

Abstract
GABA signaling molecules are critical for both human brain development and the pathophysiology of schizophrenia. We examined the expression of transcripts derived from three genes related to GABA signaling [GAD1 (GAD67 and GAD25), SLC12A2 (NKCC1), and SLC12A5 (KCC2)] in the prefrontal cortex (PFC) and hippocampal formation of a large cohort of nonpsychiatric control human brains (n = 240) across the lifespan (from fetal week 14 to 80 years) and in patients with schizophrenia (n = 30–31), using quantitative RT-PCR. We also examined whether a schizophrenia risk-associated promoter SNP in GAD1 (rs3749034) is related to expression of these transcripts. Our studies revealed that development and maturation of both the PFC and hippocampal formation are characterized by progressive switches in expression from GAD25 to GAD67 and from NKCC1 to KCC2. Previous studies have demonstrated that the former leads to GABA synthesis, and the latter leads to switching from excitatory to inhibitory neurotransmission. In the hippocampal formation, GAD25/GAD67 and NKCC1/KCC2 ratios are increased in patients with schizophrenia, reflecting a potentially immature GABA physiology. Remarkably, GAD25/GAD67 and NKCC1/KCC2 expression ratios are associated with rs3749034 genotype, with risk alleles again predicting a relatively less mature pattern. These findings suggest that abnormalities in GABA signaling critical to brain development contribute to genetic risk for schizophrenia.
Introduction
The most widely replicated postmortem brain findings in schizophrenia are related to GABA signaling (Akbarian et al., 1995; Lewis and Hashimoto, 2007; Charych et al., 2009). Most schizophrenia studies focus on adults, but the pathogenesis probably involves early brain development (Marenco and Weinberger, 2000). Genetic variation in GAD1, the gene for the GABA synthetic enzyme GAD67, may be an etiologic factor in schizophrenia (Addington et al., 2005; Straub et al., 2007; Du et al., 2008). The possibility that the pattern of GABA abnormalities associated with schizophrenia might reflect genetically regulated developmental processes has not been explored.
GABA is synthesized by glutamic acid decarboxylase, derived from two genes, GAD1 and GAD2. GAD1 has been studied by measuring GAD67 expression in normal human brain development (Chan et al., 1997; Huang et al., 2007). Another GAD1 transcript, GAD25, lacking an enzymatic domain and predominantly expressed fetally, has not been studied extensively (Bond et al., 1990; Szabo et al., 1994). GAD25 may play a role in developmental processes such as cell proliferation, migration, and/or synaptogenesis (Popp et al., 2009). Moreover, the switch from GAD25 to GAD67 is accompanied by increased GABA synthesis.
The transition of GABA from excitation to inhibition is critical in brain development (Owens and Kriegstein, 2002; Blaesse et al., 2009). Functionally linked to postsynaptic GABAA receptors in brain, the cation-chloride cotransporters NKCC1 and KCC2 help mediate the biophysical effects of GABA. Regulating intracellular Cl− concentration, these cotransporters help determine the electrophysiological effects of GABAA receptor activation. The direction and degree of GABAA receptor-mediated Cl− current depend, among other things, on the chloride gradient across the neuronal membrane, which in turns depends on the relative activity of NKCC1 (by itself resulting in higher Cl− “in”) and KCC2 (by itself resulting in higher Cl− “out”). Accordingly, NKCC1 activity helps make GABAA receptor activation depolarizing, whereas KCC2 activity has an inverse effect, making GABAA receptor activation hyperpolarizing (Kaila, 1994; Owens and Kriegstein, 2002; Yamada et al., 2004; Blaesse et al., 2009). Early on, NKCC1 expression predominates; as brain development proceeds, KCC2 expression rises. NKCC1/KCC2 expression ratio change parallels the change of GABA from an excitatory to an inhibitory neurotransmitter (Owens and Kriegstein, 2002; Blaesse et al., 2009).
NKCC1 is expressed in neurons and nonneuronal cells, including somatic tissues (Plotkin et al., 1997; Yan et al., 2001; Mikawa et al., 2002). KCC2 is brain-specific in humans (Song et al., 2002) and rats (Payne et al., 1996) and preferentially expressed in mature neurons (Payne et al., 1996). KCC2 also contributes to mature dendritic spine formation and functional excitatory synapses (Li et al., 2007).
We examined NKCC1, KCC2, GAD67, and GAD25 expression across the lifespan in the PFC and hippocampal formation of controls and separately in controls and schizophrenics, with an additional analysis of expression conditioned on the genotype at a schizophrenia-associated promoter SNP in GAD1 (rs3749034). We studied rs3749034 because of its previous association with GAD67 expression (Straub et al., 2007). GABA itself may play a role in the induction of KCC2 expression (Ganguly et al., 2001; Aguado et al., 2003; Carmona et al., 2006; Liu et al., 2006), although a strong GABA signal may not be necessary for increased KCC2 (Sipilä et al., 2009). Because postsynaptic stimulation of GABAA receptors may induce KCC2 expression, we hypothesized that schizophrenics with the rs3749034 risk genotype—associated with lower levels of GAD67 expression—would have lower levels of KCC2 and an elevated NKCC1/KCC2 ratio, a pattern reflecting immature GABA physiology.
Materials and Methods
Human postmortem brain tissue collection.
Male and female postmortem human brains from the Clinical Brain Disorders Branch (CBDB cases) were obtained at autopsy primarily from the Washington, D.C., and Northern Virginia Medical Examiners' Offices, all with informed consent from the legal next of kin (protocol #90-M-0142 approved by the NIMH/NIH Institutional Review Board). Additional postmortem fetal, infant, child, and adolescent brain tissue samples (designated UMD cases) were provided by the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders (http://medschool.umaryland.edu/BTBank) under contracts NO1-HD-4-3368 and NO1-HD-4-3383. The Institutional Review Board of the University of Maryland at Baltimore and the State of Maryland approved the protocol, and the tissue was donated to the NIMH under the terms of a Material Transfer Agreement. Clinical characterization, diagnoses, and macroscopic/microscopic neuropathological examinations were performed on all CBDB cases using a standardized paradigm (Lipska et al., 2006). The UMD cases were handled in a similar fashion (http://medschool.umaryland.edu/BTBank/ProtocolMethods.html). Toxicological analysis (including both prescription and illicit drugs) was performed on every case. All control subjects had no history of psychiatric illness, alcohol abuse, or drug abuse. Positive toxicology was not an exclusion criterion for the schizophrenic cases. The demographic characteristics of each subject group are summarized in Table 1.
Table 1.
Cohort Demographics and Characteristics
| Cohort | n | Race | Sex | Agea | PMI (h) | pH | RIN |
|---|---|---|---|---|---|---|---|
| Fetal | |||||||
| DLPFC | 36 | 34AA/2C | 16F/20M | 17.6 ± 1.7 | 2.4 ± 2.1 | N/A | 9.64 ± 0.26 |
| Hippocampus | 22 | 20AA/2C | 13F/9M | 17.7 ± 1.6 | 2.6 ± 2.6 | N/A | 9.24 ± 0.57 |
| Lifespan | |||||||
| DLPFC | 204 | 102AA/4A/5H/93C | 63F/141M | 30 ± 20 | 30 ± 6 | 6.5 ± 0.31 | 8.2 ± 0.95 |
| Hippocampus | 216 | 125AA/4A/5H/82C | 72F/144M | 36 ± 20 | 31 ± 15 | 6.5 ± 0.31 | 7.8 ± 0.82 |
| Normal controls versus schizophrenic subjects | |||||||
| Hippocampus | |||||||
| Controls | 62 | 38AA/3A/2H/19C | 18F/44M | 43 ± 16 | 32 ± 15 | 6.6 ± 0.26 | 5.8 ± 1.08 |
| Schizophrenic patients | 30 | 20AA/1H/9C | 10F/20M | 48 ± 16 | 36 ± 15 | 6.6 ± 0.33 | 5.3 ± 0.93 |
| DLPFC | |||||||
| Controls | 73 | 44AA/3A/4H/22C | 25F/48M | 43 ± 15 | 32 ± 14 | 6.6 ± 0.28 | 8.0 ± 0.71 |
| Schizophrenic patients | 31 | 17AA/2H/12C | 13F/18M | 48 ± 16 | 37 ± 18 | 6.5 ± 0.27 | 8.0 ± 0.54 |
AA, African American; C, Caucasian; A, Asian; H, Hispanic; F, female; M, male; PMI, postmortem interval; RIN, RNA integrity number.
aFetal cohort gestational age in weeks; lifespan and normal controls vs schizophrenic patients ages in years.
Tissue retrieval and processing.
For the CBDB cases, brains were hemisected, cut into 1.0–1.5-cm-thick coronal slabs, flash frozen, and stored at −80°C. The hippocampal formation (hippocampus proper and subiculum) and dorsolateral prefrontal cortex (DLPFC) gray matter (Brodmann's areas 9 and 46) were microdissected as previously described for all the CBDB cases and the UMD infant, child, and adolescent cases (Lipska et al., 2006). For the UMD fetal cases, the PFC consisted of a wedge of frontal cortex dissected from the dorsal convexity of the frontal lobes, midway between the frontal pole and the anterior temporal pole, under visual guidance using a dental drill. For the fetal hippocampal formation, a wedge of tissue was taken from the medial temporal lobe, along the rostral-caudal extent of the temporal lobe. Pulverized cerebellum was used for pH measurement.
RNA extraction, quantitative real-time PCR, GAD1 (GAD67) genotyping and analysis.
Human brain specimens were processed, RNA was extracted, and quantitative RT-PCR (qRT-PCR) was performed using a paradigm described previously (Mathew et al., 2007). Species-specific probes and primer sets (Applied Biosystems) were used; for human NKCC1 (SLC12A2): Catalog No. Hs00169032_m1 (spanning exons 4–5); for human KCC2 (SLC12A5): Catalog No. Hs01110928_m1 (spanning exons 8–9); for GAD67: Catalog No. Hs00241471_m1 (spanning exons 11–12); for GAD25: Catalog No. Hs00247564_m1 (spanning exons 6–7). For normalization purposes, the geometric mean of three control genes was used: β-2-microglobulin (B2M), β-glucuronidase, and β-actin (Applied Biosystems, Assays on Demand: Hs99999907, Hs99999908, and Hs99999903, respectively) (Lipska et al., 2006). GAD67 expression and rs3749034 genotypes were generated as described by Straub et al. (2007).
Statistical analysis comparing normal controls with patients with schizophrenia.
For analysis of transcripts across the lifespan, the data were plotted using the log2 of the normalized quantitative RT-PCR results from each subject. The lifespan curve was generated using LOESS fit (local polynomial regression fitting), using an R package with default parameters (span = 0.75, degree = 2).
From the CBDB postmortem brain collection, analyses in the hippocampus were performed on 62 normal controls and 30 schizophrenic patients and in the DLPFC on 73 normal controls and 31 schizophrenic patients (see Table 1 for detailed demographics). Outliers that were 2 SDs beyond the control mean were eliminated. Statistical analyses were performed using Statistica version 7.1 (StatSoft; www.statsoft.com). The data were normally distributed (Kolmogorov–Smirnoff test); therefore, parametric analyses were used for all comparisons. Covariates were chosen for each ANCOVA from multiple regression analyses. Multiple regression analyses were performed to assess the contributions of age, sex, pH, postmortem interval, RNA integrity number, and smoking history on endogenous control genes, KCC2, NKCC1, GAD67, and GAD25 mRNA expression. Multiple regression analyses were also performed to assess the effects of age of onset of illness, age of hospitalization, lifetime neuroleptic exposure, average daily dose, and final neuroleptic dose on mRNA expression in the patients with schizophrenia. Estimates of lifetime neuroleptic exposure, average daily dose, and final neuroleptic dose were all converted to chlorpromazine equivalents for statistical comparisons. Comparisons between diagnostic groups were made using univariate ANCOVA for mRNA expression with diagnosis as an independent variable. All experiments were conducted blind to diagnosis. The relationship between expression levels was tested using Spearman's coefficient of correlation.
Neuroleptic treatment in rats.
Chronic neuroleptic administration to rats was performed to test for drug effects on expression of the cotransporters. Male Sprague–Dawley rats (weight ∼250 g) were on a 12 h light/dark cycle (lights on/off 6:00 A.M./6:00 P.M.) in a temperature-controlled environment and with ad libitum access to food and water. Rats were randomly assigned to drug treatment groups (8–10 per dose) and administered intraperitoneal injections of haloperidol (0.08, 0.6, 2 mg/kg), clozapine (0.5, 5, 10 mg/kg) or vehicle (0.02% lactic acid) once daily for 28 d. The dosage regimen was chosen to emulate the therapeutic range of doses given to patients (Kapur et al., 2000). Haloperidol (Research Biochemicals) (20 mg/ml) was prepared in 1% lactic acid, diluted with water, and neutralized with 1 m NaOH to obtain pH 5.3. Clozapine (a gift from Sandoz Research Institute Berne) (100 mg) was dissolved in 0.1 m HCl, diluted with water, and neutralized to pH 5.2. Rats were killed 7 h after the last injection. Brains were sectioned into 20 μm slices using a cryostat and frozen at −80°C. All procedures were performed in accordance with the National Institutes of Health Guidelines for Use and Care of Laboratory Animals.
RNA extraction and qRT-PCR: rat brain.
To determine the effect of neuroleptic treatment on the expression of the cotransporters in rat brain, RNA was extracted from slide-mounted coronal tissue sections at the level of the mid-hippocampus: NKCC1, Catalog No. Rn00582505_m1, and KCC2, Catalog No. Rn00592624_m1, were used. To normalize the rat qRT-PCR data, the geometric means of glyceraldehyde-3-phosphate dehydrogenase, porphobilinogen deaminase, and B2M (Catalog Nos. Rn99999916_s1, Rn00565886_m1, Rn00560865_m1, respectively) were used. The qRT-PCR was performed using ABI Prism 7900 sequence detection system with 384-well format (Applied Biosystems). Transcription expression was measured using an ABI Prism 700 sequence detection system with a 384-well format (Applied Biosystems), as described previously (Lipska et al., 2006).
Results
Expression of cotransporters in the prefrontal cortex and hippocampal formation
In DLPFC, KCC2 and NKCC1 mRNA expression increased with fetal gestational age (GA) across the second trimester (r = 0.71, p = 1.6 × 10−6, and r = 0.49, p = 0.002, respectively) (Fig. 1A,B). Expression of both genes continued to increase after birth: KCC2, r = 0.2446, p = 0.0005; NKCC1, r = 0.3564, p = 2 × 10−7 (Fig. 2A,B). Moreover, there was a significant decline in the fetal period in the ratio of NKCC1 to KCC2 mRNA transcript levels as a function of GA (r = −0.35, p = 0.04) (Fig. 1C), reaching a nadir in early childhood (Fig. 2C). In the hippocampal formation, KCC2 mRNA expression again increased with fetal GA across the second trimester (r = 0.52, p = 0.014). However, NKCC1 mRNA expression did not increase with GA (r = 0.040, p = 0.86). Despite this, consistent with the DLPFC, both KCC2 and NKCC1 mRNA transcript levels increased after birth: KCC2, r = 0.334, p = 8.3 × 10−8 (Fig. 2D); NKCC1, r = 0.292, p = 3.12 × 10−6 (Fig. 2E). Fetal NKCC1/KCC2 expression ratio exhibited a significant decline as a function of increasing second trimester GA (r = −0.466, p = 0.03) and continued to fall after birth (Fig. 2F).
Figure 1.
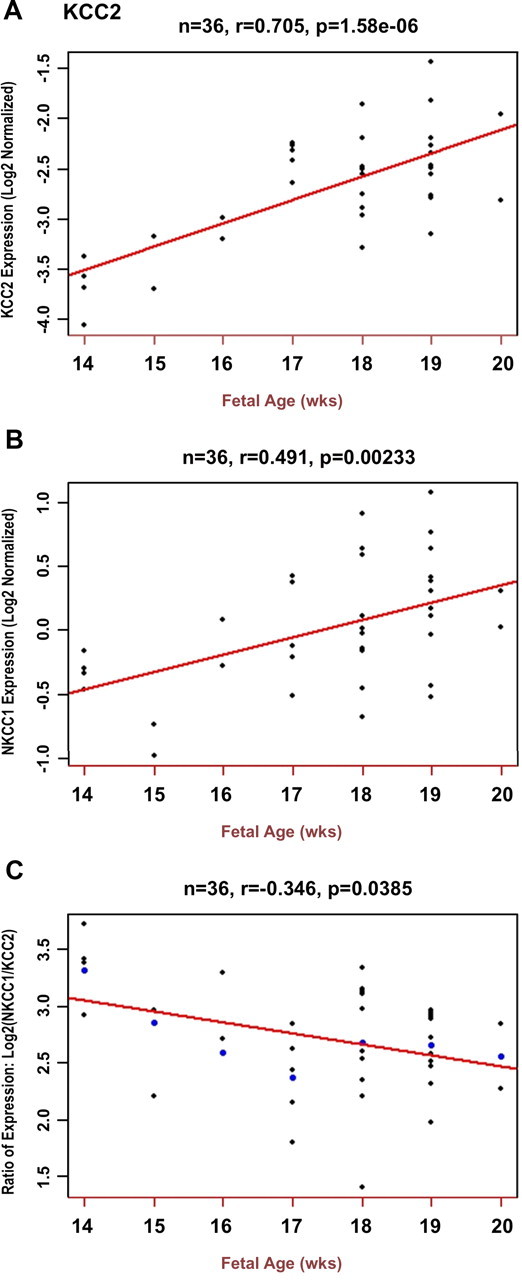
Fetal expression patterns of KCC2 and NKCC1 in prefrontal cortex (qRT-PCR). A, KCC2 expression (y-axis) shows a positive correlation with fetal gestational age expressed in weeks during the second trimester (x-axis). Each small dot represents an expression value for a single brain. B, NKCC1 expression also increases with fetal gestational age during the second trimester, with and without inclusion of the 39 week fetal sample. C, The ratio of NKCC1/KCC2 mRNA transcript levels decreases with increasing fetal gestational age. A decreasing ratio indicates that the rate of increase of the “mature” cotransporter, KCC2, is greater than that of the “immature” cotransporter, NKCC1.
Figure 2.
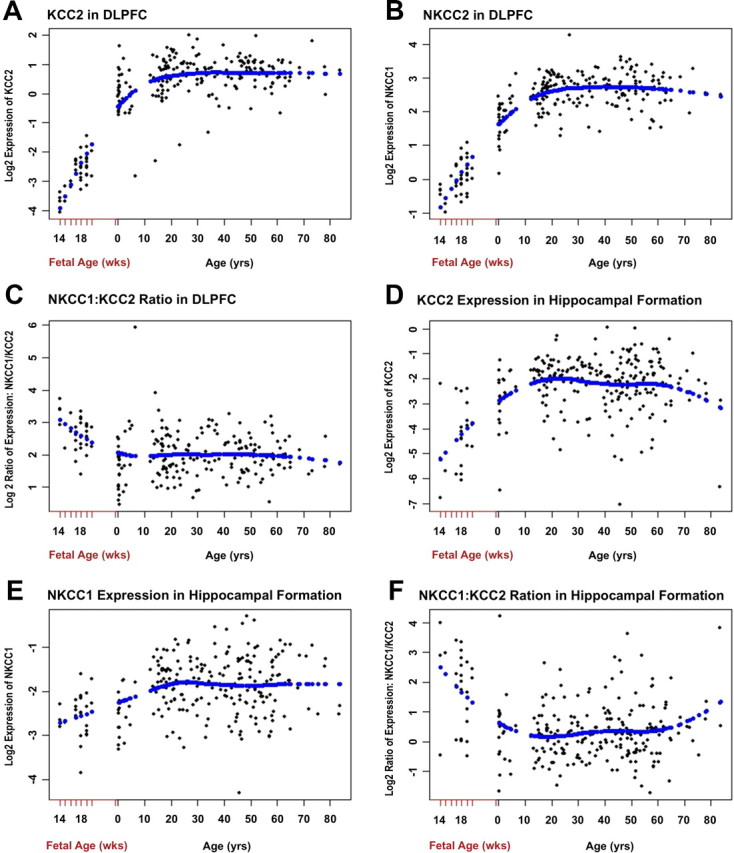
Cotransporter expression patterns in DLPFC and hippocampal formation across the lifespan (qRT-PCR). A, KCC2 expression in the DLPFC increases throughout the lifespan from birth to approximately 20 years of age and then levels off. B, In the DLPFC, NKCC1 also increases after birth and levels off at approximately 20 years of age. C, The DLPFC NKCC1/KCC2 ratio steadily declines during fetal development, reaching a plateau around 3 years of age that is maintained throughout the lifespan. D, KCC2 expression in the hippocampal formation increases from birth to approximately 20 years of age and then levels off, before declining around 60 years of age. E, NKCC1 also increases after birth in the hippocampal formation and levels off around 23 years of age. F, The hippocampal NKCC1/KCC2 ratio steadily declines during fetal development, reaching a plateau around 14 years of age that is maintained until approximately 53 years of age, before starting to rise.
Expression of GAD67, GAD25, and the GAD25/GAD67 mRNA ratio across the lifespan
In the DLPFC, prefrontal GAD67 mRNA expression increased with fetal GA and continued to increase into the first decade of life (Fig. 3A). In contrast, GAD25 expression was highest in the fetal brain and declined gradually into the first decade of life, attaining a stable expression pattern thereafter (Fig. 3B). Accordingly, the prefrontal GAD25/GAD67 expression ratio declined during early brain development (Fig. 3C). In marked contrast to the expression profile in the DLPFC, hippocampal GAD67 expression was stably expressed across development (Fig. 3D), but GAD25 expression trajectories were similar between the two brain regions, being highest early in fetal GA and steadily declining (Fig. 3E). Notwithstanding the differences in GAD67 patterns, the overall trajectory for the GAD25/GAD67 ratio was similar between the DLPFC and hippocampal formation, again being highest during early fetal development and declining with age (Fig. 3F).
Figure 3.
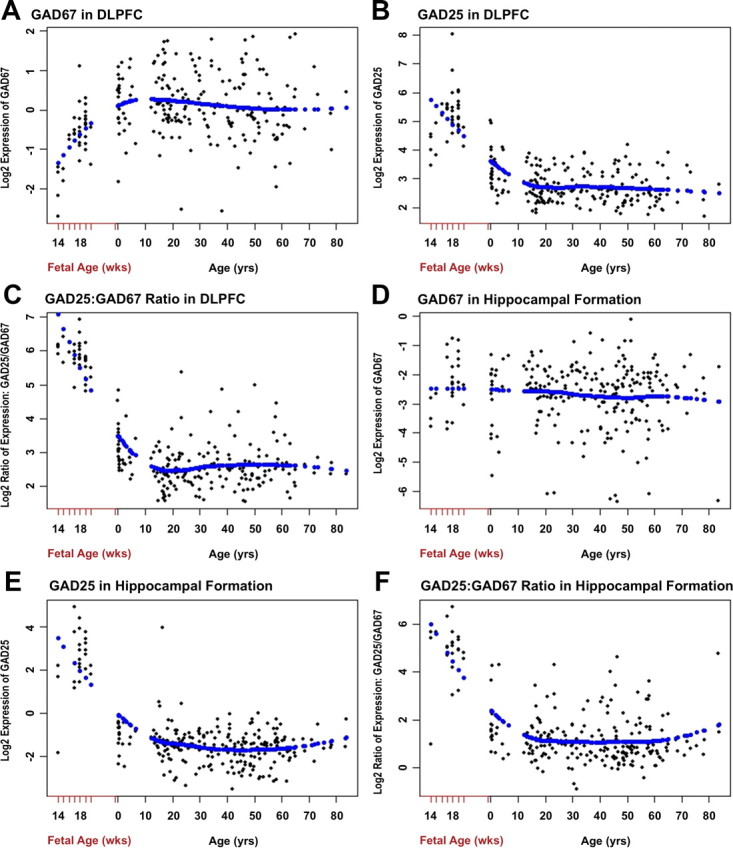
Expression patterns of GAD67, GAD25, and the GAD25/GAD67 ratio in prefrontal cortex and hippocampal formation across the lifespan (qRT-PCR). A, In the DLPFC, GAD67 expression increases from 14 weeks of gestational age until approximately 10 years of age and then levels off throughout the rest of the lifespan. B, DLPFC GAD25 expression is highest around 14 weeks of gestational age and steadily declines thereafter, reaching a stable level of expression at approximately 10 years of age. C, The DLPFC GAD25/GAD67 ratio rapidly declines during fetal development through the first decade of life after birth and then is stable across the lifespan. D, In marked contrast to the DLPFC expression pattern, in the hippocampus, GAD67 expression is relatively stable from 14 weeks of gestational age across the lifespan. E, As in the DLPFC, hippocampal GAD25 expression is highest around 14 weeks of gestational age and steadily declines thereafter, reaching a stable level of expression around 10 years of age that is maintained for the rest of the lifespan. F, The hippocampus pattern of the GAD25/GAD67 expression ratio is similar to that of the DLPFC, rapidly falling until 15 years of age and then is stable in adulthood.
Effect of diagnosis on cotransporter expression levels
In the DLPFC, no significant differences were observed for either KCC2 or NKCC1 mRNA expression or their ratio in patients with schizophrenia compared with controls (both F values <1.9; both p values >0.1). In contrast, in the hippocampal formation, patients with schizophrenia had a 27% lower level of KCC2 mRNA (F(1,86) = 4.45, p = 0.04) compared with normal controls (Fig. 4A). There was no difference in NKCC1 expression between diagnostic groups. Patients also had a trend toward an increased NKCC1/KCC2 ratio (F(1,78) = 3.08, p = 0.08) (Fig. 4B).
Figure 4.

KCC2 expression and the NKCC1/KCC2 expression ratio in the hippocampal formation in schizophrenia. A, KCC2 mRNA expression is significantly lower in the hippocampus of adult schizophrenic subjects compared with normal controls. *p = 0.04. B, The NKCC1/KCC2 ratio shows trend towards an increase in the hippocampus of adult schizophrenic subjects compared with normal controls. †p = 0.08. Error bars indicate SD.
Multiple regression analyses revealed that in the DLPFC, KCC2 mRNA levels positively correlated with lifetime neuroleptic exposure expressed as chlorpromazine equivalents and NKCC1 positively correlated with age of onset of illness (adjusted R2 > 0.13, F > 2.0, p < 0.03). Within the hippocampus, duration of illness negatively correlated with KCC2 levels (adjusted R2 = 0.27, F(1,21) = 9.3, p = 0.006). None of the clinical variables significantly correlated with NKCC1 levels in the hippocampus (adjusted R2 = 0.02, F(1,21) = 1.6, p = 0.2). Each positive variable was included as covariates in the ANCOVA.
In the DLPFC, females had a significantly higher expression of both cation chloride cotransporters (p < 0.05). In the hippocampus, females had significantly higher levels of KCC2 expression (p = 0.007).
Hippocampal cotransporter expression in neuroleptic-treated rats
The expression of KCC2 and NKCC1 mRNA was examined in rats chronically treated either with vehicle or with low, medium, or high doses of haloperidol or clozapine. There was no significant effect of neuroleptic treatment on either NKCC1 or KCC2 mRNA expression or their ratio compared with vehicle and there were no dosage effects of the two drugs (p > 0.05).
Relationships between GAD67, GAD25, and GAD1 genotype and cotransporter expression
GAD67 and KCC2 mRNA correlated positively in the hippocampus in a combined cohort of normal controls and schizophrenia patients (r = 0.76, p < 10−5), in normal controls alone (r = 0.75, p < 10−5), and in schizophrenia patients alone (r = 0.79, p < 10−5). There was a negative correlation between GAD67 and NKCC1 mRNA levels in all three groups. There was also a negative correlation between levels of GAD67 and the NKCC1/KCC2 mRNA ratio in the combined cohort (r = −0.53, p < 10−5), in normal controls (r = −0.62, p < 10−5), and in schizophrenia patients (r = −0.71, p < 4 × 10−5).
Given the correlation between GAD67 and KCC2 levels and the prior association of rs3749034 genotype with GAD67 levels in patients with schizophrenia (Straub et al., 2007), we examined KCC2 expression in the hippocampus in the schizophrenia cohort, conditioned on rs3749034 genotype. As predicted, patients with the risk-associated G/G genotype at rs3749034 had a significant decrease in KCC2 mRNA expression in the hippocampus (F(1,25) = 12.5, p < 0.004), compared with minor allele carriers (Fig. 5A). This also held true when analyzing the Caucasian schizophrenic subjects alone (F(1,7) = 20.8, p = 0.002). Whereas there was no association between the risk genotype and NKCC1 expression in either the hippocampus or the DLPFC, the NKCC1/KCC2 ratio was significantly higher in the hippocampus of patients with the G/G genotype (F(1,23) = 4.29, p = 0.05) compared with minor allele carriers (Fig. 5B).
Figure 5.

GAD1 genotypic variation and KCC2 and the NKCC1/KCC2 ratio. A, KCC2 mRNA expression levels are significantly and selectively lower in the hippocampus of schizophrenic subjects who are homozygous (1/1 = G/G) for the major allele of the GAD1 schizophrenia-associated risk SNP rs3749034. *p < 0.004. There was no association between homozygosity for this SNP and KCC2 expression levels in normal controls in hippocampus or in normal controls or schizophrenic subjects in the DLPFC. B, The NKCC1/KCC2 ratio also was significantly higher in the hippocampus of G/G schizophrenic subjects. *p = 0.05. There was no association between the 1/1 (G/G) genotype for rs3749034 and the cotransporter ratio in normal controls in the hippocampus or in normal controls or schizophrenic subjects in the DLPFC. Error bars indicate SD.
We also tested the relationship between the “fetal” GAD1 transcript, GAD25, and KCC2 mRNA expression. In the hippocampus of the combined cohort of control and schizophrenic subjects, there was an inverse correlation between GAD25 and KCC2 expression (r = −0.21, p = 0.05), also seen in the schizophrenic subjects alone (r = −0.40, p = 0.03).
GAD25 mRNA expression did not differ between diagnostic groups in the DLPFC or hippocampus. However, GAD1 rs3749034 genotype predicted GAD25 expression; those subjects with risk-associated G/G genotype had higher levels compared with minor allele carriers (F(1,85) = 4.94, p = 0.03) (Fig. 6A). There were trends toward significance in controls (F(1,59) = 3.48, p = 0.07) and schizophrenics (F(1,23) = 3.33, p = 0.08) analyzed separately. The GAD25/GAD67 expression ratio was significantly elevated in the schizophrenic subjects homozygous for the major allele at rs3749034 (F(1,20) = 4.78, p = 0.04) (Fig. 6B), but not in the controls.
Figure 6.

GAD1 genotypic variation and GAD25 expression and the GAD25/GAD67 expression ratio. A, In a combined cohort of schizophrenic and control subjects, those with a 1/1 (G/G) genotype for the schizophrenia-associated GAD1 risk allele rs3749034 had significantly higher expression of GAD25 compared with carriers of the 2 (A) allele in the hippocampus. *p = 0.03. B, In a cohort of schizophrenic subjects, those with a G/G genotype for the schizophrenia-associated GAD1 risk allele rs3749034 had a significantly higher GAD25/GAD67 expression ratio compared with carriers of the A allele in the hippocampus *p = 0.04. Error bars indicate SD.
Discussion
The maturation of GABA signaling in PFC and hippocampus of human brain is characterized by progressive switches in expression from GAD25 to GAD67 and from NKCC1 to KCC2. The former leads to the synthesis of GABA; the latter leads to GABA switching from an excitatory to an inhibitory neurotransmitter (Kaila, 1994; Ganguly et al., 2001; Blaesse et al., 2009). Ratios of GAD25/GAD67 and NKCC1/KCC2 reflect the maturational state of GABA function in the human PFC and hippocampus (Mikawa et al., 2002).
In the hippocampus of patients with schizophrenia, GAD25/GAD67 and NKCC1/KCC2 ratios are increased and KCC2 levels are decreased, reflecting a potentially immature state of the GABA system. Remarkably, these increased GAD25/GAD67 and NKCC1/KCC2 expression ratios in hippocampus are associated with the GAD1 genotype, with risk alleles predicting the relatively less mature pattern. These findings suggest that abnormalities in GABA signaling that are critical for human brain development and maturation also contribute to genetic risk for schizophrenia. These findings also implicate a genetic mechanism underlying the well established abnormalities in GABA function associated with the clinical diagnosis and provide a developmental context to these associations.
One previous study examined the expression of these two cotransporters in subjects with schizophrenia (Arion and Lewis, 2011). In keeping with our findings, they did not find a change in NKCC1 or KCC2 expression in the DLPFC of patients with schizophrenia. However, Arion and Lewis (2011) did not examine the expression of these two cotransporters in the hippocampal formation.
As is the case in any postmortem human brain study of schizophrenia, there are a number of possible confounding factors, not the least of which is antemortem neuroleptic treatment. To address this issue, the expression of NKCC1 and KCC2 was measured in rats treated with haloperidol, a typical neuroleptic, and clozapine, an atypical agent. Neither drug affected the expression of NKCC1, KCC2, or the NKCC1/KCC2 ratio. Although extrapolating medication effects from rats to humans has limitations, this finding supports the notion that changes in hippocampal KCC2 expression and the NKCC1/KCC2 ratio are not due to prior treatment. Likewise, in the hippocampus of patients with schizophrenia, there was no relationship to prior treatment with neuroleptics as measured with toxicology or with estimates of prior cumulative neuroleptic dosing. This suggests that the increases in NKCC1/KCC2 seen in patients with schizophrenia in hippocampus are unlikely to be artifacts of prior antemortem treatment with neuroleptics. The association of the immature GABA state with a genetic risk factor for schizophrenia further suggests that this is a trait association.
Positive toxicology was not an exclusion criterion for patients with schizophrenia. In the overwhelming majority of cases, positive toxicology was due to the presence of psychotropic medications (as expected). In a small subset of schizophrenia cases, toxicology studies also revealed the presence of a variety of illicit substances. There is a possibility, albeit remote, that the changes noted in the patients with schizophrenia are attributable to substance abuse. Nonpsychiatric controls were excluded if toxicology was positive for psychotropic medications or illicit substances.
The developmental patterns of human prefrontal and hippocampal KCC2 expression during development found in our study agree with and expand on previous reports in both humans (Dzhala et al., 2005; Vanhatalo et al., 2005; Bayatti et al., 2008) and other species (Balakrishnan et al., 2003). This pattern of protracted postnatal change in cortical function is in agreement with the seminal work of Yakovlev (Yakovlev, 1967), who showed that association cortex myelination is not completed until the early part of the third decade of life. It also is consistent with in vivo imaging assessments of brain development (Pfefferbaum et al., 1994). Among the frontal subdivisions, the DLPFC appears to mature last (Yakovlev, 1967). Prefrontal neuropsychological test performance also improves during childhood and adolescence, in parallel with the rise of KCC2 expression (Somsen, 2007). KCC2 mRNA expression is both brain-specific (Song et al., 2002; Balakrishnan et al., 2003) and neuron-specific (Payne et al., 1996; Karadsheh and Delpire, 2001; Mikawa et al., 2002; Wang et al., 2002; Balakrishnan et al., 2003; Gagnon et al., 2007; Blaesse et al., 2009). The progressive increase in KCC2 expression in the DLPFC may be a molecular correlate of the late maturation of this brain region and is consistent with other evidence that GABA tuning of cortical circuitry is critical for higher order prefrontal cortical cognition (Rao et al., 2000).
The patterns of expression of NKCC1 in human PFC and hippocampus are consistent with variations in the developmental expression pattern of NKCC1 in rodent brain. Although most rodent studies have reported an increase in NKCC1 mRNA and/or protein levels after birth (Clayton et al., 1998; Yan et al., 2001; Mikawa et al., 2002; Wang et al., 2002; Balakrishnan et al., 2003), there have been three reports of a decrease in expression (Hübner et al., 2001; Wang et al., 2002; Yamada et al., 2004). The latter, however, found an increase in hippocampus after birth (Wang et al., 2002). Our findings are consistent with the majority of animal studies that describe a rise in NKCC1 mRNA expression in both the neocortex and hippocampal formation after birth, but this rise is less steep than that of KCC2.
This is the first report on the developmental trajectory of both GAD67 and GAD25 expression in human brain. Chan and colleagues (1997) detected GAD67 mRNA in human fetal frontal pole at 12 weeks gestational age. The DLPFC GAD67 developmental expression pattern from this report replicates a previous study (Huang et al., 2007). Chessler and Lernmark (2000) found GAD67 in human brain but not GAD25. GAD25 mRNA during human PFC development mirrors findings in the rat and mouse. In the mouse brain, GAD67 is expressed at low levels during early fetal development and steadily rises. In contrast, GAD25 was highest in the fetal mouse brain, dropped rapidly after birth, and was almost undetectable in adult mice (Szabo et al., 1994). A similar developmental pattern was found in the rat brain (Bond et al., 1990; Popp et al., 2009). However, in rats, embryonic GAD1 transcripts had comparatively high levels of expression into adulthood in the subventricular zone, rostral migratory stream, and olfactory bulb, regions associated with active neurogenesis in adulthood (Popp et al., 2009). Our findings suggest that GAD25 is involved in the early phases of brain development, and the ratio of GAD25/GAD67 mRNA expression falls as a structure reaches full maturity. The early decline in GAD25 expression and the concomitant changes in the GAD25/GAD67 ratio suggest that there may be a developmental switch in presynaptic GABA signaling.
GAD25 may play an indirect role in regulating the expression of KCC2. There is an inverse correlation between GAD25 and KCC2 expression during early prefrontal development. As the GAD67/KCC2 ratio rises, the GAD25/KCC2 ratio falls. In the adult hippocampus, whereas there is a strong positive correlation between GAD67 and KCC2 mRNA levels, there is a negative correlation between GAD25 and KCC2 levels. GAD25 may indirectly suppress KCC2 expression early in cortical development, presumably by altering presynaptic GABA synthesis. However, it is important to recognize that these correlations do not prove causality. The same schizophrenia-associated GAD1 risk allele (at rs3749034) that was associated with decreased GAD67 expression also was associated with increased GAD25 expression. The risk genotype association with relative overexpression of GAD25 suggests that the mechanism of genetic risk involves an immature developmental profile in GABA neurons. We speculate based on these data that the putative GABA deficiency in schizophrenia may involve relatively less chloride conductance and therefore less subsequent inhibition for a given quanta of GABA signaling via GABA chloride channels.
Several putative schizophrenia-susceptibility genes help regulate KCC2 expression, including CHRNA7, GAD1, and BDNF (Harrison and Weinberger, 2005; Zintzaras, 2007). Stimulation of α-7- and α-3-subunit containing nicotinic receptors increases expression of KCC2 (Liu et al., 2006), as does activation of GABAA receptors (Ganguly et al., 2001; Aguado et al., 2003; Carmona et al., 2006; Liu et al., 2006), possibly through the promotion of calcium influx. However, a strong depolarizing GABA signal is not necessary for the developmental up-regulation of KCC2 (Ludwig et al., 2003; Sipilä et al., 2009). BDNF has opposing effects on KCC2 expression in different stages of development (Ganguly et al., 2001; Rivera et al., 2002; Aguado et al., 2003; Carmona et al., 2006). Early on, BDNF up-regulates KCC2 expression, whereas in the adult it reduces it (Aguado et al., 2003; Rivera et al., 2004). BDNF overexpression in transgenic mice leads to increased levels of KCC2 in both the developing forebrain and hippocampus (Aguado et al., 2003). BDNF, working through trkB receptors and CREB, presumably alters expression of KCC2 as part of its effects on transcriptional activity (Rivera et al., 2004). Moreover, BDNF also increases levels of glutamic acid decarboxylase and the density of GABA synapses early in development (Bolton et al., 2000; Marty et al., 2000) via trkB signaling (Carmona et al., 2006). Mice deficient in trkB express lower levels of KCC2 in the hippocampus (Carmona et al., 2006). These studies suggest that KCC2 expression might be a nexus for the action of several schizophrenia-susceptibility genes, and allelic variation in these genes may alter KCC2 expression and thus GABA function. Allelic variations in genes regulating KCC2 expression also have been associated with schizophrenia, including CHRNA7 (coding the α-7 nicotine receptor), GAD1 (coding GAD67, a primary synthetic enzyme for GABA) (Addington et al., 2005; Straub et al., 2007), and BDNF (Nicodemus et al., 2008).
Footnotes
We thank Amy Deep-Soboslay of the Clinical Brain Disorders Branch, GCAP, IRP, and NIMH for efforts in clinical diagnosis and demographic characterization; Dr. Michael Wininger for statistical consultations; David Hauptman for editorial assistance; and Vesna Imamovic, Yeva Snitkovsky, Jonathan Sirovatka, and Bianca Iglesias for excellent technical assistance. Special gratitude also is extended to Dr. H. Ronald Zielke, Robert D. Vigorito, and Robert M. Johnson of the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland for their provision of fetal, child, and adolescent brain specimens for this study.
The authors declare no competing financial interests.
References
- Addington AM, Gornick M, Duckworth J, Sporn A, Gogtay N, Bobb A, Greenstein D, Lenane M, Gochman P, Baker N, Balkissoon R, Vakkalanka RK, Weinberger DR, Rapoport JL, Straub RE. GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol Psychiatry. 2005;10:581–588. doi: 10.1038/sj.mp.4001599. [DOI] [PubMed] [Google Scholar]
- Aguado F, Carmona MA, Pozas E, Aguiló A, Martínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Huntsman MM, Kim JJ, Tafazzoli A, Potkin SG, Bunney WE, Jr, Jones EG. GABAA receptor subunit gene expression in human prefrontal cortex: comparison of schizophrenics and controls. Cereb Cortex. 1995;5:550–560. doi: 10.1093/cercor/5.6.550. [DOI] [PubMed] [Google Scholar]
- Arion D, Lewis DA. Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch Gen Psychiatry. 2011;68:21–31. doi: 10.1001/archgenpsychiatry.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan V, Becker M, Löhrke S, Nothwang HG, Güresir E, Friauf E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci. 2003;23:4134–4145. doi: 10.1523/JNEUROSCI.23-10-04134.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayatti N, Moss JA, Sun L, Ambrose P, Ward JF, Lindsay S, Clowry GJ. A molecular neuroanatomical study of the developing human neocortex from 8 to 17 postconceptional weeks revealing the early differentiation of the subplate and subventricular zone. Cereb Cortex. 2008;18:1536–1548. doi: 10.1093/cercor/bhm184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bolton MM, Pittman AJ, Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond RW, Wyborski RJ, Gottlieb DI. Developmentally regulated expression of an exon containing a stop codon in the gene for glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1990;87:8771–8775. doi: 10.1073/pnas.87.22.8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona MA, Pozas E, Martínez A, Espinosa-Parrilla JF, Soriano E, Aguado F. Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb Cortex. 2006;16:47–63. doi: 10.1093/cercor/bhi083. [DOI] [PubMed] [Google Scholar]
- Chan SO, Lyman WD, Chiu FC. Temporal and spatial expression of glutamic acid decarboxylases in human fetal brain. Brain Res Mol Brain Res. 1997;46:318–320. doi: 10.1016/s0169-328x(97)00031-4. [DOI] [PubMed] [Google Scholar]
- Charych EI, Liu F, Moss SJ, Brandon NJ. GABA(A) receptors and their associated proteins: implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology. 2009;57:481–495. doi: 10.1016/j.neuropharm.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessler SD, Lernmark A. Alternative splicing of GAD67 results in the synthesis of a third form of glutamic-acid decarboxylase in human islets and other non-neural tissues. J Biol Chem. 2000;275:5188–5192. doi: 10.1074/jbc.275.7.5188. [DOI] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Brain Res Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- Du J, Duan S, Wang H, Chen W, Zhao X, Zhang A, Wang L, Xuan J, Yu L, Wu S, Tang W, Li X, Li H, Feng G, Xing Q, He L. Comprehensive analysis of polymorphisms throughout GAD1 gene: a family-based association study in schizophrenia. J Neural Transm. 2008;115:513–519. doi: 10.1007/s00702-007-0844-z. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, Adragna NC, Fyffe RE, Lauf PK. Characterization of glial cell K-Cl cotransport. Cell Physiol Biochem. 2007;20:121–130. doi: 10.1159/000104160. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. image 5. [DOI] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner CA, Lorke DE, Hermans-Borgmeyer I. Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech Dev. 2001;102:267–269. doi: 10.1016/s0925-4773(01)00309-4. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kapur S, Wadenberg ML, Remington G. Are animal studies of antipsychotics appropriately dosed? Lessons from the bedside to the bench. Can J Psychiatry. 2000;45:241–246. doi: 10.1177/070674370004500302. [DOI] [PubMed] [Google Scholar]
- Karadsheh MF, Delpire E. Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J Neurophysiol. 2001;85:995–997. doi: 10.1152/jn.2001.85.2.995. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T. Deciphering the disease process of schizophrenia: the contribution of cortical GABA neurons. Int Rev Neurobiol. 2007;78:109–131. doi: 10.1016/S0074-7742(06)78004-7. [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE. Critical factors in gene expression in postmortem human brain: focus on studies in schizophrenia. Biol Psychiatry. 2006;60:650–658. doi: 10.1016/j.biopsych.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Marenco S, Weinberger DR. The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol. 2000;12:501–527. doi: 10.1017/s0954579400003138. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrlé R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew SV, Law AJ, Lipska BK, Dávila-García MI, Zamora ED, Mitkus SN, Vakkalanka R, Straub RE, Weinberger DR, Kleinman JE, Hyde TM. Alpha7 nicotinic acetylcholine receptor mRNA expression and binding in postmortem human brain are associated with genetic variation in neuregulin 1. Hum Mol Genet. 2007;16:2921–2932. doi: 10.1093/hmg/ddm253. [DOI] [PubMed] [Google Scholar]
- Mikawa S, Wang C, Shu F, Wang T, Fukuda A, Sato K. Developmental changes in KCC1, KCC2 and NKCC1 mRNAs in the rat cerebellum. Brain Res Dev Brain Res. 2002;136:93–100. doi: 10.1016/s0165-3806(02)00345-0. [DOI] [PubMed] [Google Scholar]
- Nicodemus KK, Marenco S, Batten AJ, Vakkalanka R, Egan MF, Straub RE, Weinberger DR. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry. 2008;13:873–877. doi: 10.1038/sj.mp.4002153. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Mathalon DH, Sullivan EV, Rawles JM, Zipursky RB, Lim KO. A quantitative magnetic resonance imaging study of changes in brain morphology from infancy to late adulthood. Arch Neurol. 1994;51:874–887. doi: 10.1001/archneur.1994.00540210046012. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E. Expression of the Na(+)-K(+)-2Cl- cotransporter BSC2 in the nervous system. Am J Physiol. 1997;272:C173–C183. doi: 10.1152/ajpcell.1997.272.1.C173. [DOI] [PubMed] [Google Scholar]
- Popp A, Urbach A, Witte OW, Frahm C. Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PLoS One. 2009;4:e4371. doi: 10.1371/journal.pone.0004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipilä ST, Huttu K, Yamada J, Afzalov R, Voipio J, Blaesse P, Kaila K. Compensatory enhancement of intrinsic spiking upon NKCC1 disruption in neonatal hippocampus. J Neurosci. 2009;29:6982–6988. doi: 10.1523/JNEUROSCI.0443-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somsen RJ. The development of attention regulation in the Wisconsin Card Sorting Task. Dev Sci. 2007;10:664–680. doi: 10.1111/j.1467-7687.2007.00613.x. [DOI] [PubMed] [Google Scholar]
- Song L, Mercado A, Vázquez N, Xie Q, Desai R, George AL, Jr, Gamba G, Mount DB. Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Brain Res Mol Brain Res. 2002;103:91–105. doi: 10.1016/s0169-328x(02)00190-0. [DOI] [PubMed] [Google Scholar]
- Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, Mayhew MB, Vakkalanka RK, Kolachana BS, Kleinman JE, Weinberger DR. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry. 2007;12:854–869. doi: 10.1038/sj.mp.4001988. [DOI] [PubMed] [Google Scholar]
- Szabo G, Katarova Z, Greenspan R. Distinct protein forms are produced from alternatively spliced bicistronic glutamic acid decarboxylase mRNAs during development. Mol Cell Biol. 1994;14:7535–7545. doi: 10.1128/mcb.14.11.7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhatalo S, Palva JM, Andersson S, Rivera C, Voipio J, Kaila K. Slow endogenous activity transients and developmental expression of K+-Cl- cotransporter 2 in the immature human cortex. Eur J Neurosci. 2005;22:2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x. [DOI] [PubMed] [Google Scholar]
- Wang C, Shimizu-Okabe C, Watanabe K, Okabe A, Matsuzaki H, Ogawa T, Mori N, Fukuda A, Sato K. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 2002;139:59–66. doi: 10.1016/s0165-3806(02)00536-9. [DOI] [PubMed] [Google Scholar]
- Yakovlev P. The myelogenetic cycles of regional maturation of the brain. In: Minkowski AO, editor. Regional development of the brain in early life. Oxford: Blackwell; 1967. pp. 3–70. [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:829–841. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Dempsey RJ, Sun D. Expression of Na+-K+-Cl− cotransporter in rat brain during development and its localization in mature astrocytes. Brain Res. 2001;911:43–55. doi: 10.1016/s0006-8993(01)02649-x. [DOI] [PubMed] [Google Scholar]
- Zintzaras E. Brain-derived neurotrophic factor gene polymorphisms and schizophrenia: a meta-analysis. Psychiatr Genet. 2007;17:69–75. doi: 10.1097/YPG.0b013e32801119da. [DOI] [PubMed] [Google Scholar]