

Abstract
Leptin regulates body weight in mice by decreasing appetite and increasing sympathetic nerve activity (SNA), which increases energy expenditure in interscapular brown adipose tissue (iBAT). Diet-induced obese mice (DIO) are resistant to the anorectic actions of leptin. We evaluated whether leptin still stimulated sympathetic outflow in DIO mice. We measured iBAT temperature as a marker of SNA. We found that obese hyperleptinemic mice have higher iBAT temperature than mice on regular diet. Conversely, obese leptin-deficient ob/ob mice have lower iBAT temperature. Additionally, leptin increased SNA in obese (DIO and ob/ob) and control mice, despite DIO mice being resistant to anorectic action of leptin. We demonstrated that neurons in the dorsomedial hypothalamus (DMH) of DIO mice mediate the thermogenic responses to hyperleptinemia in obese mammals because blockade of leptin receptors in the DMH prevented the thermogenic effects of leptin.
Peripheral Melotan II (MTII) injection increased iBAT temperature, but it was blunted by blockade of DMH melanocortin receptors (MC4Rs) by injecting agouti-related peptide (AgRP) directly into the DMH, suggesting a physiological role of the DMH on temperature regulation in animals with normal body weight. Nevertheless, obese mice without a functional melanocortin system (MC4R KO mice) have an increased sympathetic outflow to iBAT compared with their littermates, suggesting that higher leptin levels drive sympathoexcitation to iBAT by a melanocortin-independent pathway.
Because the sympathetic nervous system contributes in regulating blood pressure, heart rate, and hepatic glucose production, selective leptin resistance may be a crucial mechanism linking adiposity and metabolic syndrome.
Introduction
Leptin is an essential endocrine signal for energy homeostasis. It communicates the level of stored fat to the CNS to regulate body weight. Activation of central leptin receptors (LRb) decreases appetite and increases the activity of the sympathetic nervous system (SNS), which stimulates energy expenditure in brown adipose tissue (BAT) (Haynes et al., 1999). BAT is innervated exclusively by the SNS (Bartness et al., 2010). In rodents, and likely in humans, BAT plays a major role in the adaptive thermogenesis response to environmental challenges (Nedergaard and Cannon, 2010). The underlying mechanism for adaptive thermogenesis involves the uncoupling of oxidative phosphorylation to generate heat via increased expression of mitochondrial uncoupling protein 1 (UCP-1) in BAT. Leptin administration increases UCP-1 expression (Scarpace and Matheny, 1998). Several studies have shown that melanocortin neurons of the arcuate nucleus of hypothalamus (ARH), proopiomelanocortin (POMC), and Neuropeptide Y/AgRP neurons mediate many of the actions of leptin (Cowley et al., 2001; Cone, 2006), including the activation of SNS and thermogenesis (Haynes et al., 1999).
Melanocortin receptors (MC4R/MC3R) are widely distributed, but highly expressed in some hypothalamic centers involved in neuroendocrine and autonomic functions, such as paraventricular nucleus of the hypothalamus (PVH), DMH, and other extrahypothalamic areas (Mountjoy et al., 1994; Liu et al., 2003). MC4Rs appear to mediate thermogenesis because the leptin-mediated induction of UCP-1 expression in rats can be blocked with SHU9119, a potent antagonist of α-MSH (Satoh et al., 1998). In addition, it was also shown that central administration of MTII, an MC4R agonist, increases iBAT UCP-1 mRNA expression. This effect was blocked by surgical sympathetic denervation (Williams et al., 2003), suggesting that SNS innervation to BAT, and probably an intact melanocortin system, is required for leptin-induced thermogenesis in physiological conditions.
A complete understanding of the hierarchical organization of these neural circuits controlling the SNS to trigger thermogenesis in iBAT remains to be determined. Several studies support the idea that PVH plays a major role in the mediation of MC4R effects on energy expenditure (Voss-Andreae et al., 2007; Song et al., 2008). On the contrary, Balthasar et al. (2005) showed that genetic reinsertion of the MC4Rs in the PVH normalized the hyperphagia of MC4R KO mice, but not the thermogenic response to MTII injection. These results strongly argue against the necessary involvement of PVH in mediating the effects of melanocortins on energy expenditure.
The DMH has been implicated in the regulation of body temperature (Zaretskaia et al., 2008), and inhibition of DMH neurons prevents BAT thermogenesis (Morrison et al., 2008). Here, we tested the hypothesis that high leptin levels in obese mice act via LRb in the DMH to increase sympathetic nervous activity to iBAT and therefore increase temperature. We studied whether the thermogenic effects of leptin were intact in DIO mice despite the fact that these mice were leptin resistant at the arcuate nucleus of the hypothalamus level. We also evaluated the importance of DMH in the activation of SNS in obese mice.
Materials and Methods
Animals
At 5–6 weeks of age, C57BL/6J mice (Monash Animal Services) were fed a regular rodent diet (Specialty Feeds) or a HFD (SF04-001; Specialty Feeds) for 20 weeks. Regular diet provided 3.3 kcal/g energy (59.8% carbohydrate, 28.0% protein, and 12.1% fat). HFD provided 4.75 kcal/g energy (35.0% carbohydrate, 20.0% protein, and 45.0% fat). ob/ob mice and MC4R KO were fed a regular diet. Mice were housed (5/cage) in a controlled environment, but they were individually caged during each experiment. Food and water were available ad libitum unless otherwise indicated. Body weights were measured weekly. All procedures were performed in accordance with the guidelines and approval of the Monash University Animal Ethics Committee.
Telemetric transponder implantation, locomotor activity, iBAT, and core temperature measurement
Remote biotelemetry was performed using precalibrated sensitive transmitters (PDT-4000 G2 E-Mitter sensors, Mini Mitter Company). iBAT temperature and locomotor activity (LA) were measured as described previously (Billes and Cowley, 2008). Under isoflurane anesthesia E-Mitters were implanted beneath the iBAT pad between the scapulae. Mice were allowed 1 week recovery before studies commenced. For core body temperature recording, E-Mitters were removed from iBAT and placed into the peritoneal cavity. Signals emitted by the E-Mitter transmitters were detected by a receiver positioned underneath the animal's home cage and analyzed using VitalView software (Mini Mitter) (Harkin et al., 2002). Locomotor activity counts are a relative measure of gross motor activity. For all experiments, activity counts and interscapular temperature measurements were taken every 1 min.
Cannulation
Intracerebroventricular injections.
The procedure was performed as described previously (Enriori et al., 2007).
Intra-DMH injections.
Mice were maintained under anesthesia with 2% isoflurane in oxygen. A sterile guide cannula (4 mm long, 26 gauge, Plastics One) was stereotaxically implanted into the DMH (1.9 mm posterior, 0.4 mm lateral to bregma, and 4 mm below the surface of the skull, in accordance with Paxinos and Franklin (2001). A 28 gauge obturator was inserted into each cannula. Cannula position was verified at the end of the experiment by dye administration and postmortem histological analysis.
iBAT temperature and locomotor activity recording during different drug treatments
Intraperitoneal injections.
Individually housed mice were acclimatized to daily injections of 100 μl of saline solution for 5 consecutive days before drug treatment. After that, each mouse received either saline or drug treatment. Three days later, treatments were crossed over. Drug treatment was as follows: (1) Mice received a single intraperitoneal (i.p.) injection of recombinant murine leptin (Peprotech, 100 μl: 6.0 μg/g body weight). (2) Mice received single i.p. injection of a highly selective β3-adrenoceptor agonist (100 μl: 1.0 μg/g, CL 316243 disodium salt, Tocris Bioscience). (3) Mice received a single i.p. injection of β-3R antagonist (100 μl: 10.0 μg/g, SR 59230A hydrochloride, Tocris Bioscience). (4) Mice received an i.p. injection of β-3R antagonist (100 μl: 10.0 μg/g) and after 20 min received an i.p. injection of the β3-adrenoceptor agonist. (5) Mice received single i.p. injection of MTII (Melotan II; 100 μl: 1.0 μg/g, Bachem). Body weight, caloric intake, iBAT temperature, and LA were measured during 24 h after the injection.
Intracerebroventricular injections.
Mice received 1 μl injection of recombinant murine leptin (Peprotech, 0.2 μg/μl) or artificial CSF (aCSF) into the lateral ventricle. Three days later, experimental treatments were crossed over. Body weight, caloric intake, iBAT temperature, and LA were measured during 24 h after the injection.
Intra-DMH injections.
Mice received intra-DMH leptin (Peprotech, 0.5 μl of 0.2 μg/μl). Solution (0.5 μl) was injected over 60 s, with the injector left in place for an additional 30 s to ensure extrusion from the tip, and to minimize distribution of drug upward on the cannula tract. Three days later, treatments were crossed over. Body weight, caloric intake, iBAT temperature, and LA were measured during 24 h after the injection.
Combined injections.
(1) Mice received single i.p. injection of β-3R antagonist (100 μl: 10.0 μg/g) immediately before a single intra-DMH leptin injection (0.5 μl of 0.2 μg/μl). (2) Mice received a single intra-DMH leptin receptor antagonist injection (mouse leptin antagonist mutant L39A/D40A/F41A, 0.5 μl of 5 μg/μl, Protein Laboratories Rehovot) followed by i.p. leptin administration (100 μl: 6 μg/g) 1 h later. (3) Mice received a single DMH injection of AgRP (0.5 μl of 1.0 μg/μl) 60 min before a single i.p. MTII (100 μl: 1.0 μg/g). Body weight, caloric intake, iBAT temperature, and LA were measured during 24 h after the injection.
pSTAT3 immunohistochemical studies
Studies were performed as described previously (Enriori et al., 2007). Briefly, mice were injected i.p. with 6 μg/g leptin or saline. After 30 min, mice were killed under pentobarbital anesthesia by cardiac perfusion with PBS and paraformaldehyde. Brains were removed, dehydrated in 25% sucrose, frozen, and stored at −80°C until sectioned coronally on a microtome (30 μm). For pSTAT3 immunohistochemistry, free-floating tissue was pretreated in 1% NaOH+1% H2O2 in H2O for 10 min, 0.3% glycine for 10 min, 0.03% SDS for 10 min, and blocked in 3% normal donkey serum in PBS/0.25% Triton X-100/0.02% NaN3. Antibodies (Phospho-STAT3 rabbit #9135, 1:1000, Cell Signaling Technology) were added in blocking solution and incubated overnight at room temperature. Sections were washed, incubated with Biotin-SP-conjugated AffiniPureF(ab1)2 Fragment Donkey Anti-Rabbit IgG secondary antibody (Jackson ImmunoResearch Laboratories) 1:1000 in blocking solution, without NaN3 for 1 h. Tissue was treated with Vectastain Elite ABC Kit (Vector Labs) for 1 h, and the signal was developed by NiSO4/DAB solution. Tissue was mounted on subbed slides, coverslipped, and photographed.
UCP1 Western blot
Brown adipocytes were washed twice in ice-cold PBS and then homogenized in a modified ice-cold RIPA lysis buffer (10 mm Tris, pH 7.4, 0.1% SDS, 0.5% deoxycholic acid, 1% Triton X-100, NaCl 150 mm, 80 μm aprotinin, 2 μm leupeptin, 1.5 μm pepstatin, and 1 mm PMSF). After clearing the homogenates by centrifugation, protein concentrations were estimated using the Bradford method (Bio-Rad). Laemmli sample buffer (6×) was then added to each sample to a final concentration of 1×. The samples were boiled for 5 min before loading them (10 μg of protein/sample) onto a 10% precast Bis-Tris gel (Invitrogen). After electrophoresis at 130 V for 2 h, the proteins were transferred for 1.5 h at 4°C onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked in 5% nonfat milk for 1 h, and then incubated overnight at 4°C with a rabbit polyclonal antibody raised against UCP1 C-terminal decapeptide 4000 (kindly provided by Dr. Natasa Petrovic, Wenner-Gren Institute, Stockholm, Sweden) followed by an anti-rabbit HRP antibody (1 h at room temperature, 1:20,000; # G21230, Invitrogen). The signal was developed by enhanced chemiluminescence using the Western lightning chemiluminescence substrate (PerkinElmer Life Sciences). To correct for procedural losses, the membrane was washed several times in Tris-buffered saline-Tween 20 (TBST; 10 mm Tris, 150 mm NaCl, pH 7.5 plus 0.2% Tween 20) before exposure (overnight at 4°C) to a mouse monoclonal antibody against β-actin (sc-47778, 1:1000; Santa Cruz Biotechnology 1:5000 dilution), followed by an anti-mouse HRP antibody (1 h at room temperature, 1:10,000; #G21040, Invitrogen).
Radioimmunoassay
Leptin levels were determined by radioimmunoassay (Linco Research) as previously reported (Enriori et al., 2007).
Data analysis
All values are expressed as mean ± SEM. Area under the curve (AUC) was calculated by trapezoid analysis and was compared by t test for iBAT temperature after DMH injection plus i.p. treatment, by one-way ANOVA for baseline LA, iBAT temperature, and total calorie intake followed by Bonferroni's test, and by two-way ANOVA for leptin levels changes, weight change over time, i.p., intracerebroventricular (i.c.v.), and intra-DMH leptin effect of LA and iBAT temperature, pStat3 IMH, and Western blot. iBAT temperature correlation to leptin levels was analyzed using nonlinear regression (sigmoidal dose–response best-fit curve, p < 0.001). For the calculation of this correlation, we arbitrary assigned 0.2 ng/ml (detection limit for the assay) to the leptin levels of ob/ob mice. p < 0.05 was considered statistically significant. Analyses were performed with GraphPad Prism 4.0 (GraphPad Software).
Results
Obese mice are not resistant to leptin effects on thermogenesis
To evaluate the effects of leptin on thermogenesis, we studied two different mouse obesity models: obese, hyperleptinemic, DIO mice (C57BL/6J mice fed a high fat diet), and obese leptin-deficient (ob/ob) mice. We measured the temperature of iBAT as a marker of both sympathoexcitation and increased energy expenditure. We have used iBAT temperature as an index of sympathetic outflow and of thermogenesis previously in other studies (Xiao et al., 2007; Billes and Cowley, 2008). We found that DIO mice have chronically elevated temperature (Δ 0.6°C), compared with C57BL/6J mice fed a regular diet (controls) over 24 h (p < 0.001), during both the light period and the dark period. On the contrary, ob/ob mice have lower temperature than controls (p < 0.001) (Fig. 1A,B). LA was similar between DIO and controls. However, ob/ob mice showed hypoactivity (p < 0.001 vs control), which along with hypothermia and hyperphagia accounts for the high rates of energy storage (Fig. 1C,D). We also found that iBAT temperature was highly related to baseline leptin levels, independent of obesity (r2 = 0.54: p < 0.001, Fig. 1E). These results suggest that baseline hyperleptinemia increases SNS activity, causing hyperthermia in DIO mice, and confirms that this model mimics the increased sympathetic outflow seen in human hypertension.
Figure 1.
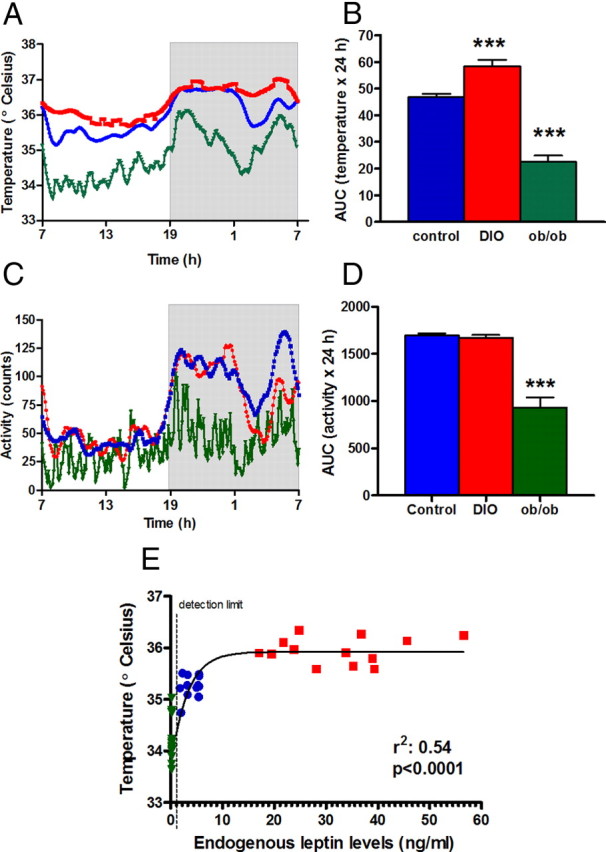
Daily baseline temperature and activity in lean control (blue circles), DIO (red squares), and ob/ob (green triangles) mice and its relationship with leptin levels. A, iBAT temperature during 24 h. Gray area represents the dark period. B, AUC of temperature (***p < 0.001 vs control). C, Locomotor activity over 24 h. D, AUC of locomotor activity. ***p < 0.001 versus control. E, iBAT temperature was highly correlated to endogenous leptin levels, sigmoidal dose–response best-fit curve, r2: 0.54, p < 0.0001. For the calculation, we arbitrarily assigned 0.2 ng/ml (detection limit for the assay) to the leptin levels of ob/ob mice. Results are mean ± SEM, n = 7–12.
To test whether the thermogenic response to leptin is intact in DIO mice, we used a high i.p. dose of leptin, which produces a systemic leptin peak of ∼15,000 ng/ml in plasma leptin after 1 h in all groups and then decreases to ∼2000 ng/ml in the second hour (Fig. 2A). After i.p. leptin administration, food intake was reduced in control mice, but not in DIO mice (p < 0.001). DIO mice were also unresponsive to the anorectic effect of central (lateral ventricle) leptin injection (p < 0.01 vs controls). Thus, at behavioral levels (food intake) leptin sensitivity was decreased in DIO mice. Nevertheless, i.p. leptin administration acutely increased iBAT temperature for at least 1 h in both control and DIO mice (Fig. 2B,C,E). As a control for nonspecific effects, such as obesity acting as a thermal insulator, we determined the thermogenic response to leptin of ob/ob mice and found that i.p. leptin significantly increased iBAT temperature (Fig. 2D,E). There were no changes in LA in any of the groups studied (data not shown).
Figure 2.
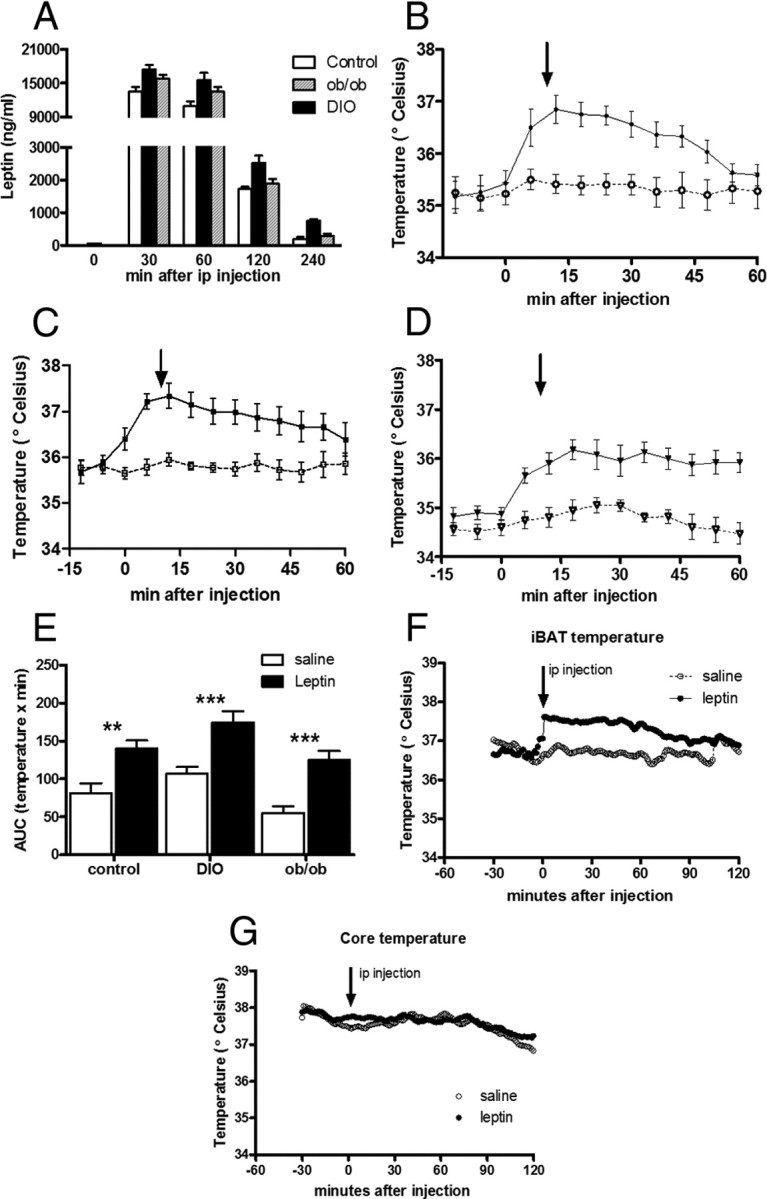
Peak of temperature after leptin treatment in lean control (circles), DIO (squares), and ob/ob (triangles) mice and comparison between the effect of leptin treatment on iBAT and core temperature in DIO mice. A, Leptin levels in control mice (open bars), DIO mice (filled bars), and ob/ob mice (striped bars) increase to similar levels at 30, 60, 120, and 240 min after peripheral leptin injection (i.p., 6 μg/g body weight). B–D, Intraperitoneal leptin (6 μg/g) increases iBAT temperature compared with i.p. saline (dashed line, open circle) in control mice (B), DIO mice (C), and ob/ob mice (D). E, AUC of iBAT temperature during 1 h after i.p. leptin. F, Peripheral leptin injection (i.p., 6 μg/g body weight) increases iBAT temperature compared with i.p. saline (dashed line, open circle). G, However, leptin treatment did not modify core temperature.
We also found that the increase of iBAT temperature after i.p. leptin was not accompanied by an increase in core body temperature in DIO mice (Fig. 2F,G), showing that the measurement of iBAT temperature is a more sensitive marker of sympathoexcitation than total body temperature.
Central leptin signaling drives iBAT effect via β3 adrenergic stimulation
To determine whether the modulation of iBAT temperature by systemic leptin injection was due to neuronal activation, we measured the effect of leptin administration directly into the brain (0.2 μg in lateral ventricle). We found an increase of temperature that was maintained for ∼8 h in DIO and control mice (Fig. 3A–C), without any changes in LA. This effect was confirmed by an increase of UCP1 protein expression in iBAT after central leptin injection (Fig. 3D–G).
Figure 3.
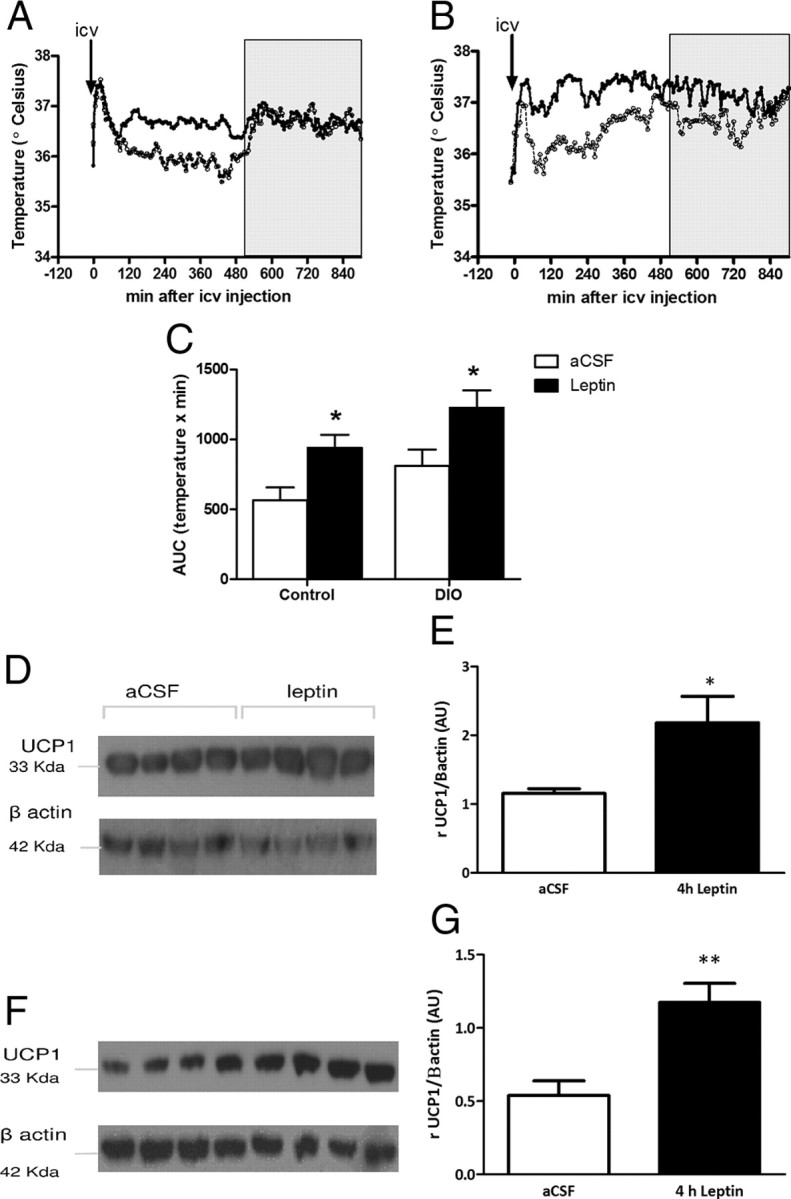
Peak of temperature and UCP1 expression after i.c.v. leptin treatment in lean and DIO mice. A, B, Intracerebroventricular leptin (0.2 μg, closed circle) increases iBAT temperature compared with i.c.v. aCSF (open circle) in control mice (A) and DIO mice (B). C, AUC of iBAT temperature during 8 h after i.c.v. leptin. The arrows represent the injections. Results are mean ± SEM, n = 8. D, F, UCP1 expression in iBAT is increased in lean (D) and DIO (F) mice 4 h after i.c.v. leptin (0.2 μg) treatment. E, G, Quantification of the UCP1 Western blot as a ratio of β-actin (housekeeping) following aCSF (white bar; n = 4) and leptin (black bar; n = 4). Results are mean ± SEM. *p < 0.05, **p < 0.01.
Because β3-R mediates the effect of sympathetic stimulation on BAT, we tested whether the capacity to produce heat from BAT was different in obese mice (DIO and ob/ob) compared with lean mice by i.p. β3-R agonist injection. We found a similar increase in temperature after β3-R agonist administration, without any changes in LA in all groups studied (Fig. 4A, β3-R agonist vs saline for each group), indicating that all mice were equally capable of robust thermogenesis. We also found that the effect of β3-R agonist was blocked by a previous systemic injection of a β3-R antagonist (Fig. 4B). Thus, there was a direct action on β3-R on BAT.
Figure 4.

Effect of peripheral β-3 agonist treatment on temperature in lean control, DIO, and ob/ob mice. A, AUC of iBAT temperature after i.p. injection of a selective β3-adrenoceptor agonist (1.0 μg/g, CL 316243 disodium salt, filled bars) or saline (open bars) in control, DIO, and ob/ob mice. B, AUC for changes of temperature during 8 h after i.p. injections of vehicle plus vehicle again 20 min later (open bar), vehicle plus β3-R agonist (filled blue bar). The increase of iBAT temperature by a single i.p. injection of β3-adrenoceptor agonist was blocked by a previous systemic injection of a β3-R antagonist (10.0 μg/g) 20 min before (red bar). Results are expressed as mean ± SEM. *p < 0.05 versus control, #p < 0.05 vehicle plus β3-R agonist versus β3-R antagonist plus β3-R agonist (n = 6).
Dorsomedial nucleus of the hypothalamus mediates the thermogenic responses to hyperleptinemia in obese mammals
In DIO mice, central leptin resistance seems to be localized to the ARH since leptin treatment neither induces STAT3 phosphorylation (Münzberg et al., 2004; Enriori et al., 2007) nor regulates α-MSH, AgRP, or NPY secretion (Enriori et al., 2007). In DIO mice, we found that the DMH neurons remained leptin responsive. Indeed, leptin was able to induce the phosphorylation of STAT3, a well known mediator of leptin activation, in the DMH but not in the ARH of DIO mice (Fig. 5A,B).
Figure 5.
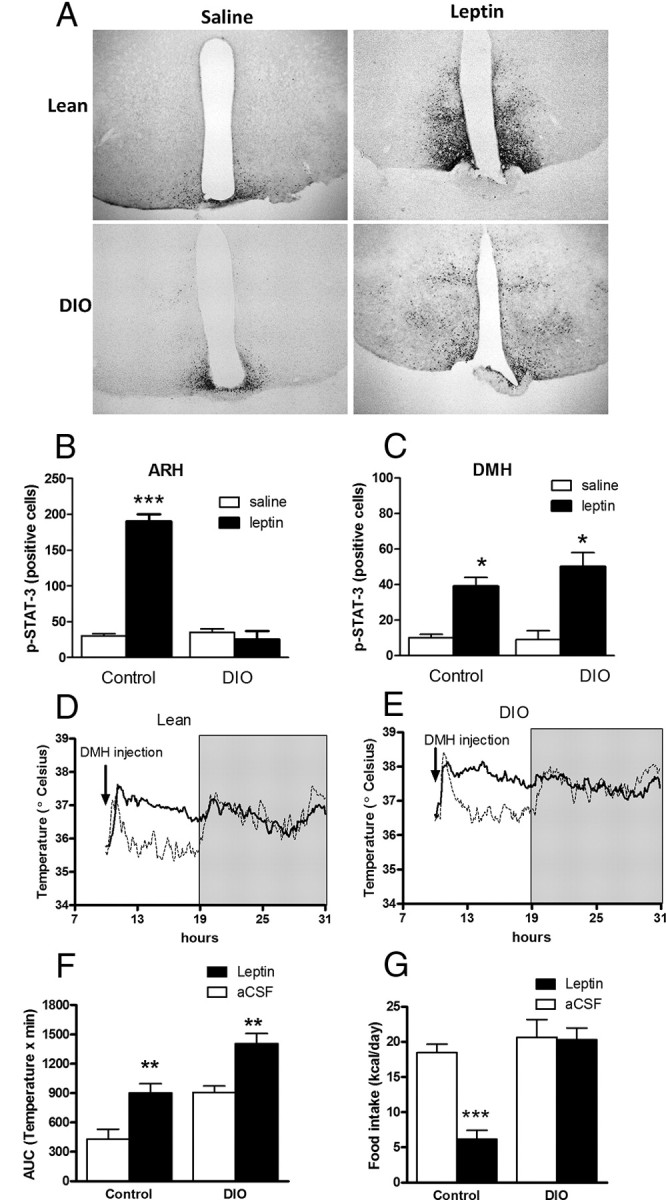
Leptin effect on DMH pSTAT3 expression and iBAT temperature. A, Representative microphotographs of hypothalamic sections from control and DIO mice showing pSTAT expression after 30 min i.p. leptin (6 μg/g) or saline. Scale bar, 100 μm. B, C, pSTAT3 expression in ARH (B) and DMH (C). D, E, iBAT temperature after intra-DMH leptin (0.2 μg, continuous line) and aCSF (dashed line) in control mice (D) and DIO mice (E). Gray area represents the dark period. F, AUC for temperature during 8 h (light period) after intra-DMH injection. G, Twenty-four hour food intake after intra-DMH injections. Results are mean ± SEM, n = 6, *p < 0.05, **p < 0.01, ***p < 0.001.
The DMH has previously been implicated in the regulation of energy balance and body temperature (Dimicco and Zaretsky, 2007). The activation of neurons in the DMH by a GABA receptor antagonist evoked increases in sympathetic vasomotor activity, arterial pressure, heart rate, and sympathetic outflow to BAT (Dimicco and Zaretsky, 2007; Zaretskaia et al., 2008). To test whether the activation of leptin-sensitive neurons in the DMH increases sympathetic activity in control and DIO mice, we injected leptin directly into the DMH. We found a persistent increase of temperature during the light period (Fig. 5C–E), without any change in LA. We also evaluated the caloric intake and found it was decreased in control but not in obese mice (Fig. 5F). Thus, there was a resistance to the anorectic effect of leptin in the DMH of obese mice, but mice were still able to respond to the thermogenic effect of leptin.
To evaluate the specific action of leptin on the DMH to increase sympathetic outflow to iBAT, we injected a β3-R antagonist (i.p.) immediately before leptin injection into the DMH of lean mice. We found that the increase of iBAT temperature was blocked by β3-R antagonist administration (Fig. 6A).
Figure 6.

Increase in iBAT temperature caused by leptin is blocked by peripheral β3-R antagonist injection or intra-DMH leptin receptor antagonist injection. A, The increase in iBAT temperature caused by intra-DMH leptin (0.2 μg, dashed line) in control mice was diminished by i.p. injection of β3-R antagonist (10.0 μg/g, continuous line). B, C, The increase of iBAT temperature caused by i.p. leptin (6 μg/g, dashed line) was blunted with an intra-DMH injection of leptin receptor antagonist (5 μg/μl, continuous line) 1 h before in control mice (B) and DIO mice (C). D, E, AUC of temperature during 2 h after i.p. leptin, with a previous intra-DMH treatment of aCSF or leptin receptor antagonist in lean mice (D) and DIO mice (E). Results are mean ± SEM, n = 7, *p < 0.05 versus aCSF.
To evaluate the effects of leptin receptors in the DMH, we took advantage of a recently developed leptin receptor neutral antagonist (Salomon et al., 2006). This antagonist binds to the LRb with a similar affinity to leptin and prevents the activation of the leptin signal cascade (Salomon et al., 2006; Zhang et al., 2007). As before, leptin i.p. increased iBAT temperature. The increase of iBAT temperature was blocked by preinjecting the leptin receptor antagonist directly into DMH in DIO and lean mice (Fig. 6B–E). These results strongly suggest that leptin receptors in the DMH are involved in the stimulation of SNS, particularly during hyperleptinemic state.
Sympathetic activation to iBAT is driven by a melanocortin-independent pathway in obese mice
A neural pathway from the central melanocortin circuitry, which stimulates the SNS and includes hypothalamic and extrahypothalamic centers of CNS, has been proposed (Voss-Andreae et al., 2007; Song et al., 2008). We analyzed the role of the DMH MC4Rs on iBAT sympathetic outflow by i.p. administration of MTII in lean mice. MTII increased iBAT temperature for at least 6 h (Fig. 7A,B), as well as decreased food intake (p < 0.001). This increase in iBAT temperature was blunted by blockade of DMH MC4Rs by a direct injection of AgRP, an endogenous melanocortin receptor antagonist, into DMH 1 h before i.p. MTII (Fig. 7C,D). It suggests a physiological role of the DMH in temperature regulation in animals with normal body weight. To evaluate the role of a possible melanocortin-independent pathway in obese mice, we studied adult obese MC4R KO mice. Surprisingly, we found that MC4R KO mice have an increased iBAT temperature compared with their littermates (Fig. 7E,F), although LA over 24 h was not different (data not shown). However, MC4R KO mice had significantly decreased LA during the dark cycle (Fig. 7G,H). Also, we found a significant increase of iBAT temperature after i.c.v. leptin treatment (Fig. 7I,J) without any change in LA. The magnitude and time course of the response was similar to the results obtained in control and DIO mice. Overall, these data suggest that higher leptin levels found in MC4R KO mice drive sympathoexcitation to iBAT by a melanocortin-independent pathway.
Figure 7.
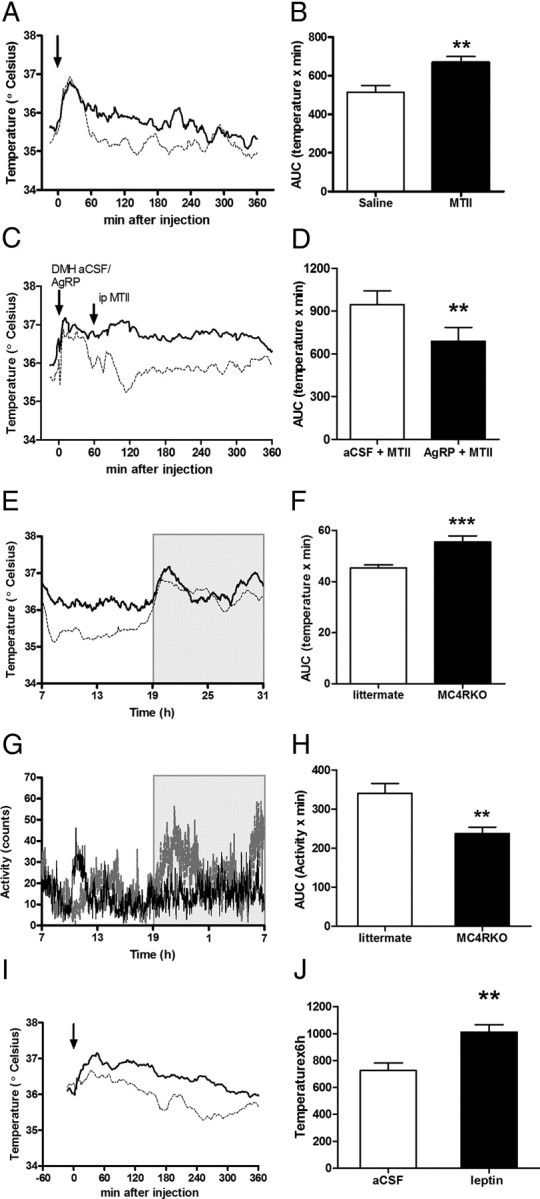
Melanocortin system modulates iBAT temperature. A, iBAT temperature after i.p. MTII (1.0 μg/g, solid line) or i.p. saline (dashed line) in control mice. B, AUC of temperature after i.p. MTII or saline. C, The increase of iBAT temperature caused by i.p. MTII was blunted with an intra-DMH injection of AgRP (0.5 μg/μl, dashed line) 1 h before. As a control aCSF (1 μl, continuous line) was injected in DMH 1 h before i.p. MTII. D, AUC for temperature after i.p. MTII, with a previous intra-DMH aCSF or AgRP treatment, n = 8. E, iBAT temperature on 24 h of MC4R KO mice (continuous line, n = 7) and littermates (dashed line, n = 6). Gray area represents dark period. F, AUC of temperature, p < 0.001 versus littermates. G, Locomotor activity over 24 h. H, AUC of locomotor activity was significantly different only in the dark cycle, p < 0.01 vs littermates. I, Intracerebroventricular leptin (0.2 μg, solid line) increases iBAT temperature compared with i.c.v. aCSF (dashed line) in MC4R KO mice. J, AUC of iBAT temperature during 6 h after i.c.v. leptin. The arrows represent the injections. Results are mean ± SEM, n = 8.
Discussion
The central melanocortin system is a physiological mediator of leptin actions on sympathetic outflow in animals of normal body weight. However, little is known about leptin actions on autonomic outflow in obese, “leptin-resistant” organisms. Here, we demonstrated that hyperleptinemia in obese mice increased SNA to iBAT by a melanocortin-independent pathway, despite these mice being resistant to anorexigenic leptin effects. We also found that leptin receptor bearing neurons in the DMH play an important role in the sympathoexcitation in obesity.
We measured iBAT temperature as a marker of sympathetic activity in BAT. We have confirmed the specificity of the thermogenic effect by measuring UCP1 protein expression in parallel experiments. We also validated the technique, showing that the increase in iBAT temperature caused by a peripheral injection of β3-R agonist was blocked by a previous injection of β3-R antagonist. The thermogenic effect of leptin was also blocked by β3-R antagonist.
We found that DIO mice have chronically increased sympathetic activation compared with control lean mice. On the contrary, leptin-deficient mice (ob/ob) have lower temperature than controls, indicating lower sympathetic nervous system activity. Our findings are consistent with other observations in both models (DIO and ob/ob mice). A significant increase in gene expression of β3-R and UCP1 in iBAT, both considered markers of sympathetic activation, was demonstrated in DIO (Shi et al., 2009). Low expression of UCP1 in ob/ob mice has also been demonstrated (Commins et al., 2000). We did not directly measure sympathetic nerve activity in these animals, but an increased renal sympathetic outflow in obese rats has been reported (Barnes et al., 2003). On the other hand, other studies failed to find an increase of sympathetic activation due to endogenous hyperleptinemia in obese mice (Rahmouni et al., 2005). However, sympathetic nerve activity is not well suited to comparing between animals, although it is useful for measuring responses to short-term stimuli (Rahmouni et al., 2005).
Overactivity of the SNS is a common feature of obesity in human and animal models (Hall et al., 2010). Several clinical studies in humans have shown strong evidence associating hyperleptinemia and sympathetic activation. Eikelis et al. (2003) have demonstrated a strong correlation between leptin plasma concentrations and renal sympathetic activity across a broad range of leptin values and adiposity. Recently, a prospective study demonstrated that circulating leptin levels were a significant predictor of the risk to develop hypertension (Galletti et al., 2008). Our results showed that sympathetic outflow to iBAT highly correlated to endogenous leptin levels, independent of obesity, suggesting that hyperleptinemia increases SNS activity causing hyperthermia in DIO mice.
We demonstrated that i.p. leptin administration acutely increased iBAT in obese (DIO and ob/ob mice) and in control mice without significant changes in LA. It has been demonstrated that treatment of ob/ob mice with leptin produces a significant increase in iBAT UCP1 mRNA comparable to increases found in lean mice (Commins et al., 2000). Our results are in agreement with those published by Rahmouni's group (Rahmouni et al., 2005; Morgan et al., 2008). These studies showed that DIO mice displayed different regional SNA responses to intravenous and central leptin administration, being higher at the renal level than at iBAT. However, in both kidney and iBAT, the response to leptin was significantly higher than that observed after saline administration. In our study, the modulation of iBAT temperature by leptin was due to neuronal activation because central leptin injection increased iBAT temperature and iBAT UCP1 protein expression in all groups. As we and others have demonstrated (Münzberg et al., 2004; Zhang and Scarpace, 2006; Enriori et al., 2007), DIO mice are unresponsive to the anorectic effect of central leptin injection, indicating that although these mice have decreased leptin sensitivity at behavioral levels, the thermogenic regulation is intact. Our results also showed that the lower baseline iBAT temperature of ob/ob mice was due to leptin deficiency and not to a diminished capacity to produce heat from BAT. In fact, ob/ob mice respond to β3 adrenergic stimulation as robustly as DIO and control mice.
Here we show that leptin increases p-STAT3 expression the DMH in obese mice as previously demonstrated (Münzberg et al., 2004), causing sympathetic activation in lean and DIO mice and a persistent increase of iBAT temperature, which was attenuated by a β3-R antagonist treatment. The stimulation produced by leptin was mediated through its own receptors because we found that leptin antagonist administration into the DMH was able to block the increase of iBAT temperature caused by peripheral leptin injection. In agreement with our results, Marsh et al. (2003) showed that DMH leptin administration increased heart rate and blood pressure in anesthetized lean rats by sympathoexcitation. Furthermore, Gautron et al. (2010) showed that neurons of the DMH region displayed a very high density of leptin receptors, which project directly to neurons located in the PVH and potentially premotor sympathetic neurons with the ability to increase SNA. Interestingly, they also found projections to the raphe pallidus (rRPa), another region that contains premotor sympathetic neurons. Moreover, Zhang et al. (2011), using a retrograde transsynaptic tracer injected into the BAT of mice, recently showed strong evidence that LRb in the DMH mediate thermoregulatory leptin actions and that LRb DMH neurons synaptically couple with neurons in the rRPa. Collectively, these results strongly support the premise that leptin-sensitive neurons in the DMH are involved in the stimulation of thermogenesis via the SNS.
We have also specifically evaluated the role of the DMH MC4R neurons on iBAT sympathetic outflow. We found that MTII produced an increase of iBAT temperature in control mice, which was blunted by blockade of the MC4R by AgRP microinjection into the DMH. It has been demonstrated that MTII increases energy expenditure (evaluated by indirect calorimetry) in lean (Satoh et al., 1998) and DIO (Pierroz et al., 2002) mice. Our results suggest a physiological role of the DMH MC4R neurons in temperature regulation in animals with normal body weight.
The finding that adult, hyperleptinemic, obese MC4R KO mice have increased iBAT temperature compared with their littermates supports our hypothesis of a melanocortin-independent pathway regulating sympathetic outflow (Fig. 8). Energy balance in MC4R KO mice has been previously studied in detail. In addition to hyperphagia, it has been reported that these mice exhibit decreased energy expenditure (Huszar et al., 1997; Butler, 2006; Cone, 2006). Energy expenditure in MC4R KO mice has been mostly based on indirect calorimetric studies, where energy expenditure was reported as VO2 per unit of body weight. These measurements were recently disputed by Butler and Kozak (2010), who showed that when the same data were expressed per mouse or normalized to lean mass, MC4R KO mice exhibited a significant increase in energy expenditure, instead of having decreased energy. Previous studies have shown that leptin injections did not modify UCP1 mRNA expression in MC4R KO mice (Ste Marie et al., 2000; Zhang et al., 2005). This suggests a decrease in energy expenditure. On the contrary, our results show that adult obese hyperleptinemic MC4R KO mice have both increased baseline iBAT temperature and a marked increase in temperature in response to i.c.v. leptin injection compared with their littermates. These findings strongly support our hypothesis of a melanocortin-independent pathway regulating sympathetic outflow (Fig. 8).
Figure 8.
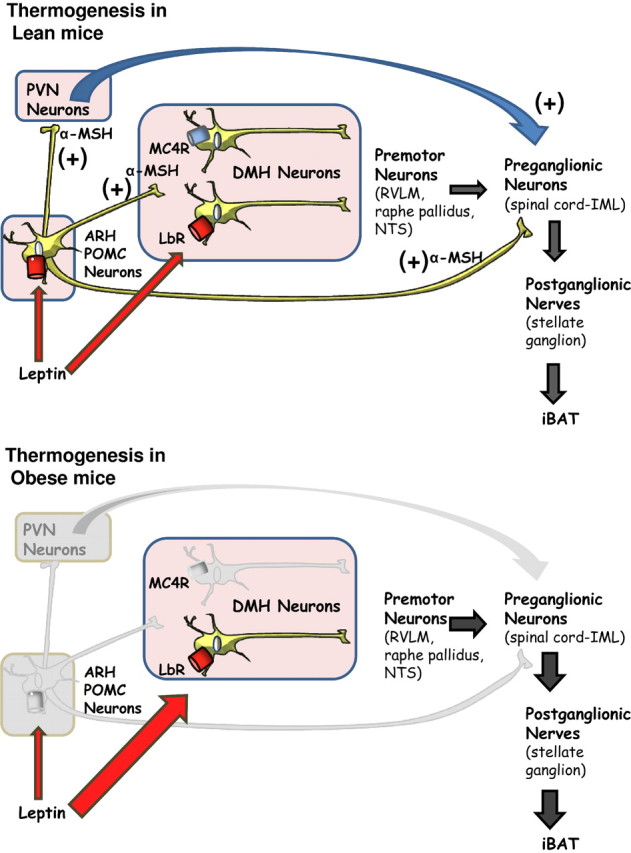
Proposed mechanism for the leptin-induced sympathetic outflow in lean and obese mice. In lean mice, leptin reaches the hypothalamus and binds to leptin receptors (LRb) in POMC neurons of the ARH to increase the secretion of α-MSH, which in turn directly activates melanocortin receptors (MC4Rs) in the PVH, DMH, and intramediolateral column (IML). MC4R activation then mediates peripheral sympathetic outflow, probably by both direct and indirect signaling processes, leading to increased expression and activity of mitochondrial UCP-1 in BAT to generate heat. When mice become obese, there is a selective leptin resistance in arcuate neurons (POMC and AgRP neurons), but other brain areas, including the DMH, are still responsive to leptin. The chronic hyperleptinemia found in obesity is able to stimulate leptin receptor expressing neurons to increase sympathetic nervous outflow to iBAT.
Several observations support the notion that melanocortin signaling mediates some, of leptin's actions. For example, MC4R KO mice, when younger and nonobese, remained highly responsive to anorexigenic effects of leptin (Marsh et al., 1999). Moreover, the yellow obese agouti mice (Ay), a model of obesity due to overexpression of agouti protein (melanocortin receptor antagonist), have an intact sympathoexcitatory response to leptin (Correia et al., 2002). In addition, the generation of transgenic (Ay/ob/ob) mice demonstrated the divergence of both pathways, since these mice were more obese and hyperinsulinemic than the two contributing strains (Boston et al., 1997). Also, pharmacological studies have demonstrated a differential acute regulation of sympathetic activation of melanocortin system and leptin (Haynes et al., 1999). Together, these results suggest that leptin mediates control of energy expenditure by anatomically distributed networks that involve functional redundancy across partially overlapping networks of LRb-expressing neurons and MC4R neurons. Further, the activation of these divergent pathways could be exacerbated in obese conditions. Melanocortin agonists have not been successful in human weight loss trials because they cause increases in cardiovascular tone (Greenfield et al., 2009). Any therapy that activates neural outflow to the SNS runs the risk of increasing cardiovascular or renal sympathetic tone, as well as tone to iBAT, which may limit the effectiveness of the therapy.
In conclusion, we have demonstrated that hyperleptinemia in obese mice increases SNA to iBAT by a melanocortin-independent pathway, despite the fact that these mice were resistant to anorexigenic effects of leptin. Our data provides strong evidence that leptin retains the ability to activate the DMH and components of the SNS through the DMH in obese mice. Selective leptin resistance could be a crucial mechanism linking adiposity and elevated sympathetic outflow.
Footnotes
This work was supported by grants from National Health and Medical Research Council of Australia (606662), Pfizer Australia, Monash University, the National Heart Foundation of Australia (G 09M 4306), and the U.S. NIH (RR0163 and DK62202). We thank Alvin Peek Lee and Weiyi Chen for their support in the laboratory, Danae Benjamin for her kind help editing the manuscript, and Joseph A Rathner for helpful discussions.
The authors declare no competing financial interests.
References
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Barnes MJ, Lapanowski K, Conley A, Rafols JA, Jen KL, Dunbar JC. High fat feeding is associated with increased blood pressure, sympathetic nerve activity and hypothalamic mu opioid receptors. Brain Res Bull. 2003;61:511–519. doi: 10.1016/s0361-9230(03)00188-6. [DOI] [PubMed] [Google Scholar]
- Bartness TJ, Vaughan CH, Song CK. Sympathetic and sensory innervation of brown adipose tissue. Int J Obes (Lond) 2010;34(Suppl 1):S36–S42. doi: 10.1038/ijo.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billes SK, Cowley MA. Catecholamine reuptake inhibition causes weight loss by increasing locomotor activity and thermogenesis. Neuropsychopharmacology. 2008;33:1287–1297. doi: 10.1038/sj.npp.1301526. [DOI] [PubMed] [Google Scholar]
- Boston BA, Blaydon KM, Varnerin J, Cone RD. Independent and additive effects of central POMC and leptin pathways on murine obesity. Science. 1997;278:1641–1644. doi: 10.1126/science.278.5343.1641. [DOI] [PubMed] [Google Scholar]
- Butler AA. The melanocortin system and energy balance. Peptides. 2006;27:281–290. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59:323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commins SP, Watson PM, Levin N, Beiler RJ, Gettys TW. Central leptin regulates the UCP1 and ob genes in brown and white adipose tissue via different beta-adrenoceptor subtypes. J Biol Chem. 2000;275:33059–33067. doi: 10.1074/jbc.M006328200. [DOI] [PubMed] [Google Scholar]
- Cone RD. Studies on the physiological functions of the melanocortin system. Endocr Rev. 2006;27:736–749. doi: 10.1210/er.2006-0034. [DOI] [PubMed] [Google Scholar]
- Correia ML, Haynes WG, Rahmouni K, Morgan DA, Sivitz WI, Mark AL. The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes. 2002;51:439–442. doi: 10.2337/diabetes.51.2.439. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Dimicco JA, Zaretsky DV. The dorsomedial hypothalamus: a new player in thermoregulation. Am J Physiol Regul Integr Comp Physiol. 2007;292:R47–R63. doi: 10.1152/ajpregu.00498.2006. [DOI] [PubMed] [Google Scholar]
- Eikelis N, Schlaich M, Aggarwal A, Kaye D, Esler M. Interactions between leptin and the human sympathetic nervous system. Hypertension. 2003;41:1072–1079. doi: 10.1161/01.HYP.0000066289.17754.49. [DOI] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Galletti F, D'Elia L, Barba G, Siani A, Cappuccio FP, Farinaro E, Iacone R, Russo O, De Palma D, Ippolito R, Strazzullo P. High-circulating leptin levels are associated with greater risk of hypertension in men independently of body mass and insulin resistance: results of an eight-year follow-up study. J Clin Endocrinol Metab. 2008;93:3922–3926. doi: 10.1210/jc.2008-1280. [DOI] [PubMed] [Google Scholar]
- Gautron L, Lazarus M, Scott MM, Saper CB, Elmquist JK. Identifying the efferent projections of leptin-responsive neurons in the dorsomedial hypothalamus using a novel conditional tracing approach. J Comp Neurol. 2010;518:2090–2108. doi: 10.1002/cne.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, Farooqi IS. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkin A, O'Donnell JM, Kelly JP. A study of VitalView for behavioural and physiological monitoring in laboratory rats. Physiol Behav. 2002;77:65–77. doi: 10.1016/s0031-9384(02)00810-7. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33:542–547. doi: 10.1161/01.hyp.33.1.542. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh AJ, Fontes MA, Killinger S, Pawlak DB, Polson JW, Dampney RA. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension. 2003;42:488–493. doi: 10.1161/01.HYP.0000090097.22678.0A. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- Morgan DA, Thedens DR, Weiss R, Rahmouni K. Mechanisms mediating renal sympathetic activation to leptin in obesity. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1730–R1736. doi: 10.1152/ajpregu.90324.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF, Nakamura K, Madden CJ. Central control of thermogenesis in mammals. Exp Physiol. 2008;93:773–797. doi: 10.1113/expphysiol.2007.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Cannon B. The changed metabolic world with human brown adipose tissue: therapeutic visions. Cell Metab. 2010;11:268–272. doi: 10.1016/j.cmet.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Pierroz DD, Ziotopoulou M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51:1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Morgan DA, Morgan GM, Mark AL, Haynes WG. Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes. 2005;54:2012–2018. doi: 10.2337/diabetes.54.7.2012. [DOI] [PubMed] [Google Scholar]
- Salomon G, Niv-Spector L, Gussakovsky EE, Gertler A. Large-scale preparation of biologically active mouse and rat leptins and their L39A/D40A/F41A muteins which act as potent antagonists. Protein Expr Purif. 2006;47:128–136. doi: 10.1016/j.pep.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Satoh N, Ogawa Y, Katsuura G, Numata Y, Masuzaki H, Yoshimasa Y, Nakao K. Satiety effect and sympathetic activation of leptin are mediated by hypothalamic melanocortin system. Neurosci Lett. 1998;249:107–110. doi: 10.1016/s0304-3940(98)00401-7. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am J Physiol. 1998;275:E259–E264. doi: 10.1152/ajpendo.1998.275.2.E259. [DOI] [PubMed] [Google Scholar]
- Shi H, Akunuru S, Bierman JC, Hodge KM, Mitchell MC, Foster MT, Seeley RJ, Reizes O. Diet-induced obese mice are leptin insufficient after weight reduction. Obesity (Silver Spring) 2009;17:1702–1709. doi: 10.1038/oby.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CK, Vaughan CH, Keen-Rhinehart E, Harris RB, Richard D, Bartness TJ. Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown adipose tissue: neuroanatomical and functional evidence. Am J Physiol Regul Integr Comp Physiol. 2008;295:R417–R428. doi: 10.1152/ajpregu.00174.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ste Marie L, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc Natl Acad Sci U S A. 2000;97:12339–12344. doi: 10.1073/pnas.220409497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss-Andreae A, Murphy JG, Ellacott KL, Stuart RC, Nillni EA, Cone RD, Fan W. Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology. 2007;148:1550–1560. doi: 10.1210/en.2006-1389. [DOI] [PubMed] [Google Scholar]
- Williams DL, Bowers RR, Bartness TJ, Kaplan JM, Grill HJ. Brainstem melanocortin 3/4 receptor stimulation increases uncoupling protein gene expression in brown fat. Endocrinology. 2003;144:4692–4697. doi: 10.1210/en.2003-0440. [DOI] [PubMed] [Google Scholar]
- Xiao XQ, Williams SM, Grayson BE, Glavas MM, Cowley MA, Smith MS, Grove KL. Excess weight gain during the early postnatal period is associated with permanent reprogramming of brown adipose tissue adaptive thermogenesis. Endocrinology. 2007;148:4150–4159. doi: 10.1210/en.2007-0373. [DOI] [PubMed] [Google Scholar]
- Zaretskaia MV, Zaretsky DV, Sarkar S, Shekhar A, DiMicco JA. Induction of Fos-immunoreactivity in the rat brain following disinhibition of the dorsomedial hypothalamus. Brain Res. 2008;1200:39–50. doi: 10.1016/j.brainres.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Matheny MK, Tümer N, Mitchell MK, Scarpace PJ. Leptin antagonist reveals that the normalization of caloric intake and the thermic effect of food after high-fat feeding are leptin dependent. Am J Physiol Regul Integr Comp Physiol. 2007;292:R868–R874. doi: 10.1152/ajpregu.00213.2006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Scarpace PJ. The role of leptin in leptin resistance and obesity. Physiol Behav. 2006;88:249–256. doi: 10.1016/j.physbeh.2006.05.038. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kilroy GE, Henagan TM, Prpic-Uhing V, Richards WG, Bannon AW, Mynatt RL, Gettys TW. Targeted deletion of melanocortin receptor subtypes 3 and 4, but not CART, alters nutrient partitioning and compromises behavioral and metabolic responses to leptin. FASEB J. 2005;19:1482–1491. doi: 10.1096/fj.05-3851com. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kerman IA, Laque A, Nguyen P, Faouzi M, Louis GW, Jones JC, Rhodes C, Münzberg H. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J Neurosci. 2011;31:1873–1884. doi: 10.1523/JNEUROSCI.3223-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]