

Abstract
Anti-ganglioside antibodies (Abs) are strongly associated with axonal forms of Guillain Barré syndrome (GBS). Some studies indicate that these Abs, including those with GD1a reactivity, are associated with poor prognosis and/or incomplete recovery. We recently demonstrated that a disease-relevant anti-ganglioside Ab with GD1a reactivity inhibits axon regeneration after PNS injury in an animal model (Lehmann et al., 2007). An implication of these findings is that anti-GD1a Abs can mediate inhibition of axon regeneration and limit recovery in some patients with GBS. The downstream inhibitory intracellular signaling that mediates anti-ganglioside Ab-induced axon inhibition remains unclear. In the current study, we show that disease-relevant and GBS patient's anti-ganglioside Abs can inhibit neurite outgrowth in dissociated primary neuronal cultures. Activation of small GTPase RhoA and its key downstream effector Rho kinase (ROCK) are critical mediators of growth cone and neurite outgrowth inhibition. Therefore, we examined the role of these intracellular signaling molecules in our primary neuronal cultures by molecular and pharmacologic approaches. Our results show that the Ab-mediated inhibition of neurite outgrowth involves the activation of RhoA and ROCK pathway and this activation is through the engagement of specific cell-surface gangliosides by Abs. In summary, these studies directly link patient autoantibodies to an intracellular inhibitory signaling pathway involved in anti-ganglioside Ab-mediated inhibition of neurite outgrowth.
Introduction
Guillain-Barré syndrome (GBS) remains a major public health burden. Most affected individuals have an uneventful recovery, but a significant proportion of patients require mechanical ventilation and 20% have severe and permanent neurologic sequelae, including 10% who cannot walk unaided (Ropper et al., 1991; Rees et al., 1998; Hughes and Cornblath, 2005). The prevalence and health costs of post-GBS cases with residual deficits are not known. Patients with residual deficits and significant disability almost always have axonal injury and target denervation; recovery thus requires regeneration from the site of axonal transection. Although the adult mammalian PNS regenerates readily after injury, the mechanisms underlying failure of axon regeneration in GBS cases with permanent neurologic sequelae are beginning to unravel (Lehmann et al., 2007; Lopez et al., 2010; Sheikh and Zhang, 2010).
Anti-ganglioside antibodies (Abs) are the most commonly recognized autoimmune markers in all forms of GBS (Willison and Yuki, 2002). Association between certain variants of GBS and specific anti-ganglioside Abs is now widely accepted (Willison and O'Hanlon, 1999; Yuki, 2001; Willison and Yuki, 2002; Hughes and Cornblath, 2005; Willison, 2005). The full spectrum of anti-ganglioside Ab-mediated pathobiologic effects and associated mechanisms remain to be defined. Several studies suggest that some GBS patients with anti-ganglioside Abs directed against GM1, GD1a, or ganglioside complexes have poor prognosis and/or incomplete recovery (Ilyas et al., 1992; Gregson et al., 1993; Simone et al., 1993; Yuki et al., 1993; Jacobs et al., 1996; Bech et al., 1997; Kuwabara et al., 1998a,b; Carpo et al., 1999; Press et al., 2001; Annunziata et al., 2003; Koga et al., 2003; Kaida et al., 2007). We recently demonstrated that experimental monoclonal and GBS patient-derived anti-ganglioside Abs can inhibit regeneration of injured axons in an animal model (Lehmann et al., 2007; Lopez et al., 2010), suggesting that Ab-mediated inhibition of nerve repair is one mechanism of delayed recovery. The cellular and molecular mechanisms of this Ab-mediated inhibition of axon regeneration are not clear.
We have now established primary neuron culture models to study molecular mechanisms involved in Ab-mediated inhibition of axon regeneration. In this study, we show that experimental and patient-derived anti-ganglioside Abs inhibit neurite outgrowth. Our studies indicate that anti-ganglioside Abs induce activation of small GTPase RhoA and its downstream effector Rho kinase (ROCK) by engaging cell-surface gangliosides/glycans. This Ab-mediated outside-in signaling modulates growth cone morphology and behavior, which is related to inhibition of neurite elongation.
Materials and Methods
Monoclonal anti-ganglioside Abs and patient sera.
Two IgG monoclonal Abs (mAbs) were used in this study because they have been tested previously in an animal model of axonal regeneration (Lehmann et al., 2007; Lopez et al., 2010). Anti-GD1a/GT1b IgG2b (GD1a/GT1b-2b) mAb is prototypic Ab, which has been extensively characterized in our previous studies (Lunn et al., 2000; Gong et al., 2002; Zhang et al., 2004), including its capacity to severely inhibit axon regeneration in an animal model (Lehmann et al., 2007). This mAb was compared with anti-GM1 IgG2b mAb (GM1-2b), which does not induce inhibition of axon regeneration in the same animal model (Lopez et al., 2010). The details of these anti-ganglioside mAbs, including generation, specificity, purification, and designation, were reported previously (Lunn et al., 2000; Gong et al., 2002). An irrelevant mouse IgG-2b mAb (Abcam) was used as a negative control Ab. Purified mAbs were used in all assays.
Sera from seven patients (six from Northern China and one from the United States) with GBS (acute motor axonal neuropathy) and high titers of IgG anti-GD1a Abs, collected during the acute phase of the disease, were used for these studies. Serum from a normal healthy volunteer without reactivity against major nerve gangliosides or intravenous Ig was used as negative control. IgG fractions were prepared from GBS or control sera by using a protein G Sepharose column (GE Healthcare) according to the instructions of the manufacturer. Purified IgG fractions were used for all studies.
Cell line.
NG108-15 is a neuroblastoma × glioma hybrid neural cell line that expresses various gangliosides, including GD1a (Dahms and Schnaar, 1983; Wu et al., 2001). The cells were cultured in a 5% CO2 humidified atmosphere at 37°C in DMEM supplemented with 5% heat-inactivated fetal bovine serum (FBS) plus a supplement of HAT (0.1 mm hypoxanthine, 0.4 μm aminopterin, and 16 μm thymidine). These cells were grown to a high density (confluence) and passaged every 2–3 d before use (see below).
Primary rat dorsal root ganglion neurons culture.
Dissociated dorsal root ganglion (DRG) neuronal cell cultures were collected from embryonic day 15 (E15) or postnatal days 5–8 Sprague Dawley rat dorsal root ganglia, as described previously (Eldridge et al., 1987). Only postnatal rat DRGs were incubated with 0.2% Collagenase I (Worthington), and both embryonic and postnatal DRGs were treated with 0.25% trypsin with EDTA (Invitrogen) after dissection. The tissue was triturated with tip-polished Pasteur pipette. After passing through a 40 μm cell strainer, these neurons were then placed onto glass coverslips (Thermo Fisher Scientific) coated with 50 μg/ml poly-d-lysine (Sigma-Aldrich) at 5000 cells per well in 24 well plates (BD Biosciences). Both embryonic and postnatal neuronal cultures were maintained in Neurobasal medium (Invitrogen) containing 0.25% (v/v) heat-inactivated FBS (Hyclone), 2 m l-glutamine, 2% B27 serum-free supplement (Invitrogen), and 50 ng/ml nerve growth factor (Sigma-Aldrich).
Dissociated adult mouse DRG neuron cultures.
Eight- to 10-week-old wild-type (C57BL/6) and Galgt1-null (UDP-N-acetyl-d-galactosamine: GM3/GM2/GD2 synthase; EC 2.4.1.92) mice were used for establishing DRG neuron cultures, as described (Malin et al., 2007). Galgt1-null mice lack complex gangliosides, including GD1a, and express only simple gangliosides GM3 and GD3 (Sheikh et al., 1999), which allowed us to examine the role of cell-surface gangliosides in anti-ganglioside Ab-mediated pathobiologic effects on neurite outgrowth. These mouse DRG neurons were cultured in F-12 culture medium (Invitrogen) containing 10% FBS plus 1% penicillin/streptomycin (Invitrogen).
Primary motor neuron culture.
Motor neurons were purified from the spinal cords of E15 Sprague Dawley rat embryos as described (Vincent et al., 2004). Briefly, the dorsal half of the cords were removed using a scalpel, and the ventral cords free of perineural membranes were chopped into little pieces. The cells were then dissociated by incubation in 0.05% trypsin with EDTA for 15 min at 37°C, followed by gentle trituration with a polished glass Pasteur pipette and differential centrifugation. Motor neuron growth culture media containing Neurobasal medium (Invitrogen) were supplemented with various additives, which were all from Sigma-Aldrich unless otherwise stated, including 2% B27 serum-free supplement (Invitrogen), 2.5 mg/ml albumin, 2.5 μg/ml catalase, 0.01 mg/ml transferrin, 15 μg/ml galactose, 6.3 ng/ml progesterone, 16 μg/ml putrescine, 4 ng/ml selenium, and 1× penicillin/streptomycin.
Neurite outgrowth assay.
The primary motor and sensory neuronal cultures were treated with different anti-ganglioside mAbs (10–100 μg/ml) or GBS patient-derived IgG fractions (300–500 μg/ml of the purified IgG) 30 min after cell plating. After 24 h incubation with the mAbs or patient IgGs, cells were fixed with 4% paraformaldehyde and stained with a neuron-specific Ab against β-III-tubulin (1:5000; Promega), followed by an IgG1-specific Alexa Fluor-conjugated Ab (1:200; Invitrogen). Molecular and pharmacological approaches were used to determine the role of RhoA GTPase and ROCK. In some studies, dissociated DRG neurons were transfected with green fluorescent protein (GFP)-tagged dominant-negative (DN) constructs of RhoA GTPase before treatment with GD1a/GT1b-2b mAb (see below). The chemical approach included treatment of DRG neurons with a RhoA inhibitor, C3-transferase (25 μg/ml; Cytoskeleton), and/or ROCK inhibitor, Y27632 (10 μm; Calbiochem), before adding anti-ganglioside mAbs or IgG fractions from sera. Images were acquired from randomly selected fields (n = 8–12), and the longest neurite of 80–120 neurons per condition (embryonic and postnatal) or neurite density (adult mouse cultures) was measured by using either Carl Zeiss software according to the manual of the manufacturer or NIH ImageJ, a public domain image processing program (http://rsb.info.nih.gov/ij). Each experimental condition was done in duplicate wells, and at least three independent experiments were conducted to acquire the final results. Data are presented as the mean ± SE neurite length.
Schwann cell-depleted DRG cultures.
Schwann cell-depleted cultures were generated by repeated AraC treatment to demonstrate that the inhibitory effect is at the level of DRG neurons. Mixed DRG cultures were plated as above for 24 h. These DRG cultures were continuously treated with AraC (10−6 m for 1 week), as described (Melli et al., 2006), and examined immunocytochemically with rabbit anti-S100 (1:500; Sigma) and mouse anti-β-III-tubulin to assess the presence of Schwann cells. Our results showed that, after AraC treatment, <5% of cells stained for S100 in DRG cultures. For inhibition assays, DRG cultures were treated with AraC as above and then incubated with GD1a/GT1b-2b overnight/24 h, and neurite length was determined as described above.
Studies with Fab fragments.
Experiments with Fab (fragment antigen binding) fragments were done to examine whether bivalent binding is necessary to induce inhibition. Fabs were prepared from mAb, GD1a/GT1b-2b, by papain digestion with a commercially available kit according to the instructions of the manufacturer (Pierce). We confirmed that Fabs do not form multimeric complexes in solution by nonreducing SDS-PAGE, which showed a single band, indicating that Fabs are monomeric in solution. Fabs (50–100 μg/ml) were compared with the parent whole IgG (50–100 μg/ml) in DRG neurite outgrowth assays as described above.
Transfection of DRG neurons.
A GFP-tagged DN construct of RhoA GTPase (Ruchhoeft et al., 1999) was used for the transfection. Embryonic DRG neurons were transfected with this DN construct by using either Lipofectamine (Invitrogen) or Nucleofector (Amaxa), according to the protocols of the manufacturers. Because of the low efficiency of transfection in primary neuronal cultures, the cationic lipid and electroporation results were combined to acquire the final results.
Determination of RhoA activation by ELISA.
DRG neuron cultures were established as described above. These cultures were serum starved before incubation with different concentrations of anti-ganglioside mAbs (1–100 μg/ml) and patient-derived IgG fractions (1:10 to 1:50 dilution) for various intervals (0–120 min). Cells were lysed, and the level of GTP-bound activated fraction of RhoA was determined according to the instructions of the manufacturer (Cytoskeleton).
Affinity precipitation of GTP-bound RhoA.
DRG cultures could not be used for these studies because a large amount of cell lysates were required for such studies. NG108-15 cell line was used for this purpose. Studies were performed to establish that GD1a/GT1b-2b mAb induces inhibition of neurite outgrowth in this cell line. GTP-bound RhoA was affinity precipitated using a RhoA activation assay kit (Millipore Corporation). NG018-15 cells were grown in DMEM containing 10% FBS and HAT supplement. Twenty-four hours before Ab treatment, cells were maintained in serum-free medium. Cells were treated for 30 min with GD1a/GT1b-2b or sham Ab. After washing, cells were lysed with lysis buffer, and GTP Rho was affinity precipitated using a Rho-binding domain of Rhotekin beads following the instructions of the manufacturer. Bound Rho proteins were detected by Western blotting using a monoclonal Ab against RhoA (Millipore Corporation).
Growth cone collapse assay.
Cells were seeded at low density (1000 cells per well) on glass coverslips in 24-well plates. After 16 h, cells were incubated for 30 min or overnight with GD1a/GT1b-2b (50 μg/ml) or sham Ab (50 μg/ml), fixed, and stained with a monoclonal IgG anti-β-III-tubulin Ab or Alexa Fluor 546-conjugated phalloidin (1:50; Invitrogen). Images were acquired, and growth cones were analyzed for collapse and simplification, i.e., number of filopodia/growth cone. Growth cone area was analyzed by creating a region of interest (ROI) that circumscribed the growth cone. The ROI area was measured with NIH ImageJ for at least 40 neurons in each experiment. The experiments were done three times in duplicate wells.
Xenopus growth cone turning assay.
These studies were done to determine whether the GD1a/GT1b-2b mAb can affect growth cone behavior directly. Ganglioside expression in growth cones of Xenopus spinal neurons (stage 22–23 embryos) was determined by double labeling with GD1a/GT1b-2b (1:100) and polymerized actin with Alexa Fluor 546-conjugated phalloidin (1:50). GD1a/GT1b-2b binding was detected with FITC-conjugated anti-mouse IgG Abs (1:200; Jackson ImmunoResearch). Some cells were treated with chloroform/methanol (1:1 v/v; 30 min at room temperature) to assess whether Ab binding was to solvent extractable glycolipids. Images were acquired with an upright fluorescent microscope (Nikon Eclipse E600) and analyzed with Open Lab software (Improvision).
Spinal neurons from stage 22–23 embryos were plated 14–20 h before turning assays as described (Ming et al., 1999, 2002; Shim et al., 2005). Microscopic gradients of GD1a/GT1b-2b mAb (1–2 mg/ml) and MAG (150 μg/ml), a known guidance cue (repellent) in this model, were generated in the micropipette as described (Shim et al., 2005), with heat-inactivated MAG (150 μg/ml) or control IgG (1–2 mg/ml) as negative controls. Turning assays were performed at 22–25°C, and the turning angle was defined by the angle between the original direction of neurite extension and a straight line connecting the positions of the center of the growth cone at the onset (0 min) and at the end of the 30 min period. Rates of neurite extension were calculated based on the net neurite extension during the assay. Only growth cones of isolated neurons with a net neurite extension of >5 μm over 30 min were included for analysis. Typically, 15–30 neurons were analyzed per condition.
Effect of GD1a/GT1b-2b on axon regeneration in p75-null mice.
Some previous studies suggest that gangliosides use tumor necrosis factor receptor p75 as an adapter molecule to induce intracellular signaling (Yamashita et al., 2002; Fujitani et al., 2005). We examined this hypothesis in our standardized sciatic nerve crush animal model, as described (Lehmann et al., 2007). Left sciatic nerves of homozygous p75-null mice (The Jackson Laboratory) were crushed 35 mm rostral to the middle toe for 30 s with fine forceps on day 0. Animals were administered six doses of GD1a/GT1b-2b (hollow fiber supernatant containing ∼2 mg of mAb per dose) or control Abs (n = 4 each) (2 mg of purified mouse IgGs per dose) on days 3, 5, 7, 9, 11, and 13 after surgery by intraperitoneal route. Nerves were harvested from these animals on day 16 after the surgery and embedded in Epon. Epon-embedded thick and thin cross sections were used for quantifying the number of regenerating myelinated and unmyelinated fibers in nerve segments 1 (sciatic) and 2 (tibial) cm distal to the crush site, as described (Lehmann et al., 2007).
Ganglioside extraction and thin-layer chromatography.
Extraction of gangliosides from embryonic DRG was done as described previously (Schnaar, 1994; Schnaar and Needham, 1994). DRGs from 20 E15 rat embryos were collected, pooled, and homogenized in 0.2 ml of water in a Microfuge tube. Methanol and then chloroform were added to give chloroform/methanol/water (4:8:3), and tissues were then extracted by vortex mixing. After removal of insoluble material by centrifugation, the supernatant was transferred to a fresh Microfuge tube and water was added to give chloroform/methanol/water (4:8:5.6). The biphasic solution was centrifuged again, and the upper phase (containing gangliosides) was loaded onto a tC18 SepPak Light Cartridge. After washing with chloroform/methanol/water (2:43:55) gangliosides were eluted with methanol, the solvent evaporated, and the purified gangliosides were dissolved in a small volume of methanol and analyzed by thin-layer chromatography (TLC) (HPTLC Silica Gel 60; Merck) with chloroform/methanol/0.25% aqueous KCl (60:35:8) as running solvent. Gangliosides were detected with a resorcinol–HCl–Cu2+ reagent. Bovine brain gangliosides were used as standards. Immune overlay with GD1a/GT1b-2b and GM1–2b was performed essentially as described previously (Lopez et al., 2008). Immediately before antibody staining, some TLC plates (as indicated) were treated with Vibrio cholerae sialidase (50 mU/ml) in PBS for 2.5 h at ambient temperature.
Statistical analyses.
Data are presented as means ± SEM. Difference between groups were compared with Student's t test or Scheffé's multiple comparison tests after one-way ANOVA. Statistical significance for growth cone turning assays was assessed using bootstrap test. p < 0.05 was considered significant.
Results
Antibodies inhibit neurite outgrowth of embryonic and postnatal neurons
We initially examined the Ab binding to DRG neurons and found that GD1a/GT1b-2b stains DRG neurons (Fig. 1A,B). Purified ganglioside fractions from embryonic DRG were extracted and analyzed by TLC. As expected for ganglion cells, the ganglioside profile was complex, with ganglioside bands migrating at positions consistent with GD1a and GT1b, as well as other complex gangliosides (GD3, GT1b, GD1b, and GD1a) (Fig. 1C). To confirm that some of these bands represented gangliosides GD1a and GT1b, TLC-resolved embryonic rat DRG gangliosides were subjected to immuno-overlay analysis. Ab GD1a/GT1b-2b stained bands migrating at the level of control GD1a and GT1b, which were greatly diminished by sialidase pretreatment of the plates. Ab GM1–2b detected only trace GM1 in DRG gangliosides but detected bands migrating at the level of GD1a, GD1b, and GT1b after sialidase treatment (which leaves only the internal branched sialic acid on GM1). These data support the conclusion that GD1a and GT1b are expressed in rat embryonic DRGs, as reported previously (Calderon et al., 1995).
Figure 1.

Ganglioside profile of DRG neurons. A, B, Rat embryonic (A) and postnatal (B) DRG neurons immunostained with GD1a/GT1b-2b. Scale bars, 50 μm. C, TLC profile of gangliosides extracted from embryonic rat DRGs (right lane) compared with bovine brain standards (GM1, GD3, GD1a, GD1b, and GT1b; left lane). Gangliosides were detected with resorcinol–HCl–Cu2+ reagent. D, TLC immuno-overlay. Standard brain gangliosides (left lanes) and embryonic DRG gangliosides (right lanes) were resolved on four replicate aluminum-backed TLC plates. After running, the plates were blocked and two plates (as indicated) were incubated for 2.5 h with 50 U/ml sialidase to cleave terminal sialic acids from complex gangliosides, leaving GM1. The other two plates were incubated with buffer alone. After washing, all plates were overlaid as indicated with GD1a/GT1b-2b or GM1–2b overnight, washed, overlaid with secondary antibody (alkaline-phosphatase-conjugated goat anti-mouse IgG), washed, and antibody binding detected with nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate.
Next we show that GD1a/GT1b-2b mAb significantly inhibited the neurite outgrowth of embryonic and postnatal DRG neurons, but GM1–2b did not induce inhibition in this assay (Fig. 2B,D,E). GD1a/GT1b-2b-induced inhibition was dependent on the concentration of the mAb (Fig. 2F). Postnatal DRG neurons show a different outgrowth pattern with multiple neurites, in contrast to the rapid unipolar or bipolar neurite outgrowth of embryonic DRG neurons. Postnatal DRG neurons also showed a decreased neurite growth when treated with GD1a/GT1b-2b, which was assessed by measuring the longest neurite of each cell (Fig. 2G) and number of branches at a defined distance from the cell body (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Neurite outgrowth assay was further conducted on the primary spinal motor neurons to evaluate the Ab effect, and similar inhibition was found on the neurite outgrowth of spinal motor neurons with the mAb GD1a/GT1b-2b (Fig. 2H).
Figure 2.
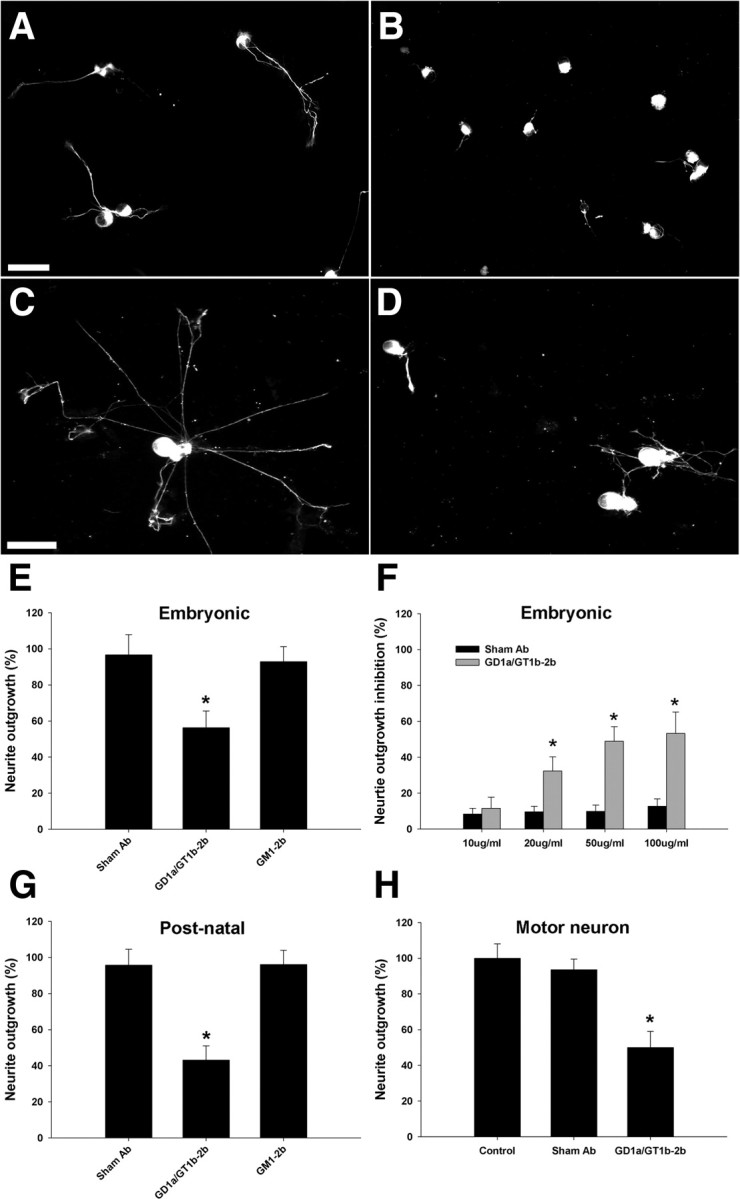
GD1a/GT1b-2b mAb inhibits neurite outgrowth in various primary dissociated neuronal cultures. A, B, Embryonic DRG neurons treated with GD1a/GT1b-2b mAb have shorter neurites (B) compared with sham Ab-treated cells (A). C, D, Postnatal DRG neurons treated with GD1a/GT1b-2b mAb have shorter neurites (D) compared with sham Ab-treated cells (C). Scale bars, 50 μm. E, GD1a/GT1b-2b mAb significantly inhibits neurite outgrowth of embryonic rat DRG neurons, whereas GM1–2b and sham Abs do not inhibit neurite outgrowth. F, GD1a/GT1b-2b mAb induces dose-dependent inhibition of neurite outgrowth in rat embryonic DRG neurons. G, H, GD1a/GT1b-2b mAb inhibits the neurite outgrowth of dissociated rat primary postnatal DRG neurons (G) and spinal motor neurons (H). *p < 0.05. Control, Cultures without sham or anti-ganglioside Ab treatment.
Because dissociated DRG cultures have both neuronal and glial cells, to address whether the Ab-mediated neurite outgrowth is directly inhibited at the level of axon or Schwann cells, Schwann cell-depleted cultures were generated by repeated AraC treatment. Mixed DRG cultures were plated as above for 24 h. These DRG cultures were continuously treated with AraC (10−6 m for 1 week), as described (Melli et al., 2006), and examined immunocytochemically with anti-S100 and anti-β-III-tubulin Abs to assess the presence of Schwann cells. Our results showed that, after AraC treatment, <5% of cells stained for S100 in DRG cultures (Fig. 3A). For inhibition assays, DRG cultures were treated with AraC as above and then incubated with GD1a/GT1b-2b (as above) for 24 h, and neurite length was determined as described above. There was a significant inhibition of neurite outgrowth in these Schwann cell-depleted cultures (Fig. 3B). Because neurite length was assessed at the termination of experiments, the possibility that GD1a/GT1b-2b induces axonal retraction rather than inhibition is not addressed by these experiments. These results support our hypothesis that Ab-mediated inhibition is at the level of the neuron and consistent with the axonal phenotype seen in our previous animal studies (Lehmann et al., 2007).
Figure 3.

GD1a/GT1b-2b mAb inhibits neurite outgrowth of DRG neurons in Schwann cell-depleted culture. A, DRG cultures with and without AraC treatment are then double labeled with neuronal (β-III-tubulin; green) and Schwann cell (S100; red) markers showing that AraC-treated cultures have almost complete elimination of Schwann cells. Scale bar, 50 μm. B, GD1a/GT1b-2b mAb induces significant inhibition of neurite outgrowth compared with sham Ab-treated neurons in Schwann cell-depleted cultures (AraC treated). *p < 0.05. Control, Cultures without sham or anti-ganglioside Ab treatment.
Antibodies engage neuronal/cell-surface gangliosides to induce inhibition
This issue was examined in comparative studies on adult DRG neuron cultures generated from wild-type mice and Galgt1-null mice, which lack complex gangliosides including GD1a and GT1b. These cultures were treated with GD1a/GT1b-2b mAb. We found that Galgt1-null DRG neurons were resistant to the inhibitory effects of GD1a/GT1b-2b mAb (Fig. 4E,F), whereas neurons from wild-type mice showed inhibition similar to rat DRG neuron cultures (Fig. 4B,C). These studies indicate that Abs engage cell-surface glycans to induce inhibition of neurite outgrowth, which is consistent with our previous in vivo results (Lehmann et al., 2007). Studies with Fab fragments showed that, compared with bivalent/native Ab, Fabs did not induce inhibition of neurite outgrowth, suggesting that ganglioside crosslinking may be required for inhibition (Fig. 5). Alternatively, lack of inhibition with Fabs could be attributable to decreased affinity of these monovalent fragments compared with the avidity of parent whole IgG antibody. We found that intracellular inhibitory signaling is not activated in Galgt1-null DRG neurons (see below), which is consistent with our phenotypic data.
Figure 4.

GD1a/GT1b-2b mAb induces inhibition of neurite outgrowth by engaging cell-surface gangliosides. A–C, Wild-type (WT) neurons treated with sham Ab have significantly longer neurites (A, C) compared with GD1a/GT1b-2b-treated group (B, C). D–F, Galgt1-null neurons treated with sham (D, F) and GD1a/GT1b-2b (E, F) Abs have similar neurite length. Scale bar, 50 μm. Control, Cultures without sham or anti-ganglioside Ab treatment.
Figure 5.

Uncleaved GD1a/GT1b-2b mAbs induce significant inhibition of neurite outgrowth in DRG neurons compared with corresponding Fab fragments. *p < 0.05. Control, Cultures without sham or anti-ganglioside Ab treatment.
p75 is not involved in Ab-mediated inhibition of axon regeneration
Because gangliosides are confined to the plasma membrane, it has been suggested that intracellular signaling induced by engaging or crosslinking gangliosides could be via recruitment of adapter proteins with cytoplasmic tails. We examined this issue in a sciatic nerve crush model in p75 knock-out animals. Our animal studies demonstrate that GD1a/GT1b-2b mAbs induce severe inhibition of myelinated nerve fibers/axon regeneration in p75 knock-out animals (Fig. 6B,C). Electron microscopic studies were performed to quantify regeneration of unmyelinated nerve fibers, and we found that GD1a/GT1b-2b mAb induced significant inhibition of these nerve fibers at tibial but not sciatic nerve level (Fig. 6E,F). These findings are consistent with our previous results in wild-type mice (Lehmann et al., 2007). Overall, these results indicate that p75-mediated signaling is not involved in Ab-mediated inhibition of axon regeneration in this model. Therefore, cell culture studies were not undertaken.
Figure 6.
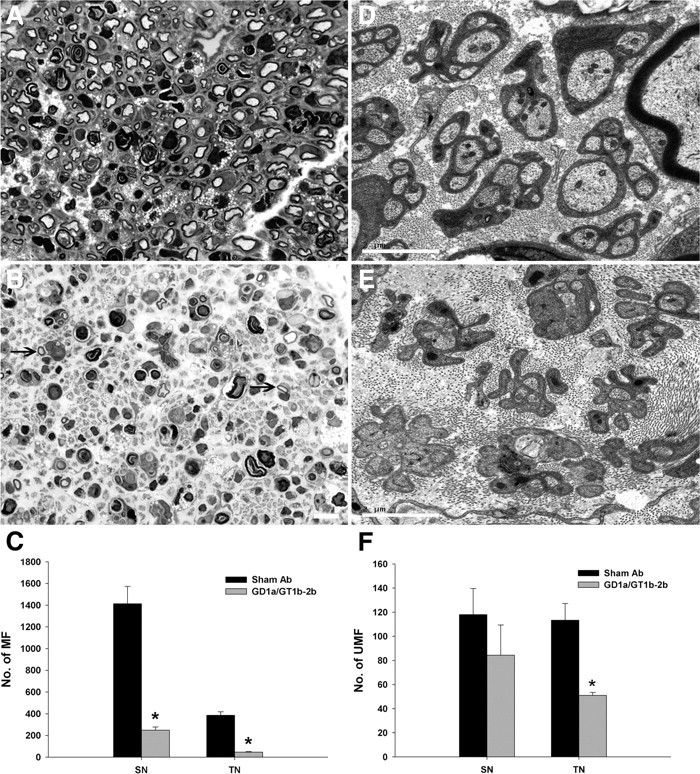
GD1a/GT1b-2b mAb induced inhibition of axon regeneration in p75 knock-out mice. A, Light micrographs showing many regenerating myelinated fibers (MF) in sham Ab-treated p75 knock-out animals. B, GD1a/GT1b-2b-treated p75 knock-out mice showed severe inhibition of axon regeneration and only rare regenerating myelinated fibers (arrows) were found. Scale bar, 20 μm. C, Numbers of regenerating myelinated fibers in GD1a/GT1b-2b-treated (gray bars; n = 4 each) sciatic (249 ± 29) (SN) and tibial (47 ± 5) (TN) nerves were significantly reduced compared with sham Ab-treated (black bars; n = 4 each) sciatic (1416 ± 166) and tibial (387 ± 32) nerves. D, Electron micrograph showing several unmyelinated fibers (UMF) are present in the tibial nerve of sham Ab-treated p75 knock-out animals. E, Electron micrograph showing both denervated Schwann cell profiles without regenerating sprouts and some Schwann cell profiles containing small number of regenerating unmyelinated fibers in GD1a/GT1b-2b-treated p75 mice. Scale bar, 2 μm. F, The number of regenerating unmyelinated fibers in GD1a/GT1b-2b-treated nerves was significantly decreased in tibial nerve and nonsignificantly reduced in sciatic nerve compared with those in sham Ab-treated nerves. *p < 0.05.
GD1a/GT1b-2b mAb activates RhoA and ROCK to induce inhibition of neurite outgrowth
The activation of RhoA and its downstream effector ROCK leads to growth cone collapse and neurite growth arrest, a mechanism that has been extensively studied during the intracellular signaling of the myelin-dependent neurite outgrowth inhibitors MAG, Nogo, and OMgp (oligodendrocyte–myelin glycoprotein) (Grados-Munro and Fournier, 2003; McGee and Strittmatter, 2003). We asked whether RhoA and ROCK signaling is involved in Ab-mediated inhibition of neurite outgrowth.
Initially, we examined the activation of RhoA in DRG neuronal lysates by ELISA. We found that the incubation of DRG neurons with GD1a/GT1b-2b mAb significantly increases the active form of RhoA (Fig. 7A,B) compared with GM1-2b and sham Abs (data not shown). This increase in RhoA was time and Ab concentration dependent (Fig. 7A,B). This was further confirmed by immunoprecipitation in a pull-down assay. For these studies, we used a neural (NG108-15) cell line because DRG neurons have relatively low levels of RhoA and extremely large quantities of cell lysates are needed for the pull-down assay. In separate studies, we confirmed that GD1a/GT1b-2b mAb inhibits extension of neurites by these cells (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material). Furthermore, NG108-15 is a cell line that can be easily cultured under serum-free conditions. Measurement of active levels of RhoA revealed that GTP-bound RhoA levels were increased by incubation of these cells with GD1a/GT1b-2b (Fig. 7C).
Figure 7.

GD1a/GT1b-2b mAbs induce activation of RhoA by engaging specific cell-surface gangliosides. A, B, GD1a/GT1b-2b mAb induce RhoA activation in DRG neurons, which is concentration (A) and time (B) dependent. RhoA activation was assessed at 30 min after antibody incubation in studies shown in A, and antibody concentration was 100 μg/ml for studies shown in B. OD, Optical density. *p < 0.05. C, Increased amount of GTP-bound RhoA was immunoprecipitated in GD1a/GT1b-2b-treated NG108–15 cells compared with sham Ab-treated cells. GST-RBD, Glutathione S-transferase/Rho-binding domain. D, GD1a/GT1b-2b mAb induces significant activation of RhoA in wild-type (black bars; WT) but not in Galgt1-null (gray bars) DRG neurons. Sham Ab did not induce RhoA activation in wild-type or Galgt1-null DRG neurons. *p < 0.05.
RhoA activation was also examined in wild-type and Galgt1-null mice to link ganglioside expression with activation of intracellular signaling. We found that there was a significant increase in the active form of RhoA in wild-type DRG neurons, whereas Galgt1-null DRG neurons did not show an increase in RhoA activation after treatment with GD1a/GT1b-2b (Fig. 7D).
To directly link the Ab-induced activation of RhoA and ROCK with inhibition of neurite outgrowth, molecular and pharmacological approaches were used to prevent the activation of these molecules to abrogate the deleterious effects of the Abs. Molecular studies with DN RhoA showed that DRG neurons transfected with this construct were not inhibited by GD1a/GT1b-2b mAb (Fig. 8A,B). To inactivate RhoA and ROCK pharmacologically, C3-transferase (a molecule that ADP ribosylates RhoA) and Y27632 (well characterized inhibitor) were used, respectively. We found that C3-transferase and Y27632 reversed the inhibitory effects of GD1a/GT1b-2b mAb. Our results showed that Y27632 abolished the inhibitory effect of GD1a/GT1b-2b completely, whereas C3-transferase reversed the inhibitory effects by ∼50% (Fig. 8C). The reversal of inhibition with C3-transferase was less than that seen with Y27632, and this is most likely attributable to relatively lower cell permeability of C3-transferase.
Figure 8.

GD1a/GT1b-2b mAbs inhibit neurite outgrowth through activation of RhoA and ROCK. A, DRG neurons transfected with GFP-tagged DN RhoA construct (green) are resistant to the inhibitory effects of GD1a/GT1b-2b mAb compared with nontransfected cells; transfected and nontransfected DRG neurons were identified by β-III-tubulin staining (red). Scale bar, 50 μm. B, GD1a/GT1b-2b-mediated inhibition of neurite outgrowth was significantly reversed in neurons transfected (gray bars) with DN RhoA compared with nontransfected cells (black bars); sham Ab had no effect on transfected and nontransfected cells. C, C3-transferase (RhoA inhibitor) partially reverses GD1a/GT1b-2b mAb induced inhibition, whereas Y27632 (ROCK inhibitor) completely reverses this inhibition. *p < 0.05.
GD1a/GT1b-2b modulates growth cone morphology and behavior
Growth cone morphology was examined in DRG cultures grown for 24 h and treated short term (30 min) with GD1a/GT1b-2b mAb. Our results show that, within 30 min, there is a significant decrease in growth cone area and the number of filopodia/growth cone in GD1a/GT1b-2b-treated group compared with controls (Fig. 9A–D). After overnight incubation with GD1a/GT1b-2b, there was a significant increase in number of collapsed growth cones (Fig. 9E).
Figure 9.

GD1a/GT1b-2b mAb induces growth cone collapse in DRG neurons. A, B, Representative images of the growth cones of DRG neurons treated with GD1a/GT1b-2b (B) and sham Abs (A) showing growth cone collapse and loss of filopodia. Scale bar, 5 μm. C, Quantitative analysis showed that GD1a/GT1b-2b mAb-treated DRG neurons (30 min) had significant decrease in growth cone area compared with sham Ab-treated group. D, The number of filopodia/growth cone in GD1a/GT1b-2b-treated group (30 min) was significantly reduced compared with sham Ab-treated neurons. E, There was a significant increase in the number of collapsed growth cones in DRG neurons treated with GD1a/GT1b-2b mAb (overnight) compared with sham Ab or control (no Ab treatment) groups. *p < 0.05.
Xenopus studies show that GD1a/GT1b-2b bound to the Xenopus spinal neurons, including their growth cones (Fig. 10A). Immunostaining with this mAb was significantly reduced with chloroform/methanol treatment, indicating that these Abs bind glycolipids moieties in Xenopus spinal neurons (data not shown). To directly test the function of gangliosides in growth cone motility, we performed a growth cone turning assay. Consistent with the notion that localized growth cone collapse leads to growth cone repulsive response, we noted that Xenopus spinal neurons had repulsive turning responses to a gradient of GD1a/GT1b-2b mAb comparable with and similar to MAG-induced repulsive responses (Fig. 10B,C). Control IgG did not affect growth cone behavior in turning assays (data not shown).
Figure 10.
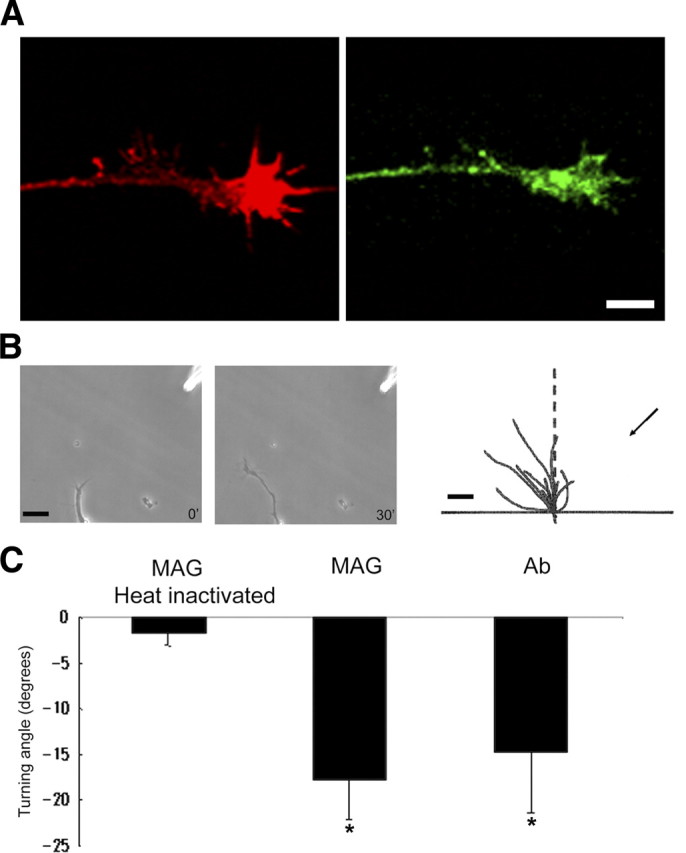
GD1a/GT1b-2b mAbs induce repulsive growth cone turning response. A, Xenopus spinal neuron growth cone staining with phalloidin (actin; red) and GD1a/GT1b-2b (green) shows that these neurons express ganglioside moieties. Scale bar, 5 μm. B, Growth cone turning responses to a gradient of GD1a/GT1b-2b mAb in the pipette (at top right corner) at the start (0 min) and end (30 min) of exposure. Superimposed trajectories of neurite extension during this period for a sample of 10 neurons are shown on the right. Scale bars: 20 and 10 μm in left two and right panels, respectively. C, Significant growth cone chemorepulsion with gradients of MAG and GD1a/GT1b-2b mAb compared with heat- inactivated MAG. *p < 0.05.
GBS IgG-mediated inhibition of neurite outgrowth is through activation of RhoA pathway
Studies with patient IgG fractions showed that five of seven patient Abs induced significant inhibition of neurite outgrowth compared with control IgGs (Fig. 11C). The extent of inhibition induced by different sera/IgG fractions was variable. The IgG fraction-mediated inhibition was not seen in DRG cultures from mice lacking complex gangliosides (Fig. 11D), indicating that anti-ganglioside Abs in the IgG fractions mediate inhibitory effects.
Figure 11.

Purified IgG fractions from GBS sera inhibit neurite outgrowth through activation of RhoA and ROCK. A, B, DRG neurons treated with GBS IgG fractions have much shorter neurites (B) compared with control IgG-treated neurons (A). Scale bar, 50 μm. C, Five of seven GBS IgG fractions induce significant (but variable) inhibition of the neurite outgrowth. D, IgG fractions purified from GBS patient (PS1; gray bar) induce significant inhibition of neurite outgrowth in wild-type (WT) DRG neurons but not in the Galgt1-null neurons; control IgG fractions (CS; black bar) do not modulate the growth behavior of wild-type and Galgt1-null neurons. E, IgG fractions from six of seven GBS sera can induce significant but variable activation of RhoA in DRG neurons compared with control IgG. F, Treatment with C3-transferase (RhoA inhibitor) and Y27632 (ROCK inhibitor) significantly reverse the inhibitory effects of GBS IgG fractions (PS1). *p < 0.05.
Short-term incubation (30 min) of DRG cultures with GBS IgG fractions induced significant but variable amount of activation of RhoA in six of seven patients compared with control IgGs as determined by ELISA (Fig. 11E). One GBS IgG fraction (PS7) that did not induce elevation of RhoA also failed to inhibit neurite outgrowth. Another GBS IgG fraction (PS5) induced statistically significant activation of RhoA but did not inhibit neurite outgrowth, but this patient's IgGs induced lowest relative activation of RhoA among the positive samples tested in RhoA activation assay (Fig. 11E).
Functional studies showed that pharmacological inhibitors of RhoA and ROCK significantly reversed patient IgG fraction-mediated inhibition of neurite outgrowth (Fig. 11F). Overall, these studies show that the inhibition induced by GBS IgG fractions is via engagement of cell-surface gangliosides and activation of RhoA and its downstream effector ROCK.
Discussion
This study shows that monoclonal and GBS patient-derived anti-ganglioside Abs inhibit neurite outgrowth in primary neuronal cultures. In our in vitro models, engagement of specific cell-surface gangliosides was necessary for Ab-mediated inhibition of neurite outgrowth. In vivo studies demonstrate that p75 was not involved in Ab-induced inhibition of axon regeneration. Our results indicate that Ab binding to cell-surface glycans leads to outside-in signaling and activation of RhoA and its downstream effector ROCK. These studies suggest that, at a subcellular level, modulation of growth cone morphology and/or growth behavior is involved in inhibition of neurite outgrowth. The current study links autoimmune Abs seen in some patients with GBS and other neuroimmunological disorders to an intracellular signaling pathway, which is important in regulation of neurite/growth cone extension.
The cell culture studies linking anti-ganglioside Abs to activation of a known inhibitory pathway that prevents axon regeneration extends the previous clinical and experimental observations. Several clinical studies indicate that anti-ganglioside Abs are associated with poor/incomplete recovery (Ilyas et al., 1992; Gregson et al., 1993; Simone et al., 1993; Yuki et al., 1993; Jacobs et al., 1996; Bech et al., 1997; Kuwabara et al., 1998a,b; Carpo et al., 1999; Press et al., 2001; Annunziata et al., 2003; Koga et al., 2003; Kaida et al., 2007). The patients with incomplete recovery almost always have some degree of failure of nerve repair/axon regeneration and target reinnervation (Brown and Feasby, 1984). These clinical observations suggest that anti-ganglioside Abs can adversely affect the nerve repair process in this disease. This hypothesis was strongly supported by our previous studies indicating that experimental and patient-derived anti-ganglioside Abs inhibit axon regeneration in an animal model (Lehmann et al., 2007; Lopez et al., 2010). The current study supports the notion that impaired nerve repair induced by autoantibodies is one mechanism of poor recovery in GBS.
Not all anti-ganglioside Abs induce inhibition in our assays as indicated by negative results with GM1-2b mAb and two patient sera with anti-GD1a Abs. We found that GM1-2b mAb did not induce inhibition of neurite outgrowth or activate RhoA (data not shown). This is consistent with our previous studies showing that this mAb binds to both motor and sensory nerve fibers (Gong et al., 2002) but does not induce inhibition in an animal model (Lopez et al., 2010). We believe that this is very likely attributable to the low affinity of this mAb (Lopez et al., 2010). In contrast, GD1a/GT1b-2b mAb induced inhibition of neurite outgrowth and associated RhoA and ROCK activation. Our previous studies show that this mAb binds to both motor and sensory fibers and severely inhibits the regeneration of these fibers in animal studies (Gong et al., 2002; Lehmann et al., 2007). We have linked this mAb to inhibition of axon regeneration via GD1a in animal studies with transgenic Siat8a-null mice that only express a-series gangliosides but not b-series gangliosides, including GT1b. The studies with patient materials show that five of seven sera with IgG anti-GD1a reactivity inhibited neurite outgrowth and activated RhoA. One patient serum did not induce inhibition or activation of RhoA, and another serum induced mild activation of RhoA but failed to inhibit neurite outgrowth. These findings indicate that activation of RhoA is central to the inhibition of neurite outgrowth. Failure of some sera to induce inhibition of neurite outgrowth is compatible with previous work indicating that anti-ganglioside Ab-mediated pathogenecity is quite complex. Anti-glycan Abs with specificity for a ganglioside are not created equally, and these Abs could be different in terms of fine specificity, tissue binding patterns, affinity/avidity, Fc-based effector functions, pathogenecity in experimental models, and other as yet undefined properties (Sheikh and Zhang, 2010;Lopez et al., 2010). Issues such as fine specificity or affinity could not be addressed in this study because of the small amount of serum available to us.
Notably, the anti-glycan Ab-induced morphological and functional changes in the growth cones of primary neurons mimicked those produced by CNS myelin inhibitors. Ab-mediated morphological changes included growth cone simplification and collapse, and similar changes are also produced by CNS myelin/inhibitors of axon regeneration in primary neuronal cultures (Jin and Strittmatter, 1997; Kuhn et al., 1999). Our in vitro findings, which showed growth cone collapse, are paradoxical to our animal studies demonstrating that anti-glycan Abs induce dystrophic bulbs reminiscent of stalled growth cones (Lehmann et al., 2007). Similar disparity of growth cone morphology in cell culture and animal studies is also seen with CNS myelin inhibitors (Silver and Miller, 2004; Tom et al., 2004). We believe that one factor that may contribute to this disparate behavior of growth cones is that, in cell culture experiments, the inhibitory ligands (Abs or CNS myelin inhibitors) are either in a soluble state or, if substrate bound, have low effective density compared with animal paradigms, in which these ligands are more likely to be substrate bound (e.g., degenerating myelin) and have higher density (multimeric) interactions with their receptors on neuronal surfaces. An alternative explanation for this paradox is different times of antibody exposure in animal and cell culture studies. We did not determine the time course of dystrophic growth cone formation in our previous animal studies (Lehmann et al., 2007). However, there are animal data indicating that, in the context of spinal cord injury, dystrophic growth cones can form within 1 d (Tom et al., 2004). Additional studies are necessary to address this interesting issue. The concept that anti-glycan Abs mimic CNS inhibitors of axon regeneration is further supported by the growth cone turning assays, showing that gradients of GD1a/GT1b-2b mAb induced repulsive turning response, which is also known to be induced by CNS myelin inhibitors (Wong et al., 2002). Overall, these findings would support the notion that inhibition of neurite outgrowth in our assays is at least partially mediated via deleterious effects of Abs on the growth cones.
It is not surprising that anti-glycan Abs induced activation of RhoA and ROCK to induce inhibitory/repulsive influences on growth cones because small GTPases of the Rho family, including RhoA, Rac1, and Cdc42, are central regulators of growth cone extension, which strictly depend on polymerization and organization of actin filaments (Dickson, 2001; Etienne-Manneville and Hall, 2002; Luo, 2000, 2002; Huber et al., 2003). There is a broad consensus that small GTPase RhoA is central to secondary signaling induced by almost all CNS inhibitors of axon regeneration (Jin and Strittmatter, 1997; Kuhn et al., 1999; Lehmann et al., 1999; Joester and Faissner, 2001; Vinson et al., 2001; Dergham et al., 2002; Ellezam et al., 2002; Monnier et al., 2003). Activation of RhoA prevents growth cone extension and it stimulates actinomyosin contractility and stress fiber formation via activation of ROCK (Dickson, 2001; Luo, 2002). Furthermore, several studies showed that either pharmacologic or genetic approaches inhibiting activation of RhoA or ROCK completely reverse CNS myelin-mediated inhibition in neurite outgrowth assays, and the pharmacologic inhibitors of RhoA and ROCK have also enhanced axonal regeneration, albeit marginally, in spinal cord injury models (Jin and Strittmatter, 1997; Kuhn et al., 1999; Lehmann et al., 1999; Vinson et al., 2001; Dergham et al., 2002; Ellezam et al., 2002; Winton et al., 2002; Fournier et al., 2003). Two previous studies, in the context of MAG, which also showed that the inhibition of neurite outgrowth through the gangliosides is via activation of RhoA and ROCK (Vinson et al., 2001; Yamashita et al., 2002; Mehta et al., 2007), support our findings. We suspect that Ab-mediated chemorepulsive response in growth cone turning assays are attributable to activation of the RhoA pathway because previous studies have shown involvement of small GTPases/RhoA in growth cone turning responses (Song and Poo, 2001; Yuan et al., 2003; Jin et al., 2005).
How is it that the signals from gangliosides in the outer leaflet of the plasma membrane are transduced to the inner leaflet of the cell membrane in which signal-transducing proteins, such as RhoA, reside or are recruited? Our studies with Galgt1-null neurons demonstrate that inhibition is dependent on engagement of cell-surface gangliosides by the Abs. The studies with Fab fragments indicate that crosslinking cell-surface gangliosides is important in inducing inhibition of neurite outgrowth. It is not clear how the crosslinking of gangliosides on outer leaflets of the plasma membrane transduces RhoA activation intracellularly because these glycolipids are confined to the outer leaflets of plasma membranes. Recruitment of transmembrane adapter protein(s) is one possible mechanism that could transduce intracellular signaling. This is well exemplified in the case of CNS myelin inhibitors, which bind to glycosylphosphatidylinositol-linked Nogo receptors requiring a neuronal coreceptor for signal transduction. So far, two members of the TNFR family, p75 and/or TROY, have been shown to be necessary for inhibitory signaling (Wang et al., 2002; Wong et al., 2002; Yamashita et al., 2002; Park et al., 2005; Shao et al., 2005). Two previous in vitro studies, in the context of CNS inhibition, reported that p75 can complex with gangliosides GD1a and/or GT1b to transduce inhibitory signaling (Yamashita et al., 2002; Fujitani et al., 2005). Another study examining the same issue did not find involvement of p75 in ganglioside-mediated inhibition of CNS and PNS neurons (Mehta et al., 2007). Our animal studies in p75 knock-out animals indicate that this molecule is not involved in GD1a/GT1b-2b mAb-mediated inhibition of PNS axon regeneration. An alternate model proposes that modulation of glycans, including gangliosides, in lipid rafts can by itself transduce intracellular signaling, a concept termed glycosynapse (Hakomori, 2002). This model, however, does not explain how the signals from gangliosides in the outer leaflet of plasma membrane are transduced to the inner leaflet of the cell membrane in which signal-transducing proteins reside. Whether or not adapter molecule(s) are involved in the anti-ganglioside Ab-mediated activation of intracellular RhoA remains to be determined.
In summary, our studies link disease-relevant and patient-derived Abs to inhibition of nerve repair in cell culture models. These studies have allowed dissection of molecular mechanisms that contribute to unsuccessful nerve repair and recovery in patients with Ab-mediated immune neuropathies. Identification of signaling molecules, which prevent successful regeneration, may allow development of novel therapeutic strategies to enhance nerve repair in patients with GBS and those with other immune neuropathies.
Footnotes
This study was supported by National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke Grants NS42888 and NS54962 and the Guillain Barré Syndrome/Chronic Inflammatory Demyelinating Polyneuropathy Foundation. R.L.S. is supported by NIH Grant NS37096.
References
- Annunziata P, Figura N, Galli R, Mugnaini F, Lenzi C. Association of anti-GM1 antibodies but not of anti-cytomegalovirus, Campylobacter jejuni and Helicobacter pylori IgG, with a poor outcome in Guillain-Barre syndrome. J Neurol Sci. 2003;213:55–60. doi: 10.1016/s0022-510x(03)00149-7. [DOI] [PubMed] [Google Scholar]
- Bech E, Orntoft TF, Andersen LP, Skinhøj P, Jakobsen J. IgM anti-GM1 antibodies in the Guillain-Barre syndrome: a serological predictor of the clinical course. J Neuroimmunol. 1997;72:59–66. doi: 10.1016/s0165-5728(96)00145-2. [DOI] [PubMed] [Google Scholar]
- Brown WF, Feasby TE. Conduction block and denervation in Guillain-Barre polyneuropathy. Brain. 1984;107:219–239. doi: 10.1093/brain/107.1.219. [DOI] [PubMed] [Google Scholar]
- Calderon RO, Attema B, DeVries GH. Lipid composition of neuronal cell bodies and neurites from cultured dorsal root ganglia. J Neurochem. 1995;64:424–429. doi: 10.1046/j.1471-4159.1995.64010424.x. [DOI] [PubMed] [Google Scholar]
- Carpo M, Pedotti R, Allaria S, Lolli F, Matà S, Cavaletti G, Protti A, Pomati S, Scarlato G, Nobile-Orazio E. Clinical presentation and outcome of Guillain-Barre and related syndromes in relation to anti-ganglioside antibodies. J Neurol Sci. 1999;168:78–84. doi: 10.1016/s0022-510x(99)00173-2. [DOI] [PubMed] [Google Scholar]
- Dahms NM, Schnaar RL. Ganglioside composition is regulated during differentiation in the neuroblastoma X glioma hybrid cell line NG108–15. J Neurosci. 1983;3:806–817. doi: 10.1523/JNEUROSCI.03-04-00806.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Rho GTPases in growth cone guidance. Curr Opin Neurobiol. 2001;11:103–110. doi: 10.1016/s0959-4388(00)00180-x. [DOI] [PubMed] [Google Scholar]
- Eldridge CF, Bunge MB, Bunge RP, Wood PM. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J Cell Biol. 1987;105:1023–1034. doi: 10.1083/jcb.105.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellezam B, Dubreuil C, Winton M, Loy L, Dergham P, Sellés-Navarro I, McKerracher L. Inactivation of intracellular Rho to stimulate axon growth and regeneration. Prog Brain Res. 2002;137:371–380. doi: 10.1016/s0079-6123(02)37028-6. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujitani M, Kawai H, Proia RL, Kashiwagi A, Yasuda H, Yamashita T. Binding of soluble myelin-associated glycoprotein to specific gangliosides induces the association of p75NTR to lipid rafts and signal transduction. J Neurochem. 2005;94:15–21. doi: 10.1111/j.1471-4159.2005.03121.x. [DOI] [PubMed] [Google Scholar]
- Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA. Localization of major gangliosides in the PNS: implications for immune neuropathies. Brain. 2002;125:2491–2506. doi: 10.1093/brain/awf258. [DOI] [PubMed] [Google Scholar]
- Grados-Munro EM, Fournier AE. Myelin-associated inhibitors of axon regeneration. J Neurosci Res. 2003;74:479–485. doi: 10.1002/jnr.10803. [DOI] [PubMed] [Google Scholar]
- Gregson NA, Koblar S, Hughes RA. Antibodies to gangliosides in Guillain-Barre syndrome: specificity and relationship to clinical features. Quart J Med. 1993;86:111–117. [PubMed] [Google Scholar]
- Hakomori SI. Inaugural article: the glycosynapse. Proc Natl Acad Sci U S A. 2002;99:225–232. doi: 10.1073/pnas.012540899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366:1653–1666. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- Ilyas AA, Mithen FA, Dalakas MC, Chen ZW, Cook SD. Antibodies to acidic glycolipids in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 1992;107:111–121. doi: 10.1016/0022-510x(92)90217-9. [DOI] [PubMed] [Google Scholar]
- Jacobs BC, van Doorn PA, Schmitz PI, Tio-Gillen AP, Herbrink P, Visser LH, Hooijkass H, van der Meché FG. Campylobacter jejuni intections and anti-GM1 antibodies in Guillain-Barré syndrome. Ann Neurol. 1996;40:181–187. doi: 10.1002/ana.410400209. [DOI] [PubMed] [Google Scholar]
- Jin M, Guan CB, Jiang YA, Chen G, Zhao CT, Cui K, Song YQ, Wu CP, Poo MM, Yuan XB. Ca2+-dependent regulation of rho GTPases triggers turning of nerve growth cones. J Neurosci. 2005;25:2338–2347. doi: 10.1523/JNEUROSCI.4889-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Strittmatter SM. Rac1 mediates collapsin-1-induced growth cone collapse. J Neurosci. 1997;17:6256–6263. doi: 10.1523/JNEUROSCI.17-16-06256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joester A, Faissner A. The structure and function of tenascins in the nervous system. Matrix Biol. 2001;20:13–22. doi: 10.1016/s0945-053x(00)00136-0. [DOI] [PubMed] [Google Scholar]
- Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M, Kusunoki S. Anti-ganglioside complex antibodies associated with severe disability in GBS. J Neuroimmunol. 2007;182:212–218. doi: 10.1016/j.jneuroim.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Koga M, Yuki N, Hirata K, Morimatsu M, Mori M, Kuwabara S. Anti-GM1 antibody IgG subclass: a clinical recovery predictor in Guillain-Barre syndrome. Neurology. 2003;60:1514–1518. doi: 10.1212/01.wnl.0000061615.77865.83. [DOI] [PubMed] [Google Scholar]
- Kuhn TB, Brown MD, Wilcox CL, Raper JA, Bamburg JR. Myelin and collapsin-1 induce motor neuron growth cone collapse through different pathways: inhibition of collapse by opposing mutants of rac1. J Neurosci. 1999;19:1965–1975. doi: 10.1523/JNEUROSCI.19-06-01965.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara S, Asahina M, Koga M, Mori M, Yuki N, Hattori T. Two patterns of clinical recovery in Guillain-Barre syndrome with IgG anti-GM1 antibody. Neurology. 1998a;51:1656–1660. doi: 10.1212/wnl.51.6.1656. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, Noda M. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barre syndrome. Ann Neurol. 1998b;44:202–208. doi: 10.1002/ana.410440210. [DOI] [PubMed] [Google Scholar]
- Lehmann HC, Lopez PH, Zhang G, Ngyuen T, Zhang J, Kieseier BC, Mori S, Sheikh KA. Passive immunization with anti-ganglioside antibodies directly inhibits axon regeneration in an animal model. J Neurosci. 2007;27:27–34. doi: 10.1523/JNEUROSCI.4017-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19:7537–7547. doi: 10.1523/JNEUROSCI.19-17-07537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez PH, Zhang G, Bianchet MA, Schnaar RL, Sheikh KA. Structural requirements of anti-GD1a antibodies determine their target specificity. Brain. 2008;131:1926–1939. doi: 10.1093/brain/awn074. [DOI] [PubMed] [Google Scholar]
- Lopez PH, Zhang G, Zhang J, Lehmann HC, Griffin JW, Schnaar RL, Sheikh KA. Passive transfer of IgG anti-GM1 antibodies impairs peripheral nerve repair. J Neurosci. 2010;30:9533–9541. doi: 10.1523/JNEUROSCI.2281-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn MP, Johnson LA, Fromholt SE, Itonori S, Huang J, Vyas AA, Hildreth JE, Griffin JW, Schnaar RL, Sheikh KA. High-affinity anti-ganglioside IgG antibodies raised in complex ganglioside knockout mice: reexamination of GD1a immunolocalization. J Neurochem. 2000;75:404–412. doi: 10.1046/j.1471-4159.2000.0750404.x. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26:193–198. doi: 10.1016/S0166-2236(03)00062-6. [DOI] [PubMed] [Google Scholar]
- Mehta NR, Lopez PH, Vyas AA, Schnaar RL. Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melli G, Keswani SC, Fischer A, Chen W, Höke A. Spatially distinct and functionally independent mechanisms of axonal degeneration in a model of HIV-associated sensory neuropathy. Brain. 2006;129:1330–1338. doi: 10.1093/brain/awl058. [DOI] [PubMed] [Google Scholar]
- Ming GL, Wong ST, Henley J, Yuan XB, Song HJ, Spitzer NC, Poo MM. Adaptation in the chemotactic guidance of nerve growth cones. Nature. 2002;417:411–418. doi: 10.1038/nature745. [DOI] [PubMed] [Google Scholar]
- Ming G, Song H, Berninger B, Inagaki N, Tessier-Lavigne M, Poo M. Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron. 1999;23:139–148. doi: 10.1016/s0896-6273(00)80760-6. [DOI] [PubMed] [Google Scholar]
- Monnier PP, Sierra A, Schwab JM, Henke-Fahle S, Mueller BK. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci. 2003;22:319–330. doi: 10.1016/s1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Press R, Matá S, Lolli F, Zhu J, Andersson T, Link H. Temporal profile of anti-ganglioside antibodies and their relation to clinical parameters and treatment in Guillain-Barre syndrome. J Neurol Sci. 2001;190:41–47. doi: 10.1016/s0022-510x(01)00580-9. [DOI] [PubMed] [Google Scholar]
- Rees JH, Thompson RD, Smeeton NC, Hughes RA. Epidemiological study of Guillain-Barre syndrome in south east England. J Neurol Neurosurg Psychiatry. 1998;64:74–77. doi: 10.1136/jnnp.64.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barre syndrome. Philadelphia: Davis; 1991. [Google Scholar]
- Ruchhoeft ML, Ohnuma S, McNeill L, Holt CE, Harris WA. The neuronal architecture of Xenopus retinal ganglion cells is sculpted by rho-family GTPases in vivo. J Neurosci. 1999;19:8454–8463. doi: 10.1523/JNEUROSCI.19-19-08454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL. Isolation of glycosphingolipids. Methods Enzymol. 1994;230:348–370. doi: 10.1016/0076-6879(94)30024-0. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Needham LK. Thin-layer chromatography of glyosphingolipids. Methods Enzymol. 1994;230:371–389. doi: 10.1016/0076-6879(94)30025-9. [DOI] [PubMed] [Google Scholar]
- Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G. An update on pathobiologic roles of anti-glycan antibodies in Guillain-Barré syndrome. F1000 Biol Rep. 2010;2:21. doi: 10.3410/B2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A. 1999;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Goh EL, Ge S, Sailor K, Yuan JP, Roderick HL, Bootman MD, Worley PF, Song H, Ming GL. XTRPC1-dependent chemotropic guidance of neuronal growth cones. Nat Neurosci. 2005;8:730–735. doi: 10.1038/nn1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Simone IL, Annunziata P, Maimone D, Liguori M, Leante R, Livrea P. Serum and CSF anti-GM1 antibodies in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 1993;114:49–55. doi: 10.1016/0022-510x(93)90048-4. [DOI] [PubMed] [Google Scholar]
- Song H, Poo M. The cell biology of neuronal navigation. Nat Cell Biol. 2001;3:E81–E88. doi: 10.1038/35060164. [DOI] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci. 2004;24:6531–6539. doi: 10.1523/JNEUROSCI.0994-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Mobley BC, Hiller A, Feldman EL. IGF-I prevents glutamate-induced motor neuron programmed cell death. Neurobiol Dis. 2004;16:407–416. doi: 10.1016/j.nbd.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Vinson M, Strijbos PJ, Rowles A, Facci L, Moore SE, Simmons DL, Walsh FS. Myelin-associated glycoprotein interacts with ganglioside GT1b. A mechanism for neurite outgrowth inhibition. J Biol Chem. 2001;276:20280–20285. doi: 10.1074/jbc.M100345200. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Willison HJ. The immunobiology of Guillain-Barre syndromes. J Peripher Nerv Syst. 2005;10:94–112. doi: 10.1111/j.1085-9489.2005.0010202.x. [DOI] [PubMed] [Google Scholar]
- Willison HJ, O'Hanlon GM. The immunopathogenesis of Miller Fisher syndrome. J Neuroimmunol. 1999;100:3–12. doi: 10.1016/s0165-5728(99)00213-1. [DOI] [PubMed] [Google Scholar]
- Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–2625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Dubreuil CI, Lasko D, Leclerc N, McKerracher L. Characterization of new cell permeable C3-like proteins that inactivate Rho and stimulate neurite outgrowth on inhibitory substrates. J Biol Chem. 2002;277:32820–32829. doi: 10.1074/jbc.M201195200. [DOI] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM. A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci. 2002;5:1302–1308. doi: 10.1038/nn975. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Xie X, Li L, Ledeen RW. Mutant NG108–15 cells (NG-CR72) deficient in GM1 synthase respond aberrantly to axonogenic stimuli and are vulnerable to calcium-induced apoptosis: they are rescued with LIGA-20. J Neurochem. 2001;76:690–702. doi: 10.1046/j.1471-4159.2001.00036.x. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Higuchi H, Tohyama M. The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol. 2002;157:565–570. doi: 10.1083/jcb.200202010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol. 2003;5:38–45. doi: 10.1038/ncb895. [DOI] [PubMed] [Google Scholar]
- Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barre and Fisher syndromes. Lancet Infect Dis. 2001;1:29–37. doi: 10.1016/S1473-3099(01)00019-6. [DOI] [PubMed] [Google Scholar]
- Yuki N, Yamada M, Sato S, Ohama E, Kawase Y, Ikuta F, Miyatake T. Association of IgG anti-GD1a antibody with severe Guillain-Barre syndrome. Muscle Nerve. 1993;16:642–647. doi: 10.1002/mus.880160610. [DOI] [PubMed] [Google Scholar]
- Zhang G, Lopez PH, Li CY, Mehta NR, Griffin JW, Schnaar RL, Sheikh KA. Anti-ganglioside antibody-mediated neuronal cytotoxicity and its protection by intravenous immunoglobulin: implications for immune neuropathies. Brain. 2004;127:1085–1100. doi: 10.1093/brain/awh127. [DOI] [PubMed] [Google Scholar]