

Abstract
The cAMP-signaling pathway has been under intensive investigation for decades. It is a wonder that such a small simple molecule like cAMP can modulate a vast number of diverse processes in different types of cells. The ubiquitous involvement of cAMP-signaling in a variety of cellular events requires tight spatial and temporal control of its generation, propagation, compartmentalization, and elimination. Among the various steps of the cAMP-signaling pathway, G-protein coupled receptors, adenylate cyclases, phosphodiesterases, the two major cAMP targets, i.e. protein kinase A and exchange protein activated by cAMP, as well as the A-kinase anchoring proteins, are potential targets for drug development. Herein we review the recent progress on the regulation and manipulation of different steps of the cAMP-signaling pathway. We end by focusing on the emerging role of cAMP-signaling in modulating protein degradation via the ubiquitin/proteasome pathway. New discoveries on the regulation of the ubiquitin/proteasome pathway by cAMP-signaling support the development of new therapeutic approaches to prevent proteotoxicity in chronic neurodegenerative disorders and other human disease conditions associated with impaired protein turnover by the ubiquitin/proteasome pathway and the accumulation of ubiquitin-protein aggregates.
Keywords: Ubiquitin/proteasome pathway, cAMP, Therapy, Protein Aggregation
Intracellular regulation of cAMP-signaling (Fig. 1, blue boxes)
Figure 1.
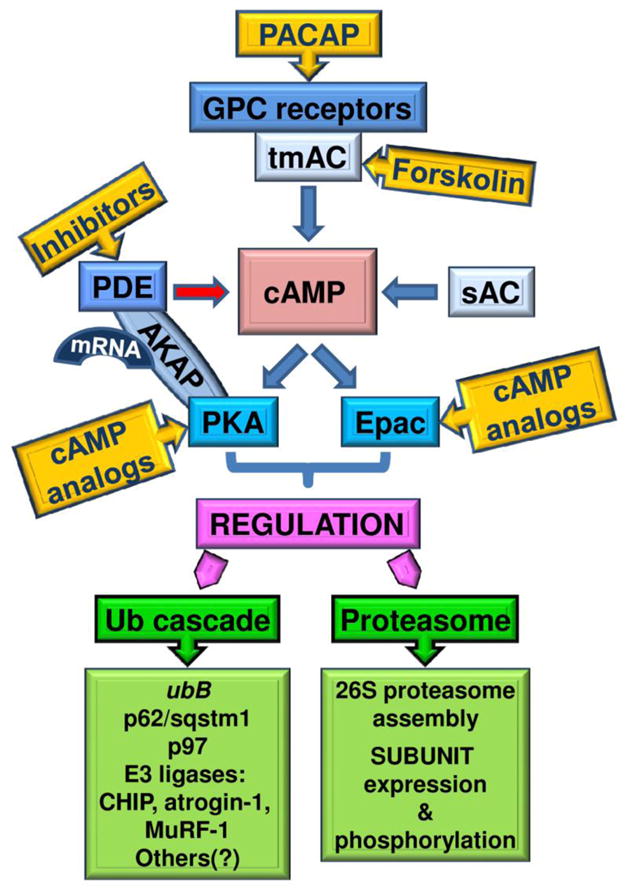
cAMP is a universal second messenger. cAMP is generated by transmembrane adenylate cyclases (tmACs) linked to and activated by G-protein coupled (GPC) receptors. These tmACs stimulate cAMP-signaling in response to extracellular factors. The soluble AC (sAC) is modulated by calcium and bicarbonate, and mediates cAMP-signaling in response to intracellular signals. Localized cAMP synthesis by the different ACs as well as the rapid hydrolysis of cAMP by phosphodiesterases (PDEs) support the cAMP microdomain model of action. Accordingly, cAMP triggers downstream events in a discrete localized manner, via its two targets: cAMP-dependent protein kinases (PKAs) and Exchange proteins activated by cAMP (Epac). Specific A-kinase anchoring proteins (AKAPs) form complex “transduceosomes” by reversibly binding GPC receptors, ACs, PDEs, PKAs, Epacs, and other molecules, such as mRNAs of several nuclear-encoded mitochondrial proteins. AKAPs provide for efficient and regulated spatiotemporal subcellular compartmentalization of cAMP signaling. Targeting the various steps within the cAMP-signaling pathway (blue boxes) with specific drugs (yellow boxes), will provide a means for regulating protein turnover by the UPP (green boxes), by manipulating levels and phosphorylation of its components, such as the ubB gene, shuttling factors (p62/sqstm1 and p97), E3 ligases (CHIP, atrogin-1, MuRF-1), and subunits of the 26S proteasome. Development of new, more specific and efficient agents to modulate cAMP-signaling will have a critical beneficial impact on human disease conditions linked to deregulated protein degradation by the UPP.
As an intracellular second messenger that can induce a wide variety of downstream effects, cAMP is under tight control in living cells. The cyclic nucleotide is generated by adenylate cyclases (ACs) and hydrolyzed by phosphodiesterases (PDEs).
1. Initiation of cAMP-signaling
Research four decades ago by Nobel Prize Laureate Paul Greengard revealed that in the caudate nucleus of rat brain, dopamine stimulated the cAMP-dependent signaling pathway via activation of the enzyme adenylate cyclase (AC) (1). These findings opened the door to new explorations focusing on the important role of cAMP in the nervous system (2).
cAMP is generated by two families of ACs, both of which belong to the class III AC superfamily (AC; E.C. 4.6.1.1). One family includes nine transmembrane ACs (tmACs AC1 to AC9) that are linked to and are activated by G-protein coupled receptors (GPCRs). These transmembrane ACs stimulate cAMP-signaling in response to extracellular factors. The second family comprises the soluble AC (sAC, AC10), which is modulated by calcium and bicarbonate, and is distributed throughout the cytoplasm and within cellular organelles (3, 4). In the human genome, there seems to be only one gene encoding for sAC, but it undergoes extensive alternative splicing (5). First discovered in the cytoplasm of rat testis (6), the N-terminus of the full length mammalian sAC (sACfl) contains two heterologous catalytic domains (C1 and C2), whereas the C-terminus exhibits several putative regulatory domains (5). The truncated form of sAC comprising C1 and C2, displays stronger activity than its full length (5). Discovery of the sAC supports a cAMP microdomain model, in which it is proposed that cAMP produced by so many different ACs, triggers downstream events locally and then cAMP is rapidly degraded by PDEs (discussed in the next section) to prevent cAMP from diffusing to other subcellular locations (5).
Different types of ACs have recently become attractive therapeutic targets for drug development (4). To explore new treatment approaches, most studies focus on tissue distribution of different ACs, particularly in the nervous system. High expression of AC1 and AC8 was detected in the brain, while AC5 and AC6 are most abundant in spinal cord (7). The phenotype of knockout mice, as well as the availability of specific inhibitors and activators for each type of ACs, are well documented in a recent review (7). Out of the nine tmACs, AC1 and AC8 have been intensively studied in neurons as they are the major Ca2+-stimulated ACs (8). AC1 and AC8 double knockout mice display defective long-term potentiation (LTP), suggesting a synergy between intracellular Ca2+ and cAMP-signaling in LTP and synaptic plasticity, and therefore in memory formation (8). Moreover, the sAC is also involved in calcium-dependent activation of the cAMP/PKA cascade in neurons (9).
The discovery of mitochondrial PKA in various rat tissues (10) was puzzling since it was not understood how cAMP molecules could travel that far into the cell, escape hydrolysis by PDEs, and get into mitochondria, which have cAMP impermeable membranes. This puzzle was solved with the discovery of a functional sAC in mitochondria (11), the existence of which was proposed ten years prior to its discovery (12). An interesting CO2-HCO3−-sAC-cAMP-PKA (mito-sAC) signaling cascade was recently proposed (11). In this cascade, bicarbonate anions synthesized by carbonic anhydrase from carbon dioxide generated in the tricarboxylic acid cycle (TCA cycle/Krebs cycle), activate the intramitochondrial sAC to produce cAMP within the mitochondrial matrix. In turn, this cAMP activates mitochondrial PKA, which then phosphorylates components of the electron transport chain therefore modulating oxidative phosphorylation to meet the requirements for intracellular metabolic homeostasis (11). The sensitivity of sAC to bicarbonate anions and calcium cations supports a role for sAC in the regulation of cellular metabolism and apoptosis (13). A recent in vitro study with adult rat cardiomyocytes subjected to simulated ischemic/reperfusion conditions, demonstrated that sAC is activated by cytosolic Ca2+ overload (14). The resulting rise in cAMP led to PKA-dependent phosphorylation of Bax at Thr167 and then to a series of downstream events culminating in activation of the mitochondrial apoptotic pathway (14). Regulation of sAC by bicarbonate was also investigated in the carotid body, which monitors pH/CO2 levels via peripheral arterial chemoreceptors (15). It was found that bicarbonate activates the sAC and up-regulates its mRNA levels in a concentration-dependent manner (15).
The compartmentalization of cAMP-signaling achieved by the complex organization and regulation of ACs at different levels, including oligomerization, lipid raft interactions, and formation of a variety of protein complexes, confers specificity to the cAMP downstream events (16). The availability of so many different ACs, considered to be promising therapeutic targets, makes cAMP a universally utilized signaling molecule (4). In addition, the existence of cAMP subcellular microdomains ensures the specificity of cAMP-signaling, albeit its widespread involvement in multiple regulatory pathways (5).
2. Termination of cAMP-signaling in cells
cAMP is hydrolyzed into adenosine monophosphate (AMP) by 3′,5′-cyclic-nucleotide phosphodiesterases (PDEs) (EC 3.1.4.17), a superfamily of 11 families encoded in mammalian cells by 21 genes (17). Decades of intensive research and the development of numerous PDE inhibitors created a multi-billion-dollar successful industry, due to the extensive tissue distribution and diverse functions of the PDEs (17–20). Moreover, coffee, cocoa and tea, which have been consumed for thousands of years, contain many effective PDE inhibitors including caffeine, the first ever known PDE inhibitor and one of the major active ingredients in these three products (17–20).
All mammalian PDEs share a conserved catalytic domain (C domain) in spite of the highly variable amino acid sequence outside of this particular region (17). A number of different types of PDEs are located in the nervous system, and each neuron usually has more than one type of PDE (18). Some neuronal PDEs have been linked to the etiology of certain neurological and psychiatric diseases, and because of their considerably distinct regulatory regions outside the C domain, PDEs are considered highly specific therapeutic targets (18).
cAMP downstream targets (Fig. 1, blue boxes)
Research by Nobel Prize Laureate Earl W. Sutherland Jr. on the mechanism by which epinephrine exerts its action, introduced the breakthrough concept of how environmental stimuli, via the second messenger cAMP, trigger intracellular signaling pathways that significantly affect diverse downstream phenomena. As a second messenger in the cell signaling transduction system, cAMP plays numerous roles in cells. Since its discovery more than half a century ago, most attention has been paid to the two major targets of cAMP, protein kinase A (PKA) (21) and Exchange protein activated by cAMP (Epac) (22, 23).
1. Protein Kinase A (PKA)
The holoenzyme of cAMP-dependent protein kinase (EC 2.7.11.11), also known as protein kinase A (PKA), is composed of two catalytic subunits (C) and two regulatory subunits (R) which form a tetramer (R2C2) (24). The activity of the catalytic subunits requires phosphorylation at threonine 197 (T197) in the activation loop, by a phosphoinositide-dependent protein kinase (25, 26). Besides T197 phosphorylation, the activity of PKA is regulated by disassociation or association of the regulatory and catalytic subunits. Without cAMP, PKA is an inactive tetrameric holoenzyme. The binding of cAMP to the regulatory subunits induces conformational changes that lead to their dissociation from the catalytic subunits, resulting in a regulatory subunit dimer with four cAMP molecules, R2cAMP4 and two active catalytic monomers (27).
The importance of PKA in so many intracellular processes increased the interest in developing PKA inhibitors to facilitate the study of its downstream mechanisms. The H-series of protein kinase inhibitors were developed by Hidaka and colleagues as early as 1977 (28). One of the most widely used PKA inhibitor is H89, but it was found to inhibit at least 8 other kinases (MAPKAP-K1b, MSK1, KBα, SGK, S6K1, ROCK II, AMPK, and CHK1) (29). Currently, there are other PKA inhibitors that appear to be more specific than H89, such as Rp-cAMPS and its analogs, as well as synthetic analogs of the endogenous PKA inhibitor (PKI) (30).
The active catalytic subunits of PKA phosphorylate serine and threonine residues on specific substrates located in different subcellular compartments, including the cytoplasm and the nucleus (31, 32). One particular family of PKA substrates, the cAMP responsive element binding protein (CREB) family of transcription factors, regulates the expression of genes with promoter regions that contain the cAMP responsive element (CRE). CREB is phosphorylated by PKA at serine 133 in the P-box, or kinase-inducible domain (KID) (33).
2. Exchange protein activated by cAMP (Epac)
The discovery of the Exchange protein activated by cAMP (Epac) presented an alternative target for cAMP. Initially described for activation of Rap1 by forskolin and cAMP, a phenomenon found to be PKA-independent, Epac provided for a mediator of many phenomena that could not be attributed to PKA (22). Two independent genes were isolated by screening clones for cAMP motifs: cAMP-GEFI and cAMP-GEFII, also known as Epac1 and Epac2, respectively (34). Both of these genes were also characterized by the presence of Ras superfamily guanine nucleotide exchange factor (GEF) domains (34). The major function of Epacs is to serve as guanine nucleotide exchange factors (GEFs) for both Rap1 and Rap2 (35). In these Rap proteins, the switch from the inactive guanosine diphosphate (GDP)-bound state to the active guanosine triphosphate (GTP)-bound state is catalyzed by the two GEF proteins, which turn on the signal pathway by exchanging GDP to GTP (36). On the other hand GAPs (GTPase-activating proteins) deactivate the small G proteins by promoting hydrolysis of GTP to GDP (36).
The regulatory region of the Epac proteins has two different types of domains: (a) cyclic nucleotide– binding (CNB) domain [one for Epac1, two (CNB-A and CNB-B) for Epac 2], and (b) Disheveled, Egl-10, and Pleckstrin (DEP) domain (37). The catalytic region is composed of three different types of domains: (a) CDC25-homology (CDC25-HD) domain containing the active site, (b) the Ras exchange motif (REM) domain, and (c) a Ras-association (RA) domain in between (37). The structure of Epac2 in complex with the cAMP analogue Sp-cAMPS and with RAP1B was solved by X-ray crystallography and single particle electron microscopy, revealing the mechanism by which Epac is activated by cAMP (38, 39). Briefly, without cAMP, Epac2 remains in an auto-inhibited state in which the CNB-B domain prevents Rap from accessing the active site. Upon cAMP binding, its interaction with the CNB-B and REM domains releases the auto-inhibition and renders the active site in the CDC25-HD domain available to Rap.
Compartmentalization of cAMP-signaling within cells (Fig. 1, blue box)
The tight spatial and temporal control of cAMP-signaling can be partially accomplished by the function of A-kinase anchoring proteins (AKAPs) also known as “transduceosomes” (40). These are a group of heterogeneous scaffold proteins sharing the same PKA-binding domain (41), although they have none or minor similarity in their primary sequences (42). The relative conservation of the PKA binding site on AKAPs, facilitates their interaction with the PKA regulatory subunits. Conversely, the wide variation among most AKAPs provides for the remarkable versatility of their association with different partners, including Epacs, to form multiprotein complexes that support their involvement in diverse processes (43, 44).
There are three ways by which AKAPs can regulate cAMP-signaling: binding to R subunits of the PKA holoenzyme, delivering PKA to the target subcellular locations, and assembling PKA with its downstream targets (45). All of these processes greatly increase the accuracy and efficiency of modulating cAMP-signaling, thus making AKAPs attractive targets for new drug development (46, 47). A recently established quantitative chemical proteomic approach, provided a means to characterize the interaction between specific isoforms of the PKA R subunits and AKAPs in various tissues and cells, therefore greatly facilitating the exploration and verification of PKA-R/AKAP interactions (48).
Manipulation of cAMP-signaling (Fig. 1, yellow boxes)
Different steps of the cAMP-signaling pathway can be targeted for manipulation, including receptors coupled to tmACs, ACs, cAMP analogues, and PDEs.
1. Pituitary adenylate cyclase-activating peptide (PACAP)
For more than twenty years, an impressive number of studies on the pituitary adenylate cyclase-activating peptide (PACAP) have been published, and they have been frequently and comprehensively reviewed. PACAP exists in two bioactive molecular forms, one of 38 residues (PACAP38) and a shorter form corresponding to the N-terminal 27 residues of PACAP38 (PACAP27) (49). PACAP is a member of the vasoactive intestinal polypeptide (VIP)-secretin-growth hormone-releasing hormone-glucagon superfamily (49). This superfamily of peptides binds to two types of receptors: (a) type I receptor (PAC1-R), which has a much higher affinity for PACAP than for VIP and activates both AC and phospholipase C (PLC); (b) type II receptors (VPAC1-R and VPAC2-R), which have a similar affinity for PACAP and VIP, and activate primarily AC (49). Gene expression profiles of the three different PACAP receptors by real time PCR, revealed that PAC1-R expression declines during later stages of development, while VPAC1-R and VPAC2-R expression increases from newborn to the later stages of development (50).
PACAP regulated genes involved in neuritogenesis, cell morphology modulation, and cell survival, have been screened in both PC12 cells and in vivo (51). A recent proteomics study conducted upon intracerebroventricular administration of PACAP to rats, further explored the mechanisms by which PACAP exerts its neurotrophic and neuroprotective effects in the brain (52). The results revealed that proteins with expression profiles altered by PACAP treatment are involved in many cellular processes including cytoskeleton modulation, synaptic plasticity, cellular differentiation, neuroprotection, neurodegeneration and apoptosis, supporting the notion that PACAP is a promising therapeutic peptide for treatment of multiple neural disorders (52).
The neuroprotective effects of PACAP are mediated by both the canonical cAMP/PKA pathway and the non-canonical cAMP/ERK1/2 pathway (53). These two downstream PACAP-mediated pathways seem to be activated in a concentration-dependent manner, as the effects of subpicomolar PACAP38 levels are mainly mediated by ERK type MAPK, whereas those of nanomolar PACAP38 levels result from activation of the cAMP/PKA pathway (54).
PACAP was found to have neuroprotective effects in several in vivo and in vitro models of neurodegenerative disorders including Parkinson disease (PD), Huntington disease (HD) and Alzheimer disease (AD) (55). The finding that genes encoding for PACAP are down-regulated in several mouse models of AD and temporal cortex samples from AD patients, strengthens the idea of using PACAP as a therapeutic drug applicable to neurodegenerative diseases (56). For instance, in a model of AD, long-term daily intranasal administration of PACAP to transgenic mice expressing the mutant form of the amyloid precursor protein (APP-V717I), stimulated its non-amyloidogenic processing (57). Moreover, combined administration of PACAP with mesenchymal stem cells (hMSCs), synergistically promoted the functional recovery of rats with severe spinal cord injury (SCI) (58). Notably, PACAP concentrations as low as 0.01 nM in combination with PDE inhibitors (rolipram, a PDE4-specific inhibitor, or IBMX, a non-specific PDE inhibitor), displayed significant neurotrophic effects against glutamate-induced excitotoxicity in motoneuron cultures (59). The latter results suggest that PACAP is a promising therapeutic approach for treatment of amyotrophic lateral sclerosis (ALS) (59).
Besides its neuroprotective actions in neurodegenerative diseases, the effects of PACAP on neurons have been studied in other pathological conditions. PACAP is also neuroprotective in the context of retinal excitotoxicity, the major etiology of glaucoma, by preventing ganglion cell death induced by N-methyl-D-aspartate acid (NMDA) (60). Likewise, ethanol-induced toxicity via the c-Jun and caspase-3 pathways is prevented by PACAP in rat cerebellar neurons (61). Moreover, PACAP controls the migration of cerebellar granule cells via the AC-cAMP and PLC-Ca2+ pathways (62). In PAC1-R−/− knockout mice, newly generated neurons in the granule cell layer of the cerebellum were eliminated before their differentiation, further supporting the role of PACAP/PAC1-R signaling in the development of cerebellar neurons (63). A comparison between wild type and PACAP knockout mice when subjected to an enriched environment, revealed an important role for endogenous PACAP in adult hippocampal neurogenesis in the dentate gyrus (64). The effects of PACAP on neural stem/progenitor cells (NPC) have also been characterized (65). PACAP significantly improved survival of postnatal mouse NPCs and decreased NPC differentiation, suggesting that PACAP may function to maintain the multipotent state of NPCs, a process that is important in self regeneration in vivo (65).
In addition to neurons, PACAP affects other cells in the nervous system. For example, PACAP prevents astrocytes from H2O2-induced oxidative stress, via PKA, PKC, and mitogen-activated protein (MAP)-kinase kinase (MEK) pathways (66). PAC1-R and VPAC2-R receptors as well as the PACAP precursor peptide were detected in a rat schwannoma cell line (CRL-2768); upon serum deprivation, treatment with PACAP and VIP prevented apoptosis in these cells (67).
Truncated forms of PACAP were developed to help understand the sequence- and conformation-requirements of its biological activity. These shorter PACAP based peptides may increase its therapeutic efficiency, not only because they are easier to synthesize and to deliver in vivo, but also because they are likely to exhibit higher stability. Notably, lipopolysaccharide-induced neurotoxicity in primary rat mesencephalic neuron-glia cultures, was prevented by a short form of PACAP containing only three amino acids [Gly-Ile-Phe, GIF or PACAP4–6), administered at an incredibly low concentration (10−13 M) (68). Failure to elevate cAMP and the inability of a PAC1-R antagonist (PACAP6-38) to abolish its neuroprotective efficacy, suggest that these GIF effects are mediated by an alternate target, such as inhibition of NADPH oxidase (68). To overcome its instability, a more stable PACAP analog was synthesized: acetyl-[Ala15, Ala20]PACAP38-propylamide (69). In a rat stroke model involving middle cerebral artery occlusion, the latter PACAP analog displayed neuroprotective effects similar to PACAP, but at remarkably lower (picomolar) levels (69).
The neuroprotective effects of a variety of PAC1-R/VPAC1-R-selective agonists were evaluated, revealing that His1, Asp3 and Phe6 are critical for activation of the PAC1-R receptor (70). This is consistent with a structure-activity relationships (SAR) study showing that Asp3 and Phe6 are primary components of the PAC1-R pharmacophore, and that the N-terminal domain (His1-Ser2-Asp3-Gly4) is crucial for PAC1-R activation (71). Using more than ten different short peptides based on the PACAP sequence, other studies addressed some of the PAC1-R activation requirements (72). It was established that several domains in PAC1-R are involved in the interactions of PACAP27 with PAC1-R (72). Furthermore, the peptide α-helical structure was not enough to activate PAC1-R, suggesting that the development of small peptidomimetic analogs exhibiting full capacity is going to be challenging (72).
2. Forskolin
More than two decades ago the natural plant product forskolin was identified as a general activator of adenylate cyclase (73). Forskolin is widely used as a standard means to increase cAMP levels to study its signaling pathways, and to test if cAMP-PKA is involved in poorly characterized mechanisms (74). Here we review a few emerging studies focusing on the neuroprotective effects of forskolin. In PC12 cells, forskolin induces neurite outgrowth via Nur77, an orphan nuclear receptor and immediate-early response gene, and protects these cells against L-3,4-dihydroxyphenylalanine (L-DOPA) toxicity (75). Activation of cAMP-PKA signaling by forskolin triggered synaptic vesicle exocytosis and recycling in NT2 cells (76). Similar to cell permeable cAMP analogs, forskolin induced neuronal differentiation of embryonal carcinoma stem cells Ntera2 (NT2), confirmed by neurite formation, extension and elaboration (77). These results underscore the role of cAMP-PKA signaling in neurite outgrowth and neuro-regeneration (77). Notably, elevating cAMP levels has been used as a successful strategy to overcome the inhibitory effect of myelin, which is one of the challenges in axonal regeneration following spinal cord injury (78).
3. cAMP analogs
For decades many cAMP analogs have been developed to explore and elucidate the functions of cAMP as a universal second messenger that regulates numerous pathways in living cells. Efforts to develop cAMP analogs date back to the 1960s, even prior to the characterization of PKA (79). Hundreds of cAMP derivatives have since been developed. The primary reasons for developing different cAMP analogs are to improve cell permeability, metabolic stability, and specificity for both activation and inhibition of downstream targets (80, 81). Structural and functional analyses provided the basis for developing cAMP analogs. In brief, the 3′ and 5′ positions in the cyclophosphate ring of cAMP are crucial for PKA affinity and therefore provide no room for modifications (82). Substitution of a halogen at position 8 of cAMP is a common strategy to render cAMP analogs resistance to PDE hydrolysis, and to increase cell permeability if the substitution groups are hydrophobic (81). Dibutyryl and 8-bromo analogs, namely, N6, O2′-dibutyryl-cAMP (Bt2-cAMP) and 8-bromo-cAMP (8-Br-cAMP) are used as standard tools for studying signal transduction and therapeutics. Their lipophilicity or hydrophobicity increase cell permeability which is slightly higher for Bt2-cAMP than for 8-Br-cAMP (83). Concerns were raised in regard to Bt2-cAMP due to a number of bioactivities associated with its metabolite butyrate, including PKC stimulation, pro-apoptotic effects, and induction of the expression of many genes (80). Therefore, control experiments with butyrate should be included when using Bt2-cAMP (80). Development of two series of phosphorothioate derivatives of cAMP, i.e. Sp- and Rp-cAMPS, which are cAMP agonists/activators and antagonists/inhibitors respectively, facilitated the purification of cAMP binding proteins including PKA, thus providing a means for their detailed biochemical characterization (84).
Identification of the new cAMP target Epac led to the development of specific Epac activators, so that the different pathways by which cAMP exerts its downstream effects can be distinguished. The availability of various cAMP analogs that selectively and specifically activate and therefore differentiate the PKA and Epac pathways, greatly improved the research on cAMP-mediated pathways (37, 85). Epac-selective analogs were obtained by replacing the 2′OH of the cAMP ribose group with 2′O-Me (86). This strategy was based on Epac lacking the glutamate that interacts with the 2′OH group of cAMP in PKA(86). Based on these findings, one of the most universally used Epac specific activator is 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8CPT-2Me-cAMP) (86). The latter cAMP analog exhibits a modification at the 2′ hydroxyl group on the ribose ring of cAMP, which is a critical site for PKA interaction and determines the selective affinity of cAMP for PKA (87). 8CPT-2Me-cAMP is considered to be the “super Epac activator” and can be found in virtually every single paper studying Epac after its discovery (88). The choice of using 8CPT-2Me-cAMP is based on the 4.6-fold increase in its affinity for Epac1 when compared to cAMP, as well as its 107-fold selectivity for Epac1 versus PKA, and its enhanced cell permeability (88). Epac activators dissimilar in structure to cAMP, were identified by high-throughput screening based on their ability to displace [3H]cAMP from the cAMP-binding site on Epac (89). Functional tests are still required prior to wide application of this new series of compounds that are not structurally related to cAMP (89).
A new approach based on fluorescence resonance energy transfer (FRET) involves the expression of a cAMP sensor that contains the cAMP binding site from human Epac1 (90). This newly synthesized cAMP sensor changes conformation upon cAMP binding, leading to alterations in detectable FRET signals that can be modulated by AC inhibitors and cAMP analogs. This approach allows for precise measurements of intracellular cAMP concentrations, and accurate evaluation of cell permeability of different cAMP analogs suitable for various cell types (90).
Currently there are no optimal specific inhibitors/antagonists for Epac (81, 91, 92). Development of Epac selective antagonists has been difficult due to the lack of high resolution structural information on cAMP bound to both Epac isoforms (88). This difficulty has delayed characterization of Epac signaling pathways (88). Moreover, the cAMP derivative Rp-cAMPS was found to also inhibit Epac, thus diminishing its value as a selective PKA inhibitor (87). Brefeldin-A, which inhibits GDP–GTP exchange on ADP-ribosylation factors (ARF), as well as the Rap-1 antagonist geranylgeranyltransferase inhibitor (GGTI) are used to block Epac signaling (93). These two drugs abolish the 8CPT-2Me-cAMP effects without directly affecting the Epac protein itself (93). Development of new Epac selective antagonists will facilitate research on Epac signaling pathways, and avoid attributing too many processes arbitrarily and unwarrantedly to Epac isoforms. Alternate strategies to investigate Epac signaling pathways include siRNA to suppress Epac expression, or mutational approaches (91).
4. PDE inhibitors
PDE inhibitors can function as neuroprotective drugs that exert their effects by elevating cAMP. Various comprehensive reviews were published recently on PDE inhibitors (17–19), but new studies are constantly emerging at a fast pace. For example, S14 (Phenyl-2-thioxo-(1H)-quinazolin-4-one), a blood brain barrier permeable PDE7 inhibitor, was shown to be neuroprotective against different insults in dopaminergic neuronal cultures (SH-SY5Y cells and primary rat mesencephalic cultures), as well as in a lipopolysaccharide-induced rat model of Parkinson disease (94). Moderate doses of rolipram, a PDE4 inhibitor, mitigated dopamine depletion in the striatum and loss of tyrosine hydroxylase-positive neurons in the substantia nigra, induced by1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mice (95). Following ischemia in mice, rolipram promoted survival of newborn neurons in the adult mouse hippocampal dentate gyrus via activation of cAMP-CREB signaling (96). Sildenafil, a PDE5 inhibitor, is considered to have therapeutic potential for treating selective neurological conditions including stroke, dementia, and neurodegenerative disorders as well as to improve learning (97).
The etiology of Huntington disease (HD) is closely associated with downregulation of CREB-modulated genes, thus PDE inhibitors have been investigated as therapeutic drugs to ameliorate the symptoms of HD (98–100). PDE4 was suggested to be one of the most critical PDEs in the brain (101). The PDE4 inhibitor rolipram was shown to ameliorate several neuropathological aspects in the quinolinic acid (QA)-induced rat model of HD (102), and in the R6/2 mouse model of HD (103, 104). Neuroprotection by rolipram, including reduction of striatal neuronal loss and improvement of motor performance, was exerted by preventing sequestration of the CREB binding protein into striatal neuronal intranuclear inclusions (NIIs) comprised of polyQ aggregates. The specific PDE10A inhibitor, TP-10 also displayed neuroprotective effects in the R6/2 mouse model of HD, by significantly reducing lesion size and elevating CREB activation in the striatal spiny neurons (105). Moreover, TP-10 tempered behavior deficits, and reduced striatal and cortical neuronal loss, NIIs formation in the striatal region, as well as microglial activation (105). Finally, TP-10 increased pCREB and BDNF levels in the striatum and cortex of R6/2 mice (105). Overall, these studies suggest that PDE4 and PDE10A inhibition by rolipram and TP-10, respectively, is a promising therapeutic approach to treat HD.
Regulation of the ubiquitin/proteasome pathway via cAMP-signaling (Fig. 1, green boxes)
There is a rising interest on the ubiquitin/proteasome pathway (UPP) as a pharmacological target to prevent/treat chronic neurodegenerative diseases, such as AD, PD, HD and ALS (106, 107), as well as cardiovascular conditions, such as hypertrophic and dilated cardiomyopathies, and ischemic heart disease (108). All of these conditions are characterized by intracellular ubiquitin-protein aggregates in the affected neurons or in the heart (106, 108). To achieve such a goal several steps of the UPP can be therapeutically targeted, including the ubiquitination cascade and/or protein degradation by the proteasome.
Recent studies focused on manipulating the UPP via cAMP-signaling, particularly in neurons and in heart. For example, in dopaminergic-like PC12 cells, the cAMP analog 8-(4-Chlorophenylthio)-cAMP or CPT-cAMP, prevented apoptosis and the intense diffuse ubiquitin staining induced by the proteasome inhibitors lactacystin or ZIE[O-t Bu]-A-leucinal (PSI) (109). Moreover, proteasome inhibition in rat cerebral cortical neurons caused the early accumulation of detergent-soluble ubiquitinated proteins, leading to caspase activation and tau pathology (110). These events were mitigated by the cAMP analog Bt2-cAMP, which was shown to stimulate proteasome activity (110). Similar protection by Bt2-cAMP against proteasome inhibition was observed in rat spinal cord neuronal cultures (111). In the latter study, Bt2-cAMP increased 26S proteasome activity and also raised the levels of various components of the UPP, including proteasome subunits Rpt6 and β5, polyubiquitin shuttling factor p62/sequestosome1 (p62/sqstm1), E3 ligase CHIP, AAA-ATPase p97 and the ubiquitin gene ubB (111). Interestingly, the scaffold protein p62/sqstm1 reversibly sequesters the phosphodiesterase PDE4A4, thus providing a means to compartmentalize cAMP-signaling within particular intracellular microdomains (112).
In mice, isoproterenol-induced cardiac hypertrophy is associated with inhibition of the 20S proteasome (113). Activation of endogenous PKA with cAMP prevented 20S proteasome inhibition under the latter condition (113). The remarkable in vivo protective effects of cAMP-PKA signaling on the proteasome were confirmed with canine hearts (114). This study demonstrated that exogenous or endogenous PKA stimulation rapidly enhanced 26S proteasome assembly and its activity, without causing changes in proteasome subunit levels (114). Structural and functional associations of PKA and protein phosphatase 2A (PP2A) with the 20S proteasome were revealed by a combination of different methods using the murine heart (115). The results from the latter study further confirmed that phosphorylation of the 20S proteasome elevates its three peptidase activities in a substrate-specific manner (115). Global characterization of the 20S proteasome phosphoproteome was carried out by combined analytical approaches in murine cardiac and hepatic tissues (116). Identification of at least 52 target sites in the 20S proteasome for PKA-mediated phosphorylation, provides guidance for investigating mechanisms by which PKA regulates proteasome activity (116). On a different note, cAMP-signaling protected pancreatic beta-cells from high glucose toxicity, by promoting proteasome-dependent degradation of the thioredoxin interacting protein (TxNIP) (117).
In conclusion, to meet the various cellular requirements, proteasome genes are subjected to tight regulation that includes both selective and concerted transcriptional induction (118) as well as post-translational modifications (119). Notably, PKA-mediated phosphorylation of proteasome subunits in its core (120, 121) and regulatory particles (122), is postulated to enhance assembly of the 26S proteasome (119).
Besides the proteasome, other components of the UPP are regulated by cAMP-signaling. As such, in vitro and in vivo elevation of cAMP in rodent skeletal muscle decreased proteasome activity, ubiquitin-protein conjugates, and the levels of atrogin-1, an E3 ubiquitin-ligase that plays a role in muscle atrophy (123). Furthermore, inhibiting PDE4 with rolipram, downregulated proteasome activity and suppressed up-regulation of the E3 ubiquitin-ligases atrogin-1 and MuRF-1, induced in rat skeletal muscle by fasting (124). These results support activating cAMP-PKA signaling to reduce proteasome activity as well as the levels of the two aforementioned E3 ubiquitin-ligases, as a potential therapeutic approach to treat skeletal muscle atrophy (124).
The A-kinase anchor protein AKAP121 complex selectively enhances the propagation of cAMP-signaling from the cell membrane to mitochondria (125). AKAP121 binds to PKA, to the phosphatase PDE4A and to several mRNAs of nuclear-encoded mitochondrial proteins (125). Upon ischemic injury, the levels of the RING domain E3 ubiquitin-ligase SIAH2 are elevated leading to accelerated degradation of AKAP121 in a proteasome-dependent manner (125). AKAP121 degradation triggers the downregulation of mitochondrial activity so that cells can adapt to low oxygen levels under hypoxic conditions (125). Therefore, the cross-talk between the UPP and PKA via AKAP121, provides a means to regulate mitochondrial dynamics and activity, so that cells can adapt to changes in oxidative metabolism (125).
Conclusions
The complexity of the UPP pathway is matched by the diversity of its regulatory mechanisms. Understanding the intricate connections between cAMP-signaling and the UPP is essential to identify relevant players and molecular mechanisms governing the function of this major intracellular proteolytic pathway, and provide the necessary knowledge to selectively overcome the challenges of protein turnover in human disease. The cAMP-signaling pathway can be manipulated at different levels as depicted in Fig. 1 and described throughout this review. The challenge rests on developing strategies that enhance degradation of misfolded and aggregation-prone proteins without compromising the normal function of the UPP. In a recent review (126), drugs that modulate specific UPP components were characterize as potential therapeutic agents. Herein we review how targeting cAMP-signaling to maintain UPP function in a sustainable manner offers an effective therapeutic approach against UPP-related proteotoxicity in chronic neurodegenerative and cardiovascular conditions associated with UPP dysfunction and the accumulation of ubiquitin-protein aggregates.
Acknowledgments
Please note that this review is not intended to be comprehensive and we apologize to the authors whose work is not mentioned. Supported by National Institutes of Health (NIH) [NINDS-NS041073 (Specialized Neuroscience Research Programs) to M.F.-P. (head of subproject) from National Institute of Neurological Disorders and Stroke; NCRR-RR003037 to Hunter College (infrastructure) from National Institute of General Medical Sciences (NIGMS)/RCMI (Research Centers in Minority Institutions)].
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Reference List
- 1.Kebabian JW, Petzold GL, Greengard P. Dopamine-sensitive adenylate cyclase in caudate nucleus of rat brain, and its similarity to the “dopamine receptor”. Proc Natl Acad Sci U S A. 1972;69:2145–2149. doi: 10.1073/pnas.69.8.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravindran S. Paul Greengard: Signals underlying moods, addictions, and brain disorders. Proc Natl Acad Sci U S A. 2011;108:18872–18874. doi: 10.1073/pnas.1116714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol. 2006;362:623–639. doi: 10.1016/j.jmb.2006.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavan B, Biondi C, Dalpiaz A. Adenylyl cyclases as innovative therapeutic goals. Drug Discov Today. 2009;14:982–991. doi: 10.1016/j.drudis.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Tresguerres M, Levin LR, Buck J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011;79:1277–1288. doi: 10.1038/ki.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braun T, Dods RF. Development of a Mn-2+-sensitive, “soluble” adenylate cyclase in rat testis. Proc Natl Acad Sci U S A. 1975;72:1097–1101. doi: 10.1073/pnas.72.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierre S, Eschenhagen T, Geisslinger G, Scholich K. Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov. 2009;8:321–335. doi: 10.1038/nrd2827. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Storm DR, Wang H. Bidirectional synaptic plasticity and spatial memory flexibility require Ca2+-stimulated adenylyl cyclases. J Neurosci. 2011;31:10174–10183. doi: 10.1523/JNEUROSCI.0009-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn TA, Storm DR, Feller MB. Calcium-dependent increases in protein kinase-A activity in mouse retinal ganglion cells are mediated by multiple adenylate cyclases. PLoS ONE. 2009;4:e7877. doi: 10.1371/journal.pone.0007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwoch G, Trinczek B, Bode C. Localization of catalytic and regulatory subunits of cyclic AMP-dependent protein kinases in mitochondria from various rat tissues. Biochem J. 1990;270:181–188. doi: 10.1042/bj2700181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009;9:265–276. doi: 10.1016/j.cmet.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papa S, Sardanelli AM, Scacco S, Technikova-Dobrova Z. cAMP-dependent protein kinase and phosphoproteins in mammalian mitochondria. An extension of the cAMP-mediated intracellular signal transduction. FEBS Lett. 1999;444:245–249. doi: 10.1016/s0014-5793(99)00070-8. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Levin LR, Buck J. Role of soluble adenylyl cyclase in the heart. Am J Physiol Heart Circ Physiol. 2012;302:H538–H543. doi: 10.1152/ajpheart.00701.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Appukuttan A, Kasseckert SA, Micoogullari M, Flacke JP, Kumar S, Woste A, Abdallah Y, Pott L, Reusch HP, Ladilov Y. Type 10 adenylyl cyclase mediates mitochondrial Bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc Res. 2012;93:340–349. doi: 10.1093/cvr/cvr306. [DOI] [PubMed] [Google Scholar]
- 15.Nunes AR, Monteiro EC, Johnson SM, Gauda EB. Bicarbonate-regulated soluble adenylyl cyclase (sAC) mRNA expression and activity in peripheral chemoreceptors. Adv Exp Med Biol. 2009;648:235–241. doi: 10.1007/978-90-481-2259-2_27. [DOI] [PubMed] [Google Scholar]
- 16.Cooper DM, Crossthwaite AJ. Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol Sci. 2006;27:426–431. doi: 10.1016/j.tips.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev. 2011;91:651–690. doi: 10.1152/physrev.00030.2010. [DOI] [PubMed] [Google Scholar]
- 18.Hebb AL, Robertson HA. Role of phosphodiesterases in neurological and psychiatric disease. Curr Opin Pharmacol. 2007;7:86–92. doi: 10.1016/j.coph.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Schudt C, Hatzelmann A, Beume R, Tenor H. Phosphodiesterase inhibitors: history of pharmacology. Handb Exp Pharmacol. 2011:1–46. doi: 10.1007/978-3-642-17969-3_1. [DOI] [PubMed] [Google Scholar]
- 20.Corbin JD, Francis SH. Molecular biology and pharmacology of PDE-5-inhibitor therapy for erectile dysfunction. J Androl. 2003;24:S38–S41. doi: 10.1002/j.1939-4640.2003.tb02744.x. [DOI] [PubMed] [Google Scholar]
- 21.Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–3765. [PubMed] [Google Scholar]
- 22.Zhou M, Fisher EA, Ginsberg HN. Regulated Co-translational Ubiquitination of Apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J Biol Chem. 1998;273:24649–24653. doi: 10.1074/jbc.273.38.24649. [DOI] [PubMed] [Google Scholar]
- 23.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 24.Dwivedi Y, Pandey GN. Elucidating biological risk factors in suicide: role of protein kinase A. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:831–841. doi: 10.1016/j.pnpbp.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng X, Ma Y, Moore M, Hemmings BA, Taylor SS. Phosphorylation and activation of cAMP-dependent protein kinase by phosphoinositide-dependent protein kinase. Proc Natl Acad Sci U S A. 1998;95:9849–9854. doi: 10.1073/pnas.95.17.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cauthron RD, Carter KB, Liauw S, Steinberg RA. Physiological phosphorylation of protein kinase A at Thr-197 is by a protein kinase A kinase. Mol Cell Biol. 1998;18:1416–1423. doi: 10.1128/mcb.18.3.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 28.Hidaka H, Watanabe M, Kobayashi R. Properties and use of H-series compounds as protein kinase inhibitors. Methods Enzymol. 1991;201:328–339. doi: 10.1016/0076-6879(91)01029-2. [DOI] [PubMed] [Google Scholar]
- 29.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–274. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 31.Wolfl S, Martinez C, Majzoub JA. Inducible binding of cyclic adenosine 3′,5′-monophosphate (cAMP)-responsive element binding protein (CREB) to a cAMP-responsive promoter in vivo. Mol Endocrinol. 1999;13:659–669. doi: 10.1210/mend.13.5.0282. [DOI] [PubMed] [Google Scholar]
- 32.Scott JD, Stofko RE, McDonald JR, Comer JD, Vitalis EA, Mangili JA. Type II regulatory subunit dimerization determines the subcellular localization of the cAMP-dependent protein kinase. J Biol Chem. 1990;265:21561–21566. [PubMed] [Google Scholar]
- 33.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 34.Springett GM, Kawasaki H, Spriggs DR. Non-kinase second-messenger signaling: new pathways with new promise. Bioessays. 2004;26:730–738. doi: 10.1002/bies.20057. [DOI] [PubMed] [Google Scholar]
- 35.de RJ, Rehmann H, van TM, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- 36.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 37.Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- 38.Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- 39.Rehmann H, rias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455:124–127. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 40.Feliciello A, Gottesman ME, Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- 41.Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E. Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol. 2010;283:235–330. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- 42.Carnegie GK, Means CK, Scott JD. A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life. 2009;61:394–406. doi: 10.1002/iub.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dessauer CW. Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott JD, Dessauer CW, Tasken K. Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pidoux G, Tasken K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J Mol Endocrinol. 2010;44:271–284. doi: 10.1677/JME-10-0010. [DOI] [PubMed] [Google Scholar]
- 46.Hundsrucker C, Klussmann E. Direct AKAP-mediated protein-protein interactions as potential drug targets. Handb Exp Pharmacol. 2008:483–503. doi: 10.1007/978-3-540-72843-6_20. [DOI] [PubMed] [Google Scholar]
- 47.Troger J, Moutty MC, Skroblin P, Klussmann E. A-kinase anchoring proteins as potential drug targets. Br J Pharmacol. 2012;166:420–433. doi: 10.1111/j.1476-5381.2011.01796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kovanich D, Aye TT, Heck AJ, Scholten A. Probing the specificity of protein-protein interactions by quantitative chemical proteomics. Methods Mol Biol. 2012;803:167–181. doi: 10.1007/978-1-61779-364-6_12. [DOI] [PubMed] [Google Scholar]
- 49.Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev. 2009;61:283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- 50.Shneider Y, Shtrauss Y, Yadid G, Pinhasov A. Differential expression of PACAP receptors in postnatal rat brain. Neuropeptides. 2010;44:509–514. doi: 10.1016/j.npep.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Eiden LE, Samal B, Gerdin MJ, Mustafa T, Vaudry D, Stroth N. Discovery of pituitary adenylate cyclase-activating polypeptide-regulated genes through microarray analyses in cell culture and in vivo. Ann N Y Acad Sci. 2008;1144:6–20. doi: 10.1196/annals.1418.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gasperini L, Piubelli C, Carboni L. Proteomics of rat hypothalamus, hippocampus and pre-frontal/frontal cortex after central administration of the neuropeptide PACAP. Mol Biol Rep. 2012;39:2921–2935. doi: 10.1007/s11033-011-1054-1. [DOI] [PubMed] [Google Scholar]
- 53.Holighaus Y, Weihe E, Eiden LE. STC1 induction by PACAP is mediated through cAMP and ERK1/2 but not PKA in cultured cortical neurons. J Mol Neurosci. 2012;46:75–87. doi: 10.1007/s12031-011-9653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakamachi T, Li M, Shioda S, Arimura A. Signaling involved in pituitary adenylate cyclase-activating polypeptide-stimulated ADNP expression. Peptides. 2006;27:1859–1864. doi: 10.1016/j.peptides.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 55.Reglodi D, Kiss P, Lubics A, Tamas A. Review on the Protective Effects of PACAP in Models of Neurodegenerative Diseases In vitro and In vivo. Curr Pharm Des. 2011;17:962–972. doi: 10.2174/138161211795589355. [DOI] [PubMed] [Google Scholar]
- 56.Wu KY, Zippin JH, Huron DR, Kamenetsky M, Hengst U, Buck J, Levin LR, Jaffrey SR. Soluble adenylyl cyclase is required for netrin-1 signaling in nerve growth cones. Nat Neurosci. 2006;9:1257–1264. doi: 10.1038/nn1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rat D, Schmitt U, Tippmann F, Dewachter I, Theunis C, Wieczerzak E, Postina R, van LF, Fahrenholz F, Kojro E. Neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) slows down Alzheimer’s disease-like pathology in amyloid precursor protein-transgenic mice. FASEB J. 2011;25:3208–3218. doi: 10.1096/fj.10-180133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang KM, Chen JK, Hung SC, Chen MC, Wu YT, Wu TJ, Lin HI, Chen CH, Cheng H, Yang CS, Tzeng SF. Effects of combinatorial treatment with pituitary adenylate cyclase activating peptide and human mesenchymal stem cells on spinal cord tissue repair. PLoS ONE. 2010;5:e15299. doi: 10.1371/journal.pone.0015299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomimatsu N, Arakawa Y. Survival-promoting activity of pituitary adenylate cyclase-activating polypeptide in the presence of phosphodiesterase inhibitors on rat motoneurons in culture: cAMP-protein kinase A-mediated survival. J Neurochem. 2008;107:628–635. doi: 10.1111/j.1471-4159.2008.05638.x. [DOI] [PubMed] [Google Scholar]
- 60.Endo K, Nakamachi T, Seki T, Kagami N, Wada Y, Nakamura K, Kishimoto K, Hori M, Tsuchikawa D, Shinntani N, Hashimoto H, Baba A, Koide R, Shioda S. Neuroprotective effect of PACAP against NMDA-induced retinal damage in the mouse. J Mol Neurosci. 2011;43:22–29. doi: 10.1007/s12031-010-9434-x. [DOI] [PubMed] [Google Scholar]
- 61.Botia B, Jolivel V, Burel D, Le JV, Roy V, Naassila M, Benard M, Fournier A, Vaudry H, Vaudry D. Neuroprotective effects of PACAP against ethanol-induced toxicity in the developing rat cerebellum. Neurotox Res. 2011;19:423–434. doi: 10.1007/s12640-010-9186-y. [DOI] [PubMed] [Google Scholar]
- 62.Cameron DB, Raoult E, Galas L, Jiang Y, Lee K, Hu T, Vaudry D, Komuro H. Role of PACAP in controlling granule cell migration. Cerebellum. 2009;8:433–440. doi: 10.1007/s12311-009-0121-9. [DOI] [PubMed] [Google Scholar]
- 63.Falluel-Morel A, Tascau LI, Sokolowski K, Brabet P, Cicco-Bloom E. Granule cell survival is deficient in PAC1−/− mutant cerebellum. J Mol Neurosci. 2008;36:38–44. doi: 10.1007/s12031-008-9066-6. [DOI] [PubMed] [Google Scholar]
- 64.Ago Y, Yoneyama M, Ishihama T, Kataoka S, Kawada K, Tanaka T, Ogita K, Shintani N, Hashimoto H, Baba A, Takuma K, Matsuda T. Role of endogenous pituitary adenylate cyclase-activating polypeptide in adult hippocampal neurogenesis. Neuroscience. 2011;172:554–561. doi: 10.1016/j.neuroscience.2010.10.044. [DOI] [PubMed] [Google Scholar]
- 65.Scharf E, May V, Braas KM, Shutz KC, Mao-Draayer Y. Pituitary adenylate cyclase-activating polypeptide (PACAP) and vasoactive intestinal peptide (VIP) regulate murine neural progenitor cell survival, proliferation, and differentiation. J Mol Neurosci. 2008;36:79–88. doi: 10.1007/s12031-008-9097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masmoudi-Kouki O, Douiri S, Hamdi Y, Kaddour H, Bahdoudi S, Vaudry D, Basille M, Leprince J, Fournier A, Vaudry H, Tonon MC, Amri M. Pituitary adenylate cyclase-activating polypeptide protects astroglial cells against oxidative stress-induced apoptosis. J Neurochem. 2011;117:403–411. doi: 10.1111/j.1471-4159.2011.07185.x. [DOI] [PubMed] [Google Scholar]
- 67.Castorina A, Tiralongo A, Giunta S, Carnazza ML, Rasi G, D’Agata V. PACAP and VIP prevent apoptosis in schwannoma cells. Brain Res. 2008;1241:29–35. doi: 10.1016/j.brainres.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 68.Yang S, Yang J, Yang Z, Chen P, Fraser A, Zhang W, Pang H, Gao X, Wilson B, Hong JS, Block ML. Pituitary adenylate cyclase-activating polypeptide (PACAP) 38 and PACAP4-6 are neuroprotective through inhibition of NADPH oxidase: potent regulators of microglia-mediated oxidative stress. J Pharmacol Exp Ther. 2006;319:595–603. doi: 10.1124/jpet.106.102236. [DOI] [PubMed] [Google Scholar]
- 69.Dejda A, Seaborn T, Bourgault S, Touzani O, Fournier A, Vaudry H, Vaudry D. PACAP and a novel stable analog protect rat brain from ischemia: Insight into the mechanisms of action. Peptides. 2011;32:1207–1216. doi: 10.1016/j.peptides.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 70.Doan ND, Bourgault S, Dejda A, Letourneau M, Detheux M, Vaudry D, Vaudry H, Chatenet D, Fournier A. Design and in vitro characterization of PAC1/VPAC1-selective agonists with potent neuroprotective effects. Biochem Pharmacol. 2011;81:552–561. doi: 10.1016/j.bcp.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 71.Bourgault S, Vaudry D, Segalas-Milazzo I, Guilhaudis L, Couvineau A, Laburthe M, Vaudry H, Fournier A. Molecular and conformational determinants of pituitary adenylate cyclase-activating polypeptide (PACAP) for activation of the PAC1 receptor. J Med Chem. 2009;52:3308–3316. doi: 10.1021/jm900291j. [DOI] [PubMed] [Google Scholar]
- 72.Bourgault S, Vaudry D, Guilhaudis L, Raoult E, Couvineau A, Laburthe M, Segalas-Milazzo I, Vaudry H, Fournier A. Biological and structural analysis of truncated analogs of PACAP27. J Mol Neurosci. 2008;36:260–269. doi: 10.1007/s12031-008-9081-7. [DOI] [PubMed] [Google Scholar]
- 73.Iwatsubo K, Okumura S, Ishikawa Y. Drug therapy aimed at adenylyl cyclase to regulate cyclic nucleotide signaling. Endocr Metab Immune Disord Drug Targets. 2006;6:239–247. doi: 10.2174/187153006778249994. [DOI] [PubMed] [Google Scholar]
- 74.Insel PA, Ostrom RS. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol. 2003;23:305–314. doi: 10.1023/A:1023684503883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maruoka H, Sasaya H, Sugihara K, Shimoke K, Ikeuchi T. Low-molecular-weight compounds having neurotrophic activity in cultured PC12 cells and neurons. J Biochem. 2011;150:473–475. doi: 10.1093/jb/mvr113. [DOI] [PubMed] [Google Scholar]
- 76.Tegenge MA, Stern M, Bicker G. Nitric oxide and cyclic nucleotide signal transduction modulates synaptic vesicle turnover in human model neurons. J Neurochem. 2009;111:1434–1446. doi: 10.1111/j.1471-4159.2009.06421.x. [DOI] [PubMed] [Google Scholar]
- 77.Tegenge MA, Roloff F, Bicker G. Rapid differentiation of human embryonal carcinoma stem cells (NT2) into neurons for neurite outgrowth analysis. Cell Mol Neurobiol. 2011;31:635–643. doi: 10.1007/s10571-011-9659-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hannila SS, Filbin MT. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp Neurol. 2008;209:321–332. doi: 10.1016/j.expneurol.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Posternak T, Sutherland EW, Henion WF. Derivatives of cyclic 3′,5′-adenosine monophosphate. Biochim Biophys Acta. 1962;65:558–560. doi: 10.1016/0006-3002(62)90475-4. [DOI] [PubMed] [Google Scholar]
- 80.Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther. 2000;87:199–226. doi: 10.1016/s0163-7258(00)00051-6. [DOI] [PubMed] [Google Scholar]
- 81.Szentandrassy N, Harmati G, Farkas V, Horvath B, Hegyi B, Magyar J, Szenasi G, Marton I, Nanasi PP. Modified cAMP derivatives: powerful tools in heart research. Curr Med Chem. 2011;18:3729–3736. doi: 10.2174/092986711796642445. [DOI] [PubMed] [Google Scholar]
- 82.Jastorff B, Hoppe J, Morr M. A model for the chemical interactions of adenosine 3′:5′-monophosphate with the R subunit of protein kinase type I. Refinement of the cyclic phosphate binding moiety of protein kinase type I. Eur J Biochem. 1979;101:555–561. doi: 10.1111/j.1432-1033.1979.tb19750.x. [DOI] [PubMed] [Google Scholar]
- 83.Krass JD, Jastorff B, Genieser HG. Determination of lipophilicity by gradient elution high-performance liquid chromatography. Anal Chem. 1997;69:2575–2581. doi: 10.1021/ac961246i. [DOI] [PubMed] [Google Scholar]
- 84.Bertinetti D, Schweinsberg S, Hanke SE, Schwede F, Bertinetti O, Drewianka S, Genieser HG, Herberg FW. Chemical tools selectively target components of the PKA system. BMC Chem Biol. 2009;9:3. doi: 10.1186/1472-6769-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Enserink JM, Christensen AE, de RJ, van TM, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 87.Rehmann H, Schwede F, Doskeland SO, Wittinghofer A, Bos JL. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:38548–38556. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- 88.Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McPhee I, Gibson LC, Kewney J, Darroch C, Stevens PA, Spinks D, Cooreman A, MacKenzie SJ. Cyclic nucleotide signalling: a molecular approach to drug discovery for Alzheimer’s disease. Biochem Soc Trans. 2005;33:1330–1332. doi: 10.1042/BST0331330. [DOI] [PubMed] [Google Scholar]
- 90.Borner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, Nikolaev VO. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc. 2011;6:427–438. doi: 10.1038/nprot.2010.198. [DOI] [PubMed] [Google Scholar]
- 91.Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc’h F. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell Signal. 2011;23:1257–1266. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 92.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 93.Ster J, De BF, Bertaso F, Abitbol K, Daniel H, Bockaert J, Fagni L. Epac mediates PACAP-dependent long-term depression in the hippocampus. J Physiol. 2009;587:101–113. doi: 10.1113/jphysiol.2008.157461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morales-Garcia JA, Redondo M, onso-Gil S, Gil C, Perez C, Martinez A, Santos A, Perez-Castillo A. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS ONE. 2011;6:e17240. doi: 10.1371/journal.pone.0017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang L, Calingasan NY, Lorenzo BJ, Beal MF. Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp Neurol. 2008;211:311–314. doi: 10.1016/j.expneurol.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sasaki T, Kitagawa K, Omura-Matsuoka E, Todo K, Terasaki Y, Sugiura S, Hatazawa J, Yagita Y, Hori M. The phosphodiesterase inhibitor rolipram promotes survival of newborn hippocampal neurons after ischemia. Stroke. 2007;38:1597–1605. doi: 10.1161/STROKEAHA.106.476754. [DOI] [PubMed] [Google Scholar]
- 97.Farooq MU, Naravetla B, Moore PW, Majid A, Gupta R, Kassab MY. Role of sildenafil in neurological disorders. Clin Neuropharmacol. 2008;31:353–362. doi: 10.1097/WNF.0b013e31815cd94c. [DOI] [PubMed] [Google Scholar]
- 98.Jiang H, Nucifora FC, Jr, Ross CA, DeFranco DB. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum Mol Genet. 2003;12:1–12. doi: 10.1093/hmg/ddg002. [DOI] [PubMed] [Google Scholar]
- 99.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 100.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 101.Castro LR, Gervasi N, Guiot E, Cavellini L, Nikolaev VO, Paupardin-Tritsch D, Vincent P. Type 4 phosphodiesterase plays different integrating roles in different cellular domains in pyramidal cortical neurons. J Neurosci. 2010;30:6143–6151. doi: 10.1523/JNEUROSCI.5851-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DeMarch Z, Giampa C, Patassini S, Martorana A, Bernardi G, Fusco FR. Beneficial effects of rolipram in a quinolinic acid model of striatal excitotoxicity. Neurobiol Dis. 2007;25:266–273. doi: 10.1016/j.nbd.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 103.Giampa C, Middei S, Patassini S, Borreca A, Marullo F, Laurenti D, Bernardi G, mmassari-Teule M, Fusco FR. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington’s disease. Eur J Neurosci. 2009;29:902–910. doi: 10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- 104.DeMarch Z, Giampa C, Patassini S, Bernardi G, Fusco FR. Beneficial effects of rolipram in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. 2008;30:375–387. doi: 10.1016/j.nbd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 105.Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS ONE. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nijholt DA, De KL, Elfrink HL, Hoozemans JJ, Scheper W. Removing protein aggregates: the role of proteolysis in neurodegeneration. Curr Med Chem. 2011;18:2459–2476. doi: 10.2174/092986711795843236. [DOI] [PubMed] [Google Scholar]
- 107.Huang Q, Figueiredo-Pereira ME. Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis. 2010;15:1292–1311. doi: 10.1007/s10495-010-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang X, Li J, Zheng H, Su H, Powell SR. Proteasome functional insufficiency in cardiac pathogenesis. Am J Physiol Heart Circ Physiol. 2011;301:H2207–H2219. doi: 10.1152/ajpheart.00714.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rideout HJ, Larsen KE, Sulzer D, Stefanis L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001;78:899–908. doi: 10.1046/j.1471-4159.2001.00474.x. [DOI] [PubMed] [Google Scholar]
- 110.Metcalfe MJ, Huang Q, Figueiredo-Pereira ME. Coordination between proteasome impairment and caspase activation leading to TAU pathology: neuroprotection by cAMP. Cell Death Dis. 2012;3:e326. doi: 10.1038/cddis.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Myeku N, Wang H, Figueiredo-Pereira ME. cAMP stimulates the ubiquitin/proteasome pathway in rat spinal cord neurons. Neurosci Lett. 2012;527:126–131. doi: 10.1016/j.neulet.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Houslay MD, Christian F. p62 (SQSTM1) forms part of a novel, reversible aggregate containing a specific conformer of the cAMP degrading phosphodiesterase, PDE4A4. Autophagy. 2010;6:1198–1200. doi: 10.4161/auto.6.8.13479. [DOI] [PubMed] [Google Scholar]
- 113.Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ Res. 2010;107:1094–1101. doi: 10.1161/CIRCRESAHA.110.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Asai M, Tsukamoto O, Minamino T, Asanuma H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, Takashima S, Hori M, Kitakaze M. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol. 2009;46:452–462. doi: 10.1016/j.yjmcc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 115.Zong C, Gomes AV, Drews O, Li X, Young GW, Berhane B, Qiao X, French SW, Bardag-Gorce F, Ping P. Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res. 2006;99:372–380. doi: 10.1161/01.RES.0000237389.40000.02. [DOI] [PubMed] [Google Scholar]
- 116.Lu H, Zong C, Wang Y, Young GW, Deng N, Souda P, Li X, Whitelegge J, Drews O, Yang PY, Ping P. Revealing the dynamics of the 20 S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach. Mol Cell Proteomics. 2008;7:2073–2089. doi: 10.1074/mcp.M800064-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shao W, Yu Z, Fantus IG, Jin T. Cyclic AMP signaling stimulates proteasome degradation of thioredoxin interacting protein (TxNIP) in pancreatic beta-cells. Cell Signal. 2010;22:1240–1246. doi: 10.1016/j.cellsig.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 118.Hanna J, Finley D. A proteasome for all occasions. FEBS Lett. 2007;581:2854–2861. doi: 10.1016/j.febslet.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang F, Paterson AJ, Huang P, Wang K, Kudlow JE. Metabolic control of proteasome function. Physiology (Bethesda) 2007;22:373–379. doi: 10.1152/physiol.00026.2007. [DOI] [PubMed] [Google Scholar]
- 120.Pereira ME, Wilk S. Phosphorylation of the multicatalytic proteinase complex from bovine pituitaries by a copurifying cAMP-dependent protein kinase. Arch Biochem Biophys. 1990;283:68–74. doi: 10.1016/0003-9861(90)90613-4. [DOI] [PubMed] [Google Scholar]
- 121.Marambaud P, Wilk S, Checler F. Protein kinase A phosphorylation of the proteasome: a contribution to the alpha-secretase pathway in human cells. J Neurochem. 1996;67:2616–2619. doi: 10.1046/j.1471-4159.1996.67062616.x. [DOI] [PubMed] [Google Scholar]
- 122.Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem. 2007;282:22460–22471. doi: 10.1074/jbc.M702439200. [DOI] [PubMed] [Google Scholar]
- 123.Goncalves DA, Lira EC, Baviera AM, Cao P, Zanon NM, Arany Z, Bedard N, Tanksale P, Wing SS, Lecker SH, Kettelhut IC, Navegantes LC. Mechanisms involved in 3′,5′-cyclic adenosine monophosphate-mediated inhibition of the ubiquitin-proteasome system in skeletal muscle. Endocrinology. 2009;150:5395–5404. doi: 10.1210/en.2009-0428. [DOI] [PubMed] [Google Scholar]
- 124.Lira EC, Goncalves DA, Parreiras-E-Silva LT, Zanon NM, Kettelhut IC, Navegantes LC. Phosphodiesterase-4 inhibition reduces proteolysis and atrogenes expression in rat skeletal muscles. Muscle Nerve. 2011;44:371–381. doi: 10.1002/mus.22066. [DOI] [PubMed] [Google Scholar]
- 125.Carlucci A, Lignitto L, Feliciello A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008;18:604–613. doi: 10.1016/j.tcb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 126.Edelmann MJ, Nicholson B, Kessler BM. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev Mol Med. 2011;13:e35. doi: 10.1017/S1462399411002031. [DOI] [PMC free article] [PubMed] [Google Scholar]