicles and show that self-association is of only modest avidity. We also measure the Kd for cholesterol binding under the same conditions and demonstrate that cholesterol binding to the C99 monomer and protein homodimerization are competitive processes, both involving the G700XXXG704XXXG708 zipper motif.
Methods
Chemicals
Sodium dodecylsulfate (SDS), lyso-myristoylphosphatidylglycerol (LMPG), 1-palmitoyl-2-oleoylphosphatidylcholine (POPC, and 1-palmitoyl-2-oleoyl-phosophatidylglycerol (POPG) were purchased from Anatrace-Affymetrix (Maumee, OH) and Avanti (Alabaster, AL). Thiol-activated spin label, 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate (MTSL), was purchased from Toronto Research Chemicals (Toronto, ON). Fluorescent labels N, N-dimethyl-N-(iodoacetyl)-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) ethylenediamine (IANBD) and 5-(((2-iodoacetyl)amino)ethyl)amino)-naphthalene-1-sulfonic acid (IAEDANS) were purchased from Invitrogen (www.lifetechnologies.com). All other chemicals were from Sigma (St. Louis, MO).
Expression, Purification, and Spin labeling of Wild Type and Mutant Forms of C99
As described previously(15, 20) human C99 was expressed in E. coli and then purified using metal ion affinity chromatography into micelles composed of either 0.05% LMPG or 0.2% SDS micelles.
Wild type C99 has no Cys residues. Selected residues were mutated to cysteine using the Quik-Change method utilizing whole-plasmid PCR (Stratagene, CA, USA). A total of eleven single-Cys C99 mutants were prepared: S697C, G700C, A701C, I702C, I703C, G704C, L705C, G708C, G709C, A713C and S730C.
Single-Cys C99 mutants were spin labeled as previously described(15), resulting in final solutions containing pure spin labeled C99 in a buffer containing 0.2% SDS and 250 mM imidazole, pH 7.8. The spin labeling efficiency for each C99 sample was quantitated by double integrating EPR resonances from each labeled sample relative to the signal from a 100 μM TEMPOL standard. The estimated spin label concentration was then compared to the protein concentration determined by UV absorbance at 280 nm using an extinction coefficient of 5960 M−1cm−1. In all cases, the spin labeling efficiency was observed to be >90%.
Fluorescent Labeling of Single-Cys C99 Mutants
The S730C C99 mutant was purified into 0.05% LMPG and treated with DTT to ensure complete reduction of the cysteines, as described previously.(15) This mutant was previously employed in FRET studies of C99 dimerization in detergent micelles using an approach similar to that used here.(35) For FRET experiments, separate batches of S703C C99 were labeled with thiol-reactive probes IANBD (as an acceptor) and IAEDANS (as a donor). The protein solution was first desalted using a PD-10 column (GE Healthcare, NJ) and equilibrated in 20 mM Tris-HCl, 150 mM NaCl, pH 7.8 containing 0.05% LMPG. Labeling was then initiated by the immediate addition of 10X molar excess of IANBD (from a 2 mM stock in acetonitrile) to the protein solution. The resulting mixture was incubated at room temperature in the dark for 1 hour and then loaded onto a PD-10 size-exclusion column equilibrated and eluted with 20 mM Tris-HCl, 150 mM NaCl, pH 7.8 containing 0.2% SDS. The labeling of S730C with IAEDANS was performed in an analogous manner. The degree of labeling was calculated based on the concentration of protein (determined from A280) and the concentration of IANBD (determined from A472 based on ε472 = 23700 M−1cm−1) or IAEDANS (determined from A337 based on ε337 = 5600 M−1cm−1).
Reconstitution of C99 into Lipid Vesicles
Reconstitution of C99 into pH 6.5 lipid vesicles was initiated using spin- or fluorescently-labeled C99 prepared as described above in a Tris-HCl buffer containing 0.2% SDS. Labeled C99 in 0.2% SDS was concentrated using an Amicon Ultra-15 centrifugal filter (molecular weight cutoff = 10 kDa, Millipore, MA) to a final concentration of 1 mM. The concentrated C99 solution was then mixed with a stock SDS/lipid mixture (400 mM SDS, 75 mM POPC, 25 mM POPG) with or without cholesterol, resulting in a clear solution, which was then subjected to >15 cycles of freeze/thaw to ensure complete conversion to mixed micelles. The C99/mixed micellar mixture was then subjected to extensive dialysis in Spectra/Pro dialysis tubing (molecular weight cut-off: 15 kDa; Spectrum Laboratories Inc., CA) at room temperature to remove all SDS, a process that is accompanied by formation of C99:POPC:POPG vesicles (with or without cholesterol). To ensure complete removal of SDS, the 4 L of dialysis buffer (100 mM imidazole and 1 mM EDTA at pH 6.5) was changed twice daily for 5 days. SDS removal was deemed completed when the C99/lipid solution became cloudy (due to vesicle formation) and the surface tension of the dialysate indicated complete removal of SDS.
Continuous Wave (CW) EPR Measurements
To assess dimerization of C99, the C99:lipid mole ratio in 3:1 POPC:POPG lipid vesicles was varied from 1:800 to 1:50. To investigate the effects of cholesterol on dimerization, one experiment was performed in which the C99:lipid mole ratio was varied from 1:800 to 1:50 in vesicles that contained not only the usual 3:1 POPC:POPG, but also 20 mol% cholesterol, where mol% cholesterol =100 X [moles cholesterol/(moles POPC + moles POPG + moles cholesterol)]. An additional experiment involved fixing the ratio of C99:lipid at 1:100 while cholesterol was varied from 0 to 30 mol%. X-band CW-EPR spectra were collected at 9.81 GHz on a Bruker EMX spectrometer with a TM110 cavity (BrukerBiospin, Billerica, MA) and using a 5 mW microwave power and 1 G field modulation at 100 kHz. Spin labeled C99 samples were prepared as described above and 20 μL of each was transferred to 50 μL glass capillaries. All EPR spectra were collected at 298 K.
Analysis of EPR Data
Most of the CW EPR spectra of the spin labeled C99 mutants exhibited two components characterized by different motional properties. If it is assumed that the narrow component arises from a population of monomer in equilibrium with a population of dimer that yields the broad component, then the dependency of the position of the momoner-dimer population as a function of the protein-to-lipid ratio can be monitored by measuring the ratio-dependent intensity of any point in the superimposed pair (monomer+dimer) of spectra that is sensitive to the monomer/dimer equilibrium. This approach is similar to that used in previous EPR studies to quantitate binding equilibria.28,29 We chose to monitor both the intensity of the signal at the field at which monomer has its highest intensity (3474 Gauss) and the intensity at the field where the dimer component is most evident (3464 Gauss). The dependency of the these intensities as a function of the mole fraction of protein relative to total lipid+protein was fit by a model for homodimerization (below).
FRET Measurements
To assess homodimerization of C99, samples were prepared in which donor IAEDANS-labeled S730C C99 was reconstituted into lipid vesicles at a fixed donor-labeled S730C C99:lipid ratio of 1:800 (mol:mol) with varying amounts of acceptor IANBD-labeled S730C C99 at donor-to-acceptor mole ratios of 1:0, 1:1, 1:2, 1:4, 1:6, 1:8, and 1:16, This results in total C99:lipid ratios of 1: 800, 1:400, 1:267, 1:160, 1:114, 1:89, and 1:47, respectively. To investigate the effects of cholesterol on dimerization, a mixture of donor- and acceptor-labeled C99 S730C (1:4 donor to acceptor) was reconstituted into vesicles with a constant C99:lipid mole ratio of 1:100 in the presence of varying levels of cholesterol (0 mol%, 5 mol%, 10 mol%, 15 mol%, 20 mol%, and 30 mol%). Steady-state fluorescence emission spectra were collected at 298 K using a FluoroMax-3 spectrometer from 400 nm to 600 nm, with the excitation wavelength at 339 nm. For both sets of experiments the fraction of dimer in each sample was calculated from the decrease in the emission intensity of the donor (at 474 nm) due to the energy transfer to the acceptor (see below for data fitting). The data were fit by the appropriate competitive binding models (below) using Origin 8.0.
Fitting of EPR and FRET Titration Data
The fraction of dimer (fD) in a C99 monomer-dimer equilibrium can be expressed as a function of the dissociation constant Kd and the total concentration of C99 as:
| (1) |
where [D] is the concentration of C99 dimer, [M] is the concentration of C99 monomer, and [T] is the total C99 concentration of C99 (equal to [M] + 2[D]), all in mol% concentration units. The dissociation constant Kd is given by:
| (2) |
where [M] is the concentration of C99 monomer and [D] = ([T] − [M])/ 2. Because the assumption cannot be made that [M] = [C99]total, [M] in equation (1) must be its analytic value:
| (3) |
Substituting this equation for [M] in equation (1) leads to:
| (4) |
fD can also be expressed with experimental FRET data is:
| (5) |
where FDA and FD are donor intensities at 474 nm in the presence and absence of acceptor-labeled C99, respectively, and FS is the donor intensity at 474 nm at saturation. However, FS is not directly observed experimentally. The combination of equation (4) and (5) gives:
| (6) |
Rearrangement of (6) leads to:
| (7) |
A similar equation for EPR intensity as a function of the concentration of C99 is:
| (8) |
where IX is the observed EPR intensity at X Gauss (where X is any field at which the spectrum varies in intensity as a function of fD), IM is the intrinsic EPR intensity of monomer at X Gauss, IS is the intrinsic EPR intensity at X Gauss at saturation, and IX is the observed EPR intensity at X Gauss. Kd values were obtained by fitting equations (4), (7), or (8) to the appropriate EPR- or FRET-monitored titration data with Origin 8.0.
When cholesterol was titrated into a sample containing 1 mol% C99, cholesterol can bind to the C99 monomer, leading to two competing equilibria:
| (9) |
which is described by Kd,dimer, and:
| (10) |
as described by Kd,chol. The combination of (9) and (10) gives:
| (11) |
where D is the C99 dimer, L is the cholesterol, and ML is the C99-cholesterol complex.
The superequilibrium constant K of reaction (11) can be written as
| (12) |
where [ML] is the concentration of the C99-cholesterol complex, Kd,dimer is the dissociation constant for C99 dimerization, and Kd,chol is the dissociation constant for the C99-cholesterol complex. In this case, the total cholesterol ([Lt] = [L] + [ML]) was a variable and the total C99, [T], equals 2[D] + [ML], which was experimentally held constant at 1 mol%. fD is a function of the superequilibrium constant K from equation 12 and the total concentration of cholesterol [Lt] as follows:
| (13) |
For EPR data with cholesterol present:
| (14) |
where IX is the observed EPR intensity at X Gauss (where X is any field at which the spectrum varies in intensity as a function of fD), I0 is the EPR intensity at X Gauss in the absence of cholesterol, and Is is the EPR intensity at X Gauss at saturation, IX is the observed EPR intensity at X Gauss, I0 is the EPR intensity at X Gauss in the absence of cholesterol, and Is is the EPR intensity at X Gauss at saturation. The combination of (13) and (14) gives:
| (15) |
For FRET data in the presence of cholesterol:
| (16) |
where Fobs is the observed intensity at any wavelength that varies due to a cholesterol-based alteration in protein donor/protein acceptor dimerization, F0 is the intensity at that wavelength without cholesterol, and Fs is the intensity at saturation. The combination of (13) and (16) gives:
| (17) |
K was obtained by fitting equation 15 (EPR data) or 17 (FRET data) to the data using Origin 8.0. Kd,chol was then calculated from equation (12) in conjunction with the value of Kd,dimer determined independently from EPR and FRET C99 titrations in the absence of cholesterol.
Results
All studies of this work were conducted using purified C99 that was reconstituted into bilayered liquid crystalline phase lipid vesicles containing 3:1 (mol:mol) POPC and POPG, the latter of which confers a net anionic charge (as present in biological membranes). This lipid composition is similar to lipid mixture commonly used in studies of mammalian membrane proteins and matches the conditions used in our previous EPR studies of the topology and structure of C99.(15) This work employed a series of single-cysteine mutant forms of C99, which has no wild type cysteine residues. It was previously shown that the Cys mutations are non-disruptive of the membrane topology and structure of C99.(15)
Use of EPR and FRET Spectroscopy to Quantitate C99 Monomer-Dimer Equilibrium
Single cysteine mutant forms of C99 were spin labeled, reconstituted into bilayered vesicles and subjected to continuous wave (CW) EPR. Spectra were acquired as a function of the C99-to-lipid mole ratio. Shown in Figure 2 are the spectra for C99 that is spin labeled at a site either in the solvent-exposed loop immediately N-terminal to the TMD (S697C, Figure 2A) or within the transmembrane domain (L705C, Figure 2B). At the lowest C99 concentration (1:800 C99:lipid) the lineshapes for both mutants appear to be dominated by a single narrow component, consistent with intermediate motion. As the protein:lipid ratio increases, the lineshapes become more complex as the contribution from a second, broader component grows. This broader component is clearly evident at the low and high-field wings of the spectrum for samples with higher protein:lipid ratios. Based on the preliminary assumption (confirmed below) that these EPR spectra represent the superimposed resonances from monomer (narrow component) and dimer (broad component) at equilibrium we selected two fields in the spectrum that were dominated by the putative monomer and dimer resonances and plotted their changes in intensities as a function of the mole% concentration of spin labeled C99 in the vesicles (Figures 2C and 2D). Each data set was then fit to a model (see Methods) describing the concentration dependence of the position of the monomer-dimer equilibrium, as shown in Figures 2C and 2D. A similar approach for using EPR data to quantitate binding titration data has previously been reported.(37, 38) In all cases, the data was well fit by this model, regardless of which region of the spectrum was used to extract intensities. Moreover, approximately the same Kd for each spin labeling site was determined regardless of whether the monitored spectral region is dominated by the loss of monomer occurring at higher protein concentrations or by the appearance of dimer. Finally, the Kd were similar when measurements were made using data from spin label either at the water-exposed S697C site or at the membrane-buried L705C site. The average Kd from these four measurements was 0.44 ± 0.15 mol%. The high degree of self-consistency of the data and the fits are consistent with a monomer-dimer equilibrium. The simplest explanation for the concentration-dependence of the EPR spectra is that C99 populates a mostly monomeric state at 1:800 protein-to-lipid and a mostly dimeric state at 1:50. There was no evidence for higher order oligomerization or aggregation, as may have been present in samples from a previous study(39) that focused on a peptide corresponding to the isolated transmembrane domain of C99 at a significantly higher level of protein (1:30 C99:lipid) than the highest level examined in this work. The breadth of the EPR signal arising from this putative dimer can be explained as arising from partial immobilization of the Cys-attached spin labels due to quaternary structure formation and/or by dipolar interaction between now-proximal pairs of spin labels. The validity of the approach taken to determine Kd from the titration spectra does not depend in the mechanism of line broadening.
Figure 2.

(A) and (B): CW EPR spectra of spin labeled C99 in lipid vesicles with different protein-to-lipid molar ratios (from 1:800 to 1:50). S697R1 (panel A) is the spin labeled S697C mutant, while L705R1 (panel B) is the spin labeled L705C mutant. Site 697 is located outside of the TMD, while 705 is located inside. The arrows in the top trace of each series of spectra indicates the peaks at 3464 (left) and 3474 (right) Gauss. (C) and (D): the EPR intensities at 3474 G and 3464 G from panels A and B, respectively, were plotted as a function of the total C99 concentration and fit by a homodimerization model (equation 8).
Use of FRET to Quantitate Homodimerization
That the structural transition detected by EPR is a reversible monomer-to-dimer event was verified by fluorescence measurements in which FRET was measured as a constant amount of donor-labeled C99 mutant in vesicles was titrated by the same mutant labeled with a FRET acceptor. In this case, the reduction of the signal from the constant donor-labeled C99 represents is due to FRET to the increasing amount of acceptor-labeled protein. A plot of the data for loss of donor fluorescence intensity as a function of the total C99 concentration can be fit very well by a by a model for homodimerization, with a derived Kd of 0.51 ± 0.13 mol% that is similar to that determined from the two EPR data sets (Figure 3). The concurrence of the C99:lipid ratio-dependence of the EPR AND FRET data sets indicates that C99 homodimerizes with a Kd of 0.47 ± 0.15 mol% (the average of all measured Kd,dimer values). We note that a similar approach was previously used to document homodimerization of C99 in detergent micelles(35) although the derived Kd was determined in bulk molarity units, which is not appropriate for thermodynamic analysis of a molecular association event that occurs between two molecules that are associated with a model membrane phase.(40)
Figure 3.

FRET spectra of IAEDANS-labeled C99 S730C (donor) and IANDB-labeled C99 S730 (acceptor) in lipid vesicles. (A) IAEDANS-labeled C99 (donor) was reconstituted into lipid vesicles with a protein-to-lipid molar ratio of 1:800 (black) with increasing amounts of IANDB-labeled C99 (acceptor). The acceptor IANDB-labeled C99 (acceptor) was increased relative to donor IAEDANS-labeled C99 over the following protein:protein molar ratios: 1:1 (red), 1:2 (blue), 1:4 (cyan), 1:6 (magenta), 1:8 (orange), and 1:16 (green), corresponding to total C99-to-lipid molar ratios of 1:400, 1:267, 1:160, 1:114, 1:89, and 1:47, respectively. (B) The intensity at 474 nm in each spectrum was plotted as a function of total C99 concentration and was fit by a model for homodimerization (equation 7).
Identification of Homodimerization Interface Sites in the TMD
Most previous studies of C99 homodimerization are in agreement that the homodimerization interface of C99 is associated with the G700XXXG704XXXG708 and/or G709XXXA713 motifs.(20, 24, 26, 28–31, 35) We therefore mutated sites 700–704, 705, 708, 709, and 713 to cysteine and attached spin labels. Replacement of each of the four Gly sites (700, 704, 708, 709) with spin labeled cysteine led to EPR spectra sets that are no longer dependent on the protein-to-lipid ratio (Figure 4A), indicating that these glycines are essential for homodimerization. For two of those sites (700 and 704) we also made Gly-to-Leu mutations in conjunction with spin labeling at site 697 (the EPR spectrum of which is shown in Figure 1 to be sensitive to dimerization). Again, the EPR spectra were invariant with changes in the protein-to-lipid ratio (Figure 4). These results indicate that all three glycines in the G700XXXG704XXXG708 zipper and also Gly709 are important for homodimerization. In contrast, replacement of Ala701, Ile702, Ile703, Leu705, and Ala713 with spin labeled Cys showed saturable curves with increasing C99 concentration (Figures 4C and 4D, Supporting Figure 1). The fitting of these curves by the model for homodimerization results in Kd,dimer values of 0.30 ± 0.18 mol% (for 701), 0.34 ± 0.16 mol% (for 702), 0.51 ± 0.15 mol% (for 703) and 0.48 ± 0.20 mol% mol% (for 713). These Kd,dimer values are in the same range with each other and with wild type (above), indicating that these sites are not essential for homodimerization (Figures 4C and 4D; data for L705 is shown in Figure 2). It is particularly interesting that mutation of Ala713 does not result in loss of dimerization as this is the alanine in the G709XXXA713 motif. This indicates that the role of Gly709 in promoting dimerization is unrelated to Ala713.
Figure 4.

CW EPR spectra of spin labeled C99 in lipid vesicles with different protein-to-lipid mol:mol ratios from 1:800 to 1:50. (A) Superimposed spectra (at 1:800, 1:400, 1:200, 1:100, and 1:50 C99:lipid) for G700R1, G704R1, G708R1 and G709R1. The overlays of 5 spectra for each mutant exhibit little variation within each series. (B) Superimposed spectra (same 5 ratios as above) of the G700L and G704L mutants containing a second mutation site (to Cys) for spin labeling (S697R1). As for panel A, the superimposed spectra demonstrate little variation within each series. (C) and (D): The EPR intensity at 3464 G was plotted as a function of the total C99 concentration and fit by a model for homodimerization.
Cholesterol Binding to Monomeric C99 Competes With Homodimerization of the Protein
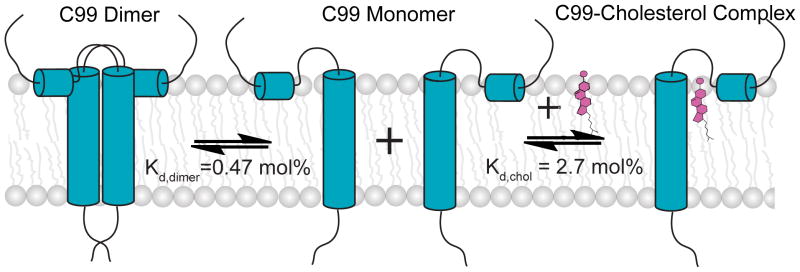
C99 was spin labeled at the L705C site in its transmembrane domain, a site that reports well on oligomeric state (Figure 2) but that is not at the dimer interface. A sample was prepared with a 1:100 C99:lipid ratio, where over 50% of the protein is homodimerized based on the above-determined Kd for homodimerization. This is evident via the broad component seen in the top trace spectrum of Figure 5A. A series of samples were then prepared in which spin labeled C99 was maintained at a constant mol% concentration (relative to lipid), but in which cholesterol was introduced into the membranes at different concentrations ranging from 5–30 mol%. This range is physiologically relevant.(36, 41–43) It can be seen that the very broad dimer component of the CW-EPR spectrum disappears in the course of this titration, suggestive of a cholesterol binding-induced shift in the monomer-dimer equilibrium towards monomer. A plot of the EPR intensity at 3474 G as a function of the cholesterol concentration (Figure 5B) exhibits a saturable curve. A model (equation 15) describing competition between homodimerization and cholesterol binding to monomeric C99 fit very well to the data, leading to an average superequilibrium constant K of 0.067 ± 0.010 mol%−1 (Figure 5B). Using the Kd,dimer of 0.47 mol% determined independently above, the Kd,chol was then calculated using equation 12 to be 2.7 ± 0.8 mol%.
Figure 5.

CW EPR spectra of C99 in the presence of cholesterol. (A) CW EPR spectra of L705R1 C99 at a protein-to-lipid molar ratio of 1:100 in vesicles containing cholesterol ranging from 0 to 30 mol%. All spectra were normalized (with respect to their double integrations) to the same amount of spin label. (B) The EPR intensities at 3474 G (right arrow) and 3464 G (left arrow) from panel A were plotted as a function of total C99 concentration and then fit by a model describing competition between homodimerization of C99 and 1:1 complex formation between C99 and cholesterol (equation 15). Note that the determined “K” describes the superequilibrium (equation 12) and is not a true dissociation or association constant. (C). CW EPR spectra of S697R1 C99 in vesicles containing 20 mol% cholesterol at protein-to-lipid mol:mol ratios varying from 1:800 to 1:50. (D) Same as C except that the spin label is attached to L705C, which is located inside the membrane.
We next took the same two spin labeled mutants used in the C99 titrations of Figure 2 and repeated the EPR-monitored protein-to-lipid ratio variation experiment, but now in membranes that included 20 mol% cholesterol, a concentration that is both 4-fold higher than its Kd for binding to C99 in bicelles(15) and that is physiologically reasonable.(36, 41–43) It can be seen that cholesterol at this level was able to suppress homodimerization of C99 at concentrations of the protein as high as 4X its Kd for homodimerization (Figures 5C and 5D).
That competition between cholesterol binding and homodimerization takes place was confirmed by FRET measurements. Shown in Figure 6 are the FRET results for a 1:100 C99:lipid (1 mol% C99) sample that contains a fixed ratio of FRET donor- and acceptor-labeled forms of C99. The FRET acceptor signal at ca. 525 nm has maximum intensity for a sample containing no cholesterol and then gradually decreases as the cholesterol mole fraction levels in the sample are increased, indicative of disruption of donor-acceptor proximity by cholesterol. In Figure 5B the intensity of the growing FRET donor signal at ca. 470 nm is plotted as a function of the cholesterol concentration. It can be seen that the intensity of the donor signal (representing the monomer) increases with increasing cholesterol. Once again, a model (equation 17) describing competition between homodimerization and cholesterol binding fits the data well, yielding a superequilibrium constant K of 0.065 ± 0.006 mol%−1. This leads to a Kd of 2.6 ± 0.9 mol%, for cholesterol binding to the monomer (equation 12), a value in excellent agreement with the value determined from the EPR data of Figure 5. Averaging the EPR result with the fluorescence result leads to the conclusion that cholesterol binds to monomeric C99 in POPC:POPG vesicles with Kd of 2.7 ± 0.3 mol%, in a manner that is competitive with homodimerization.
Figure 6.

(A): FRET spectra of fluorescently labeled C99 S730C in lipid vesicles with a donor-to-acceptor ratio of 1:4 and a total C99-to-lipid mol:mol ratio of 1:100, containing different amounts of cholesterol: 0 mol% (black), 5 (red), 10 (orange), 15 (blue), 20 (purple), and 30 mol% (cyan). (B): The intensity of the spectrum at 474 nm was plotted as a function of the cholesterol mol% and then fit by a model (equation 17) describing competition between C99 homodimerization and 1:1 complex formation between monomeric C99 and cholesterol. (Note that 1 mol% cholesterol corresponds to 1 molecule of cholesterol for every 100 molecules of lipid and protein in the vesicles).
Discussion
Energetics of Homodimerization of C99 in Lipid Vesicles
The full length amyloid precursor protein is thought to dimerize in a manner that is driven primarily by interactions involving its large extracellular domain.(44–48) The C99 domain (APP672–770) of APP does not appear to significantly contribute to dimerization of the full length protein(28). However, it is clear from a variety of biochemical and biophysical studies of the C99 protein and its fragments that this protein does have a propensity to dimerize, although the avidity of homodimerization has not been thermodynamically quantified.(20, 21, 23–26, 28–30, 32, 33, 35) Of particular importance is the NMR-based determination of the structure (in dodecylphosphocholine micelles) of homodimeric APP686–724,(30) a TMD-domain containing fragment of C99. This structure has interesting similarities to the NMR-based structure of the C99 monomer in lyso-phospholipid micelles.(15) In particular, both structures highlight a hinge role for the G708G709 segment in facilitating flexible bending of the C99 TMD, as previously proposed based on simulations.(49) The APP686–724 dimer structure is surprising in that the transmembrane segments interact as left-handed coiled-coil helices, with the G709XXXA713 motif being central to the dimer interface. While the possible importance of this motif in driving homodimerization had previously been proposed,(24) most earlier studies had concluded that it is the glycine zipper (G700XXXG704XXXG708), particularly interactions involving the G700 and G704 sites, that drives dimerization of C99.(20, 26, 28, 29, 31, 35) There is a clear imperative to quantitate the equilibrium between the monomer and dimer forms of C99 and also to verify which residues are critical to the dimer interface. Moreover, because biophysical and biochemical measurements made in micelles or other non-bilayered model membrane conditions are invariably subject to questions regarding possible distortion of results relative to more native-like bilayer conditions it is important that these issues be addressed in lipid bilayers.
Here we used EPR and FRET methods to interrogate the homodimerization of C99 in POPC:POPG (3:1 mol:mol) lipid vesicles at pH 6.5 and temperatures where the bilayers are in the liquid crystalline phase (Lα), which closely matches the properties of bulk phase biological membranes. It was observed using both EPR and FRET that homodimerization occurs with a Kd of 0.47 ± 0.15 mol%. This means that in POPC:POPG vesicles at C99 concentrations lower than 0.47 mol% (ca. 1:200 C99:lipid) the protein will be primarily monomeric, while the homodimer will dominate at C99 concentrations higher than 1 C99 for every 200 lipids. A Kd of 0.47 mol% corresponds to an equilibrium free energy in favor of dimerization of only −3.2 kcal/mol. Therefore, C99 does have a propensity to dimerize in POPC:POPG vesicles, but only to a modest degree. Assuming that the 0.47 mol% Kd measured for dimerization in POPC:POPG bilayers approximates the Kd in the bulk phase of native membranes, this indicates that homodimerization of C99 is likely to occur to a significant extent only if physiological concentrations of C99 are in the range of 1 or more C99 molecules for every ca. 200 lipids. Is such a concentration likely to ever be attained under physiological conditions? One C99 molecule per 200 lipids means the membranes would have to contain 10% or higher by weight C99. While C99 does not seem to have been ever been quantitated in cells, it is known that it has a much more rapid turnover rate than its full length APP precursor, and can therefore be assumed to have a lower concentration than APP.(50–52) APP has been purified to near homogeneity from the membrane protein fraction of brain membranes, with the reported(52, 53) fold-purification being in the range of 103–104. This suggests the physiological concentration of C99 in neuronal membranes is orders of magnitude lower than 0.47 mol%. One might expect to find C99’s abundance to be relatively high in the detergent-resistant (“lipid raft”) fraction of neuronal membranes(50) (particularly hippocampal neurons(54)), but it is clear from SDS-PAGE of this fraction (Figure 5 in reference(55)) that C99 is, at most, only a minor component of the neuronal raft proteome. Neurons appear to generate amyloid-β at a rate of a few molecules per second(56)—inconsistent with a high (>0.1 mol%) level of its C99 precursor in neuronal membranes, particularly since C99 is thought to be rapidly converted to amyloid-β. While it cannot be absolutely ruled out that local membrane concentrations of C99 under physiological conditions ever reaches the order of 0.5 mol%, this seems unlikely. It should also be noted that in cell biological studies that involve expression of C99 in model mammalian cell lines, the concentration of the protein may often be much higher than under true physiological conditions. This fact should be taken into account when interpreting or designing experiment experimental studies of C99 dimerization that involve the use of vector-based expression of this protein in model cell lines.
That C99 is rich in GXXXG motifs and yet homodimerizes with only modest affinity is not unprecedented for membrane proteins containing these motifs. For example the homodimerization of the isolated transmembrane domains of receptor tyrosine kinases is typically weak, despite the presence of GXXXG motifs.(17, 57–59).
Roles of the Glycine Zipper and GXXXA Motifs in Homodimerization of C99
The results of this work indicate significantly reduced dimerization following replacement of any of the three Gly sites in the G700XXXG704XXXG708 zipper with spin labeled cysteine. The same was also true for Gly709 in the G709XXXA713 motif. For G700XXXG704 it was previously observed that mutation of either glycine to alanine was sufficient to suppress dimerization.(15) On the other hand, replacement of Ala713 with spin labeled cysteine in this study had no impact on dimerization. The same is true for replacements at sites 701–703 and 705. While our results cannot rule out the possibility that other (untested) sites in C99 contribute significantly to homodimerization this work shows that the glycine zipper is a critical structural element, with the Gly sites on the face of the helix allowing two interacting C99 molecules to form an intimate and stabilizing interface. These results are generally consistent with previous TOXCAT, mutagenesis, and biophysical studies that concluded the Gly zipper is key for dimerization.(24, 30) Possible specific structural modes consistent with a central role for the Gly zipper have previously been forwarded based on computer modeling.(31, 33, 35) Our results are somewhat less consistent with a previous study from our lab and one other group that concluded that only the G700XXXG704 motif and not the entire zipper is important for homodimerization.(20, 28)
It is interesting that Gly709 is important for dimerization but that A713 is not. This disfavors the conclusions of certain previous studies that the G709XXXA713 motif is the central element of the C99 dimer.(24, 30) Most striking among previous studies is the NMR-based structure of the APP686–724 (fragment of C99) dimer in dodecylphosphocholine micelles.(30) The disagreement of our data with that structure may reflect the very different model membrane conditions used in the NMR study (DPC micelles) than in this work. Alternatively, removal for the NMR study of most of the N-terminal ectodomain and all of the C-terminal cytosolic domain of C99 might have resulted in a different dimerization mode than pertains for the full length C99. This seems plausible in light of previous data that C99 may adopt an alternative G709XXXA713-centric mode of dimerization if the glycine zipper is disrupted via mutagenesis,(26) although this phenomenon was not observed in this work.
Cholesterol Binding to the C99 Monomer Competes with Homodimerization
In previous work we demonstrated that cholesterol forms a 1:1 complex with monomeric C99 in dihexanoylphosphatidylcholine-dimyristoylphosphatidylcholine (DHPC-DMPC) bicelles, with an observed Kd of 5 mol%.(15) In this work we confirmed that cholesterol binds to C99 in bilayered POPC:POPG lipid vesicles with a Kd of 2.7 ± 0.3 mol%, corresponding to a free energy of −2.1 kcal/mol in favor of complex formation. Moreover, this binding event was seen to be competitive with homodimerization. The fact that binding of cholesterol to C99 in lipid vesicles is stronger than in bicelles is not surprising. While bicelles more closely approximate membranes than do micelles, they do contain detergent and are a less ideal membrane mimic than lipid vesicles. At membrane cholesterol concentrations higher than the 2.7 mol% value for Kd determined in this work, a majority of C99 will be in complex with cholesterol. 2.7 mol% is on the low end of the concentration range for cholesterol in the membranes of mammalian cells (especially the Golgi, plasma, and endosomal membranes).(36, 41–43) Given this and in light of the fact that the concentration of C99 in native cell membranes is probably usually far below its Kd for homodimerization (discussed above), the available data suggest that the most common form of C99 under physiological conditions will be monomer in complex with cholesterol. Of course, we cannot rule out the possibility that other factors pertaining to physiological conditions could alter the Kd values for dimerization and/or cholesterol binding; for example, the presence of other proteins that may interact with C99 and alter its monomer-dimer equilibrium.
That cholesterol binding to monomeric C99 competes with homodimerization is not surprising in light of the fact that the binding interfaces on C99 for these two types of interactions are highly overlapped. Here, we found that all four Gly sites in C99’s TMD are essential to homodimerization. Previously, we showed that these very same Gly were either critical (G700 and G704) or important (G708 and G709) for cholesterol binding.(15) The remainder of the cholesterol binding site is located in the N-terminal loop leading into the transmembrane domain, which includes the residues that form hydrogen bonds with cholesterol’s hydroxyl headgroup. The 3-D structure of the C99 monomer determined by NMR in LMPG micelles revealed that the three glycine residues of the zipper confer a flat surface to the face of TM helix near the ectoplasm.(15) This surface provides the basis for either an intimate homodimerization interface or for a favorable interface between C99 and the steroid ring system of cholesterol, which also is flat and rigid. For both homodimer and monomer-cholesterol interfaces, Van der Waals interactions should be favorable. Formation of these interfaces is also likely to be driven in part by entropy: when two flat surfaces interact with each other in a fluid phase membrane, their interface is entropically favored because the surfaces no longer have to be solvated by the normally dynamic chains of surrounding lipids, whose motions would be dampened somewhat by interacting with the rigid and flat surfaces.(60, 61)
The work of this paper consolidates the notion of a bifunctional role for the key GXXXG motifs of C99 in promoting either cholesterol binding or homodimerization. In this regard it is interesting to recognize a very recent paper by Octave and co-workers that describes an apparent role for APP and C99 in regulating the biosynthesis and turnover of cholesterol in neurons(62), a paper that follows previous studies suggesting roles for the amyloidogenic pathway in regulating cellular cholesterol homeostasis (review in(63) ). The Octave paper provides evidence for direct binding of C99 and APP to sterol regulatory element binding protein 1 (SREBP1), a key protein in the signaling pathway that induces transcriptional response when cellular cholesterol levels are low. Binding of C99/APP to SREBP1 suppresses this signaling pathway. Moreover, C99 was seen to be unable to bind SREBP1 when either of the Gly in the G700XXXG704 motif were mutated. It is an interesting and yet unaddressed question as to whether the mutations suppress binding because Gly700 and Gly704 are directly involved in the binding interface with SREBP1 (which would represent yet another functional role for these glycines!) or whether binding of C99 to SREBP1 is activated when C99 forms its 1:1 complex with cholesterol. This latter possibility would suggest that one function of C99 (and possibly of APP) is to directly serve as a cholesterol sensor in pathways that control cell cholesterol levels, as previously hypothesized.(20)
Dimerization of C99 and Cleavage by γ-Secretase
There has been much interest in the oligomeric state of γ-secretase and in the question of how it relates to the oligomeric state of C99. The γ-secretase complex is comprised of four different integral membrane proteins, of which presenilin is the catalytic subunit. A number of early reports suggest that the γ-secretase complex forms dimers or higher oligomers.(64–66) However, recent medium resolution cryo-EM results and related biochemical studies appear to clearly demonstrate that the complex is both monomeric and active.(67–69) In addition, the recently-published crystal structure of an archaebacterial homolog of presenilin shows that while this enzyme crystallizes as a homotetramer, each subunit appears to contain its own active site located on the far side of the transmembrane domain from the 4-fold symmetry axis.(70) These studies disfavor the notions that the active site of γ-secretase is comprised of residues from more than one subunit and that active sites on adjacent subunits could be sufficiently proximal such that both components of a C99 dimer can simultaneously occupy adjacent active sites without disrupting homodimer contacts.
γ-Secretase does appear to be able to cleave both dimeric and monomeric substrates.(22, 34) Disulfide-linked C99 homodimers have been shown to be cleaved to generate disulfide-bonded amyloid-β dimers.(34) On the other hand, mutations of the Gly residues critical for the weak homodimerization of C99 do not dramatically reduce overall γ-secretase processing of C99, although these mutations do alter the distribution of amyloid-β peptides that are generated.(26, 28) It has been proposed that γ-secretase has a substrate docking site that is located next to the active site. It therefore seems possible that the enzyme may be able to bind one C99 molecule at its active site and a second at the docking site.(71–74) Whether the Gly zipper-associated homodimerization interface of C99 could be maintained with one subunit at the active site and the other at the docking site is not yet clear. It is important to note that γ-secretase has many other single span membrane protein substrates and many do not have a Gly zipper, GXXXG-class motifs, or a known propensity to homodimerize.(75) It is also revealing that in studies of a NOTCH chimera in which its normal transmembrane domain was replaced with that of the avidly-homodimerizing glycophorin A, the version of the chimera with the wild type glycophorin A sequence was processed by γ-secretase significantly less efficiently than a mutant form in which the GXXXG motif was disrupted.(76) Another study also concluded that possible homodimerization of the NOTCH TMD is unrelated to its cleavage by γ-secretase.(77) Our conclusion in this paper that C99 dimerizes with only modest affinity and may have little tendency to form dimers at physiological concentrations does not conflict with what is currently believed about its interactions with γ-secretase.
Cholesterol Binding By C99 and Cleavage by γ-Secretase
There is considerable evidence that elevated levels of cholesterol promote the amyloidogenic pathway, resulting increased levels of amyloid-β production.(5–14) How might cholesterol binding to C99 explain this phenomenon?
This work confirms our previous suggestion that C99 will very often be complexed with cholesterol under physiological conditions.(15) We have previously hypothesized that this complex (and the corresponding complex of cholesterol with full length APP) may promote amyloidogenesis by increasing partitioning of C99 and APP into cholesterol-rich membrane domains known as “lipid rafts”.(15, 20) This would promote formation of amyloid-β because, as reviewed elsewhere, there is much evidence that β- and γ-secretase preferentially associate with rafts.(78–83)
A second possible mechanism by which cholesterol binding to C99 could promote amyloid-β formation is by directly promoting C99 binding to γ-secretase (to either the putative docking site or the active site or both) and subsequent cleavage. Studies with purified γ-secretase that has been reconstituted into lipid vesicles have shown a 2- to 4-fold enhancement in the rates of production of both Aβ40 and Aβ42 by the presence of cholesterol in the membranes, with optimal levels of cholesterol being in the range of 5–20 mol%, depending on vesicle composition.(67) It is possible that the rate increase may simply reflect the impact of membrane cholesterol on γ-secretase function as a result of the effect of cholesterol on bulk membrane properties and/or direct interactions with the enzyme.(84–86) However, it is interesting that the maximally-activating levels of cholesterol correspond to concentrations at which near saturation of cholesterol binding to C99 is expected based on the Kd of 2.7 mol% determined in this work. One wonders if cholesterol could remain associated with C99 at the γ-secretase substrate docking site and/or in the active site. The glycine zipper of C99 is associated with empty space on the flat Gly-rich face of the transmembrane helix that is normally occupied by non-Gly amino acid side chains in most other γ-secretase substrates.(75) This suggests the possibility of a cleft between the face of the C99 Gly zipper and γ-secretase sidechains at the binding site. Whether such a cleft is present following C99 binding, whether cholesterol could remain bound in such a cleft, and how this would impact cleavage of C99 may be worthy of future investigation.
Cholesterol may under some conditions compete with γ-secretase modulators (GSMs) for binding to C99 when bound to γ-secretase. GSMs are small molecules that modulate cleavage of C99 by γ-secretase to alter the ratio between the short forms of amyloid-β and the more pathogenic long forms.(87, 88) GSM action is C99-selective in the sense that the cleavage of other γ-secretase substrates is not impacted. While there has been controversy as to whether GSMs can bind directly to free C99,(21, 32, 89–91) most recent evidence suggests that GSMs act by forming a ternary complex with C99 and γ-secretase.(87, 88, 92–96) Remarkably, there are a couple of reports that mutation of the Gly in the zipper motif of C99 results in elimination of GSM action, suggesting that the face of the zipper may interact directly with GSMs in ternary complexes with γ-secretase.(91, 97) While this is not without contention,(98) it seems plausible that the cleft hypothesized above between the face of the C99 Gly zipper and the surrounding γ-secretase residues might represent the GSM binding site. Whether GSMs might compete with cholesterol for C99-mediated binding to γ-secretase is an interesting possibility that may be worthy of future testing.
Finally, we note the possibility that cholesterol might also bind to the full length APP and promote its amyloidogenic cleavage by β-secretase through mechanisms analogous to those outlined above for C99 and γ-secretase. The structure and membrane interactions of the C99 domain of full length APP is likely to be similar to the structure of isolated C99, such that cholesterol binding to APP is likely to be of similar affinity as for C99. Moreover, there are persistent reports that β-secretase is also found to be enriched in cholesterol-rich membrane domains (reviews in(78, 81)).
Conclusions
This work extends recent studies by showing that cholesterol binds to C99 in bilayered lipid vesicles in a manner that is competitive with homodimerization and by confirming observations made in bicellar model membranes that the dissociation constant is well within the physiological concentration range of cholesterol in mammalian cells. We also found that the quantitative homodimerization affinity of C99 is modest to the point where it is difficult to argue that dimerization is likely to be physiologically relevant. On the other hand, C99 forms a 1:1 complex with cholesterol that appears to be of clear physiological relevance. This raises questions for future study regarding the significance of this binding event for C99 function, membrane trafficking, and amyloidogenic proteolytic processing. Finally, we note that the involvement of the glycine zipper in the C99 TMD in cholesterol binding, homodimerization, and interactions with GSMs (in the C99/γ-secretase complex) suggests extraordinary importance for this motif, the possession of which sets C99 apart from most other γ-secretase substrates.
Supplementary Material
Figure 7.
Competition between homodimerization and cholesterol binding to free C99 in lipid vesicles. The Kd values were determined in this work.
Acknowledgments
We thank Arina Hadziselimovic for technical assistance, Brett Kroncke for proofreading, and Paul Barrett for much helpful discussion.
Funding This work was supported by US NIH grants RO1 GM106672 and PO1 GM080513.
Footnotes
Supporting Information Supporting Figure 1, which shows the dependence of the EPR spectra from C99 that is spin labeled at sites 701, 702, and 713 on the protein mol% in vesicles. The material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.De SB, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2011;3:a004457. doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teich AF, Arancio O. Is the amyloid hypothesis of Alzheimer’s disease therapeutically relevant? Biochem J. 2012;446:165–177. doi: 10.1042/BJ20120653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- 6.Fonseca AC, Resende R, Oliveira CR, Pereira CM. Cholesterol and statins in Alzheimer’s disease: current controversies. Exp Neurol. 2010;223:282–293. doi: 10.1016/j.expneurol.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 7.Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem. 2008;283:11302–11311. doi: 10.1074/jbc.M801520200. [DOI] [PubMed] [Google Scholar]
- 8.Guardia-Laguarta C, Coma M, Pera M, Clarimon J, Sereno L, Agullo JM, Molina-Porcel L, Gallardo E, Deng A, Berezovska O, Hyman BT, Blesa R, Gomez-Isla T, Lleo A. Mild cholesterol depletion reduces amyloid-beta production by impairing APP trafficking to the cell surface. J Neurochem. 2009;110:220–230. doi: 10.1111/j.1471-4159.2009.06126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci U S A. 2001;98:5815–5820. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simons M, Keller P, De SB, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 12.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiology of disease. 2001;8:890–899. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 14.Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci. 2002;22:1679–1689. doi: 10.1523/JNEUROSCI.22-05-01679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S, Jeon TJ, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci U S A. 2005;102:14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacKenzie KR, Fleming KG. Association energetics of membrane spanning alpha-helices. Curr Opin Struct Biol. 2008;18:412–419. doi: 10.1016/j.sbi.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 19.Kleiger G, Grothe R, Mallick P, Eisenberg D. GXXXG and AXXXA: common alpha-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry. 2002;41:5990–5997. doi: 10.1021/bi0200763. [DOI] [PubMed] [Google Scholar]
- 20.Beel AJ, Mobley CK, Kim HJ, Tian F, Hadziselimovic A, Jap B, Prestegard JH, Sanders CR. Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): does APP function as a cholesterol sensor? Biochemistry. 2008;47:9428–9446. doi: 10.1021/bi800993c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botev A, Munter LM, Wenzel R, Richter L, Althoff V, Ismer J, Gerling U, Weise C, Koksch B, Hildebrand PW, Bittl R, Multhaup G. The amyloid precursor protein C-terminal fragment C100 occurs in monomeric and dimeric stable conformations and binds gamma-secretase modulators. Biochemistry. 2011;50:828–835. doi: 10.1021/bi1014002. [DOI] [PubMed] [Google Scholar]
- 22.Eggert S, Midthune B, Cottrell B, Koo EH. Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J Biol Chem. 2009;284:28943–28952. doi: 10.1074/jbc.M109.038646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goo JH, Park WJ. Elucidation of the interactions between C99, presenilin, and nicastrin by the split-ubiquitin assay. DNA Cell Biol. 2004;23:59–65. doi: 10.1089/104454904322745934. [DOI] [PubMed] [Google Scholar]
- 24.Gorman PM, Kim S, Guo M, Melnyk RA, McLaurin J, Fraser PE, Bowie JU, Chakrabartty A. Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer’s disease mutants. BMC Neurosci. 2008;9:17. doi: 10.1186/1471-2202-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khalifa NB, Van HJ, Tasiaux B, Huysseune S, Smith SO, Constantinescu SN, Octave JN, Kienlen-Campard P. What is the role of amyloid precursor protein dimerization? Cell Adh Migr. 2010;4:268–272. doi: 10.4161/cam.4.2.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kienlen-Campard P, Tasiaux B, Van HJ, Li M, Huysseune S, Sato T, Fei JZ, Aimoto S, Courtoy PJ, Smith SO, Constantinescu SN, Octave JN. Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J Biol Chem. 2008;283:7733–7744. doi: 10.1074/jbc.M707142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao G, Tan J, Cui MZ, Chui D, Xu X. The GxxxG motif in the transmembrane domain of AbetaPP plays an essential role in the interaction of CTF beta with the gamma-secretase complex and the formation of amyloid-beta. J Alzheimers Dis. 2009;18:167–176. doi: 10.3233/JAD-2009-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munter LM, Botev A, Richter L, Hildebrand PW, Althoff V, Weise C, Kaden D, Multhaup G. Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J Biol Chem. 2010;285:21636–21643. doi: 10.1074/jbc.M109.088005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nadezhdin KD, Bocharova OV, Bocharov EV, Arseniev AS. Dimeric structure of transmembrane domain of amyloid precursor protein in micellar environment. FEBS Lett. 2012;586:1687–1692. doi: 10.1016/j.febslet.2012.04.062. [DOI] [PubMed] [Google Scholar]
- 31.Pester O, Barrett PJ, Hornburg D, Hornburg P, Probstle R, Widmaier S, Kutzner C, Durrbaum M, Kapurniotu A, Sanders CR, Scharnagl C, Langosch D. The Backbone Dynamics of the Amyloid Precursor Protein Transmembrane Helix Provides a Rationale for the Sequential Cleavage Mechanism of gamma-Secretase. J Am Chem Soc. 2013;135:1317–1329. doi: 10.1021/ja3112093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter L, Munter LM, Ness J, Hildebrand PW, Dasari M, Unterreitmeier S, Bulic B, Beyermann M, Gust R, Reif B, Weggen S, Langosch D, Multhaup G. Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc Natl Acad Sci U S A. 2010;107:14597–14602. doi: 10.1073/pnas.1003026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato T, Tang TC, Reubins G, Fei JZ, Fujimoto T, Kienlen-Campard P, Constantinescu SN, Octave JN, Aimoto S, Smith SO. A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc Natl Acad Sci U S A. 2009;106:1421–1426. doi: 10.1073/pnas.0812261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Barreyro L, Provasi D, Djemil I, Torres-Arancivia C, Filizola M, Ubarretxena-Belandia I. Molecular determinants and thermodynamics of the amyloid precursor protein transmembrane domain implicated in Alzheimer’s disease. J Mol Biol. 2011;408:879–895. doi: 10.1016/j.jmb.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antoniou C, Lam VQ, Fung LW. Conformational changes at the tetramerization site of erythroid alpha-spectrin upon binding beta-spectrin: a spin label EPR study. Biochemistry. 2008;47:10765–10772. doi: 10.1021/bi800840p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao H, Ellena J, Liu L, Szabo G, Cafiso D, Castle D. Secretory carrier membrane protein SCAMP2 and phosphatidylinositol 4,5-bisphosphate interactions in the regulation of dense core vesicle exocytosis. Biochemistry. 2007;46:10909–10920. doi: 10.1021/bi701121j. [DOI] [PubMed] [Google Scholar]
- 39.Lu JX, Yau WM, Tycko R. Evidence from solid-state NMR for nonhelical conformations in the transmembrane domain of the amyloid precursor protein. Biophys J. 2011;100:711–719. doi: 10.1016/j.bpj.2010.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Ann Rev Biophys Biomolec Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 41.Calderon RO, Attema B, DeVries GH. Lipid composition of neuronal cell bodies and neurites from cultured dorsal root ganglia. J Neurochem. 1995;64:424–429. doi: 10.1046/j.1471-4159.1995.64010424.x. [DOI] [PubMed] [Google Scholar]
- 42.Calderon RO, DeVries GH. Lipid composition and phospholipid asymmetry of membranes from a Schwann cell line. J Neurosci Res. 1997;49:372–380. [PubMed] [Google Scholar]
- 43.Wood WG, Cornwell M, Williamson LS. High performance thin-layer chromatography and densitometry of synaptic plasma membrane lipids. J Lipid Res. 1989;30:775–779. [PubMed] [Google Scholar]
- 44.Gralle M, Ferreira ST. Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol. 2007;82:11–32. doi: 10.1016/j.pneurobio.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Kaden D, Munter LM, Reif B, Multhaup G. The amyloid precursor protein and its homologues: Structural and functional aspects of native and pathogenic oligomerization. Eur J Cell Biol. 2011 doi: 10.1016/j.ejcb.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 46.Lee S, Xue Y, Hu J, Wang Y, Liu X, Demeler B, Ha Y. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry. 2011;50:5453–5464. doi: 10.1021/bi101846x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devauges V, Marquer C, Lecart S, Cossec JC, Potier MC, Fort E, Suhling K, Leveque-Fort S. Homodimerization of amyloid precursor protein at the plasma membrane: a homoFRET study by time-resolved fluorescence anisotropy imaging. PLoS One. 2012;7:e44434. doi: 10.1371/journal.pone.0044434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gralle M, Botelho MG, Wouters FS. Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem. 2009;284:15016–15025. doi: 10.1074/jbc.M808755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyashita N, Straub JE, Thirumalai D. Structures of beta-amyloid peptide 1-40, 1-42, and 1-55-the 672-726 fragment of APP-in a membrane environment with implications for interactions with gamma-secretase. J Am Chem Soc. 2009;131:17843–17852. doi: 10.1021/ja905457d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abramowski D, Wiederhold KH, Furrer U, Jaton AL, Neuenschwander A, Runser MJ, Danner S, Reichwald J, Ammaturo D, Staab D, Stoeckli M, Rueeger H, Neumann U, Staufenbiel M. Dynamics of Abeta turnover and deposition in different beta-amyloid precursor protein transgenic mouse models following gamma-secretase inhibition. J Pharmacol Exp Ther. 2008;327:411–424. doi: 10.1124/jpet.108.140327. [DOI] [PubMed] [Google Scholar]
- 52.Potempska A, Styles J, Mehta P, Kim KS, Miller DL. Purification and tissue level of the beta-amyloid peptide precursor of rat brain. J Biol Chem. 1991;266:8464–8469. [PubMed] [Google Scholar]
- 53.de La Fournière-Bessueille L, Grange D, Buchet R. Purification and spectroscopic characterization of beta-amyloid precursor protein from porcine brains. Eur J Biochem. 1997;250:705–711. doi: 10.1111/j.1432-1033.1997.00705.x. [DOI] [PubMed] [Google Scholar]
- 54.Guo Q, Li H, Gaddam SS, Justice NJ, Robertson CS, Zheng H. Amyloid precursor protein revisited: neuron-specific expression and highly stable nature of soluble derivatives. J Biol Chem. 2012;287:2437–2445. doi: 10.1074/jbc.M111.315051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williamson R, Thompson AJ, Abu M, Hye A, Usardi A, Lynham S, Anderton BH, Hanger DP. Isolation of detergent resistant microdomains from cultured neurons: detergent dependent alterations in protein composition. BMC Neurosci. 2010;11:120. doi: 10.1186/1471-2202-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moghekar A, Rao S, Li M, Ruben D, Mammen A, Tang X, O’Brien RJ. Large quantities of Abeta peptide are constitutively released during amyloid precursor protein metabolism in vivo and in vitro. J Biol Chem. 2011;286:15989–15997. doi: 10.1074/jbc.M110.191262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobus FJ, Fleming KG. The GxxxG-containing transmembrane domain of the CCK4 oncogene does not encode preferential self-interactions. Biochemistry. 2005;44:1464–1470. doi: 10.1021/bi048076l. [DOI] [PubMed] [Google Scholar]
- 58.Stanley AM, Fleming KG. The transmembrane domains of ErbB receptors do not dimerize strongly in micelles. J Mol Biol. 2005;347:759–772. doi: 10.1016/j.jmb.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 59.Li E, Hristova K. Receptor tyrosine kinase transmembrane domains: Function, dimer structure and dimerization energetics. Cell Adh Migr. 2010;4:249–254. doi: 10.4161/cam.4.2.10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Popot JL, Engelman DM. Helical membrane protein folding, stability, and evolution. Annu Rev Biochem. 2000;69:881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- 61.Helms V. Attraction within the membrane. Forces behind transmembrane protein folding and supramolecular complex assembly. EMBO Rep. 2002;3:1133–1138. doi: 10.1093/embo-reports/kvf245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pierrot N, Tyteca D, D’Auria L, Dewachter I, Gailly P, Hendrickx A, Tasiaux B, Haylani LE, Muls N, N’Kuli F, Laquerriere A, Demoulin JB, Campion D, Brion JP, Courtoy PJ, Kienlen-Campard P, Octave JN. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med. 2013;5:608–625. doi: 10.1002/emmm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimm MO, Rothhaar TL, Hartmann T. The role of APP proteolytic processing in lipid metabolism. Exp Brain Res. 2012;217:365–375. doi: 10.1007/s00221-011-2975-6. [DOI] [PubMed] [Google Scholar]
- 64.Cervantes S, Saura CA, Pomares E, Gonzalez-Duarte R, Marfany G. Functional implications of the presenilin dimerization: reconstitution of gamma-secretase activity by assembly of a catalytic site at the dimer interface of two catalytically inactive presenilins. J Biol Chem. 2004;279:36519–36529. doi: 10.1074/jbc.M404832200. [DOI] [PubMed] [Google Scholar]
- 65.Clarke EE, Churcher I, Ellis S, Wrigley JD, Lewis HD, Harrison T, Shearman MS, Beher D. Intra- or intercomplex binding to the gamma-secretase enzyme. A model to differentiate inhibitor classes. J Biol Chem. 2006;281:31279–31289. doi: 10.1074/jbc.M605051200. [DOI] [PubMed] [Google Scholar]
- 66.Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, Lewis HD, Saxena MT, De SB, Coonrod A, Tomita T, Iwatsubo T, Moore CL, Goate A, Wolfe MS, Shearman M, Kopan R. A presenilin dimer at the core of the gamma-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci U S A. 2003;100:13075–13080. doi: 10.1073/pnas.1735338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Renzi F, Zhang X, Rice WJ, Torres-Arancivia C, Gomez-Llorente Y, Diaz R, Ahn K, Yu C, Li YM, Sisodia SS, Ubarretxena-Belandia I. Structure of gamma-secretase and its trimeric pre-activation intermediate by single-particle electron microscopy. J Biol Chem. 2011;286:21440–21449. doi: 10.1074/jbc.M110.193326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De SB, Steiner H, Haass C, Wolfe MS. Active gamma-secretase complexes contain only one of each component. J Biol Chem. 2007;282:33985–33993. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- 70.Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- 71.Li H, Wolfe MS, Selkoe DJ. Toward structural elucidation of the gamma-secretase complex. Structure. 2009;17:326–334. doi: 10.1016/j.str.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Imamura Y, Umezawa N, Osawa S, Shimada N, Higo T, Yokoshima S, Fukuyama T, Iwatsubo T, Kato N, Tomita T, Higuchi T. Effect of Helical Conformation and Side Chain Structure on gamma-Secretase Inhibition by beta-Peptide Foldamers: Insight into Substrate Recognition. J Med Chem. 2013 doi: 10.1021/jm301306c. [DOI] [PubMed] [Google Scholar]
- 73.Kornilova AY, Bihel F, Das C, Wolfe MS. The initial substrate-binding site of gamma-secretase is located on presenilin near the active site. Proc Natl Acad Sci U S A. 2005;102:3230–3235. doi: 10.1073/pnas.0407640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watanabe N, Image I, Takagi S, II, Image I, Tominaga A, II, Image I, Tomita T, I, Image I, Iwatsubo T, II, Image II. Functional analysis of the transmembrane domains of presenilin 1: participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of gamma-secretase. J Biol Chem. 2010;285:19738–19746. doi: 10.1074/jbc.M110.101287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beel AJ, Sanders CR. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci. 2008;65:1311–1334. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vooijs M, Schroeter EH, Pan Y, Blandford M, Kopan R. Ectodomain shedding and intramembrane cleavage of mammalian Notch proteins is not regulated through oligomerization. J Biol Chem. 2004;279:50864–50873. doi: 10.1074/jbc.M409430200. [DOI] [PubMed] [Google Scholar]
- 77.Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000;6:625–636. doi: 10.1016/s1097-2765(00)00061-7. [DOI] [PubMed] [Google Scholar]
- 78.Beel AJ, Sakakura M, Barrett PJ, Sanders CR. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? Biochim Biophys Acta. 2010;1801:975–982. doi: 10.1016/j.bbalip.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheng H, Vetrivel KS, Gong P, Meckler X, Parent A, Thinakaran G. Mechanisms of disease: new therapeutic strategies for Alzheimer’s disease--targeting APP processing in lipid rafts. Nat Clin Pract Neurol. 2007;3:374–382. doi: 10.1038/ncpneuro0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Di PG, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hicks DA, Nalivaeva NN, Turner AJ. Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front Physiol. 2012;3:189. doi: 10.3389/fphys.2012.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martins IJ, Berger T, Sharman MJ, Verdile G, Fuller SJ, Martins RN. Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J Neurochem. 2009;111:1275–1308. doi: 10.1111/j.1471-4159.2009.06408.x. [DOI] [PubMed] [Google Scholar]
- 83.Rushworth JV, Hooper NM. Lipid Rafts: Linking alzheimer’s amyloid-beta production, aggregation, and toxicity at neuronal membranes. Int J Alzheimers Dis. 2010;2011:603052. doi: 10.4061/2011/603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, van DA, Wang R, Selkoe DJ, Wolfe MS. Purification and characterization of the human gamma-secretase complex. Biochemistry. 2004;43:9774–9789. doi: 10.1021/bi0494976. [DOI] [PubMed] [Google Scholar]
- 85.Wrigley JD, Schurov I, Nunn EJ, Martin AC, Clarke EE, Ellis S, Bonnert TP, Shearman MS, Beher D. Functional overexpression of gamma-secretase reveals protease-independent trafficking functions and a critical role of lipids for protease activity. J Biol Chem. 2005;280:12523–12535. doi: 10.1074/jbc.M413086200. [DOI] [PubMed] [Google Scholar]
- 86.Holmes O, Paturi S, Ye W, Wolfe MS, Selkoe DJ. Effects of membrane lipids on the activity and processivity of purified gamma-secretase. Biochemistry. 2012;51:3565–3575. doi: 10.1021/bi300303g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bulic B, Ness J, Hahn S, Rennhack A, Jumpertz T, Weggen S. Chemical Biology, Molecular Mechanism and Clinical Perspective of gamma-Secretase Modulators in Alzheimer’s Disease. Curr Neuropharmacol. 2011;9:598–622. doi: 10.2174/157015911798376352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wolfe MS. gamma-Secretase inhibitors and modulators for Alzheimer’s disease. J Neurochem. 2012;120(Suppl 1):89–98. doi: 10.1111/j.1471-4159.2011.07501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barrett PJ, Sanders CR, Kaufman SA, Michelsen K, Jordan JB. NSAID-based gamma-secretase modulators do not bind to the amyloid-beta polypeptide. Biochemistry. 2011;50:10328–10342. doi: 10.1021/bi201371j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beel AJ, Barrett P, Schnier PD, Hitchcock SA, Bagal D, Sanders CR, Jordan JB. Nonspecificity of binding of gamma-secretase modulators to the amyloid precursor protein. Biochemistry. 2009;48:11837–11839. doi: 10.1021/bi901839d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, Cottrell BA, Torpey J, Rosenberry TL, Fauq A, Wolfe MS, Schmidt B, Walsh DM, Koo EH, Golde TE. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Crump CJ, Fish BA, Castro SV, Chau DM, Gertsik N, Ahn K, Stiff C, Pozdnyakov N, Bales KR, Johnson DS, Li YM. Piperidine acetic acid based gamma-secretase modulators directly bind to Presenilin-1. ACS Chem Neurosci. 2011;2:705–710. doi: 10.1021/cn200098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jumpertz T, Rennhack A, Ness J, Baches S, Pietrzik CU, Bulic B, Weggen S. Presenilin is the molecular target of acidic gamma-secretase modulators in living cells. PLoS One. 2012;7:e30484. doi: 10.1371/journal.pone.0030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ohki Y, Higo T, Uemura K, Shimada N, Osawa S, Berezovska O, Yokoshima S, Fukuyama T, Tomita T, Iwatsubo T. Phenylpiperidine-type gamma-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 2011;30:4815–4824. doi: 10.1038/emboj.2011.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shelton CC, Zhu L, Chau D, Yang L, Wang R, Djaballah H, Zheng H, Li YM. Modulation of gamma-secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci U S A. 2009;106:20228–20233. doi: 10.1073/pnas.0910757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zettl H, Weggen S, Schneider P, Schneider G. Exploring the chemical space of gamma-secretase modulators. Trends Pharmacol Sci. 2010;31:402–410. doi: 10.1016/j.tips.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 97.Sagi SA, Lessard CB, Winden KD, Maruyama H, Koo JC, Weggen S, Kukar TL, Golde TE, Koo EH. Substrate sequence influences gamma-secretase modulator activity, role of the transmembrane domain of the amyloid precursor protein. J Biol Chem. 2011;286:39794–39803. doi: 10.1074/jbc.M111.277228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Page RM, Gutsmiedl A, Fukumori A, Winkler E, Haass C, Steiner H. Beta-amyloid precursor protein mutants respond to gamma-secretase modulators. J Biol Chem. 2010;285:17798–17810. doi: 10.1074/jbc.M110.103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.