

Abstract
Serotonin (5-HT)-selective reuptake inhibitors (SSRIs) are widely administered for the treatment of depression, anxiety, and other neuropsychiatric disorders, but response rates are low, and side effects often lead to discontinuation. Side effect profiles suggest that SSRIs inhibit dopaminergic activity, but mechanistic insight remains scarce. Here we show that in mice, chronic 5-HT transporter (5-HTT) blockade during adulthood but not during development impairs basal ganglia-dependent behaviors in a dose-dependent and reversible fashion. Furthermore, chronic 5-HTT blockade reduces striatal dopamine (DA) content and metabolism. A causal relationship between reduced DA signaling and impaired basal ganglia-dependent behavior is indicated by the reversal of behavioral deficits through l-DOPA administration. Our data suggest that augmentation of DA signaling would reduce side effects and increase efficacies of SSRI-based therapy.
Introduction
The serotonin (5-HT) and dopamine (DA) systems modulate numerous brain functions and are implicated in the pathophysiology of affective, anxiety, and movement disorders. Both systems interact by mutually innervating brain regions/circuits and reciprocally modulating each other. This 5-HT/DA interaction plays a critical role in the action of antidepressant drugs. 5-HT-selective reuptake inhibitors (SSRIs) for example, block 5-HT transporter (5-HTT) function to increase 5-HT signaling, but may also induce side effects typically associated with reduced DA signaling, such as anhedonia, apathy, reduced libido, akathisia, and extrapyramidal motor symptoms (EPSs) (Settle, 1998; Cassano and Fava, 2004; Damsa et al., 2004; Preskorn et al., 2004). These side effects can lead to discontinuation even when treatment is efficacious (Kaplan, 1997; Chelben et al., 2001) and reduce response rates (Rush et al., 2006). Increased mechanistic understanding of 5-HT/DA interaction should allow for improved treatment strategies and drug design.
5-HT neurons of the dorsal and median raphe nuclei densely innervate DAergic nuclei and their projection areas (Michelsen et al., 2007). In mouse and rat, the densest afferent 5-HT innervation is present in the substantia nigra pars compacta (SNc) and pars reticulata (SNr)—both integral parts of the basal ganglia (BG) (Miller et al., 1975; Dray et al., 1978; Imai et al., 1986; Vertes, 1991; Moukhles et al., 1997). The SNc DAergic neurons project to the striatum and provide DAergic modulatory input into the direct and indirect pathways of the BG. The SNr is the main output region of the BG and contains mostly GABAergic neurons projecting to the thalamus and the SNc (Bolam et al., 2000). The latter projection provides feedback inhibition to DAergic activity (Tepper and Lee, 2007). Based on this circuitry, we investigated the impact of 5-HTT blockade on the nigrostriatal DA system and BG function. We focused our studies on BG-dependent motor behavior as a proxy for BG function because of its well-characterized sensitivity to alterations in DAergic activity (Brooks and Dunnett, 2009).
Here we show that 5-htt−/− mice exhibit impaired BG-dependent motor behavior. This finding is congruent with motor deficits in maoa−/− mice (Salichon et al., 2001), because both manipulations increase extracellular 5-HT levels (Cases et al., 1996; Wellman et al., 2007; Popa et al., 2008). We investigated the origins of this behavioral phenotype using genetic and pharmacological tools to probe behavior, neuroanatomy, and neurochemistry and conclude that chronic 5-HTT blockade impairs motor behavior by reducing DAergic input into the BG.
Materials and Methods
Transgenic mice.
Mice heterozygous for the 5-htt mutation (5-htt+/−; 10th to 12th generations; 129S6/SvEv) were crossed to produce 5-htt+/+, 5-htt+/−, and 5-htt−/− offspring (Lira et al., 2003). Independent groups of male subjects weighing 25–45 g were experimentally tested at ages 2–13 months. Subjects were genotyped with PCR of genomic DNA isolated from tissue samples harvested at 18–21 d of age.
Animal husbandry.
Animals were housed in groups of five per cage and provided with food and water ad libitum (except as noted). Animals were maintained on a 12 h light/dark cycle (8:00 A.M./8:00 P.M.) Animal testing was conducted in accordance with the National Institutes of Health guidelines (Principles of Laboratory Animal Care) and the institutional animal committee guidelines.
Drugs.
Fluoxetine (FLX; ANAWA Trading) was dissolved in 0.9% NaCl or drinking water to achieve 2.5, 5, or 10 mg/kg when administered intraperitoneally or through the drinking water. The peripherally acting aromatic l-amino acid decarboxylase inhibitor benserazide (Sigma-Aldrich) was dissolved in 0.9% NaCl to achieve 12.5 mg/kg when administered intraperitoneally. l-DOPA (Sigma-Aldrich) was dissolved in 0.1 m hydrochloric acid and then neutralized with 0.1 m NaOH to achieve 100 mg/kg when administered intraperitoneally. Benserazide and l-DOPA were coadministered by consecutive intraperitoneal injections separated by 20 min with the benserazide injection preceding the l-DOPA injection. All intraperitoneal injections were performed at 10 ml/kg except when noted otherwise.
Postnatal drug treatment.
Postnatal drug treatments were performed as described previously (Ansorge et al., 2008). In short, litters were culled to five pups. On postnatal day 4 (P4), pups were tattooed and randomly assigned to vehicle (VEH; 0.9% NaCl, 5 ml/kg, i.p.) and FLX (10 mg/kg, 5 ml/kg, i.p.) treatment. Mice were treated once per day from P4 to P21. After weaning, mice were housed separately according to sex and randomly mixed for treatment.
Bioamine levels.
We measured tissue DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) levels using high-performance liquid chromatography (HPLC). Mice were injected with anesthetic and killed via cervical dislocation. Brains were dissected on ice. Frontal brain pieces containing the striatum and frontal cortex but not the hippocampus or brain pieces containing only the dorsal striatum or frontal cortex were dissected and placed into Eppendorf tubes containing 250 μl HeGA buffer [catecholamine/monoamine preservative solution containing 0.1 m glacial acetic acid, 0.1 mm EDTA (American Chemical Society grade reagent; 99.4–100.06%), and 0.12% oxidized l-glutathione, pH adjusted with filtered NaOH to 3.70]. Brains were homogenized manually using a pestle. Tissue was placed in dry ice and stored temporally at −20°C. For archival storage, tubes were stored at −80°C, ready to be processed by HPLC. The supernatant was filtered using a 0.2 μm nylon filter, and a 50 μl aliquot was prepared for HPLC processing. The HPLC program was a 27 L pickup (Model 582 Solvent Delivery System and Model 542 Autosampler Model 5600A, Esa; ChromSep HPLC SS 100 × 3.0 mm including guard column, Varian; ODS-3, Intersil), using electrochemical detection (CoulArray Detector Microdialysis Cell 5014A, Esa). The mobile phase was a 75, mm sodium phosphate buffer containing 10% acetonitrile, 25 mm citric acid, 2.2 mm 1-octanesufonic acid sodium salt, 2% methanol, 25 μm EDTA, and 100 μl/l tetraethylammonium, and pumped at a rate of 0.400 ml/min (∼1500 psi). CoulArray software was used for data acquisition and analysis using external standards of 1 μm, 100 nm, 10 nm, and 1 nm. Sample concentrations were normalized for tissue weight.
Immunohistochemistry and stereology.
Brains were fixed and sectioned (40 μm) as described previously (Lira et al., 2003). Immunohistochemical detection of tyrosine hydroxylase (TH) was performed on floating sections. Sections were rinsed in 0.1 m TBS, incubated in 0.02% H2O2/0.1 m TBS for 10 min, and washed with 0.1 m TBS (three times, 5 min each). Sections were then blocked for 30 min (3% normal goat serum, 1% bovine serum albumin, 0.4% Triton X-100, 0.1 m TBS) and subsequently incubated with rabbit anti-TH affinity-purified polyclonal antibody (AB152; Millipore Bioscience Research Reagents; 1:10,000) in blocking solution overnight at 4°C. After washing (three times, 5 min each; 0.1 m TBS), sections were incubated with biotinylated goat anti-rabbit IgG (heavy and light chains; BA-1000; Vector Laboratories; 1:400) in blocking solution for 2 h at room temperature and subsequently washed again (three times; 0.1 m TBS). Biotinylated residues were detected using the Vectastain Elite ABC system (Standard; Vector Laboratories). Sections were mounted and coverslipped using DPX (Sigma). Sections from pairs of 5-htt+/+ and 5-htt−/− mice were processed and reacted simultaneously to reduce assay variability. Antibody and reaction specificity was determined in control sections omitting the primary antibody.
SNc/ventral tegmental area (VTA) volume, total number of SNc/VTA DAergic neurons, and their densities were estimated using the fractionator approach (West et al., 1991) under bright-field microscopy (Leitz Ergolux light microscope with a Ludl motorized stage) with computer-assisted sampling (Stereo Investigator version 5.04.3; MircroBrightField). The microscope image was captured by a CCD camera (Sony PowerHAD DXC-970MD) and displayed on a computer monitor. Using the Preliminary Population Estimate feature of the software, we set the counting frame at 30 × 30 μm and the sampling grid size at 100 × 100 μm. These parameters yielded between 200 and 300 counts per brain, with a Schmitz–Hof intrasample coefficient of error averaging 0.075 ± 0.006 SD (Schmitz and Hof, 2000). There were no significant differences in coefficient of error values between genotypes. The SNc/VTA area was initially identified and traced at 4× magnification and then taken to 40× magnification, where the optical fractionator was used to perform estimates. Neurons were identified by shape and staining. Upon analyzing all the sections within a mouse, an estimated SNc/VTA volume, total TH-positive cell count, and TH-positive cell density were calculated.
Behavioral testing.
The open-field test was performed as described previously (Lira et al., 2003). In short, motor activity was quantify in Plexiglas activity chambers (Model ENV-520; Med Associates; length, 43.2 cm; width, 43.2 cm; height, 30.5 cm) equipped with infrared beams located 1.5 cm above the chamber floor and spaced 2.5 cm apart to detect horizontal activity. Vertical activity was detected using another set of infrared beams affixed 6 cm above the chamber floor and spaced 2.5 cm apart. Locomotor activity was measured as total ambulatory distance and total ambulatory time.
The footprint test was performed as described previously (Carter et al., 1999) with slight modifications. To obtain footprints, the hind feet of the mice were dipped in nontoxic black paint. The animals were then allowed to walk on a paper track in a 1-m-long, 10-cm-wide runway with 10-cm-high walls into an enclosed box. Hind base width and stride length were assessed.
The beam-walking test was performed as described previously (Carter et al., 1999). The beam was made of wood, 8 mm in diameter, and 1 m long. The beam was placed horizontally, 50 cm above the bench surface, with one end mounted on a 10 × 10 cm illuminated support and the other end attached to a walled-in 20 × 20 cm box into which the mouse could escape. During training, mice were placed at the start of the beam and trained over 3 d (four trials per day) to traverse the beam to the enclosed box. After the third day, all mice had traversed the beam in 20 s at least once. During testing, mice received three consecutive trials, which were videotaped. The videos were scored blind to genotype. The latency to traverse each beam and the number of times the hind feet slipped off were recorded for each trial. Analysis was based on the mean scores of the three trials.
The rotarod apparatus (accelerating model; Ugo Basile) was used to measure motor coordination and balance. Mice were placed individually on the rotating rotarod system and trained for 6 consecutive days, first in accelerating speed mode at 0–20, 0–30, and 0–40 rpm, and then in constant speed mode at 20, 30, and 40 rpm. Mice received three trials per day. During training period, all mice attained a stable baseline level of performance. Testing was performed at 20 and 30 rpm in constant speed mode. Overall rotarod performance was expressed as latency to fall.
Statistical analysis was performed using StatView 5.0 software (SAS Institute). Data were analyzed using Student's t test or one- or two-way ANOVA. The criterion for significance for all analyses was p < 0.05. The criterion for a trend was 0.05 < p < 0.1. Post hoc comparisons were conducted using the Student–Newman–Keuls test. Results from data analyses are expressed as mean ± SEM.
Results
Genetic 5-htt ablation impairs BG-dependent motor behavior
To study the consequences of increased 5-HT signaling on BG function, we first investigated the effect of 5-htt−/− ablation on motor behavior using tests that depend on BG function (Brooks and Dunnett, 2009).
In tests of gait, 5-htt−/− mice displayed reduced stride length and increased hind base width in the footprint test compared to 5-htt+/+ littermates (F(1,33) = 12.142, p = 0.0014 and F(1,33) = 6.015, p = 0.0196, respectively) (Fig. 1a). The within-mouse variability of stride length and hind base width, a feature commonly associated with cerebellar dysfunction, did not differ between genotypes (5-htt+/+, 0.155 ± 0.014; 5-htt−/−, 0.183 ± 0.015; F(1,33) = 1.858, p = 0.1820; and 5-htt+/+, 0.123 ± 0.011; 5-htt−/−, 0.114 ± 0.009; F(1,33) = 0.375, p = 0.5446, respectively). The beam-walking test required mice to traverse an elevated beam from a lit to a dark compartment. 5-htt−/− mice displayed an increased latency to cross the beam and an increased number of foot slips while crossing the beam compared to 5-htt+/+ littermates (F(1,20) = 9.444, p = 0.0060 and F(1,20) = 83.814, p < 0.0001, respectively) (Fig. 1b). In the rotarod test, mice were trained to remain atop a rod rotating at 20, 30, and 40 rpm. We observed that 5-htt−/− mice were quicker to fall in both the accelerating and constant speed modes (F(1,46) = 52.486, p < 0.0001 and F(1,46) = 14.568, p = 0.0004, respectively) (Fig. 1c). Hindlimb clasping behavior is a motor dysfunction observed in mice with reduced DAergic innervation of the striatum and abnormal striatal development (Cuellar et al., 2008; Baydyuk et al., 2011; Homma et al., 2011). 5-htt−/− mice exhibited a higher degree of hindlimb clasping compared to 5-htt+/+ littermates (F(1,47) = 21.446, p < 0.0001), with increased clasping over time as revealed by an interaction between genotype and time (F(5,235) = 22.485, p < 0.0001) (Fig. 1d). To rule out potential confounds of body weight and muscular strength, we assessed each parameter in 5-htt+/+ and 5-htt−/− mice and found no differences in either weight or grip strength (weight in footprint test group, F(1,33) = 3.517, p = 0.0696; weight in beam-walking test group, F(1,20) = 3.348, p = 0.0823; weight in rotarod test group, F(1,45) = 3.397, p = 0.0719; grip strength, F(1,19) = 0.366, p < 0.5526) (Fig. 1e,f).
Figure 1.
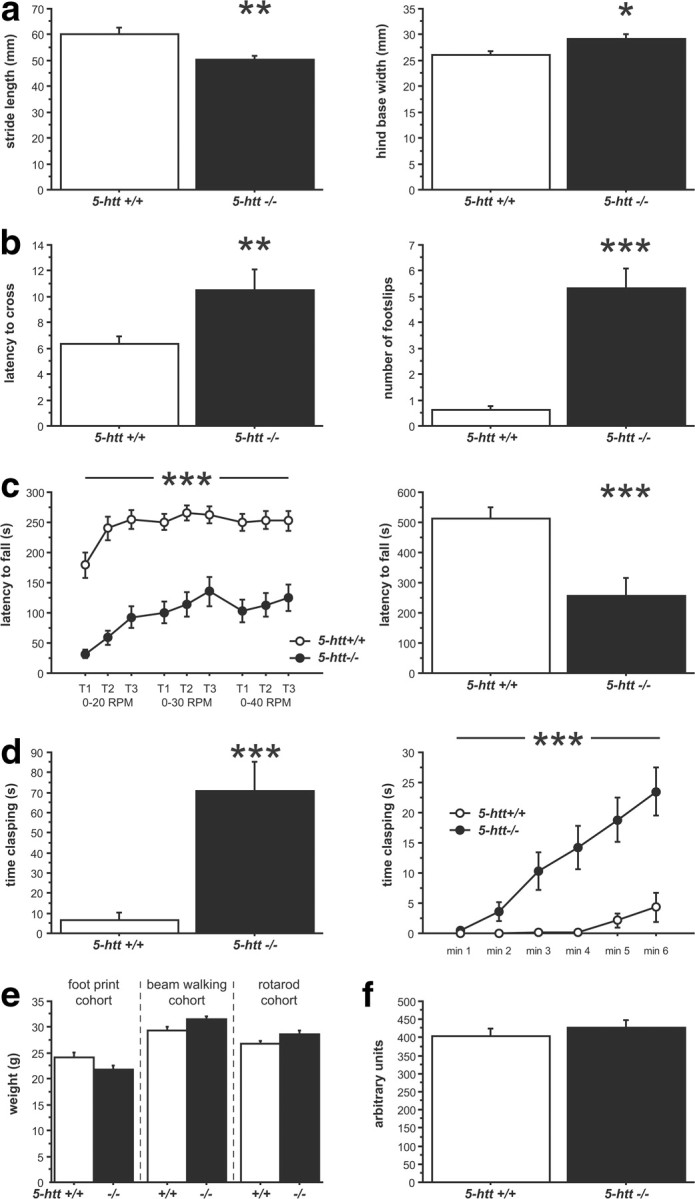
Impaired motor behavior in 5-htt−/− mice. a–d, BG-dependent motor behavior in 5-htt−/− mice was assessed using the footprint test (a), the beam-walking test (b), the rotarod test (c), and the clasping test (d). a, 5-htt−/− mice display reduced stride length (left) and increased hind base width (right) compared to 5-htt+/+ control mice (n = 17–18 mice per group). b, 5-htt−/− mice displayed an increased latency to cross the beam (left) and an increased number of foot slips while crossing the beam (right) compared to 5-htt+/+ control mice (n = 6–16 mice per group). c, In the rotarod test, 5-htt−/− mice were quicker to fall in both, the accelerating (left) and constant speed (20 rpm, right) modes compared to 5-htt+/+ control mice (n = 22–26 mice per group). d, 5-htt−/− mice display increased clasping behavior compared to 5-htt+/+ control mice (left), with increased clasping over time (right) (n = 23–26 mice per group). e, Body weights did not differ between genotypes in the behavioral cohorts. f, Muscular strength as assessed in the grip strength test did not differ between genotypes (n = 6–15 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001.
All testing was done in adult mice, but given the constitutive nature of genetic ablations, we sought to assess the ontogeny of the 5-htt−/− motor deficit. In the rotarod test, we performed longitudinal and cross-sectional studies of 5-htt−/− mice at various ages. In the longitudinal study, we tested one cohort of mice repeatedly at increasing ages and detected a main effect of genotype (F(1,32) = 14.986, p = 0.0005) but behavioral differences manifested themselves only at 14 weeks of age (as identified by post hoc comparisons) (Fig. 2a). Motor performance worsened with age for both genotypes, as revealed by a main effect of time (F(9,288) = 13.124, p < 0.0001), with a more rapid and severe decrease in performance seen in 5-htt−/− mice compared to 5-htt+/+ mice, as revealed by an interaction between genotype and age (F(9,288) = 5.155, p < 0.0001) (Fig. 2a). In the cross-sectional study, we tested multiple independent cohorts of mice at different ages. Again, progressive worsening of motor function in 5-htt−/− mice compared to 5-htt+/+ mice was observed, with no effect of genotype at 12 weeks of age (F(1,56) = 0.324, p = 0.5712), an effect of genotype at 16, 38, and 56 weeks of age (F(1,56) = 5.531, p = 0.0222; F(1,30) = 19.99, p = 0.0001; and F(1,20) = 3060.399, p < 0.0001, respectively), and effect sizes increasing with age (Fig. 2a). Because both genotypes were equally trained before the onset of motor dysfunction, motor learning deficits are unlikely to account for this phenotype.
Figure 2.

Progressive motor deficit in 5-htt−/− mice. a–c, BG-dependent motor behavior in 5-htt−/− mice as a function of age was assessed using the rotarod test (a), the beam-walking test (b), and the clasping test (c). a, Rotarod performance progressively worsened with age in 5-htt−/− mice compared to 5-htt+/+ control mice in a longitudinal study (20 rpm; top; n = 17 per group). Four independent cohorts of mice were tested at 12 weeks of age (n = 29–29 mice per group), 16 weeks of age (n = 29–29 per group), 38 weeks of age (n = 15–17 per group), and 56 weeks of age (n = 6–16 mice per group) at 20 rpm as indicated (bottom). No effect was detected at 12 weeks of age. An effect of genotype was detected at 16, 38, and 56 weeks of age, and effect sizes increase with age. b, Beam-walking performance progressively worsened with age in 5-htt−/− mice compared to 5-htt+/+ control mice (n = 14–17 per group). c, Clasping behavior progressively increased with age in 5-htt−/− mice compared to 5-htt+/+ control mice (n = 22–27 per group). The dotted line indicates reduced n compared to previous ages. *p < 0.05; **p < 0.01; ***p < 0.001.
In a longitudinal beam-walking study, we tested one cohort of mice repeatedly at increasing ages and detected a main effect of genotype (F(1,29) = 23.654, p < 0.0001), with behavioral differences manifesting themselves at 10 weeks of age (as identified by post hoc comparisons) (Fig. 2b). Motor performance worsened with age for both genotypes, as revealed by a main effect of time (F(13,377) = 4.653, p < 0.0001), with a more rapid and severe decrease in performance seen in 5-htt−/− mice compared to 5-htt+/+ mice, as revealed by an interaction between genotype and age (F(13,377) = 2.294, p < 0.0063) (Fig. 2b).
In the longitudinal assessment of clasping behavior, we tested one cohort of mice repeatedly at increasing ages and detected a main effect of genotype (F(1.47) = 7.578, p < 0.0084), with behavioral differences manifesting themselves at 9 weeks of age (as identified by post hoc comparisons) (Fig. 2c). Clasping behavior increased with age for both genotypes as revealed by a main effect of time (F(6,282) = 8.67, p < 0.0001) (Fig. 2c).
5-HTT inhibition during early postnatal development does not impair BG-dependent motor behavior
Similar to the ontogeny of motor deficits in 5-htt−/− mice, developmental 5-HTT blockade from P4 to P21 alters emotional behavior with a delayed onset at 3 months of age (Ansorge et al., 2004, 2008). To test whether motor deficits in adult 5-htt−/− mice also result from developmental inhibition of 5-HTT function, we first investigated the effect of FLX treatment from P4 to P21 in 5-htt+/+ and 5-htt−/− mice on motor behavior in the rotarod test. Postnatal FLX (PNFLX) treatment did not affect motor behavior in 5-htt+/+ or 5-htt−/− mice before or after the onset of the genotype effect (F(1,53) = 0.153, p = 0.6971 and F(1,53) = 0.459, p = 0.501, respectively) (Fig. 3a).
Figure 3.

Transient developmental 5-HTT blockade does not produce motor deficits. a, b, BG-dependent motor behavior in PNFLX and 5-htt−/− mice was assessed using the rotarod test (a) and the clasping test (b). a, No effect of P4–P21 FLX treatment was detected on the latency to fall before (12 week old; 20 rpm) or after the onset of the genotype effect (16 weeks old; 20 rpm; n = 13–16 mice per group). b, No effect of P4–P21 FLX treatment was detected on clasping behavior at 3 months of age, with an effect of genotype present (n = 13–14 mice per group). **p < 0.01.
Next, we first investigated the effect of FLX treatment from P4 to P21 in 5-htt+/+ and 5-htt−/− mice on clasping behavior. PNFLX treatment did not affect clasping behavior in 5-htt+/+ or 5-htt−/− mice at 3 months of age (F(1,51) = 0.007, p = 0.9346), with a genotype effect present (F(1,51) = 7.446, p = 0.0087) (Fig. 3b).
Together, these findings partially dissociate the affective phenotype present in 5-htt−/− mice (Holmes et al., 2003; Lira et al., 2003) from their motor phenotype.
Chronic 5-HTT blockade during adulthood impairs BG-dependent motor behavior
Our data imply that the motor phenotype is elicited by 5-HTT blockade during adulthood. To test this hypothesis, we analyzed motor behavior on the rotarod test in adult mice as a function of pharmacologic 5-HTT blockade. Mice treated acutely with FLX (10 mg/kg) displayed normal rotarod performance compared to VEH-treated controls (F(1,58) = 0.955, p = 0.3326) (Fig. 4a). Mice treated continuously with FLX (10 mg/kg/d) displayed impaired rotarod performance compared to VEH-treated controls (F(1,28) = 22.335, p < 0.0001) (Fig. 4a). We also detected an interaction between treatment and treatment duration (F(9,252) = 3.263, p = 0.0009) (Fig. 4a). Post hoc analysis revealed that chronic (at least 2 weeks) FLX treatment reduced the latency to fall and that impaired rotarod performance was reversed after cessation of chronic FLX treatment (Fig. 4a).
Figure 4.

Chronic FLX treatment during adulthood produces motor deficits. a, b, Motor behavior was evaluated using the rotarod test (a) and the clasping test (b). a, No effect of acute FLX treatment was detected on the latency to fall in a cross-sectional (box; n = 30 mice per group; 20 rpm) or longitudinal study (n = 15 mice per group; 20 rpm). Chronic FLX treatment reduced the latency to fall starting after 2 weeks of treatment, and performance normalized after cessation of FLX treatment (n = 15 mice per group; 20 rpm). b, No effect of acute FLX treatment was detected on clasping behavior (n = 5–8 mice per group). Chronic FLX treatment increased clasping behavior starting after 2 weeks of treatment, and cessation of FLX treatment reduced clasping behavior (n = 15 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001.
Similarly, clasping behavior was not elicited by acute FLX treatment (F(1,11) = 0.008, p = 0.9307) but by chronic FLX treatment (F(1,28) = 19.245, p = 0.0001) (Fig. 4b). Again, we detected an interaction between treatment and treatment duration (F(3,84) = 3.006, p = 0.0348) (Fig. 4b). Post hoc analysis revealed that chronic (at least 2 weeks) FLX treatment elicited clasping behavior and that motor impairments are ameliorated after cessation of chronic FLX treatment (Fig. 4b).
In addition, chronic FLX treatment elicited motor impairments in the rotarod test in a dose-dependent fashion at 20 and 30 rpm (F(3,34) = 3.254, p = 0.0335 and F(3,34) = 5.444, p = 0.0036, respectively) (Fig. 5a). To test whether chronic FLX elicits motor impairments through 5-HTT blockade as opposed to off-target effects, we treated 5-htt+/+ and 5-htt−/− mice with FLX (10 mg/kg/d, drinking water) and investigated rotarod performance. We detected an interaction between genotype and treatment at 20 and 30 rpm (F(1,44) = 9.801, p = 0.0031 and F(1,44) = 12.171, p = 0.0011, respectively) (Fig. 5b). Post hoc analysis revealed that chronic FLX treatment reduced the latency to fall in 5-htt+/+ mice without worsening rotarod motor behavior in 5-htt−/− mice (Fig. 5b), demonstrating that the effect of FLX is mediated via 5-HTT blockade. In addition, motor deficits do not resemble a serotonin syndrome-like phenotype—which is characterized by head weaving, forepaw treading, backward movement, hindlimb abduction, and Straub tail—and are present in multiple genetic background strains (C57BL/6, VEH, 552.167 ± 22.813; C57BL/6, FLX, 444.13 ± 38.395, F(1,59) = 10.17, p = 0.0023; BALB/C, VEH, 243.603 ± 13.672; BALB/C, FLX, 196.824 ± 16.916; F(1,36) = 4.498, p = 0.0409). Together, these findings demonstrate that chronic 5-HTT blockade during adulthood elicits motor deficits and that the emotional and motor phenotypes of the 5-htt−/− mice are fully dissociable.
Figure 5.

Chronic 5-HTT blockade during adulthood produces motor deficits. Motor behavior was evaluated using the rotarod test. a, Chronic FLX treatment reduces the latency to fall in a dose-dependent manner at 20 and 30 rpm (n = 9–10 mice per group). b, Chronic FLX treatment does not reduce the latency to fall in 5-htt−/− mice (n = 10–14 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001.
Chronic 5-HTT inhibition reduces striatal DA and DA metabolite levels
The progressive deterioration of motor behavior in 5-htt−/− mice suggests that neurodegeneration of DAergic neurons might underlie this behavioral phenotype. But because motor behavior normalizes after cessation of 5-HTT blockade, we hypothesized that reduced nigrostriatal DA signaling without neurodegeneration constitutes the underlying mechanism generating motor deficits after increased 5-HT signaling. In support of this hypothesis, electrolytic lesions of the dorsal and median raphe nuclei increase striatal levels of the DA metabolites DOPAC and HVA in rats (Juorio and Greenshaw, 1986).
To test this hypothesis, we first quantified TH-positive neurons in the SNc and VTA of 5-htt+/+ and 5-htt−/− mice. Indeed, stereologic analysis revealed no differences in SNc and VTA volume, number of TH-positive neurons, and density of TH-positive neurons in the SNc and VTA between 5-htt+/+ and 5-htt−/− mice (F(1,10) = 2.207, p = 0.1682; F(1,10) = 0.817, p = 0.3873; and F(1,10) = 0.133, p = 0.7228, respectively) (Table 1).
Table 1.
Normal SNc/VTA volume, DAergic cell number, and DAergic cell density in 5-htt−/− mice
| Volume (mm3) | Total number | Density (mm−3) | |
|---|---|---|---|
| 5-htt+/+ | 1.041e8 ± 0.062e8 | 3289 ± 157 | 3.179e5 ± 0.118e5 |
| 5-htt−/− | 0.944e8 ± 0.020e8 | 3074 ± 179 | 3.247e5 ± 0.150e5 |
Tyrosine hydroxylase-positive neurons were quantified using stereology. n = 6 mice per group.
Next, we analyzed DA and DA metabolite levels (DOPAC and HVA) in frontostriatal tissue of 5-htt+/+ and 5htt−/− mice. HPLC measurements revealed that mice with constitutive 5-htt ablation (5-htt−/−) exhibited significantly lower DOPAC and HVA levels in frontostriatal regions and a trend toward reduced DA compared to 5-htt+/+ littermate controls (F(1,12) = 8.684, p = 0.0122; F(1,12) = 17.808, p = 0.0012; and F(1,12) = 4.563, p = 0.054, respectively) (Fig. 6a). To investigate a potential dissociation of this phenotype from the impact of 5-htt ablation on adult emotional behaviors (Ansorge et al., 2004), we next analyzed the consequences of PNFLX treatment on adult DA and DA metabolite levels. Indeed, we detected a dissociation, with PNFLX treatment not affecting DA, DOPAC, or HVA levels compared to VEH-treated littermate controls (Fig. 6b). Finally, we analyzed DA, DOPAC, and HVA content in frontostriatal tissue of mice treated with FLX for 3 weeks (oral 10 mg/kg/d) and found reduced DA and DOPAC levels in FLX-treated mice compared to their VEH-treated littermates (F(1,14) = 6.376, p = 0.0243 and F(1,14) = 6.712, p = 0.0214, respectively) (Fig. 6c), indicating a reduction in the readily releasable pool of DA and reduced DA release/reuptake after chronic 5-HTT blockade.
Figure 6.

5-HTT blockade reduces DA and DA metabolite levels in frontostriatal regions. a–e, DA, DOPAC, and HVA levels were measured by high-performance liquid chromatography in 5-htt−/− mice (a), after transient developmental 5-HTT blockade (FLX; 10 mg/kg/d, i.p.; P4–P21; b), and after chronic fluoxetine treatment (FLX; 10 mg/kg/d; drinking water; c–e). Total frontostriatal (a–c), frontal cortex (d), and dorsal striatal (e) levels were assessed. DOPAC and HVA levels, normalized by tissue weight, decrease after constitutive genetic 5-htt ablation (a). No changes of DA, DOPAC, or HVA levels, normalized by tissue weight, were detected after transient developmental 5-HTT blockade (b). DA and DOPAC levels, normalized by tissue weight, decrease after chronic FLX treatment in frontal–striatal (c) and dorsal–striatal (e) tissue samples. n = 6–13 mice per group. *p < 0.05; **p < 0.01.
To discriminate effects of increased 5-HT signaling on the mesolimbic and nigrostriatal system, we next assessed DA, DOPAC, and HVA levels separately in the frontal cortex and dorsal striatum of mice treated with VEH or FLX for 3 weeks (oral 10 mg/kg/d). DA, DOPAC, and HVA levels in the frontal cortex were not affected by treatment (F(1,13) = 0.4, p = 0.757; F(1,13) = 0.5541, p = 0.369; and F(1,13) = 0.5699, p = 0.34, respectively) (Fig. 6d). However, chronic FLX treatment reduced DA and DOPAC levels in the dorsal striatum compared to VEH treatment (F(1,14) = 5.684, p = 0.0318 and F(1,14) = 7.252, p = 0.0175, respectively) (Fig. 6e). These data demonstrate that chronic 5-HTT blockade during adulthood preferentially inhibits DA signaling in the nigrostriatal system in mice.
l-DOPA treatment reverses motor deficits elicited by chronic 5-HTT inhibition
We reasoned that if reduced 5-HTT activity impairs motor function by reducing the pool of readily releasable DA and subsequent DA signaling, we should be able to augment DA release with l-DOPA and normalize motor behaviors. Mice treated with VEH or FLX for 3 weeks were acutely administered l-DOPA/benserazide (15/12.5 and 100/12.5 mg/kg, i.p.) or VEH. Four hours after the benserazide injection, rotarod performance was assessed, and we detected a main effect of l-DOPA/benserazide for the latency to fall at 20 and 30 rpm (F(2,26) = 3.647, p = 0.0402 and F(2,26) = 3.445, p = 0.0471, respectively) (Fig. 7a,b). Post hoc analysis revealed that chronic FLX impairs motor performance, and l-DOPA/benserazide rescues this impairment at both doses (Fig. 7a,b). Next, we treated 5-htt+/+ and 5-htt−/− mice with l-DOPA/benserazide (100/12.5 mg/kg, i.p.) or VEH and observed a significant improvement of rotarod performance in 5-htt−/− mice treated with l-DOPA. Specifically, we detected a genotype by treatment interaction on the latency to fall for 20 rpm and a treatment effect for 30 rpm (F(1,30) = 14.133, p = 0.0007 and F(1,30) = 7.2, p = 0.0177, respectively) (Fig. 7c,d). Post hoc comparisons revealed an effect of genotype in VEH-treated mice but not in benserazide/l-DOPA-treated mice and an effect of treatment in 5-htt−/− but not in 5-htt+/+ mice at both 20 and 30 rpm (Fig. 7c,d). To investigate whether benserazide/l-DOPA treatment impacts general locomotor activity, we analyzed behavior in the open-field test. While we detected the previously described effect of genotype (F(1,74) = 4.46, p = 0.0381) with 5-htt−/− mice displaying a reduction in total ambulatory distance, no effect of benserazide/l-DOPA treatment or genotype by treatment interaction was detected (F(1,74) = 1.043, p = 0.3105 and F(1,74) = 0.036, p = 0.8495, respectively) (Fig. 7e). Likewise, benserazide/l-DOPA did not alter stereotypic activity, as we detected no treatment effect or genotype by treatment interaction (F(1,74) = 1.766, p = 0.188 and F(1,74) = 0.264, p = 0.6087, respectively) (Fig. 7f). These data support our hypothesis that 5-HT-induced motor dysfunction is mediated by reduced nigrostriatal DA signaling and rule out cerebellar dysfunction, because cerebellar ataxias respond poorly to anti-Parkinson drugs (Gilman, 2004).
Figure 7.

l-DOPA treatment reverses motor deficits elicited by 5-HTT blockade. a–f, Motor behavior was evaluated using the rotarod test (a–d) and the open-field test (e, f). FLX-treated mice displayed a reduced latency to fall compared to VEH-treated controls only when treated with VEH but not when treated with l-DOPA/benserazide (15/12.5 and 100/12.5 mg/kg, i.p) at 20 rpm (a) and at 30 rpm (b; n = 7–8 mice per group). 5-htt−/− mice displayed a reduced latency to fall compared to 5-htt+/+ littermates only when treated with VEH but not when treated with l-DOPA/benserazide (100/12.5 mg/kg, i.p) at 20 rpm (c) and at 30 rpm (d; n = 14–18 mice per group). e, f, l-DOPA/benserazide treatment did not affect behavior in the open-field test (n = 13–24 mice per group). *p < 0.05; **p < 0.01.
Discussion
Our data demonstrate that chronic 5-HTT blockade impairs BG-dependent motor behavior and reduces striatal DA and DA-metabolite levels. Behavioral rescue through l-DOPA treatment indicates a causal relationship, where 5-HTT blockade results in reduced nigrostriatal DA signaling to consequently impair motor function.
5-HT signaling and DAergic control of BG function
In the classic model, DA regulates BG function by modulating the direct and indirect pathway via projections from the SNc to the striatum. The direct pathway is under positive control through D1 receptors, whereas the indirect pathway is under negative control through D2 receptors. Increased striatal DA signaling disinhibits thalamocortical circuits both through activation of the direct pathway and inhibition of the indirect pathway; conversely, reduced DA tone inhibits thalamocortical circuits both through reduced activation of the direct pathway and reduced inhibition of the indirect pathway (Kravitz et al., 2010).
We found that increased 5-HT signaling through chronic blockade of 5-HTT function reduces the pool of readily releasable DA in the striatum. Reduced striatal DOPAC levels after chronic 5-HTT blockade further suggest reduced striatal DA signaling as a consequence. Consistent with this conclusion, we find BG-dependent motor behaviors to be impaired after chronic 5-HTT blockade and demonstrate reversibility of motor deficits through l-DOPA treatment. Because l-DOPA treatment is generally not efficacious for disorders based on selective dysfunction in either the direct or indirect pathway (Watts and Koller, 2004), our data indicate that chronic 5-HTT blockade leads to increased 5-HT signaling, modulating BG function at the level of the DAergic tone provided to both the direct and indirect pathway. Interestingly, wild-type animals usually metabolize l-DOPA to DOPAC without affecting DA levels, but l-DOPA can restore DA transmission in DA-depleted animals by replenishing vesicular pools (Buu, 1989; Zhou and Palmiter, 1995). Thus, our data indicate that chronic 5-HTT blockade partially depletes vesicular DA levels, which can be restored by systemic l-DOPA treatment. This vesicular DA depletion hypothesis is consistent with the crowding-out effect described after 5-HTT blockade, when 5-HT is taken up by the dopamine transporter (DAT) into DAergic neurons (Cases et al., 1998; Zhou et al., 2002, 2005). Promiscuous 5-HT reuptake by the DAT can also explain the specific effect of 5-HTT blockade on striatal but not frontal cortex DA and DOPAC levels, because DAT immunoreactivity is high in the striatum and sparse in the frontal cortex (Sesack et al., 1998). Thus, we predict that under conditions of reduced 5-HTT function, l-DOPA treatment shifts the intracellular DA/5-HT ratio of SNc DAergic neurons back toward DA and reverses its vesicular displacement.
Yet, the relatively small reduction in striatal DA and DOPAC levels is unlikely to explain the quantitative extent of motor impairment observed after 5-HTT blockade. 5-HT receptors located in the BG likely play a role in conjunction with vesicular DA depletion to elicit BG dysfunction. Because many different 5-HT receptors are expressed in the BG, identification of their contribution to 5-HTT blockade-elicited motor impairments will require a screening-like approach, potentially yielding therapeutic targets for SSRI supplementation. The total reversal of behavioral deficits by l-DOPA indicates that such treatment might not only reverse vesicular DA depletion, but also decrease 5-HTergic signaling through specific 5-HT receptors in the BG. Another “false neurotransmitter”-related mechanism could accomplish such feat: “false DA” generated from l-DOPA by the aromatic amino-acid decarboxylase in 5-HTergic neurons (Carta et al., 2008) could reduce 5-HTergic signaling and contribute to the behavioral recovery. Thus, l-DOPA might replenish vesicular DA and reduce vesicular 5-HT.
Abnormal motor and affective behaviors in 5-htt−/− mice are dissociable
5-htt−/− mice display increased anxiety- and depression-like behaviors (Holmes et al., 2003; Lira et al., 2003; Zhao et al., 2006; Wellman et al., 2007). Here we report that 5-htt−/− mice also display BG-dependent motor deficits. Because dependent measures in behavioral tasks often are influenced by multiple factors, behavioral data can be vulnerable to misinterpretation. We further report that BG-dependent motor deficits are phenocopied by chronic adult 5-HTT blockade, but not by transient developmental 5-HTT blockade. Conversely, increased anxiety- and depression-like behaviors of 5-htt−/− mice are phenocopied by transient developmental 5-HTT blockade, but not by chronic adult 5-HTT blockade (Ansorge et al., 2004, 2008; Popa et al., 2008). This double dissociation supports our interpretation of the motor-related phenotypes reported here and corroborates the interpretation of published affective phenotypes for 5htt−/− and PNFLX-treated mice.
Clinical implications
Reduced DA signaling following chronic SSRI administration may underlie several SSRI induced side effects and might lower therapeutic response rates (Damsa et al., 2004). Our findings reported here provide substantial support for this hypothesis. The impaired BG-dependent motor function after chronic 5-HTT blockade that we observed directly models EPSs elicited by 5-HTT-blocking drugs in humans (Arya, 1994; Leo, 1996; Caley, 1997; Gill et al., 1997; Lane, 1998; Hedenmalm et al., 2006; Madhusoodanan et al., 2010). Our data suggest that concurrent treatment with drugs augmenting nigrostriatal DA signaling might ameliorate EPSs.
Using motor function as a behavioral proxy for nigrostriatal DA signaling and BG function, our results provide insight into potential treatment strategies for other BG-related diseases. At a macrocircuit level, striatonigrostriatal pathways from the nucleus accumbens shell to the dorsolateral striatum form an ascending network interconnecting different functional regions of the striatum in a feedforward organization (Haber et al., 2000). Through respective parallel BG outputs to thalamocortical neurons, this connectivity allows information to flow from limbic to cognitive to motor circuits (Middleton and Strick, 2000; Haber, 2003; Haber and Calzavara, 2009). This architecture explains how BG function is not only involved in the control of motor coordination, but also in the modulation of goal-directed motor function through emotions, motivation, and cognition. Hence, SSRI treatment might impact these processes analogously to its consequences on motor coordination described here. While this hypothesis remains to be tested, it is supported by the efficacy of SSRIs in the treatment of disorders with impaired inhibition of thoughts and behaviors, such as obsessive-compulsive disorder, where increased DAergic tone and frontostriatal hyperactivity are thought to cause symptoms (Marsh et al., 2009).
Because 5-HT/DA interaction in BG circuitry controls motor actions as well as thoughts and emotions, it will be important to identify and target molecules that augment nigrostriatal DA signaling to provide improved pharmacotherapy for patients suffering from diseases as diverse as movement disorders, disorders with impaired inhibition of thoughts and/or behaviors, and depression and/or anxiety.
Footnotes
This work was supported by the Sackler Institute for Developmental Psychobiology, a Sackler Award (M.S.A.), and the National Institute of Mental Health. We thank R. Hen, M. Milekic, N. Bivens, and C. Kellendonk for their critical review of this manuscript, and K. Krueger for help with the HPLC-related analytical work.
References
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Ansorge MS, Morelli E, Gingrich JA. Inhibition of serotonin but not norepinephrine transport during development produces delayed, persistent perturbations of emotional behaviors in mice. J Neurosci. 2008;28:199–207. doi: 10.1523/JNEUROSCI.3973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya DK. Extrapyramidal symptoms with selective serotonin reuptake inhibitors. Br J Psychiatry. 1994;165:728–733. doi: 10.1192/bjp.165.6.728. [DOI] [PubMed] [Google Scholar]
- Baydyuk M, Russell T, Liao GY, Zang K, An JJ, Reichardt LF, Xu B. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc Natl Acad Sci U S A. 2011;108:1669–1674. doi: 10.1073/pnas.1004744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat. 2000;196:527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user's guide. Nat Rev Neurosci. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- Buu NT. Vesicular accumulation of dopamine following L-DOPA administration. Biochem Pharmacol. 1989;38:1787–1792. doi: 10.1016/0006-2952(89)90413-9. [DOI] [PubMed] [Google Scholar]
- Caley CF. Extrapyramidal reactions and the selective serotonin-reuptake inhibitors. Ann Pharmacother. 1997;31:1481–1489. doi: 10.1177/106002809703101208. [DOI] [PubMed] [Google Scholar]
- Carta M, Carlsson T, Munoz A, Kirik D, Bjorklund A. Involvement of the serotonin system in l-dopa-induced dyskinesias. Parkinsonism Relat Disord. 2008;14(Suppl 2):S154–S158. doi: 10.1016/j.parkreldis.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Cases O, Lebrand C, Giros B, Vitalis T, De Maeyer E, Caron MG, Price DJ, Gaspar P, Seif I. Plasma membrane transporters of serotonin, dopamine, and norepinephrine mediate serotonin accumulation in atypical locations in the developing brain of monoamine oxidase A knock-outs. J Neurosci. 1998;18:6914–6927. doi: 10.1523/JNEUROSCI.18-17-06914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassano P, Fava M. Tolerability issues during long-term treatment with antidepressants. Ann Clin Psychiatry. 2004;16:15–25. doi: 10.1080/10401230490281618. [DOI] [PubMed] [Google Scholar]
- Chelben J, Strous RD, Lustig M, Baruch Y. Remission of SSRI-induced akathisia after switch to nefazodone. J Clin Psychiatry. 2001;62:570–571. doi: 10.4088/jcp.v62n07a13. [DOI] [PubMed] [Google Scholar]
- Cuellar TL, Davis TH, Nelson PT, Loeb GB, Harfe BD, Ullian E, McManus MT. Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc Natl Acad Sci U S A. 2008;105:5614–5619. doi: 10.1073/pnas.0801689105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsa C, Bumb A, Bianchi-Demicheli F, Vidailhet P, Sterck R, Andreoli A, Beyenburg S. “Dopamine-dependent” side effects of selective serotonin reuptake inhibitors: a clinical review. J Clin Psychiatry. 2004;65:1064–1068. doi: 10.4088/jcp.v65n0806. [DOI] [PubMed] [Google Scholar]
- Dray A, Davies J, Oakley NR, Tongroach P, Vellucci S. The dorsal and medial raphe projections to the substantia nigra in the rat: electrophysiological, biochemical and behavioural observations. Brain Res. 1978;151:431–442. doi: 10.1016/0006-8993(78)91077-6. [DOI] [PubMed] [Google Scholar]
- Gill HS, DeVane CL, Risch SC. Extrapyramidal symptoms associated with cyclic antidepressant treatment: a review of the literature and consolidating hypotheses. J Clin Psychopharmacol. 1997;17:377–389. doi: 10.1097/00004714-199710000-00007. [DOI] [PubMed] [Google Scholar]
- Gilman S. Clinical features and treatment of cerebellar disorders. In: Watts RL, Koller WC, editors. Movement disorders: neurological principles and practice. Ed 2. Columbus, OH: McGraw-Hill; 2004. pp. 723–736. [Google Scholar]
- Haber SN. The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat. 2003;26:317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Haber SN, Calzavara R. The cortico-basal ganglia integrative network: the role of the thalamus. Brain Res Bull. 2009;78:69–74. doi: 10.1016/j.brainresbull.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20:2369–2382. doi: 10.1523/JNEUROSCI.20-06-02369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedenmalm K, Guzey C, Dahl ML, Yue QY, Spigset O. Risk factors for extrapyramidal symptoms during treatment with selective serotonin reuptake inhibitors, including cytochrome P-450 enzyme, and serotonin and dopamine transporter and receptor polymorphisms. J Clin Psychopharmacol. 2006;26:192–197. doi: 10.1097/01.jcp.0000203200.96205.34. [DOI] [PubMed] [Google Scholar]
- Holmes A, Murphy DL, Crawley JN. Abnormal behavioral phenotypes of serotonin transporter knockout mice: parallels with human anxiety and depression. Biol Psychiatry. 2003;54:953–959. doi: 10.1016/j.biopsych.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Homma D, Sumi-Ichinose C, Tokuoka H, Ikemoto K, Nomura T, Kondo K, Katoh S, Ichinose H. Partial biopterin deficiency disturbs postnatal development of the dopaminergic system in the brain. J Biol Chem. 2011;286:1445–1452. doi: 10.1074/jbc.M110.159426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Steindler DA, Kitai ST. The organization of divergent axonal projections from the midbrain raphe nuclei in the rat. J Comp Neurol. 1986;243:363–380. doi: 10.1002/cne.902430307. [DOI] [PubMed] [Google Scholar]
- Juorio AV, Greenshaw AJ. The effect of raphe nuclei lesions on striatal tyramine concentration and dopamine turnover in the rat. Neurochem Res. 1986;11:687–693. doi: 10.1007/BF00965337. [DOI] [PubMed] [Google Scholar]
- Kaplan EM. Antidepressant noncompliance as a factor in the discontinuation syndrome. J Clin Psychiatry. 1997;58(Suppl 7):31–35. discussion 36. [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RM. SSRI-induced extrapyramidal side-effects and akathisia: implications for treatment. J Psychopharmacol. 1998;12:192–214. doi: 10.1177/026988119801200212. [DOI] [PubMed] [Google Scholar]
- Leo RJ. Movement disorders associated with the serotonin selective reuptake inhibitors. J Clin Psychiatry. 1996;57:449–454. doi: 10.4088/jcp.v57n1002. [DOI] [PubMed] [Google Scholar]
- Lira A, Zhou M, Castanon N, Ansorge MS, Gordon JA, Francis JH, Bradley-Moore M, Lira J, Underwood MD, Arango V, Kung HF, Hofer MA, Hen R, Gingrich JA. Altered depression-related behaviors and functional changes in the dorsal raphe nucleus of serotonin transporter-deficient mice. Biol Psychiatry. 2003;54:960–971. doi: 10.1016/s0006-3223(03)00696-6. [DOI] [PubMed] [Google Scholar]
- Madhusoodanan S, Alexeenko L, Sanders R, Brenner R. Extrapyramidal symptoms associated with antidepressants—a review of the literature and an analysis of spontaneous reports. Ann Clin Psychiatry. 2010;22:148–156. [PubMed] [Google Scholar]
- Marsh R, Maia TV, Peterson BS. Functional disturbances within frontostriatal circuits across multiple childhood psychopathologies. Am J Psychiatry. 2009;166:664–674. doi: 10.1176/appi.ajp.2009.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelsen KA, Schmitz C, Steinbusch HW. The dorsal raphe nucleus–from silver stainings to a role in depression. Brain Res Rev. 2007;55:329–342. doi: 10.1016/j.brainresrev.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Strick PL. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev. 2000;31:236–250. doi: 10.1016/s0165-0173(99)00040-5. [DOI] [PubMed] [Google Scholar]
- Miller JJ, Richardson TL, Fibiger HC, McLennan H. Anatomical and electrophysiological identification of a projection from the mesencephalic raphe to the caudate-putamen in the rat. Brain Res. 1975;97:133–136. doi: 10.1016/0006-8993(75)90920-8. [DOI] [PubMed] [Google Scholar]
- Moukhles H, Bosler O, Bolam JP, Vallee A, Umbriaco D, Geffard M, Doucet G. Quantitative and morphometric data indicate precise cellular interactions between serotonin terminals and postsynaptic targets in rat substantia nigra. Neuroscience. 1997;76:1159–1171. doi: 10.1016/s0306-4522(96)00452-6. [DOI] [PubMed] [Google Scholar]
- Popa D, Lena C, Alexandre C, Adrien J. Lasting syndrome of depression produced by reduction in serotonin uptake during postnatal development: evidence from sleep, stress, and behavior. J Neurosci. 2008;28:3546–3554. doi: 10.1523/JNEUROSCI.4006-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Ross R, Stanga CY. Selective serotonin reuptake inhibitors. In: Preskorn SH, Feighner HP, Stanga CY, Ross R, editors. Antidepressants: past, present and future. Berlin: Springer; 2004. pp. 241–262. [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Salichon N, Gaspar P, Upton AL, Picaud S, Hanoun N, Hamon M, De Maeyer E, Murphy DL, Mossner R, Lesch KP, Hen R, Seif I. Excessive activation of serotonin (5-HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase a and 5-HT transporter knock-out mice. J Neurosci. 2001;21:884–896. doi: 10.1523/JNEUROSCI.21-03-00884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hof PR. Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J Chem Neuroanat. 2000;20:93–114. doi: 10.1016/s0891-0618(00)00066-1. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Hawrylak VA, Matus C, Guido MA, Levey AI. Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci. 1998;18:2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settle EC., Jr Antidepressant drugs: disturbing and potentially dangerous adverse effects. J Clin Psychiatry. 1998;59(Suppl 16):25–30. discussion 40–22. [PubMed] [Google Scholar]
- Tepper JM, Lee CR. GABAergic control of substantia nigra dopaminergic neurons. Prog Brain Res. 2007;160:189–208. doi: 10.1016/S0079-6123(06)60011-3. [DOI] [PubMed] [Google Scholar]
- Vertes RP. A PHA-L analysis of ascending projections of the dorsal raphe nucleus in the rat. J Comp Neurol. 1991;313:643–668. doi: 10.1002/cne.903130409. [DOI] [PubMed] [Google Scholar]
- Watts RL, Koller WC. In: Movement disorders: neurological principles and practice. Ed 2. Watts RL, Koller WC, editors. Columbus, OH: McGraw-Hill; 2004. [Google Scholar]
- Wellman CL, Izquierdo A, Garrett JE, Martin KP, Carroll J, Millstein R, Lesch KP, Murphy DL, Holmes A. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci. 2007;27:684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Zhao S, Edwards J, Carroll J, Wiedholz L, Millstein RA, Jaing C, Murphy DL, Lanthorn TH, Holmes A. Insertion mutation at the C-terminus of the serotonin transporter disrupts brain serotonin function and emotion-related behaviors in mice. Neuroscience. 2006;140:321–334. doi: 10.1016/j.neuroscience.2006.01.049. [DOI] [PubMed] [Google Scholar]
- Zhou FC, Lesch KP, Murphy DL. Serotonin uptake into dopamine neurons via dopamine transporters: a compensatory alternative. Brain Res. 2002;942:109–119. doi: 10.1016/s0006-8993(02)02709-9. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Salas R, Zhang L, De Biasi M, Dani JA. Corelease of dopamine and serotonin from striatal dopamine terminals. Neuron. 2005;46:65–74. doi: 10.1016/j.neuron.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]