

Summary
Modeling the in vivo microenvironment typically involves placing cells in a three-dimensional (3D) extracellular matrix (ECM) in physiologically relevant context with respect to other cells. The mechanical and chemical features of 3D microenvironments play important roles in tissue engineering, tumor growth and metastasis, and in defining stem cell niches, and it is increasingly recognized that cells behave much differently when surrounded by a 3D ECM than when anchored to a 2D substrate. To create microenvironments that more closely mimic in vivo settings, here we describe a novel microfluidic device that allows multiple discrete constructs of 3D cell-laden hydrogels to be patterned in a sequence of simple steps. The microfluidic platform allows for real time imaging of the interactions between multiple cell types exposed to both autocrine and paracrine signaling molecules, all within a 3D ECM environment. Detailed modeling determined that surface tension, hydrophobic interactions, and spatial geometry were important factors in containing the gels within distinct separate channels during the filling process. This allowed us to pattern multiple gel types side-by-side and pattern 3D gels spatially with tight dimensional control. Cells embedded in gels could be patterned by culturing MDA-MB-231 metastatic breast cancer cells and RAW 264.1 macrophage cells within distinct collagen type I and Matrigel ECM environments, respectively. Over a 7 day culture experiment, RAW cells invaded into neighboring gels containing MDA-MB-231 cells, but not into gels lacking cells. These studies demonstrate the versatility and potential of this new microfluidic platform to engineer 3D microscale architectures to investigate cell-cell and cell-matrix interactions.
Introduction
Driven in part by independent research efforts in the fields of tissue engineering and cancer, there is an increased emphasis to move cell biology away from homotypic two dimensional cell cultures towards three dimensional (3D) multicellular culture studies 1–4. Multicellularity and three-dimensionality have both been shown to be important features of the microenvironment that can influence cell function 1, 2, 4. In the body, 3D microenvironments not only surround most cells and provide a scaffolding structure with which these cells interact, they also involve multiple cell types that interact directly via heterotypic cell-cell junctions or via paracrine-mediated signaling mechanisms. Nevertheless, the ability to study these types of interactions in well-controlled 3D culture systems remains a challenge.
There are many examples in which multicellular culture systems have been used to provide physiologic cues and reveal insights into how cells may behave in vivo. For example, hepatocytes were shown to be more viable and retain their differentiated phenotype when cultured in the presence of 3T3 fibroblasts 5. Embryonic stem cells are typically grown on a monolayer of irradiated fibroblasts to provide nutrients and paracrine factors that maintain pluripotency and support growth 6.
In tumor biology specifically, the metastatic spread of cancer cells involves a dynamic interaction and remodeling of the microenvironment that is often facilitated by other cell types present in the surrounding stroma. It was recently reported that the invasion of carcinoma cells is enhanced when cultured with stromal fibroblasts, and that the tumor cells may actually follow the fibroblasts 7. Other co-culture studies have shown that tumor cells recruit macrophages, which in turn produce growth factors and nutrients to permit a more migratory phenotype 8, and that endothelial cells display enhanced capillary morphogenesis when cultured in the presence of mesenchymal stem cells or fibroblasts 8, 9.
The extracellular microenvironment associated with tumors is vastly different from normal healthy tissues 10, and it is widely believed this abnormal environment plays an important instructive role in tumor morphogenesis. The loss of normal tissue structure and function observed in most cancers coincides with, and may be causal for, changes in cellular phenotype 10. The microenvironment minimally provides sites for integrin-mediated cell attachment, sequesters soluble growth factors and chemokines, and influences diffusive and interstitial transport 4. Cell attachment to the extracellular matrix (ECM) allows cells to exert tractional forces upon their microenvironment, which in turn is critical for their ability to migrate along and/or through the ECM. Cell-mediated remodeling of the ECM through both force generation and proteolysis is also believed to play a key role in tumor cell invasion and metastatic spread, although there is also evidence that tumor cells can literally squeeze their way through openings in the ECM independent of proteolysis 11.
To date, most studies of 3D tumor cell invasion involve some variation of a transwell assay 13–15. These transwell invasion assays, a modification of the Boyden chamber, typically measure cell migration through a 3D gel (typically Matrigel) into a filter under the influence of a chemoattractant. These assay systems are available commercially, but do not provide a real time assessment of the cell activity in the 3D environment. Furthermore, despite insights gleaned from these and other studies employing 3D cultures, most systems typically lack the ability to spatially pattern multiple cell types in 3D in a consistent manner. To solve these issues, many groups have turned to microfluidic systems, which allow the patterning of cells in specific configurations 3, 16–18 and allow precise control over cell-cell or cell-matrix interactions. They also allow the incorporation of biomaterial hydrogels (either natural or synthetic) to create 3D environments 16, 19–23. However, past microfluidic devices either required extensive pumping systems to pattern gels 12, complicated fabrication processes involving etching of substrates 13, syringe micromanipulators,15, 16 or complex polymers to confine the hydrogel in a specific channel 14. These devices require experimental setups and equipment not readily available in most labs. In addition, the forces involved in containment of the hydrogels within these microfluidic devices have not been adequately characterized and/or modeled.
To address these limitations and to better mimic the complex nature of the microenvironment, here we present a new microfluidic device that allows the precise patterning of 3D biopolymer gels into distinct layers. Key features of this microfluidic environment are the ability to observe cell-ECM and cell-cell interactions in 3D in real time, and to engineer precise structures with well-defined geometries. In addition, we developed a quantitative model to predict the process of filling the microfluidic system with biopolymer gel precursor solutions in order to better understand how the gels are fabricated and constrained by the design specifications of our system. We demonstrate the utility of this device to systematically investigate multiple cell types and multiple ECM substrates in a controlled microenvironment. Combined with our prior work demonstrating the ability to apply gradients across these 3D environments 15, this integrated platform allows for the study of multicellular morphogenic processes in a dynamic microenvironment.
Results and Discussion
Device design and fabrication
This paper describes a novel microfluidic platform that was designed to permit patterning of gels and cells in a 3D microenvironment. Although a number of approaches have been described for patterning cells on 2D surfaces, it has been a challenge to embed multiple cell types in a precisely controlled manner in 3D gels. We recently described an approach to pattern microscale gels within a microfluidic device and generate gradients under flow-free conditions 15, 16. The system described here advances the capability to pattern multiple gels reliably. Similar to the our prior “ladder chamber” device15, 16, the basic design of the device consists of two parallel channels that are connected to reservoirs, but with important modifications that allow different gels to be placed in precise spatial orientations. Instead of a series of narrow microchannels, the main channels were separated by a series of juxtaposed channels partitioned by linear arrays of regularly spaced posts. Using this design, the multiple channels can be independently filled with distinct hydrogels, either through thermally or photo-crosslinked polymerization. Gel composition and dimensions can be engineered for specific applications.
The basic design of the device with a single gel channel configuration is shown schematically in Figure 1a and supplementary movie 1. The top view and cross sectional view illustrate the three main structural components of the device: a gel channel (pink), two main channels (blue), and an array of post structures (white) separate them. The design of the device can be readily adapted to include multiple gel channels that can be independently and sequentially filled with gels of desired mechanical and chemical properties. The gel channel (highlighted in pink) can also be designed with varying dimensions. We have successfully tested channels with widths from 100–1000 μm and heights of 50–300 μm. The gel channel is flanked by main channels that act as media and growth factor reservoirs (highlighted in blue). Each gel channel is partitioned using a series of regularly spaced posts that are 50–100 μm apart. Cylindrical and hexagonal posts with diameters ranging from 50–100 μm were used. The purpose of the post structures is to separate gel compartments and prevent leakage into neighboring channels. Instead of a solid barrier that would prevent diffusion and movement of molecules and cells, the use of post structures allows better control of the interface between gel compartments and main channels (interface area can be varied according to need). Using this approach, it is possible to associate (independent) gels with controlled dimensions.
Figure 1.

(A) Device schematic of a single channel device. Two parallel reservoir channels provide media and nutrients to the gel channels contained between them. Arrays of hexagonal posts provide support and contain the gels during the injection process. Each post is 100 μm in diameter and each gel channel is 400 μm across. (B) Gel suspensions can be injected into the inlets via manual pipetting or syringe pumps. Filling multichannel devices involves injecting hydrogels in alternating channels, polymerizing, and repeating for remaining channels.
Successful filling of the gels depends on balancing the capillary forces and surface tension within the microfluidic gel channel. If the surfaces within the channel were hydrophilic, we found that the gel or fluid leaked into adjacent channels as soon as the fluid was injected. However, if the surfaces within the channel were hydrophobic, the entire length of the gel channel (≥ 2 cm in our experiments) could be filled without leakage (Please see supplemental movie 1). A simple two-step process was applied to generate multiple juxtaposed gels, in which cell-laden gels are formed by filling alternate gel channels followed by polymerization in the first step, with subsequent filling of the remaining channels in the second step (Figure 1b). These two steps can be achieved by simply filling the gel channels with a pipette in order to produce well-defined 3D gels that are spatially patterned. Alternatively, a syringe pump can be used to infuse gels in a more controlled manner if desired. Importantly, the versatility of our design and fabrication processes allow for the dimensions of each individual gel channel to be tailored, and for each to contain a distinct biomaterial. This may be particularly useful to address specific biological questions regarding the spatial orientation of one cell population with respect to another, or the interface separating one matrix from another. Such control could be useful, for example, to facilitate a better understanding of the roles of soluble diffusible factors in the interactions between tumor cells and their surrounding stromal environment. Ultimately, these approaches may enable construction of custom microenvironments for multicellular co-cultures, the microscale equivalent of master-planned communities.
The effects of surface tension and injection pressures on gel entrapment
Currently there are a variety of microfluidic devices that are being used to study 3D behavior of cells.12–14, 17–19 A distinction of our system is the presence of the interface between juxtaposed gel channels, a critical feature to permit cell-cell and/or cell-gel interactions between adjacent compartments. Creation of this interface was made possible by a series of regularly spaced posts, which function as geometric capillary burst valves20. We used hexagonal posts to that assured the interfaces were pinned between the designated vertices as explained below. In the following, we describe how the posts prevent liquid from leaking into adjacent channels since this feature underlies the central working principle of our system. Creation of this interface was made possible by a series of regularly spaced posts (50–100 μm diameter), and empirically found to depend on three different variables: the spacing between posts, the surface properties of the device, and the viscosity of the hydrogel precursor solutions. With respect to these three variables, we first systematically tested various gap spacings (ranging from 50–200 μm) to optimize this interface so that the structural integrity of the gels within each individual gel channel was maintained (i.e., there was no leakage between adjacent channels). Gap spacings up to 200 μm were found to contain gels within a 400 μm wide gel channel. Second, the surface properties of the device were equally important to prevent the gels from leaking into neighboring channels. When the surfaces of the gel channels were made relatively hydrophobic, the gel precursor solutions did not cross the boundary defined by the posts, thus maintaining the integrity of the resulting gels. If the devices were left hydrophilic, the hydrogels would not be reliably contained within their designated channels and escape into adjacent regions. Finally, hydrogel viscosity was also found to play a crucial role in optimizing the interface between gel channels, with higher density hydrogels exhibiting higher viscosity and displaying an increased chance of leakage between adjacent channels. Collagen gels up to 6 mg/mL were successfully filled into the system with minimal leakage (<30%), while 8 mg/mL collagen gels could not be injected reliably without compromising the integrity of the interface. Matrigel concentrations of 8 mg/mL were also observed to induce leakage into adjacent compartments with a high probability (>80%), while 6 mg/mL concentrations generated minimal leakages (<10%). Fibrin precursor solutions with concentrations up to 20 mg/mL could also be injected and polymerized within the device (data not shown). These observations indicate that hydrogels of various identities and densities are compatible with our platform.
To better understand our empirical observations and determine the critical factors that affect the success of the gel filling process within the channels of our device, a modified capillary burst valve model20 was introduced and adapted to our system to investigate the effects of different geometric dimensions during the gel filling process. As described in the Experimental section, the model equation describes the pressure drop across the liquid interface (Pi −Po) as a function of the surface tension (γ), contact angles of the gel channels (θs and θv), and the width (w) and height (h) of the gel channels (Figure 2A–2C). We hypothesized that the pressure differences exerted at the liquid interface are critical in determining if leakage may occur, and that a balance between surface tension forces and capillary forces during the gel injection process is necessary to maintain the integrity of the gel interface between adjacent channels. During the injection process, a pressure difference (ΔP) is created at the interface by the liquid (assumed to be water) which drives the liquid into the channel. As the gel fills the channel and gap spacings, pressure differences are generated at the multiple leading interfaces of the hydrogel. The pressure differences decrease as the interface moves away from the injection site. From this model we would predict that if the pressure differential (ΔPgap − ΔPwidth) exceeds a minimum threshold, then leakage will not occur. Experimentally, if and when the fluid interface passes the first gap spacing, our model would predict that the above minimum threshold condition was satisfied and thus leakages further down the channel would be rare. It was desirable to determine the channel and post dimensions, i.e. w and s (Figure 2C), such that the difference between ΔPwidth and ΔPgap was large. ΔPwidth is the minimum amount of pressure required to drive the liquid to overcome the pressure drop along the fluid channel, while ΔPgap is the maximum pressure threshold that the system can withstand before leakage occurs. The injection pressure has to be finely tuned so that it lies within the range between ΔPgap and ΔPwidth and will vary depending on the viscosity of the liquid (which correlates with our observations). For example, in our PDMS channels with h=50 μm, w=400 μm, ΔPwidth =2480 Pa when the liquid is assumed to be water (γ =0.072 N/m and θA =140°). When the post spacing was s=100 μm, ΔPgap =3310 Pa, then the pressure differential, ΔPwidth − ΔPgap =830 Pa, which was above the threshold to prevent liquid leakage into adjacent channels while the liquid completely filled the straight channel. However, we experimentally observed that the liquid interface pinned between the posts nearest to the fluid inlet started to burst when the gap spacing (s) increased past 200 μm. In this case, the corresponding ΔPgap = 2750 Pa, which exceeds ΔPwidth by 270 Pa (~2 cmH2O) (Figure 2D). To provide a consistent threshold for future designs, a differential pressure limit of 500 Pa was set as a guide for designing geometries to maximize the interface between channels and provide containment of hydrogels. Although this model assumes a filling liquid of water, these predictions are consistent with our empirical observation and provide a guide to designing geometries to entrap hydrogels.
Figure 2.

(A–C) Diagram of a filling process within a channel. During injection, a difference in pressure occurs at the air-liquid interface (ΔP) which depends on surface tension forces (γ), channel geometry (w and s), and contact angles with the wall of the advancing liquid (θA). (B) When the liquid interface encounters a gap spacing (point AB), a new contact angle (θm) develops. (C) The difference between the gap spacing (ΔPgap) and channel (ΔPwidth) need to be maximized to prevent gel leakage. (D–G) Graphs of calculated pressure differences across a variety of parameters: height (E–F), width (E), contact angles (F), and surface tension (G). (D) Graph of pressure differences of increasing gap spacing. A threshold of 500 Pa was set empirically for criteria to prevent leakages and drawn as a guide for reference.
In addition, changes in the surface hydrophobicity (characterized by the contact angle), geometry of the channels will influence the magnitude of the pressure difference. Under hydrophobic conditions (assuming a θs ≈ θv = 140° between PDMS and water) 21, and varying the geometries of the channel width and height, we calculated higher pressure differences (Figure 2E) which scaled linearly if either parameter was changed. As height or width increased from 50–100 μm, the pressure differences dropped drastically but eventually leveled off when past 200 μm. The same trends were observed when plotting with varying contact angles (Figure 2F) and surface tensions (Figure 2G). With increasing contact angles from 100° to 150°, higher pressures were calculated which correlates with empirical data that indicates that hydrophobicity promotes gel integrity.
In summary, these modeling results confirm that the magnitude of the surface tension forces can influence the filling process, and will vary with the type and concentration (i.e., the viscosity) of the hydrogel precursor solution. Furthermore, the relative wettability of the PDMS channels, characterized by the contact angle, will also influence the likelihood of maintaining the integrity of the gel channel interfaces within our device. In general terms, if the pressure difference of the liquid interfaces between the channel and gap spacing is minimal (<~250Pa), then the gel precursor solution will leak into the neighboring channels. Therefore, while several other studies have reported similar designs to create 3D cell cultures within microfluidic devices 12, 16–19, the combination of experimental data and simulation presented here provides a unique fundamental perspective to understand the rationale behind the operation of this microfluidic platform.
Patterning juxtaposed gels of distinct ECM compositions
To demonstrate the capability to spatially pattern distinct and separate gels, type I collagen and Matrigel were used to fill multiple adjacent channels (Figure 3, top and bottom rows). Initially, 4 mg/mL type I collagen hydrogels were mixed with FITC and patterned within a 5-gel channel device to demonstrate that the gel could be contained within single channels without leakage into adjacent channels (Figure 3, top). The collagen-filled channels are highlighted in green whereas the empty adjacent channels are in black. These data illustrate that the gels were entrapped in separate compartments and did not leak into neighboring channels. Each of the gel channels and gap spacings were filled in a homogenous fashion and created uniform 100 μm boundaries between adjacent gel channels. Using this technique, precursor solutions of collagen type I, Matrigel, or fibrin were also successfully infused into the device and polymerized in adjacent channels for subsequent experiments. Type I collagen gels (4 mg/ml) polymerized adjacent to Matrigel (4 mg/ml) in a 5-gel channel device could be easily distinguished from the Matrigel channels by the fibrillar nature of the collagen. Patterning different hydrogels in adjacent gel channels in this fashion demonstrates that the formation of multi-layered microenvironments within our versatile device is straightforward and relatively simple, without the need for complicated materials, pumps, or other equipment. Such multilayered environments may yield new insights regarding how the migration and invasion of cells in 3D changes as they encounter different ECM ligands and different soluble cues sequestered within the different ECM layers.
Figure 3.

Collagen gels with FITC dye were patterned in alternating gel channels to show the ability to pattern separate distinct channels without any leakage (top row). Collagen and Matrigel hydrogels (each 4 mg/ml) were polymerized in alternating gel channels (bottom row). GFP tagged MDA-MB-231 cells were suspended in the collagen and cultured for up to two weeks. Shown is a combined phase and fluorescent micrograph taken at day 6. The magnified insert shows a cell crossing the boundary of the Matrigel-collagen interface. Each gel channel can be patterned independently. Scale bars are 100 μm.
To demonstrate the validity of this system for cell culture applications, MDA-MB-231 cells (a metastatic breast cancer cell line), stably expressing GFP, were suspended in the collagen gels (Figure 3 bottom row). These cells were observed for up to one week within the devices, and largely remained confined within the collagen gel channels for the duration of the experiment. In one case, a single cell was observed to cross the collagen-Matrigel boundary (Figure 3, inset) only to return to collagen gel the following day (not shown). This illustrates the utility of this device to perform live-cell imaging on single cells as they navigate across different microenvironments, and indicates that the interfaces between gels are permeable to cells and soluble factors. Cancer cells are known to have unique extracellular environments which differ from normal pathologies 2, 10, and the extracellular environment plays a role in the migratory behavior and invasiveness 12, 15. Using the methods and systems we have developed here, we can now construct different microscale cellular niches for cellular studies using a variety of ECM materials and thereby explore how tumor cell behavior depends on microscale architecture and matrix identity in 3D.
Second Harmonic Generation (SHG) imaging of collagen remodeling
Like most cells, the ability of cancer cells to migrate through 3D environments likely depends on their ability to exert tractional forces. It has also been presumed for some time that tumor cells use a battery of proteases to degrade the 3D ECM in order to migrate through it. However, recent evidence suggests that may adopt an amoeboid morphology to slip through the pores present within the ECM network 15. Thus, to better understand the dynamic nature of the 3D microenvironment surrounding cells, it is also important to observe how the cells remodel their environment. To observe the cell-matrix interface, DS-red labeled MDA-MB-231 cells were suspended in type I collagen (2 mg/mL) and visualized via two-photon microscopy, with the second harmonic generation signals used to visualize the collagen fibrils (highlighted in blue). In an experiment in which two different adjoined collagen gels were observed, an acellular gel remained intact while a gel channel containing the MDA-MB-231 cells displayed empty areas devoid of collagen fibrils (Figure 4). As expected, seeding cells at higher densities increased the degradation and remodeling of the collagen matrix compared to lower cell seeding densities (data not shown). These data show that the MDA-MB-231 cells likely remodeled the collagen matrix through a combination of proteolysis and tractional stress (shown in supplementary movie 2), both of which will depend on the cell density and the ECM density.
Figure 4.

Collagen fibers were imaged via Second Harmonic Generation (SHG) within a device. DS-Red labeled MDA-MB-231 cells were suspended in type I collagen and placed adjacent to another layer of collagen gel. Matrix degradation and remodeling are observed as cells cluster at the boundary of the gels shown in the image. Scale bar represents 100 μm.
Constructing multicellular niches for real-time observation of co-cultures
The tumor microenvironment is complex, and recreating it for in vitro studies is a daunting task. Besides the complexities imposed by a 3D microenvironment, cancer cells also interact with a variety of cells types as they grow and metastasize in physiological conditions. For example, recent work has shown that cancer cells actively recruit fibroblasts and macrophages to remodel and produce growth factors to enable the cancer cells to be more invasive 2, 7, 8. By dissecting the tumor microenvironment into discrete, well-defined compartments, our system offers a viable strategy to understand the complexities. To show the versatility of the system for potential multicellular culture studies, DS-Red labeled MDA-MB-231 cells and GFP-labeled RAW macrophages (a tumor-associated macrophage cell line) were suspended in collagen (4 mg/mL) and Matrigel (4 mg/mL) respectively and polymerized within the gel channels. Over a one-week time course, the MDA-MB-231 cells (in red) did not invade the neighboring gels, which contained the RAW cells (green) (Figure 5a). Instead, the RAW cells invaded into the neighboring gels containing the breast cancer cells beginning at day 3. [RAW cells were less invasive into the neighboring gels when MDA-MB-231 cells were not present (n=3) (Figure 5b).] The invasion of the RAW cells was more extensive after 7 days. RAW cells also appeared to multiply in the presence of MDA-MB-231 cells. These observations may represent a normal macrophage response in which the presence of the foreign tumor cells mobilizes the RAW cells. Alternatively, the MDA-MB-231 cells may also secrete soluble chemoattractant factors that induce the macrophages to proliferate and invade. Further investigation of these phenomena in the system we have described here could therefore potentially provide an improved mechanistic understanding of the interactions between tumor cells and stromal cells in their 3D microenvironment. While such details are beyond the scope of this current work, the system described here nevertheless offers the potential to study the relative contributions of these effects in a systematic and reasonably high-throughput manner.
Figure 5.
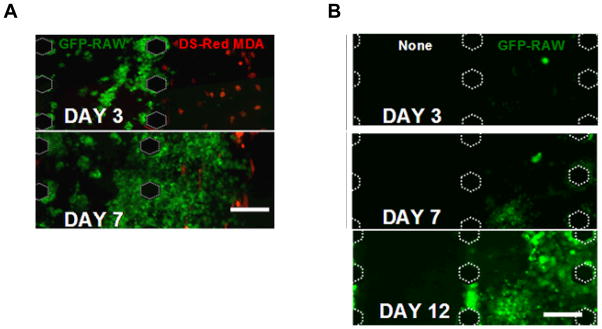
(A) A co-culture system consisting of 4 mg/mL collagen containing DS-red-labeled MDA-MB-231 patterned adjacent to 4 mg/mL Matrigel containing GFP-labeled RAW cells demonstrates the potential of the system to pattern multilayers of gels as well as multiple cell types. (B) GFP labeled RAW cells were cultured independently from MDA-MB-231 cells in a multi gel channel device and observed for over 12 days. Scale bars are 200 μm.
Experimental
Fabrication and preparation of the device
Silicon wafers (Silicon Inc., Boise, ID) were coated with SU-8 (MicroChem, Newton MA) to generate master molds with 50 μm or 100 μm heights. The wafers were baked at 65°C and then 95°C to evaporate any solvent, leaving only the epoxy resin and photo acid generator. When exposed to UV light, an acid reaction occurred which cross links the epoxy resins to form the patterned structures. The SU-8 was then baked at 95°C. PDMS (polydimethyl siloxane) was then cast on this mold, cured at 65°C, and then cut out with appropriately sized holes punched out using cylindrical biopsy punches of various sizes. The devices, along with #2 cover glass slips, were then plasma treated for 5 minutes using an oxygen plasma generator (Harrick Plasma, NY). The PDMS devices and the glass cover slips were then bonded together irreversibly. To minimize gel leakage from the devices, they were incubated in an oven set at 100°C for 1–2 hours. The devices were then UV treated for 15 minutes to sterilize prior to cell culture applications.
Gel fabrication and injection protocols
High concentration rat tail type I collagen was purchased from BD Biosciences (Cat #354249). To make gel precursor solutions, the collagen stock (solubilized in 0.02 N acetic acid) was diluted in ddH2O, 0.1N NaOH, and 5x DMEM media. Different concentrations of collagen were made by adjusting the amount of collagen and adding the appropriate amount of 0.1N NaOH (to neutralize the acetic acid). The solution was then placed on ice to impede the polymerization and allow time to prepare the cells along with the devices. High concentration Matrigel was purchased from BD Biosciences (Cat #354248). Gels of 4 mg/mL Matrigel concentrations were prepared by diluting the Matrigel stock with DMEM.
Suspensions of cells were mixed with collagen or Matrigel solutions and then carefully injected (10μL total volume) into each PDMS device using a P20 pipetteman. The devices were then left in the cell culture hood for 20 min to 1 hour to allow for in situ polymerization. After the gel polymerized, media was added to the inlet reservoirs of the device and gently suctioned using a glass micropipette attached to a house vacuum. The device was then placed in a Petri dish to maintain sterility and transferred to a 37°C incubator.
Co-culture and multi-channel experiments
MDA-MB-231 was purchased through ATCC. (#HTB-26) and transfected with GFP using Lipofectamine 2000 (Invitrogen). DS-red labeled MDA-MB-231 cells and GFP labeled RAW 264.1 macrophages were obtained as a gift from the Klemke lab (UCSD). All cells were cultured in DMEM with high glucose (Sigma-Aldrich) supplemented with 10% (v/v) fetal bovine serum (Gibco) at 37°C in a 5% CO2 humid incubator. For the multigel experiments, a 5-gel multichannel device was used to pattern the different hydrogels. GFP labeled MDA-MB-231 cells (2.5×105) were suspended in 100 μL of 4 mg/mL collagen and then injected into alternating gel channels and polymerized at 37°C for 40 minutes. Matrigel (100 μL of 4 mg/mL) was then injected into the two remaining channels and polymerized at 37°C for 40 minutes. Media was then flowed into the main channels and the device was cultured in an incubator at 37°C with 5% CO2. The media was changed every other day before image acquisition.
For co-culture experiments, a 5-gel multichannel device was employed. DS-red labeled MDA-MB-231 cells (2.5×105) were suspended in 100 μL of 4 mg/mL collagen precursor solution and then injected into the three alternating gel channels. Devices were then incubated at 37°C for 40 minutes to allow for gel polymerization. GFP labeled RAW cells (2.5×105) were then suspended in 100 μL of 4 mg/mL Matrigel and injected into the remaining channels. The device was incubated at 37°C for another 40 minutes to ensure the Matrigel solidified before filling the main channels with media.
Fluorescent and phase micrographs images were acquired every two days using an Olympus 1X-51 inverted microscope. Multigel and co-culture experiments were captured at 10X magnification using a Q-Imaging QICAM 12 bit Color Fast 1394 camera (QImaging, Surrey, BC) and Qcapture Pro (QImaging, BC, Canada) imaging software. The images were then stitched and merged using ImageJ (NIH) and Adobe Photoshop (Adobe Systems Inc., CA).
Multiphoton microscopy
Second harmonic generation (SHG) and two photon fluorescence (TPF) images used to visualize the collagen matrix and cells were imaged on a Zeiss LSM510 meta multiphoton system (Jena, Germany) as previously described 22. Images generated using 543 nm and 800 nm excitation wavelengths were visualized with a 40X Achroplan0.8 numerical aperture water immersion objective (Zeiss). Z-stacks were compiled by imaging 1–2 μm thick optical sections. Each 12 bit image consists of a 225×225 μm2 area and pinhole size and laser power were kept constant throughout the imaging. A 675 μm × 1350 μm montage was generated by stitching the acquired images.
A triple-gel channel device was prepared by polymerizing a 2 mg/mL collagen suspension containing 2.5×104 DS-Red MDA-MB-231 cells in the center gel channel. Adjacent channels were then filled and polymerized with 2 mg/mL collagen. The device was cultured in growth media and imaged on the third day. Areas surrounding the interface between the acellular and cell containing collagen were imaged at 40X. Fibers were highlighted by back scattering of the second harmonic signal while the cells were fluorescently excited.
Capillary Burst Valve Injection Model
To understand the physical basis of our gel filling process and the forces generated on the gap spacings, a two dimensional model of a single gel channel within the device was constructed from a modified capillary burst valve. We assume the critical point during the gel injection occurs when the gel front approaches the first gap spacing between post structures. To simplify the calculations as the hydrogel solution approaches, a single channel with hexagonal posts and gap spacings of 100 μm was created. When an advancing liquid interface meets the straight section of the posts at equilibrium (Figure 2a), the difference between the pressure inside the liquid Pi and atmospheric pressure Po is given by the Young-Laplace equation (which states that the pressure drop is a function of the surface tension γ and the interface curvature), Pi −Po = −2γ (cosθs/w + cosθv/h), where w and h are the width and the height of the channel, respectively, θs is the advancing contact angle formed between the liquid interface and the side wall, and θv is the advancing contact angle of the liquid with the top and bottom walls. Here it should be noted that the liquid interface can only move forward when the contact angles with the solid walls exceed the critical advancing contact angle, θA, which is determined by the liquid/solid combination. When the contact line speed is sufficiently low that the capillary number, Ca =μU/γ <10−3, where μ being the liquid viscosity and U the contact line velocity, we may set θs ≈ θv ≈ θA. When the interface meets the edge A of the post as in Figure 2b, the contact angle with a new wall (A–B) is reduced to θm =θA − α, where α =60°, which causes the contact line to stop because θm < θA. For the contact line to resume advancing, the interface should bulge until the contact angle with the new wall increases to θA, leading to the increase of the contact angle with the old wall (before A) from θA to min{θA +α, 180°} (The contact angle never exceeds 180°). Therefore, for the liquid interface to move beyond the point A, the driving pressure difference Pi − Po should be greater than ΔP1 = −2γ (cosθ1/w + cosθA/h), where θ1 =min{θA +α, 180°}. Assuming that a sufficient pressure is supplied to overcome the foregoing barrier A, the liquid fills the straight channel until its interface meets a new wall (B–C) with which it makes a new contact angle θn =θA − α, as shown in Figure 2c. Unless the driving pressure increases to bulge the interface till θn reaches θA, the liquid interface is pinned at B. The critical pressure causing the interface pinned at B to resume advancing (or the liquid to leak into adjacent channels) is again given by the Young-Laplace equation,
, where θ2 =min{θA +α, 180°}. Stopping the liquid interface at B, where the diverging section begins, is the major role of the posts as the geometric capillary burst valve. It should be noted that the interface pinning location is precisely determined by hexagonal posts (between nearest vertexes of neighboring posts) while the rectangular posts as previously used are unable to guarantee pinning of liquid interface at pre-defined locations.17
Conclusions
This paper describes an approach to engineer 3D microenvironments for investigating cell-cell and cell-ECM interactions using a microfluidic platform. This approach is based on a set of parallel microfluidic channels separated by an array of posts that allow selective filling of individual channels, yet permits continuous interfaces between adjacent channels for free diffusion of molecules as well as movement of cells across distinct regions. The advantages of this system when compared to current models presently available are: (i) it allows the patterning of different combinations of hydrogels and culturing of multiple cell types in a relatively easy and straightforward manner, (ii) it can be scaled dimensionally to mimic multicellular in vivo structures, and (iii) it can be used to generate chemically and mechanically tailored 3D cellular niches in a reproducible manner. We successfully patterned adjacent channels filled with ECM hydrogels (collagen, fibrin, Matrigel) of various concentrations to create distinct layers of different composition with a range of dimensions (100–300 μm in height, 100–400 μm in width). We also provided a simulation model to better understand the mechanics of the injection process. Finally, the utility of this system for multicellular 3D cultures was demonstrated by investigating the behaviors of metastatic breast cancer cells and tumor-derived macrophages in spatially well-defined geometries. In these experiments, distinct phenotypes were exhibited by the macrophages when cultured next to the tumor cells, possibly due to paracrine signals that diffuse between adjacent gel channels. In future studies, this system will enable these paracrine effects on normal and tumor cell functions to be investigated further, in concert with changes in ECM ligand identity and density and gradients of soluble diffusible factors.
Supplementary Material
Acknowledgments
Financial support for this study was partially provided by the California Institute for Regenerative Medicine (RN1-00556 to A.J.P.) and the Susan B. Komen Foundation (#BCTR0601235 to N.L.J.). Thanks to Tatianna Krasieva and the BLI at UCI for their technical support and help with confocal microscopy. Thanks to the Klemke and Tsien labs for their expertise, cell lines, and plasmid constructs. Thanks to Dr. Segall for his advice and helpful discussions.
References
- 1.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, Polverini PJ, Mooney DJ. Nat Methods. 2007;4:855–860. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 2.Yamada KM, Cukierman E. Cell. 2007;130:601–610. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Albrecht DR, Underhill GH, Wassermann TB, Sah RL, Bhatia SN. Nat Methods. 2006;3:369–375. doi: 10.1038/nmeth873. [DOI] [PubMed] [Google Scholar]
- 4.Griffith LG, Swartz MA. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 5.Bhandari RN, Riccalton LA, Lewis AL, Fry JR, Hammond AH, Tendler SJ, Shakesheff KM. Tissue Eng. 2001;7:345–357. doi: 10.1089/10763270152044206. [DOI] [PubMed] [Google Scholar]
- 6.Khademhosseini A, Ferreira L, Blumling J, 3rd, Yeh J, Karp JM, Fukuda J, Langer R. Biomaterials. 2006;27:5968–5977. doi: 10.1016/j.biomaterials.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 7.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 8.Ghajar CM, Chen X, Harris JW, Suresh V, Hughes CC, Jeon NL, Putnam AJ, George SC. Biophys J. 2008;94:1930–1941. doi: 10.1529/biophysj.107.120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghajar CM, Blevins KS, Hughes CC, George SC, Putnam AJ. Tissue Eng. 2006;12:2875–2888. doi: 10.1089/ten.2006.12.2875. [DOI] [PubMed] [Google Scholar]
- 10.Nelson CM, Bissell MJ. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf K, Muller R, Borgmann S, Brocker EB, Friedl P. Blood. 2003;102:3262–3269. doi: 10.1182/blood-2002-12-3791. [DOI] [PubMed] [Google Scholar]
- 12.Toh YC, Zhang C, Zhang J, Khong YM, Chang S, Samper VD, van Noort D, Hutmacher DW, Yu H. Lab Chip. 2007;7:302–309. doi: 10.1039/b614872g. [DOI] [PubMed] [Google Scholar]
- 13.Frisk T, Rydholm S, Liebmann T, Svahn HA, Stemme G, Brismar H. Electrophoresis. 2007;28:4705–4712. doi: 10.1002/elps.200700342. [DOI] [PubMed] [Google Scholar]
- 14.Wong AP, Perez-Castillejos R, Christopher Love J, Whitesides GM. Biomaterials. 2008;29:1853–1861. doi: 10.1016/j.biomaterials.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosadegh B, Huang C, Park JW, Shin HS, Chung BG, Hwang SK, Lee KH, Kim HJ, Brody J, Jeon NL. Langmuir. 2007;23:10910–10912. doi: 10.1021/la7026835. [DOI] [PubMed] [Google Scholar]
- 16.Saadi W, Rhee SW, Lin F, Vahidi B, Chung BG, Jeon NL. Biomed Microdevices. 2007;9:627–635. doi: 10.1007/s10544-007-9051-9. [DOI] [PubMed] [Google Scholar]
- 17.Chung S, Sudo R, Mack PJ, Wan CR, Vickerman V, Kamm RD. Lab Chip. 2009;9:269–275. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- 18.Ng CP, Pun SH. Biotechnol Bioeng. 2008;99:1490–1501. doi: 10.1002/bit.21698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vickerman V, Blundo J, Chung S, Kamm R. Lab Chip. 2008;8:1468–1477. doi: 10.1039/b802395f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho H, Kim HY, Kang JY, Kim TS. J Colloid Interface Sci. 2007;306:379–385. doi: 10.1016/j.jcis.2006.10.077. [DOI] [PubMed] [Google Scholar]
- 21.Engländer T. Journal of colloid and interface science. 1996;179:635–636. [Google Scholar]
- 22.Raub CB, Suresh V, Krasieva T, Lyubovitsky J, Mih JD, Putnam AJ, Tromberg BJ, George SC. Biophys J. 2007;92:2212–2222. doi: 10.1529/biophysj.106.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.