

Abstract
The biochemical and structural characterization of ubiquitin-conjugating enzymes (E2s) over the past 30 years has fostered important insights into ubiquitin transfer mechanisms. Although many of these enzymes share high sequence and structural conservation, their functional roles in the cell are decidedly diverse. Here, we report that the mono-ubiquitinating E2 UBE2W forms a homodimer using two distinct protein surfaces. Dimerization is primarily driven by residues in the ß-sheet region and Loops 4 and 7 of the catalytic domain. Mutation of two residues in the catalytic domain of UBE2W is capable of disrupting UBE2W homodimer formation, however, we find that dimerization of this E2 is not required for its ubiquitin transfer activity. In addition, residues in the C-terminal region, although not compulsory for the dimerization of UBE2W, play an ancillary role in the dimer interface. In all current E2 structures, the C-terminal helix of the UBC domain is at least 15Å away from the primary dimerization surface shown here for UBE2W. This leads to the proposal that the C-terminal region of UBE2W adopts a noncanonical position that places it closer to the UBC ß-sheet, providing the first indication that at least some E2s adopt C-terminal conformations different from the canonical structures observed to date.
Keywords: Ubiquitin, Ubiquitin-conjugating enzyme, UBE2W, Dimerization, NMR
Introduction
The attachment of ubiquitin (Ub) is a highly regulated cellular process that requires a cascade of three enzymes. The E1 Ub-activating enzyme first forms an ATP-dependent, high-energy thiolester bond with Ub through an active site cysteine. The E2 Ub-conjugating enzyme next receives Ub from the E1 in a transthiolation reaction forming an E2 ~ Ub conjugate. Finally, an E3 Ub-ligase binds E2 ~ Ub using either a HECT domain or RING-finger motif and facilitates the transfer of Ub from the E2 ~ Ub onto a target substrate [1]. In humans two E1s, about 35 E2s, and hundreds of E3s are responsible for attaching Ub to proteins [2]. For transfer that involves RING-type E3s, the E2 plays a role in dictating the type of Ub modification [3, 4]. Depending on the required cellular cue, some substrates are modified by the addition of a single Ub molecule (mono-ubiquitination) while others are covalently attached to a chain of Ub molecules which can be linked via any of the seven lysine residues in Ub or via its N-terminal end (poly-ubiquitination) [1]. At present, features and properties that determine different E2 functions, especially as they relate to monoversus poly-Ub transfer, are not well understood. Here, we report biochemical and structural features of human UBE2W, a mono-ubiquitinating E2.
UBE2W is one of ten E2s that can bind to the BRCA1 RING domain of the BRCA1/BARD1 E3 complex [3]. In vitro, UBE2W adds only a single Ub to its target substrate, however, chain-building E2s such as the UBE2N/UBE2V2 (Ubc13/Mms2) complex can extend poly-ubiquitin chains from sites mono-ubiquitinated by UBE2W [3]. This E2 has been found to mono-ubiquitinate FANCD2, an essential step in the activation of the Fanconi anemia tumor suppressor pathway that is important in the DNA damage response [5]. More recently, Ataxin-3, a specialized deubiquitinating enzyme (DUB) important for protein quality control, has been shown to be mono-ubiquitinated by UBE2W [6]. In both cases, UBE2W can directly attach Ub to substrate lysines in the absence of an E3 in an as yet unknown mechanism, although ubiquitination of these substrates by UBE2W was enhanced by the presence of a RING-type E3 ligase [5, 6].
To better understand the structure and function of UBE2W, we began to characterize it biochemically. During size exclusion chromatography (SEC), we unexpectedly observed that monomeric UBE2W is in equilibrium with a dimeric species. Here, we show that homodimerization of UBE2W is driven by contacts involving its ß-sheet surface, a region used by other E2s for noncovalent protein–protein interactions with Ub and non-RING E3 domains. Mutations that disrupt the dimer have only a modest effect on the rate of Ub transfer, revealing that dimerization of UBE2W is not required for its ubiquitination activity. Surprisingly, the dimerization interface also involves residues in the C-terminal region of UBE2W. Based on the distance of the C-terminus from the ß-sheet in other known E2 structures, we conclude that the C-terminal region of UBE2W must adopt a noncanonical position to make this contribution. Furthermore, a C-terminally truncated version of UBE2W maintained dimeric properties similar to WT indicating the C-terminus is not required for dimerization.
Materials and Methods
Plasmids, Protein Expression, and Purification
All constructs of UBE2W (WT, KK (V30K, D67K), 131Δ (truncation following Y131), and 131Δ-KK) were expressed from the pET24 vector without affinity tags. UBE2W constructs were transformed into Escherichia coli (BL21 DE3) cells and protein expression was induced with 0.4 mM isopropyl-β-D-thio-galactoside (IPTG) at OD600 of 0.6, followed by growth at 16 °C for 16 h. Cells were lysed by French press in 25 mM sodium phosphate (pH 7.0), 1 mM EDTA for full-length constructs, or 25 mM 2-(N-morpholino)ethanesulfonic acid (pH 6.0), 1 mM EDTA for 131Δ constructs. Following centrifugal clarification, the lysate was subjected to cation exchange chromatography with an elution gradient of 0–0.5 M NaCl. E2-rich fractions were identified by UV absorbance, pooled, and further purified by SEC on a Superdex 75 (GE Healthcare) column in 25 mM sodium phosphate (pH 7.0) and 150 mM NaCl, the buffer used for all NMR and SEC experiments. Wheat Uba1, human Ub, and human FLAG–BRCA1304/BARD1327 used for ubiquitination activity assays were purified as previously described [3, 7]. SEC for Fig. 1 and Supplementary Fig. 2 was performed on purified samples using a Superdex 75 column (GE Healthcare). 250 μL of 250, 25, or 5 μM protein was applied to the column.
Fig. 1.

UBE2W exists in a monomer–dimer equilibrium. a Size exclusion elution profiles for UBE2W-WT (blue) and UBE2D3 (red) detect both a monomer and dimer for UBE2W, but only a monomer of UBE2D3. b Mutation of V30 and D67 to lysine in UBE2W (UBE2W-KK) (red) shifts the population of E2 toward a single monomeric peak as compared to UBE2W-WT (blue) c A C-terminal truncation mutant (UBE2W-131D) (red) elutes as both a monomer and dimer, similar to UBE2W-WT (blue). d The ‘KK’ mutation in the UBE2W-131Δ (UBE2W-131Δ-KK) (red) truncation background shifts the E2 population to a single monomeric peak when compared with the UBE2W-131Δ (blue). All size exclusion injections were performed on a column with a relative void volume of −5 mL
Autobiquitination Activity Assay
Reactions for BRCA1-directed ubiquitination assays contained 20 μM Ub, 0.5 μM wheat Uba1, 2 μM UBE2W-WT, or UBE2W-KK, 2 μM FLAG–BRCA1304/BARD1327 (residues 1–304 or 1–327, respectively), and 10 mM MgCl2. Reactions were initiated by the addition of 5 mM ATP and incubated at 37 °C. Samples were collected at 0 and 60 min after the addition of ATP. Products were visualized by western blotting for FLAG–BRCA1.
NMR Spectroscopy
NMR spectra were collected on a 500 MHz Bruker Avance II (University of Washington). Spectra were recorded at 25 °C in 25 mM sodium phosphate (pH 7.0), 150 mM NaCl, and 10 % D2O. All datasets were collected using 15N-HSQC-TROSY experiments with 200 μM 15N-isotopically labeled protein. Data were processed using NMR-Pipe/NMRDraw [8] and visualized with NMRView [9]. Chemical shift perturbations (CSPs) were calculated using the formula Δδj = [(Δδj 15N/5)2 + (Δδj 1H)2]1/2.
UBE2W Homology Model
The homology model for UBE2W was created using the secondary structure prediction algorithim PHYRE2 [10]. The model is based on the structure of Ub-conjugated UBE2D3 in complex with the human deubiquitinating enzyme OTUB1 (PDB: 4DDG) [11].
Structure Visualization
All structural figures were created using the Pymol Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
Results
UBE2W Exists in a Monomer–Dimer Equilibrium
In the process of purifying recombinant human UBE2W, we observed properties that differ from other common, well-characterized ubiquitin-conjugating enzymes. In particular, resolving 250 μM UBE2W using a Superdex 75 size exclusion column resulted in the separation of two distinct peaks, corresponding in size to a dimeric and monomeric species (Fig. 1a). The ability to resolve the two species by SEC indicates that their interconversion is slow. SDS-PAGE analysis of the UBE2W dimer and monomer peaks under reducing (+ß-mercaptoethanol) and nonreducing conditions indicated that dimerization of UBE2W is not dependent on disulfide bond formation, implying a noncovalent interaction (Sup. Fig. 1). Reapplication of the monomeric UBE2W fraction showed a re-equilibration into dimeric and monomeric forms (Sup. Fig. 2b), and as predicted for a self-associating system, dimer formation displays a clear concentration dependence. Ten- and 50-fold dilution of UBE2W prior to SEC shift the equilibrium in favor of the monomeric species, consistent with an estimated affinity in the mid-micromolar range (Sup. Fig. 2).
Other biochemically characterized, class I E2s1 do not show evidence for self-association at similarly high-protein concentrations. For example, the well-studied E2 UBE2D3 (UbcH5c) elutes as a single peak at an elution volume consistent with its being a monomer (Fig. 1a) as do UBE2L3 (UbcH7) and UBE2N (Ubc13) (data not shown). Thus, the observation that UBE2W forms a dimer is not due simply to our use of high-protein concentrations (required for NMR spectroscopy, see later), but rather indicates that UBE2W differs from canonical class I E2s. Serendipitously, we found two residues (V30 and D67) during the course of our early studies with UBE2W and its activity with BRCA1/BARD1, that, when mutated to lysine, disfavor the dimer and shift the equilibrium to a monomeric species (termed the UBE2W–KK mutant) (Fig. 1b). This allowed us to (1) ask whether dimerization is required for UBE2W activity and (2) assign and compare resonances from the 1H, 15N-HSQC spectra of UBE2W to obtain insights regarding the structural source for dimer formation.
UBE2W transfers a single Ub to its target substrate resulting in a mono-ubiquitinated product [3, 5]. In the in vitro assay shown, BRCA1304/BARD1327 functions as both the RING E3-ligase and as proxy substrate, with mono-ubiquitin products formed on BRCA1304/BARD1327 serving as a readout for activity. UBE2W–KK showed only a modest decrease in its ability to transfer Ub compared with wildtype, indicating dimerization of this E2 is not a strict requirement for Ub transfer activity (Fig. 2).
Fig. 2.
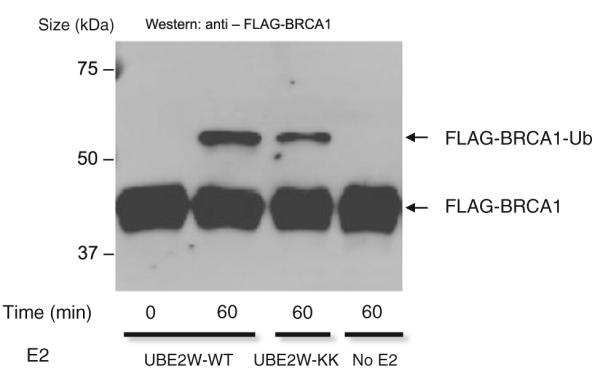
Disruption of the UBE2W dimer does not abrogate its ubiquitin transfer activity. Autoubiquitination of BRCA1 by UBE2W-WT and UBE2W-KK (1 h reaction) is detected in a western blot for FLAG–BRCA1
Identification of the Surfaces Involved in UBE2W Dimerization
NMR spectroscopy was used to understand the structural consequences of the V30K/D67K double mutation and to obtain insights regarding the nature of UBE2W self association. For these experiments, we used 1H, 15N-HSQC NMR spectroscopy. This type of spectrum provides a distinct signal for every amino acid (except proline) that corresponds to that residue’s backbone amide group. The precise position (i.e., the “chemical shift”) and intensity of each peak are highly sensitive to the local environment and dynamics around the residue. Comparison of spectra for two related species allows easy identification of differences at an amino acid residue level. In this case, comparison of the 1H, 15N-HSQC spectra of UBE2W-WT and UBE2W-KK revealed some remarkable differences. Foremost among these, the ‘KK’ mutant spectrum contains roughly 120 peaks (of the 139 peaks expected) whereas the spectrum of the WT has fewer than 100 peaks (Fig. 3a). As expected, three-dimensional NMR spectra of UBE2W–WT also contained many fewer resonances than did datasets of the monomeric mutant (unpublished data). The simplest explanation for missing peaks in the UBE2W-WT spectrum is that residues involved in the interface experience different environments in a monomer versus a dimer and are exchanging between these states on the timescale of NMR data collection. To identify residues affected by the mutation, we assigned the spectrum of monomeric UBE2W-KK using conventional heteronuclear 3D-NMR approaches. We could identify most of the residues in the UBE2W-WT spectrum with high confidence by comparison with the ‘KK’ spectrum.2
Fig. 3.
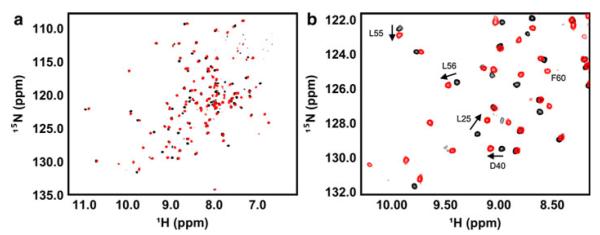
Comparison of the NMR spectra of UBE2W-WT and UBE2W-KK. a Full 1H, 15N-HSQC spectrum of UBE2W-WT (black) overlayed with the 1H, 15N-HSQC spectrum of UBE2W-KK (red). b Region of 1H, 15N-HSQC spectrum showing representative resonances shifted between UBE2W-WT (black) and UBE2W-KK (red) (i.e., L25, D40, L55, and L56) and new resonances in the UBE2W-KK spectrum (i.e., F60)
Differences observed between the 1H, 15N-HSQC spectra of UBE2W-KK and UBE2W-WT fell into two distinct classes: (1) signals whose positions are shifted between the WT and mutant spectra and (2) signals that appear in the mutant ‘KK’ spectrum that are not present in the WT (Fig. 3b). It is generally expected that peaks arising from residues that are proximal to a site of mutation will have small CSPs due to the change of side chain and, therefore, of the chemical environment due to the mutation. In the double ‘KK’ mutant, we expect peaks from residues near V30 or D67 in the E2 structure to be shifted (i.e., in the first class). CSPs are also expected to arise due to environmental changes in residues involved in the dimerization interface. For the second class, in the WT protein, some residues are interconverting between two states on a similar timescale to NMR data collection (~100 ms); one exposed to solvent (monomer) and one buried in a protein interface (dimer). This behavior splits the intensity of a single resonance over two states causing broadening and a loss of intensity, often to a point below the level of detection. These “missing” or unobserved resonances reappear in the ‘KK’ spectrum because it exists predominantly as a monomer, resulting in accumulation of peak intensity at the resonance position that corresponds to the monomer.
Although a majority of human E2 structures have been determined experimentally, the only such structure for UBE2W is a crystal structure of a truncated form in which all C-terminal residues after S116 are missing (PDB: 2A7L) [12]. The structure adopted in this crystal is unlike any other E2 structure solved to date. In the crystal lattice, a symmetry mate from one unit cell domain swaps its N-terminal helix with that of another UBE2W molecule in an adjacent unit cell (Fig. 4a). This structure might imply that UBE2W dimers form using their N-terminal helices, but our NMR results are not consistent with such an inference as we do not detect perturbations in the N-terminal helix. Furthermore, because the C-terminus is absent in the crystal, it is impossible to visualize its potential role in dimerization in this context. Given the disparities between the crystal structure and our NMR observations, we chose to map resonances that differ between the UBE2W-WT and ‘KK’ spectra onto a UBE2W sequence homology model based on the structure of UBE2D3 (PDB: 4DDG) (Fig. 5b) [11]. This model is more consistent with our NMR data and is therefore suitable for mapping the observed NMR perturbations.
Fig. 4.

Surface 1 on UBE2W is involved in crystal contacts. a Domain-swapped UBE2W (truncated at S116) crystal structure (2A7L). The N-terminal helix from one UBE2W subunit (green) noncovalently interacts with the UBC domain of another subunit (cyan), and vice versa. Only one half of the dimer is shown for clarity. b Surface 1 resonances mapped onto the dimerswapped structure (red). In blue are positions V30 and D67, and in yellow is the active site cysteine (C91)
Fig. 5.

a Domain architecture and nomenclature of an E2 based on the structure of UBE2D3, the active site C85 is colored in yellow (PDB: 2FUH). b Observed NMR perturbations identify two surfaces on UBE2W. UBE2W homology model based on the structure of UBE2D3 (4DDG) is shown. Positions V30 and D67 (mutated to lysine in the ‘KK’ mutant background) are colored in blue, while red represents both significantly shifted (top 10 % of shifted resonances) resonances between WT and ‘KK’ spectra and new peaks that appear in the ‘KK’ spectrum. Surface 1 includes Loops 4 and 7, the ß-sheet region, and the 310 helix. Surface 2 is comprised entirely of the C-terminal region. The active site cysteine (C91) is colored yellow. For perturbed residues, only those resonances with unambiguous assignments are colored red
Substitutions of V30 and D67 with lysine result in perturbations on two distinct surfaces on UBE2W. One surface is made of residues in Loops 4 and 7, the ß-sheet (“backside”) region, and the 310 helix (“surface 1” in Fig. 5). The other surface corresponds to the C-terminal region of UBE2W (“surface 2” in Fig. 5). Both surfaces have some resonances that are shifted from their WT position and some that are exchange broadened in the WT spectrum and appear in the ‘KK’ mutant. On surface 1, residues L25, D40, L56, and L57 in the ß-sheet are shifted compared with the WT spectrum (Fig. 3b). These resonances are shifted due to their proximity to the V30K mutation and/or because they are close to the dimerization interface. F60, however, is an example of a residue that is exchange broadened in the WT, but appears in the ‘KK’ spectrum, and can therefore be assigned unambiguously as being located within the dimer interface (Fig. 3b).
The sites of both mutations, V30 and D67 (blue), are near surface 1, implying that this region of the protein dictates dimerization of UBE2W. However, several C-terminal residues are also perturbed suggesting that residues in this region experience different environments in monomeric and dimeric states. To assess the possible contribution of the C-terminal region to self-association, a UBE2W construct truncated after residue Y131 (UBE2W-131Δ), was expressed and purified. This truncation removes the final 20 amino acids of UBE2W that are predicted to form the terminal helix and creates a construct of this E2 that is slightly longer than the previously crystallized structure [7]. UBE2W-131Δ is remarkably well behaved as evidenced by SEC and is stably folded as seen by NMR spectroscopy (Figs. 1c, 6). Underthe same conditions as UBE2W-WT, UBE2W-131Δ shows a similar monomer–dimer equilibrium indicating that the C-terminal region is affected by monomer–dimer exchange, it is not required for the formationof UBE2W homodimers (Fig. 1c). Furthermore, the ‘KK’ mutation in the UBE2W-131Δ truncation background shifts the E2 population toward a monomeric state in the same manner as in the full-length protein (Fig. 1d). Altogether, these results indicate that residues in surface 1 constitute the driving force for UBE2W dimerization.
Discussion
UBE2W displayed dimeric properties by SEC purification at the high concentration required for NMR studies (Fig. 1a). This property proved problematic for detailed structural characterization by NMR spectroscopy, as multimeric proteins with mid-micromolar dissociation constants often suffer from loss of peak intensity if multiple interconverting states exist. We serendipitously discovered a monomer-favoring ‘KK’ (V30K/D67K) double mutation in early studies aimed at creating an E2/E3 specificity-switch-pair. The mutant proved particularly useful for structural characterization by NMR spectroscopy, as the in vitro E2 activity was retained with a modest reduction at most (Fig. 2).
Comparison of NMR spectra of ‘KK’ and WT UBE2W reveal two distinct surfaces that participate in homodimerization. We note that although the C-terminally truncated and domain-swapped UBE2W crystal structure lack the residues that compose surface 2, the structure observed in the crystal does appear to use residues in surface 1 to pack neighboring UBE2W molecules. In Fig. 4b, the surface 1 NMR perturbations are mapped onto the crystal structure, revealing reasonable concordance, although our surface implicates residues not entirely congruent with the current domain-swapped dimer interface. The role of the C-terminal region cannot be addressed by the crystal structure, as removal of these residues was apparently required to obtain crystals. Though the C-terminal region is not required for UBE2W dimerization, its NMR resonances are clearly perturbed upon disruption of the dimer interface (Fig. 5b). In all experimentally determined structures of E2 UBC domains, the C-terminal helix is at least 15Å from the UBC ß-sheet surface. It is therefore difficult to reconcile the observed effects in the C-terminus unless it adopts a noncanonical conformation and/or location in UBE2W to position it closer to the ß-sheet and Loop 4 regions. We propose that UBE2W dimer formation through surface 1 induces ancillary contacts between surface 1 residues and C-terminal residues which, while not be required for induction of the dimer, might stabilize the species once formed. Our observations are not consistent with an alternative possibility that UBE2W forms a dimer via heteromeric interactions in which the C-terminus of one protomer interacts with surface 1 residues of the other and rather supports a model in which surface 1 to surface 1 contacts drive homodimerization.
The nature of the monomer-favoring mutations, as well as the identification of the dimeric interface provide insights regarding UBE2W self-association. V30 is a hydrophobic residue on the surface of the ß-sheet region of UBE2W. On its own, the V30K mutation almost completely disrupts the formation of UBE2W homodimers as detected by SEC (data not shown), indicating that it is a key residue in the dimer interface. Substitution of a lysine for D67 is not as effective in disrupting UBE2W dimerization on its own, but the double mutation further pushes the monomer–dimer equilibrium of UBE2W toward the monomeric state and reveals more resonances in the 1H, 15N-HSQC spectrum. Intriguingly, D67 is in Loop 4, a part of the canonical E2–E3 binding interface. This position is strongly conserved as lysine in many E2s, which is why we originally designed the D67K-UBE2W mutant. In our homology model of UBE2W, V30 rests on a turn immediately following the first ß-strand in the UBC domain; the analogous position is conserved as valine in all isoforms of UBE2D (i.e., V26), as well as numerous other E2s. The surface created by the ß-sheet consists of few charged residues and notably, an analogous surface on UBE2D3 interacts noncovalently with Ub, enabling formation of large multimeric assemblies of UBE2D3 ~ Ub conjugates [13–15]. Introduction of a single charged residue into the UBE2D3 surface (S22R) is sufficient to abrogate the noncovalent E2–Ub interaction. Substitution of a positively charged lysine in place of V30 in UBE2W can disrupt the E2–E2 interaction similar to the S22R mutation in UBE2D3. Other E2s use their UBC ß-sheet region in noncovalent interactions. For example, the E2s UBE2G2 and Rad6 bind non-RING portions of the E3 ligases, gp78 and Rad18, respectively, in positions very similar to the surface used by UBE2D3 to interact noncovalently with Ub [16–18]. Finally, we note that surface 1 utilized by UBE2W for self-association is closer to the active site region than the surfaces used by other E2s that use their UBC ß-sheet region to form noncovalent interactions [13–18]. In this respect, the UBE2W surface more closely resembles the surface used by UBE2N to interact with the E2-like protein UBE2V2 (Mms2), although this heterodimeric interaction is significantly stronger than the UBE2W homomeric E2–E2 interaction [19, 20].
Dimerization of E2s in vitro has been reported sporadically in ubiquitination literature for more than 20 years [21–24]. Studies have also shown that crosslinking reagents enable the formation of E2 homodimers and increase ubiquitination activity, presumably by raising the local concentration of E2 ~ Ub conjugates [25, 26]. Among the putative dimer-forming E2s, the best characterized is the S. cerevisiae E2, Cdc34 [24]. Dimerization of Cdc34 ~ Ub conjugates has been shown to be required for E3-independent poly-ubiquitin chain formation by this E2 [27]. Cdc34 also participates in E3-dependent poly-ubiquitination reactions mediated by the Skp1-CUL1-F-box (SCF) E3 ligase [28]. In this case, a model was proposed in which the high-local concentrations of Cdc34 ~ Ub conjugates form dimers that are recruited to the Rbx1/Roc1 (RING) domain of the SCF complex to poly-ubiquitinate the S. cerevisiae substrate Sic1 in vitro [29]. Currently, there is no concrete evidence that E2 dimerization is a requirement for activity in vivo.
Cellular concentrations of UBE2W are currently unknown, however, given a Kd estimated to be in the mid-micromolar range, a small fraction of UBE2W is in the dimeric state even at the submicromolar concentrations. Dimerization is not a requirement for activity, but may play a role in E3 recruitment or in determining E3-independent substrate specificity. It seems unlikely that dimerization of UBE2W functions similarly to S. cerevisiae Cdc34 so as to raise the local concentration of E2 at sites of ubiquitination, as the benefit of this property for an E2 that mono-ubiquitinates its substrates is unclear. However, weak protein–protein interactions are a hallmark of the ubiquitination machinery in the cell and regulate critical aspects of Ub transfer and signaling. With this in mind, the identified surface could play a role in mediating as an yet unknown protein–protein interaction that is important for UBE2W function in cells. The results presented here also suggest that the C-terminus of UBE2W adopts a noncanonical position in UBE2W, placing it close to the ß-sheet of the UBC domain. This result makes UBE2W unique among other characterized class I E2s. Although a functional significance for UBE2W self-association remains to be determined, there are clearly structural aspects of this protein that point to unique function and properties, the details of which await further elucidation.
Supplementary Material
{kind=link}
{kind=link}
Fig. 6.
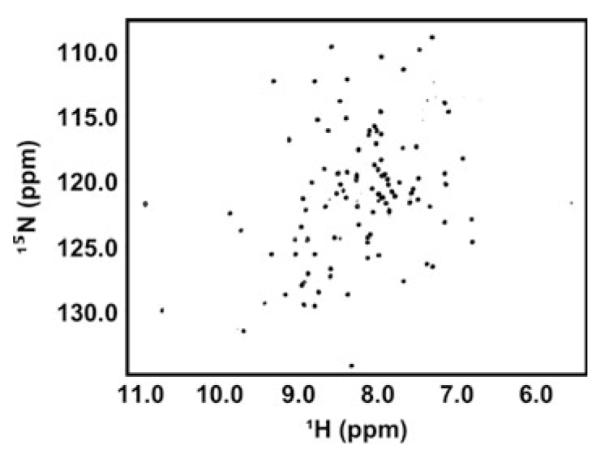
UBE2W lacking its C-terminal region is stable and retains its ability to form a dimer. Dispersion of resonances in the UBE2W-131Δ spectrum reveals a folded protein
Acknowledgments
The authors acknowledge D. Christensen and C. Eakin for their initial observations on the UBE2W-KK mutant. This study was supported by the National Institute of General Medical Sciences Grants R01 GM088055 (R.E.K.). V. V. was supported in part by the Hurd Fellowship Fund and PHS NRSA 2T32 GM007270.
Footnotes
E2s are organized into classes: Class I E2s contain only the ~150-residue catalytic domain (“UBC” domain); Class II, III, and IV E2s also contain N-terminal, C-terminal extension, or both. Currently, little is known regarding the function of such extensions and there is little structural information available on any extensions. All available E2 structures, regardless of the class they belong to, are of the UBC domain.
Of the ~120 resonances in the UBE2W-‘KK’ 1H, 15N-HSQC spectrum, we were able to unambiguously assign 115. From this dataset, we were able to transfer assignments to the ‘WT’ spectrum and identify 80 of the ~90 resonances present.
Electronic supplementary material The online version of this article (doi:10.1007/s12013-013-9633-5) contains supplementary material, which is available to authorized users.
Contributor Information
Vinayak Vittal, Department of Biochemistry, University of Washington, Seattle, WA 98195, USA.
Dawn M. Wenzel, Department of Biochemistry, University of Washington, Seattle, WA 98195, USA; Department of Biochemistry, University of Utah, Salt Lake City, UT 84112, USA
Peter S. Brzovic, Department of Biochemistry, University of Washington, Seattle, WA 98195, USA
Rachel E. Klevit, Department of Biochemistry, University of Washington, Seattle, WA 98195, USA
References
- 1.Pickart CM. Mechanisms underlying ubiquitination. Annual Review of Biochemistry. 2001;2001(70):195–201. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 2.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nature Reviews Molecular Cell Biology. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nature Structural and Molecular Biology. 2007;14:941–948. doi: 10.1038/nsmb1295. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigo-Brenni MC, Foster SA, Morgan DO. Catalysis of lysine 48-specific ubiquitin chain assembly by residues in E2 and ubiquitin. Molecular Cell. 2010;39:548–559. doi: 10.1016/j.molcel.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Molecular Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodríguez-Lebrón E, et al. Ube2w and Ataxin-3 Coordinately Regulate the Ubiquitin Ligase CHIP. Molecular Cell. 2011;43:599–612. doi: 10.1016/j.molcel.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickart CM, Raasi S. Controlled synthesis of polyubiquitin chains. Methods in Enyzmology. 2005;399:21–36. doi: 10.1016/S0076-6879(05)99002-2. [DOI] [PubMed] [Google Scholar]
- 8.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 9.Johnson BA, Blevins RA. NMR view: A computer program for the visualization and analysis of NMR data. Journal of Biomolecular NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 10.Kelley LA, Sternberg MJE. Protein structure prediction on the web: A case study using the Phyre server. Nature Protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 11.Juan YC, Landry MC, Sanches M, Vittal V, Leung CC, Ceccarelli DF, et al. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Molecular Cell. 2012;45:384–397. doi: 10.1016/j.molcel.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheng Y, Hong JH, Doherty R, Srikumar T, Shloush J, Avvakumov GV, et al. A human ubiquitin conjugating enzyme (E2)-HECT E3 ligase structure-function screen. Molecular Cell Proteomics. 2012;11:329–341. doi: 10.1074/mcp.O111.013706. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, Klevit RE. A UbcH5c/ubiquitin noncovalent complex is required for processive BRCA1-direct ubiquitination. Molecular Cell. 2006;21:873–880. doi: 10.1016/j.molcel.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Sakata E, Satoh T, Yamamoto S, Yamaguchi Y, Yagi-Utsumi M, Tanaka K, et al. Crystal structure of UbcH5b ~ ubiquitin intermediate: Insight into the formation of the self-assembled E2 ~ Ub conjugates. Structure. 2010;18:138–147. doi: 10.1016/j.str.2009.11.007. 2010. [DOI] [PubMed] [Google Scholar]
- 15.Page RC, Pruneda JN, Amick J, Klevit RE, Misra S. Structural insights into the conformation and oligomerization of E2 ~ ubiquitin conjugates. Biochemistry. 2012;51:4175–4187. doi: 10.1021/bi300058m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das R, Mariano J, Tsai YC, Kalathur RC, Kostova Z, Li J, et al. Allosteric activation of E2-RING finger-mediate ubiquityation by a structurally defined specific E2-binding region of gp78. Molecular Cell. 2009;34:674–685. doi: 10.1016/j.molcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, Tu D, Li L, Wollert T, Ghirlando R, Brunger, et al. Mechanistic insights into active site-associated polyubiquitination by the ubiquitin-conjugating enzyme Ube2g2. Proceedings of the National Academy of Sciences of the United States of America; 2009. pp. 3722–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hibbert RG, Huang A, Boelens R, Sixma TK. E3 ligase Rad18 promotes monubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proceedings of the National Academy of Sciences of the United States of America; 2011. pp. 5590–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.VanDermark AP, Hofmann RM, Tsui C, Pickart CM, Wolberger C. Molecular insights into polyubiquitin chain assembly: Crystal structure of the Mms2/Ubc13 heterodimer. Cell. 2001;105:711–720. doi: 10.1016/s0092-8674(01)00387-7. [DOI] [PubMed] [Google Scholar]
- 20.Moraes TF, Edwards RA, McKenna S, Pastushok L, Xiao W, Glover JN, et al. Crystal structure of the human ubiquitin conjugating enzyme complex, hMms2-hUbc13. Natural Structural Biology. 2001;8:669–673. doi: 10.1038/90373. [DOI] [PubMed] [Google Scholar]
- 21.Pickart CE, Rose IA. Functional heterogeneity of ubiquitin carrier proteins. Journal of Biological Chemistry. 1985;260:1573–1581. [PubMed] [Google Scholar]
- 22.Silver ET, Gwozd TJ, Ptak C, Goebl M, Ellison MJ. A chimeric ubiquitin conjugating enzyme that combins the cell cycle properties of CDC34 (UBC3) and the DNA repair properties of RAD6 (UBC2): Implications for the structure, function and evolution of the E2s. EMBO Journal. 1992;11:3091–3098. doi: 10.1002/j.1460-2075.1992.tb05381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girod PA, Vierstra RD. A major ubiquitin conjugation system in wheat germ extracts involves a 15-kDa ubiquitin-conjugating enzyme (E2) homologous to the years UBC4/UBC5 gene products. Journal of Biological Chemistry. 1993;268:955–960. [PubMed] [Google Scholar]
- 24.Ptak C, Prendergast JA, Hodgins R, Kay CM, Chau V, Ellison MJ. Functional and physical characterization of the cell cycle ubiquitin-conjugating enzyme CDC34 (UBC3). Identification of a functional determinant within the tail that facilitates CDC34 self-association. Journal of Biological Chemistry. 1994;269:26539–26545. [PubMed] [Google Scholar]
- 25.Haldeman MT, Xia G, Kasperek EM, Pickart CM. Structure and function of ubiquitin conjugating enzyme E2–25K: The tail is a core-dependent activity element. Biochemistry. 1997;36:10526–10537. doi: 10.1021/bi970750u. [DOI] [PubMed] [Google Scholar]
- 26.Gazdoiu S, Yamoah K, Wu K, Escalante CR, Tappin I, Bermudez V, et al. Proximity-induced activation of human Cdc34 through heterologous dimerization. Proceedings of the National Academy of Sciences of the United States of America; 2005. pp. 15053–15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varelas X, Ptak C, Ellison MJ. Cdc34 self-association is facilitated by ubiquitin thiolester formation and is required for its catalytic activity. Molecular and Cellular Biology. 2003;23:5388–5400. doi: 10.1128/MCB.23.15.5388-5400.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skowyra D, Graig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 29.Deffenbaugh AE, Scaglione KM, Zhang L, Moore JM, Buranda T, Sklar LA, et al. Release of ubiquitin-charged Cdc34-S – Ub from the RING domain is essential for ubiquitination of the SCF(Cdc4)-bound substrate Sic1. Cell. 2003;114:611–622. doi: 10.1016/s0092-8674(03)00641-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.