

Abstract
Detection of weak ligand binding to membrane-spanning proteins, such as receptor proteins at low physiological concentrations, poses serious experimental challenges. Saturation transfer difference nuclear magnetic resonance (STD-NMR) spectroscopy offers an excellent way to surmount these problems. As the name suggests, magnetization transferred from the receptor to its bound ligand is measured by directly observing NMR signals from the ligand itself. Low-power irradiation is applied to a 1H NMR spectral region containing protein signals but no ligand signals. This irradiation spreads quickly throughout the membrane protein by the process of spin diffusion and saturates all protein 1H NMR signals. 1H NMR signals from a ligand bound transiently to the membrane protein become saturated and, upon dissociation, serve to decrease the intensity of the 1H NMR signals measured from the pool of free ligand. The experiment is repeated with the irradiation pulse placed outside the spectral region of protein and ligand, a condition that does not lead to saturation transfer to the ligand. The two resulting spectra are subtracted to yield the difference spectrum. As an illustration of the methodology, we review here STD-NMR experiments designed to investigate binding of ligands to the human sweet taste receptor, a member of the large family of G-protein-coupled receptors. Sweetener molecules bind to the sweet receptor with low affinity but high specificity and lead to a variety of physiological responses.
Keywords: Saturation transfer difference (STD), Saturation transfer double difference (STDD), Membrane-bound receptors, G-protein-coupled receptor (GPCR), Sweet taste receptor, T1R2, T1R3, Neotame, Dextrose
1. Introduction
Nuclear magnetic resonance (NMR) is a powerful tool for directly monitoring ligand binding to protein receptors. Saturation transfer difference (STD) NMR can be used to monitor weak ligand binding (Kd ∼ mM–μM) via non-scalar magnetization transfer from a large protein (receptor, >20 kDa, example used here ∼190 kDa for the heterodimeric sweet receptor) to smaller ligands (1–3). STD-NMR does not require expensive stable isotope or radioisotope labeling, and the experiment requires only a low receptor concentration (nM–pM) in the presence of 20–1,000 times excess ligand in a sample of ∼200 μl. Hence it offers an economical method to assay the function of proteins in an isolated form or when expressed on a cell surface (2, 4). Membrane proteins studied by this approach can be present in a cell surface membrane or in isolated membranes or liposomes. Transferred Nuclear Overhauser Enhancement (TrNOE) (5, 6) and saturation transfer methods (3), which both utilize transfer of magnetization between receptor and bound ligand, have been applied successfully in vivo (7, 8) to monitor oligomerization (9) and to measure dissociation constants for complexes (10). Because signals from small ligands are monitored, the molecular weight of the protein is not an issue with STD. In fact, spin diffusion is more efficient for a large or membrane-bound protein than for a small protein in solution. These experiments are particularly useful for monitoring weak (mM scale) binding. Further, when a protein (or receptor) has more than one binding site, competition STD experiments can be used to distinguish between competitive and noncompetitive ligand binding (11). The binding of multiple ligands can be analyzed simultaneously as long as signals from the different ligands do not overlap.
An alternative approach to investigating ligand binding by NMR spectroscopy is to observe signals from the protein itself as ligands are added. Binding is followed by monitoring changes in the chemical shifts of signals from the protein. This approach requires a high concentration of protein (>100 μM) labeled with stable isotopes. In addition, membrane proteins studied by this approach in solution must be investigated in detergent micelles or small bicelles so that they tumble rapidly enough to yield resolvable NMR signals.
Membrane proteins are notoriously difficult to produce and isolate in functional form. Over-expression in bacterial systems often requires refolding of the membrane proteins, and a functional assay is needed to determine whether the protein has refolded properly. The STD experiment is sensitive enough to detect ligand binding at protein concentration levels frequently found in cells (pM–nM). This relieves the need for high protein yields from expression systems making the use of bacterial expression systems unnecessary. Furthermore, samples of membrane-bound proteins from cells can be obtained under nondestructive conditions without unfolding and refolding the protein.
The initial step in the STD experiment is the saturation of proton spins on the receptor, which is achieved by applying a selective radiofrequency (RF) signal to a spectral region that contains signals from the receptor but not the ligand(s). Magnetization transferred from the receptor to the bound ligand serves to saturate its NMR signals. Upon dissociation, the saturated ligand contributes a decrease in the intensity of the NMR signal from the pool of free ligand until the ligand loses the saturation condition by longitudinal (R1) relaxation. Because the R1 relaxation rate is much slower than the rate of dissociation of ligands from the saturated protein, one protein has the capacity to saturate many ligand molecules. The kon, koff, and R1 rates in conjunction with the power and duration of the selective RF determine the observed strengths of the ligand signals. Several experimental parameters can be varied to achieve optimal signal-to-noise ratios (S/N). If koff is slow (tight binding, nM), the net saturation transferred to the pool of free ligand may be low. If koff is very fast (extremely weak binding, i.e., >mM), the residence time of the ligand in the complex may be insufficient to achieve full saturation. Excess ligand is required to ensure that the receptor molecules are saturated sufficiently with ligand, but very high ligand concentrations dilute out the effects of saturation. Temperature and pH can be varied to modify kon and koff. As a rule of thumb, when the ligand:receptor ratio is increased from 20:1 to 1,000:1, the magnitude of observed saturation follows a sigmoidal curve. This information can be used to choose the optimal conditions for the STD experiment. To test for competitive binding to a single site on the receptor, the concentration of one ligand is changed at a time. In order to correct for effects of nonspecific binding, a control solution lacking the receptor or containing a receptor with blocked binding site can be used in a parallel STD experiment. Subtraction of the control STD from the experimental STD yields a double difference STD (STDD) spectrum (Figs. 5 and 6).
Fig. 5.
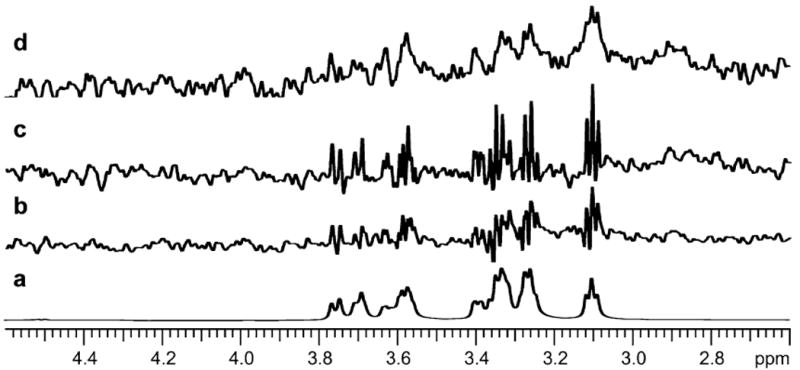
(a) 1H 1D spectrum of dextrose; (b) dextrose binding to parental membrane; (c) dextrose binding to mT1R2; (d) dextrose binding to hT1R2. Data sets were processed by VNMRJ software in the time domain.
Fig. 6.
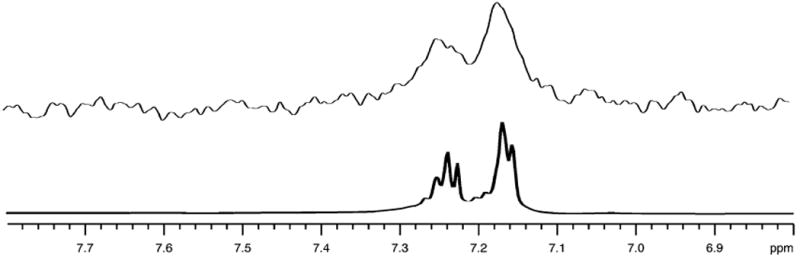
(a) 1H 1D spectrum of neotame; (b) STD signal resulting from binding of neotame to the VFTM of T1R2. Only resonances at lower frequencies from the water signal are shown; these signals arise from binding of neotame to the purified amino terminal domain of the hT1R2 subunit. Data were processed by NMRPipe software, and subtraction was performed by Newton (in-house) software in the frequency domain.
We have successfully used STD and STDD methods to monitor ligand binding to the sweet receptor (2, 12). By optimizing concentrations of receptor-to-ligand ratios and the temperature, we were also able to distinguish between ligands that bind to mutant receptors but fail to activate and those that do not bind at all (see Note 1) (Figs. 5 and 6).
The sweet receptor is a member of the family of Class C G-protein-coupled receptors. The heterodimeric sweet receptor consists of the two subunits: T1R2 and T1R3. Each T1R subunit contains a large extracellular domain called the Venus FlyTrap Module (VFTM), linked via a Cysteine-Rich Domain (CDR) to a seven helix Trans-Membrane Domain (TMD). A schematic of the sweet receptor is shown in Fig. 1. A large number of small molecule ligands are known to interact with the sweet receptor, and these interactions can be investigated in cells transfected with sweet receptors by means of a calcium flux assay. This has enabled comparison of the properties of sweet receptors from different species, such as mouse and human. Results have shown that dipeptide sweeteners, aspartame and its analog neotame, while active with human receptor, do not activate the mouse receptor (12). On the other hand, both human and mouse receptors are activated by natural sugars, such as dextrose and some small artificial sweeteners such as sucralose or sweet amino acids such as d-tryptophan. We prepared membranes from transfected cells expressing the sweet receptor and from the parental cell line for negative control (Fig. 1). The control parental cell line was used to detect and correct for nonspecific binding in the STDD spectrum.
Fig. 1.
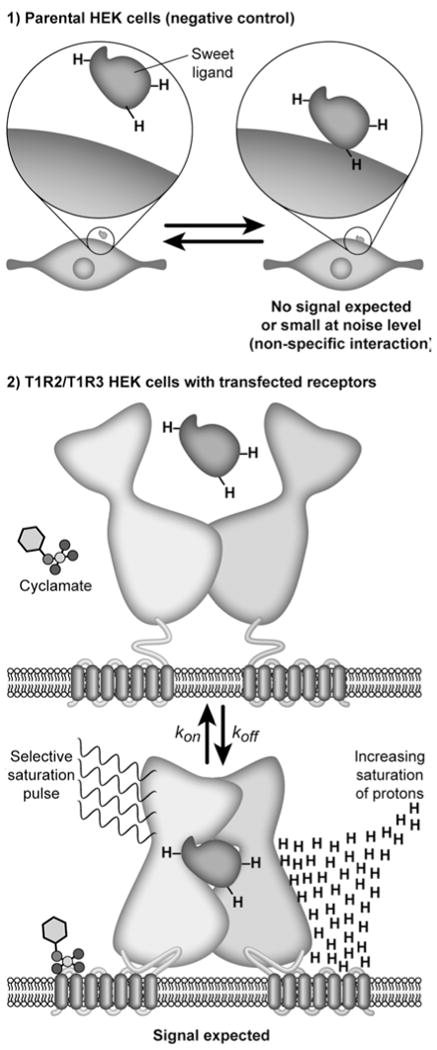
Schematic illustrating the use of STD NMR for monitoring binding interactions of non-expressing cells and cell-expressed receptors (2): (1) negative control: due to possible nonspecific binding of ligand to parental membrane background parental cells that are not transfected and do not contain receptor of interest, e.g., parental HEK cells, are used for preparing membranes from these cells as negative control membranes; (2) ligand binding to the sweet receptor is expected for T1R2/T1R3 transfected HEK cells where the receptors are expressed and displayed leading to STD signals. STD signals depend on both equilibrium constants (kon/koff rates), which describe kinetic interactions between ligand and receptor. A selective saturation NMR pulse is applied to the receptor to transfer magnetization from receptor through spin diffusion to the nearby (bound) ligand (bottom). Effects are detected as STD signals on the pool of free ligands by rapid exchange of the saturation transfer only if binding occurs.
We describe here our protocol for STD-NMR studies of neotame and dextrose binding to human sweet receptor subunits, which includes experimental procedures used in acquiring and analyzing STD and STDD data.
2. Materials
Most of the chemicals were purchased from Sigma Aldrich (http://www.sigmaaldrich.com/sigma-aldrich/home.html). Deuterated water was obtained from Cambridge Isotopes Limited (http://www.isotope.com/cil/index.cfm?123&CFID=25580810&CFT OKEN=89808295). Shigemi tubes were obtained from Shigemi Inc. (http://www.shigeminmr.com/main.html). Neotame was obtained from NutraSweet Company (http://www.nutrasweet.com/).
3. Methods
In the following four sections, we describe (Subheading 3.1) our method for preparing membrane samples containing proteins of interest, (Subheading 3.2) sample preparation for NMR, (Subheading 3.3) NMR data collection methods, and (Subheading 3.4) data analysis.
3.1. Preparation of Membranes for STD-NMR
This method uses transfected human embryonic kidney 293 E cells, grown in 175 cm2 flasks. Transfect 293 E cells grown to 60–70% confluence in 175 cm2 flasks (∼1.2 × 107 cells/flask) with Lipofectamine-2000, per Invitrogen protocol, http://tools.invitrogen.com/content/sfs/manuals/lipofectamine2000_man.pdf; 48 h after transfection, harvest the cells using trypsin-free cell dissociation buffer (0.5 mM EDTA in phosphate-buffered saline—PBS). Suspend the cells in PBS after dissociating them from the flask and spin at 800 × g for 10 min to pellet the cells as described below.
Day 1: 24 h prior to transfection, seed 6.2 × 106 cells in OPTI-MEM containing 10% fetal bovine serum with no antibiotics in a 175 cm2 flask.
Day 2: Make sure that cells are 60–70% confluent before transfection. Dilute 75 μg DNA (37.5 μg DNA each of T1R2 and T1R3 plasmid) in 2.0 ml OPTI-MEM (serum free) and mix gently. Make sure that Lipofectamine 2000 is completely suspended by gently shaking it before use and then dilute 150 μl of it into 2.0 ml serum-free OPTI-MEM. Mix gently and let sit for 5 min at room temperature. Combine the diluted Lipofectamine 2000 with the diluted DNA and incubate for 30 min at room temperature. Add 4 ml DNA-Lipofectamine 2000 mixture to each flask. Distribute the DNA–lipofectamine solution over the cells by gently rocking the plate back and forth. Incubate the cells at 37°C in a CO2 incubator.
Day 3: Replace growth medium with regular OPTI-MEM containing antibiotics and serum. This is an optimized protocol and works well with 293 cells. Usually, transfection makes cells come off the plate more easily, resulting in loss. Gentle aspiration and addition of new media are required for good yields.
Day 4: Remove media completely from the flasks and add 5 ml trypsin-free cell dissociation rinse buffer (0.5 mM EDTA in PBS). Rock the rinse buffer over the cells thoroughly but only once, and then gently aspirate the medium. Leave at room temperature for 5 min to allow the cells to detach. Gentle tapping on the side of flask will help to dislodge them. Once detached, add 10 ml PBS lacking Ca2+/Mg2+ and harvest the cells using a broken, fire-polished pipette. Avoid harsh handling of the cells, because if they lyse at this point, nuclear DNA can be released, spoiling the membrane preparation. To pellet the cells, spin at 800 × g for 10 min. Do not spin any faster, to avoid breaking the cells (see Note 1).
3.2. Membrane Preparation from Cell Pellet
Add 4 ml homogenization buffer, 20 mM Tris–HCl pH 7.4, 10% glycerol, and complete protease inhibitor (Roche, Indianapolis, IN) to the cell pellet and homogenize with a Polytron® homogenizer. This buffer composition and protease inhibitor cocktail work very well. Allow only eight strokes of the Polytron at half-maximal speed while the cells are kept on ice (see Note 2).
Centrifuge homogenate at 1,500 × g for 15 min at 4°C to remove unbroken cells, cell debris, and nuclei. The resulting supernatant contains cytosol and total cellular membranes. Transfer the supernatant to a new centrifuge tube.
Ultracentrifuge the supernatant at 100,000 × g for 1 h at 4°C. The resulting pellet is referred as the membrane pellet. Remove the supernatant without disturbing the membrane pellet.
Add 8 ml homogenization buffer (20 mM Tris–HCl pH 7.4, 10% glycerol lacking protease inhibitor) to further wash, and gently resuspend the membrane pellet using a 1 ml pipette tip. Ultracentrifuge at 100,000 × g for 30 min at 4°C, and carefully remove the supernatant without disturbing the resulting membrane pellet.
To the pellet add 200 ml homogenization buffer lacking protease inhibitor and resuspend by 20 passages through a 25-gauge needle. This renders an even suspension of membranes in the buffer (see Note 3). Protease inhibitor-free homogenization buffer is preferred to avoid interference from inhibitors in future experiments.
Membranes can be stored in this buffer at −80°C. Total membrane protein concentration is checked by the Lowry protein assay (13). Total membrane protein (including the receptor) is found to be 10–13 μg/ml when following this protocol.
For STD-NMR studies, prepare 50–75 μg total protein/160 μl NMR PBS consisting of 137 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, and 1 mM KH2PO4 at pH 7.2–7.6 in D2O (see Note 4).
3.3. Preparation of the Receptor–Ligand Complex (the Same Procedure is Used to Prepare Control Membrane Without Receptor)
The total amount of membrane protein to be used in an NMR sample is 50–75 μg. We found that membrane protein preparations were stable in PBS buffer. Buffers for small molecule ligands were prepared in 99.98% D2O; buffers for peptide or small protein ligands were prepared in H2O (see Note 5).
To 50 μl of membrane (with its incorporated receptor), 150 μl of ice-cold PBS buffer is added, and the membrane is resuspended by gentle pipetting (using 250 μl tip size) up and down 2–3 times. The sample is spun in a tabletop centrifuge at 800 × g for 3 min.
The supernatant is removed; to the washed pellet containing the receptor, 160 μl of ligand solution (at the appropriate concentration: e.g., 5 mM for neotame, obtained from http://www.nutrasweet.com/) in deuterated PBS buffer is added, and the pellet is resuspended by gentle vortexing at 5,000 × g for 5 min (see Note 6).
A well-dispersed sample has an opaque color (Fig. 2). If membranes are not prepared correctly, they can include genomic DNA, which will prevent proper dispersal. The sample turns to a solid clump without any dispersion (see Note 7). In such a situation, the sample should be discarded and replaced with freshly made membrane.
The ligand concentrations varied from 1 mM for the sweet proteins (brazzein and other protein ligands) to 5–10 mM for small molecule ligands (see Note 8).
The membrane sample (150 μl with opaque color) is placed into a 3 mm outer diameter (o.d.) Shigemi tube (Fig. 2), and NMR data are recorded immediately. We also use NMR tubes of other sizes (with different volume requirements): 5 mm o.d. regular or Shigemi tube (500 μl), or 1.7 mm o.d. NMR tube (30 μl) (see Note 9).
Fig. 2.

(Left) A 1.7 mm o.d. capillary tube (30 ml) used in a 1.7 mm cryogenic probe when membrane or protein samples are severely limited. (Right) Membrane sample (150 ml) placed into a 3 mm o.d. Shigemi tube with opaque color showing the appearance of a properly dispersed membrane sample prepared for NMR. We have used 3 and 5 mm Shigemi tubes in 5 mm cryogenic probes.
3.4. STD NMR Spectroscopy
3.4.1. NMR Data Collection
NMR data are collected at 25°C on a Varian (Agilent Technologies) Avance 600 NMR spectrometer equipped with a cryogenic probe (see Note 10). The pulse programs are written in house and can be obtained from nmrfam.wisc.edu.
Selective saturation of the receptor is achieved by a train of Gaussian-shaped pulses of about 30 ms each, saturating a bandwidth of about 20 Hz, at −2 ppm (where the receptor has signals, but the ligands do not) for a saturation time of approximately 3 s. This ensures full saturation of the receptor (see Note 11). The on-resonance irradiation of the protein can be varied depending on the type of ligand present (normally around −1.0 ppm to 1.0 ppm).
Watergate 3-9-19 pulse sequence with gradients is employed prior to acquisition of the FID signal for solvent water suppression (see Note 12).
Data are collected with 12 k complex points in the direct dimension for 1.5 s (see Note 13). Small molecules relax slowly and are in a dynamic equilibrium in solution between the free and in complex states. A delay of 3 s is applied between each FID to ensure complete relaxation of the ligand.
256–1,024 scans are accumulated for each of the STD experiments depending on signal intensity. The pulse sequence used to record the experiments is shown in Fig. 3. STD pulse sequence is available in Varian or Bruker pulse libraries which require their own specific pulse sequences set up for this section please consult your NMR pulse library and its setup. For an experiment of 256 scans, the experiment time is about 160 min.
Off-resonance irradiation is set at ∼50 ppm, a region where no receptor or ligand signals are present. The spectra for both on-resonance and off-resonance saturation are collected interleaved (see Note 14). The decrease in signal intensity, resulting from the transfer of saturation from the protein to the ligand, is evaluated by subtracting the on-resonance spectrum from the off-resonance spectrum. This subtraction yields a positive signal from a bound ligand.
An in-house script is used to separately process and add on-and off-resonance FIDs in Varian (Agilent Technologies) VNMRJ software (http://www.varianinc.com/cgi-bin/nav?products/nmr/apps/corner&cid=LPHKNLKJFL), and the peak intensities are manually inspected. Usually the difference between on and off reference spectra is between 10 and 200 times less than the 1H signal intensity. If the STD signal is very weak or negligible it can be at noise level or 1,000 times less than the 1D intensity. If any FID has values differing significantly from the average, the FID is excluded from further analysis (see Note 15).
To remove any possible nonspecific binding contribution of the ligand to the parental membrane itself, the experiment is repeated with a membrane preparation lacking the receptor of interest; this is referred to as a negative control. The STD signal from the negative control is subtracted from that of the receptor-containing preparation to yield the STDD spectrum (see Notes 16 and 17).
With a 15N-labeled peptide or 15N–2H-labeled protein samples, either 1D or 2D STD edited 1H–15N HSQC NMR spectra are collected, with 80–100 increments in the indirect dimension and with 2 k sampling points in the direct dimension. The in-house written pulse program is available by request at nmrfam.wisc.edu. The resulting data are processed and analyzed by NMRPipe (http://spin.niddk.nih.gov/NMRPipe/) and SPARKY (http://www.cgl.ucsf.edu/home/sparky/) software.
Fig. 3.
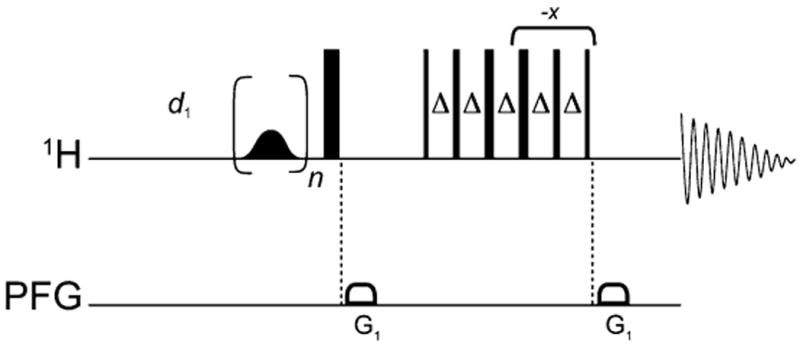
Pulse sequence used in the STD-NMR experiment. The shaped pulse in the beginning is for saturation; the 90° 1H pulse is followed by gradients and 3-9-19 pulses for water suppression by Watergate.
3.4.2. NMR Data Analysis and Interpretation
The subtraction of signals arising from nonspecific interaction with the parental membrane from total specific and nonspecific binding signals to membrane expressing the receptor protein can be achieved in the time domain or the frequency domain (see Note 18). VNMR provides simple and elegant steps that enable time domain subtraction. All spectra are processed and analyzed using the VNMRJ (version 2.1B) software in the time domain mode (see Note 19). The steps involved in data processing using VNMRJ are the following:
Process the interleaved 1D data for receptor and parental membrane samples to obtain the resulting STD signals. By default, all the operations are done on arbitrary experiment number 5 (see Note 20). Save the 1D STD spectrum, which is the third increment with the appropriate file name.
In two new experiments other than number 5, load the two saved spectra.
Go to the experiment containing receptor STD data. Process data with the “wft” command. Apply shifted sine multiplication and line broadening parameters as per requirement. Routinely sbs = sb and lb = 5 are used for processing. Reference the peaks of interest (see Note 20).
Command “clradd add” deletes and adds the experiment on experiment number 5.
Go to the experiment containing the parental (negative control) STD data. Process as in step 3. Use command “add(−1.0)” to subtract the data on experiment number 5.
If normalization is required for the parental membrane, use the coefficient c in add(c) instead of −1.
Go to experiment number 5 and process with command “wft”.
Data processing and subtractions in the frequency domain can be achieved by a combination of NMRPipe software for data processing and in-house-developed Newton software for subtractions. Newton is an application for performing spectral deconvolution. The program contains libraries for performing generalized addition and subtraction of NMR matrices in NMRPipe and SPARKY data formats and is available at nmrfam.wisc.edu. The frequency domain data are matched by chemical shifts in the two spectra, by grid interpolation before subtraction. First, the data from the spectrometer are converted to NMRPipe format. The data are further Fourier transformed with required processing parameters and correct phases (see details in VNMRJ manual; http://www.varianinc.com/). Samples of NMRPipe conversion scripts are given in Fig. 4. To add spectra, the command “newton add-matrix −m1 file1.ft2 −m2 file2.ft2 −c2 coefficient 2” is used, where coefficient 2 is the scaling factor for the second matrix.
Fig. 4.
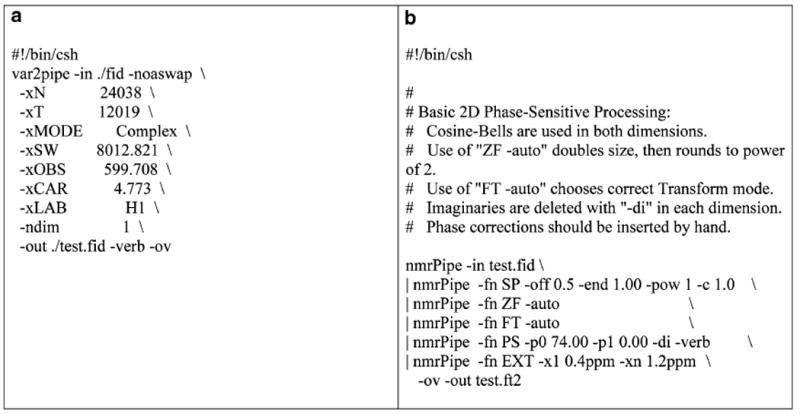
(a) Example script for Varian (Agilent) to NMRPipe format conversion; (b) example script for processing the converted NMRPipe data.
Examples of STD spectra produced by time domain analysis are shown in Fig. 5. The STD results show binding of dextrose to human (hT1R2) and mouse (mT1R2) sweet receptor subunits. A control 1D 1H spectrum of dextrose is shown in panel a. Dextrose binds weakly to parental membrane alone, as seen in panel b. This signal is subtracted to obtain the STDD signal for receptor binding. Dextrose binding to mT1R2 is shown in panel c, and dextrose binding to hT1R2 is shown in panel d. Notice the significant line broadening of the ligand peaks upon binding to the receptor.
Previous mutational and chimeric studies have reported several binding sites on the heterodimeric sweet receptor (T1R2 + T1R3) (14). Jiang et al. have demonstrated that the TMD of hT1R3 is the binding site for lactisole (a sweet receptor antagonist) (15), whereas the extracellular domain of hT1R2 is the binding site for neotame (a dipeptide sweet receptor agonist) (Max and Maillet, submitted). However, binding evaluation of the T1R2 (and T1R3) subunit has remained elusive because homodimeric form or interspecies form of the receptor mT1R2 + hT1R3 cannot be activated by the sweet ligand (16). Previous studies indicated that neotame does not activate to mT1R2 + mT1R3 (12); however, it is unknown whether this reflects lack of binding or binding that does not lead to activity. STD experiments may prove to be useful to test the binding properties of sweet ligands such as dipeptide sweeteners (for example: neotame) to domains for which species differences in activity cannot be used.
Examples of STD spectra produced by frequency domain processing are illustrated in Fig. 6, which shows results for neotame binding to the purified VFTM (the extracellular domain) of hT1R2 as is consistent with indirect biological chimera studies (12). This example shows that STD NMR also can serve as a powerful method to detect binding to isolated domains of receptor proteins which cannot be determined from activity assays.
4. Notes
800 × g for 10 min is just sufficient to spin the cells down and will not lyse cells. Centrifugal speeds or longer times may result in cell lysis, so take care in maintaining the recommended limits.
To protect the receptor-expressing cells from trypsin digestion during cell harvest, it is important to harvest the cells in PBS containing 0.5 mM EDTA. While homogenizing the cells, great care has to be taken to restrict homogenization to only eight strokes. Exceeding this number may result in DNA breaks resulting in contamination of the preparation. DNA contamination leads to string-like clumps in the supernatant and ruins the membrane preparation.
At the stage of final suspension, the membrane has to be thoroughly mixed by pulling up and down with a syringe. Incomplete suspension may result in clumping of suspension. Aliquot membrane preparations in smaller volumes in Eppendorf tubes and keep frozen at −80°C for long-term storage.
To make PBS buffer for 1-l volume, weigh the following: 8 g NaCl (137 mM), 1.15 g Na2HPO4 (10 m M), 0.2 g KCl (2.7 mM), and 0.2 g KH2PO4 (1 mM). Dissolve in 1-l double-distilled water; the pH should be around 7.4.
Small molecule ligands are normally detected by conventional one-dimensional (1D)1H STD-NMR experiments and do not require labeling. With ligands that are peptides or small proteins, uniform 15N-labeling (or uniform 2H, 15N-labeling with back exchange in H2O to protonate NH-groups) can be used along with 1H–15N edited 2D-STD detection to determine what parts of the ligand bind to the receptor (12).
When the receptor is membrane-spanning, it is crucial for the success of the experiment to obtain stable membrane preparations at optimal pH and ionic strength. In small molecule ligand-binding experiments, the signal-to-noise ratio of the detected signal can be improved by exchanging the solvent from H2O to D2O to improve the suppression of the solvent signal.
It is easier and cleaner to transfer samples to and from NMR tubes with a pipette tip that reaches the bottom of the tube. Commercially such tips are not available for purchase. For transfers to and from 3-mm NMR tubes, we added a 1 mm polyethylene capillary tube (Intramedic, http://vwrlabshop.com/intramedic-polyethylene-tubing-clay-adams/p/0013744/) running along the length of the NMR tube.
However, only 7–10% D2O solvent (as required for the NMR frequency lock) should be used for 15N-labeled peptide or protein ligands to minimize the loss of backbone amide signals. Further, if a dual sample spinner of 2.7 mm is available, samples can be prepared in 100% H2O to increase S/N. In this case, the first tube contains the sample and the second tube is filled with deuterated buffer to provide the frequency lock for acquisition.
The bottom of the Shigemi tube is constructed of glass whose magnetic susceptibility matches that of an aqueous solution. The sample is placed in the tube, and a plunger made of susceptibility-matched glass is lowered to the top of the solution. Care must be taken to avoid air bubbles to ensure good shimming quality. The solid base and the plunger together limit the sample volume to the most sensitive region of the NMR receiver coil. We used 3 mm Shigemi tubes to reduce the sample volume to 150 μl. If sample volume is not limiting, a 5 mm Shigemi tube be used, which requires 250–300 μl sample volume. Although the 3 mm Shigemi tube reduces the sample volume, the S/N is lower than that from a solution of the same concentration in a 5 mm Shigemi tube.
Cryogenic probes increase S/N by about two when compared to room-temperature probes. If the spectrometer is not equipped with a cryogenic probe, data collection times are nearly four times longer to obtain similar S/N.
It should be noted that if the receptor or the ligand is completely deuterated, there is no saturation transfer, because there are no protons present to saturate. If the ligand is a larger protein, it may not be possible to selectively saturate the receptor, and artifacts will result from joint saturation of receptor and ligand.
Without water suppression, the spectrum may exhibit a large baseline roll. This can give rise to anomalies in subtraction resulting in peaks in the vicinity of water. For small ligands, the use of >98% deuterated PBS buffer removes most of the water signal. However, the residual (1–2%) water peak is still larger than the ligand signals. Thus, water suppression ensures cleaner subtraction of the on- and off-resonance spectra.
The long acquisition time ensures detection of magnetization that is transferred upon binding to small molecules. If the ligand is a protein with larger molecular weight, shorter acquisition time may be employed.
Owing to long data collection times and low S/N, any instability during a single FID is sufficient to cause discrepancies in the subtracted data. Hence, on-resonance and off-resonance data are collected in an interleaved fashion.
A 1H 1D spectrum is to be collected to decide the required offset for on-resonance irradiation. The irradiation is performed at least 1–2 ppm away from the nearest ligand resonances. If the on-resonance irradiation is too close to ligand resonances, ligand signals will be excited and will be seen in the spectrum even if there is no binding. Several NMR standardization experiments are required to ensure that the STD NMR experiment is set up properly on a given spectrometer. Examples of such experiments consist of ligand only in the sample buffer to ensure no excitation of the ligand by itself and no STD signal. Further, a sample of protein–ligand complex known to exhibit an STD can be used as a positive standard control. We recommend using 1:20 aldehyde dehydrogenase:NADH as positive STD standard control. This sample can be used to optimize the S/N ratio by varying ligand concentration. Some of the ligands under investigation exhibited STD signals in the presence of parental membrane (negative control) in which no receptor was expressed. In this case the STD spectrum of the parental membrane preparation is subtracted from that for membrane containing the expressed receptor to obtain the double difference saturation transfer (STDD spectrum containing specific signals arising from receptor–ligand binding).
It is important to have the receptor sample and the negative control membrane under similar concentrations and buffer conditions. All experimental conditions, including protein and ligand concentrations, sample volume, temperature, and number of scans, should be kept the same for both samples.
In time-domain subtraction of data we assume that the two data sets have the same offset, sweep width, and number of points. If there is a shift in the frequencies between the sample and control spectrum, it can be adjusted in the frequency domain before subtraction.
With data collected on Bruker spectrometers running TopSpin™ or XWIN-NMR, built-in processing scripts can be used for data processing and analysis. For instructions on using the above programs, consult the manuals that come with the specific version of the program and the spectrometer. Currently, the pointers to manuals can be reached at http://www.brukerbiospin.com/software_nmr.html.
If the VnmrJ version used has a default experiment number other than 5, then the processed data usually go to that experiment number. The user should verify this experiment number in advance. In our example, we set the default experiment to number 5.
In VnmrJ, wft refers to Fourier transformation of the raw NMR data (to transform time domain data to frequency domain). A window function multiplication is normally used after transformation. Routinely, a sine-bell window function is used and is denoted as sb. Other window functions applied prior to Fourier transformation are sbs (shifted sine bell shifted) and lb (line broadening). The command “clradd” clears any previously added data from experiment 5, and “add” adds the present data to newly created default experiment 5. Because the control experiment is to be subtracted, a coefficient of −1.0 is added in parentheses and the command “add (−1.0)” is used to subtract the control signals from the sample. For referencing, 0.5–1 mM DSS may be used as internal standard.
Acknowledgments
This research was supported by NIH grants R21 DC008805 to MM and FAP, R01 DC009018 and Wisconsin Institute of Discovery Grant (WID-135A039) to FAP, R01 DC006696, R01 DC008301 to MM, and P41 RR02301 which funds the National Magnetic Resonance Facility at Madison. We thank Drs. Marco Tonelli and Roger Chylla for their helpful discussions and assistance with the Newton program developed by Dr. Chylla. We thank the NutraSweet Company for providing the sample of neotame.
Contributor Information
Rani Parvathy Venkitakrishnan, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI, USA.
Outhiriaradjou Benard, Department of Neuroscience, Mount Sinai School of Medicine, New York, NY, USA.
Marianna Max, Department of Neuroscience, Mount Sinai School of Medicine, New York, NY, USA.
John L. Markley, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI, USA; National Magnetic Resonance Facility at Madison, University of Wisconsin-Madison, Madison, WI, USA
References
- 1.Wang YS, et al. Competition STD NMR for the detection of high-affinity ligands and NMR-based screening. Magn Reson Chem. 2004;42:485–489. doi: 10.1002/mrc.1381. [DOI] [PubMed] [Google Scholar]
- 2.Assadi-Porter FM, et al. Direct NMR detection of the binding of functional ligands to the human sweet receptor, a heterodimeric family 3 GPCR. J Am Chem Soc. 2008;130:7212–7213. doi: 10.1021/ja8016939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayer MMayer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 4.Assadi-Porter FM, et al. Interactions between the human sweet-sensing T1R2-T1R3 receptor and sweeteners detected by saturation transfer difference NMR spectroscopy. Biochim Biophys Acta. 2010;1798:82–86. doi: 10.1016/j.bbamem.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balaram P, et al. Negative nuclear Overhauser effects as probes of macromolecular structure. J Am Chem Soc. 1972;94:4015–4017. doi: 10.1021/ja00766a063. [DOI] [PubMed] [Google Scholar]
- 6.Megy S, et al. STD and TRNOESY NMR studies on the conformation of the oncogenic protein beta-catenin containing the phosphorylated motif DpSGXXpS bound to the beta-TrCP protein. J Biol Chem. 2005;280:29107–29116. doi: 10.1074/jbc.M501628200. [DOI] [PubMed] [Google Scholar]
- 7.Claasen B, et al. Direct observation of ligand binding to membrane proteins in living cells by a saturation transfer double difference (STDD) NMR spectroscopy method shows a significantly higher affinity of integrin alpha(IIb)beta3 in native platelets than in liposomes. J Am Chem Soc. 2005;127:916–919. doi: 10.1021/ja044434w. [DOI] [PubMed] [Google Scholar]
- 8.Mari S, et al. 1D saturation transfer difference NMR experiments on living cells: the DC-SIGN/oligomannose interaction. Angew Chem Int Ed Engl. 2004;44:296–298. doi: 10.1002/anie.200461574. [DOI] [PubMed] [Google Scholar]
- 9.Di Micco S, et al. Differential-frequency saturation transfer difference NMR spectroscopy allows the detection of different ligand-DNA binding modes. Angew Chem Int Ed Engl. 2005;45:224–228. doi: 10.1002/anie.200501344. [DOI] [PubMed] [Google Scholar]
- 10.Angulo J, et al. Ligand-receptor binding affinities from saturation transfer difference (STD) NMR spectroscopy: the binding isotherm of STD initial growth rates. Chemistry. 2010;16:7803–7812. doi: 10.1002/chem.200903528. [DOI] [PubMed] [Google Scholar]
- 11.Feher K, et al. Competition saturation transfer difference experiments improved with isotope editing and filtering schemes in NMR-based screening. J Am Chem Soc. 2008;130:17148–17153. doi: 10.1021/ja804468k. [DOI] [PubMed] [Google Scholar]
- 12.Assadi-Porter FM, et al. Key amino acid residues involved in multi-point binding interactions between brazzein, a sweet protein, and the T1R2-T1R3 human sweet receptor. J Mol Biol. 2010;398:584–599. doi: 10.1016/j.jmb.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowry OH, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 14.Cui M, et al. The heterodimeric sweet taste receptor has multiple potential ligand binding sites. Curr Pharm Des. 2006;12:4591–4600. doi: 10.2174/138161206779010350. [DOI] [PubMed] [Google Scholar]
- 15.Jiang P, et al. Lactisole interacts with the transmembrane domains of human T1R3 to inhibit sweet taste. J Biol Chem. 2005;280:15238–15246. doi: 10.1074/jbc.M414287200. [DOI] [PubMed] [Google Scholar]
- 16.Jiang P, et al. The cysteine-rich region of T1R3 determines responses to intensely sweet proteins. J Biol Chem. 2004;279:45068–45075. doi: 10.1074/jbc.M406779200. [DOI] [PubMed] [Google Scholar]