

Abstract
Far from now are the days when investigators raced to identify the proteolytic system responsible for the degradation of their favorite protein. Nowadays, it is well accepted that the same protein can be degraded by different systems depending on factors such as cell type, cellular conditions, or functionality of each proteolytic pathway. The realization of this sharing of substrates among pathways has also helped to unveil deeper levels of communication among the different proteolytic systems. Thus, cells often respond to blockage of one degradative mechanism by upregulating any of the other available pathways. In addition, effectors and regulators of one proteolytic system can be degraded by a different proteolytic pathway that exerts, in this way, a regulatory function. In this mini-review, we describe the different levels of cross-talk among autophagic pathways and the ubiquitin/proteasome system. We also provide examples of how this proteolytic communication is used for compensatory purposes in different pathological conditions and discuss the possible therapeutic potential of targeting the modulators of the cross-talk among proteolytic pathways.
1. Introduction
In the field of intracellular protein degradation considerable efforts have been dedicated in the past to identifying the proteolytic pathway followed by specific proteins. This emphasis on mixing and matching seems to now fade as it becomes clear that the contribution of the proteolytic systems to the maintenance of a functional proteome is highly dynamic and capable of rapidly adapting to the cellular requirements at particular times. Although a large percentage of a specific protein may undergo degradation through a particular proteolytic system in a given cell type, this may not be the case in a different cell type, where other pathways may have a more prevalent role. In addition, if a protein cannot be degraded by its “usual” pathway, other proteolytic systems will now chip in and take care of its removal. This apparent redundancy in the way in which proteins are degraded in the cell speaks to the importance of maintaining proteostasis and the detrimental consequences that poor handling of altered or damaged proteins has for the cell.
The discovery of this sharing of degradative tasks by different proteolytic systems has brought to light two new concepts: the idea of cross-talk among the degradative pathways and the need for a strict regulation of the “laws” of this cross-talk to assure a coordinated functioning of these systems. Growing evidence supports that the cellular degradation systems not only share substrates, but also effectors and molecular regulators, which allows for one system to modulate functioning of the others.
As part of this special issue dedicated to the ubiquitin/proteasome system, we focus in this mini-review on its interplay with the autophagic system and the possible consequences of their cooperative functioning. We first briefly introduce the different autophagic pathways and then focus on their cross-communication among themselves and with the ubiquitin/proteasome system. The purpose of this review is not to provide a comprehensive list of every instance in which interactions among these pathways have been described, but rather, to highlight common emerging themes on the characteristics of the cross-talk and to provide representative examples of the different layers of interaction that are now becoming evident. Our intention is to stimulate future analysis and discussion of the molecular mechanisms that govern the talk among the different proteolytic systems.
2. Autophagy
Autophagy refers to “self-eating” of the cell through a process by which cellular components undergo degradation within lysosomes. Lysosomes are specialized organelles full of luminal hydrolases that include proteases, lipases, nucleotidases, and glycases, and are thus capable of degrading a wide range of substrates, ranging from macromolecular cellular components to entire organelles [1]. The lysosomal function in autophagy extends beyond cellular removal of unwanted cellular contents by proteolysis or degradation because the breakdown products are for the most part, recycled and utilized as new building blocks or for the generation of energy. Several types of autophagy coexist in all mammalian cells and differ largely in their mode of cargo delivery to the lysosomes, which gives rise to macroautophagy (MA), microautophagy and chaperone-mediated autophagy (CMA) (Fig. 1) [2, 3].
Figure 1. Intracellular proteolytic systems cross-talk.

Recent studies support many levels of functional interaction and cross-talk among the main proteolytic systems in the cell: the ubiquitin proteasome system (UPS) and the different autophagic pathways (macroautophagy, microautophagy and chaperone-mediated autophagy (CMA)). Some of the already identified levels of cross-talk are highlighted and discussed in the text.
Macroautophagy is a highly-conserved pathway that involves more than 30 autophagy-related genes (Atgs) [4] whose protein products organize into functional complexes and act in a multi-step manner in this process. Cytosolic cargo is sequestered by elongation of a double limiting membrane that seals to form an autophagosome (Fig. 1). Degradation occurs when the lysosome and autophagosome membranes fuse and hydrolases from the lysosomal lumen gain access to the sequestered cargo [5]. At the molecular level, the process is initiated by the release of autophagy-inhibitory complexes (such as the mammalian target of rapamycin or mTOR, one of the best characterized autophagy inhibitors), in response to a wide range of activating stimuli, such as scarcity of nutrients or growth factors, and endogenous and exogenous sources of cellular stress (e.g. endoplasmic reticulum (ER) stress, oxidative stress, hypoxia, inflammatory conditions, etc.) [6]. Upon activation, nucleation of the limiting membrane (or phagophore) is initiated by phosphorylation by a second kinase complex of membrane lipids in specific cellular locations from where the autophagosome will form that include the ER, mitochondria, Golgi, and most recently, the plasma membrane [7–10]. This activating kinase complex (known as the initiation complex) is comprised of the core proteins Beclin-1, Vps34 and Vps15, and additional proteins such as Atg14, UVRAG and Ambra-1 that determine the location and activity of the complex [11, 12]. Elongation of the limiting membrane is achieved by two ubiquitin-like conjugation events, namely the one mediated by the Atg5-Atg12-Atg16 protein-to-protein conjugation system and the LC3-PE (phosphatidyl ethanolamine) protein-to-lipid conjugation system [5]. Further maturation and sealing of this limiting membrane and fusion of the autophagosome with late endosomes and lysosomes involve specific subsets of SNARE proteins and their GTP-binding modulators as well as additional lipid-modifying enzymes [13, 14]. Although for a long time macroautophagy was considered to be an “in bulk” degradation process in which cytosolic materials were randomly sequestered, recent studies support that although a small fraction of macroautophagy may still occur in a non-selective manner, in most cases, cargo is specifically recognized by a growing number of cargo-recognition molecules or autophagy receptors that physically interact with the material tagged for degradation and with effectors of the autophagy process [15] (see more details in the next section).
In microautophagy, formation of an intermediary cytosolic vesicle for cargo delivery is not required, as cargo is sequestered by invaginations of the lysosomal membrane itself that engulf entire regions of the cytosol (Fig. 1). The invaginating membrane seals in the form of small vesicles that then pinch off into the lysosomal lumen where they are rapidly degraded. This process has been well characterized in yeast and a dedicated machinery along with a subset of Atgs has been shown to mediate recognition of cargo, sequestration and internalization in a step-wise manner [16–18]. Although this type of vesicle-mediated internalization of cytosolic material into lysosomes has not been identified yet in mammals, a microautophagy-like process (termed endosomal-microautophagy) has been recently described to take place in late endosomes through the small vesicular invaginations that the ESCRT machinery forms in the surface of these organelles. Cargo can be trapped by the vesicles “in bulk” or selectively through the action of a cytosolic chaperone that binds specific cytosolic proteins and delivers them to the forming vesicles [19].
In chaperone-mediated autophagy, the cytosolic chaperone hsc70 (heat shock cognate protein of 70kDa), a constitutive member of the heat shock protein 70 family of proteins, along with several co-chaperones, functions in the identification and lysosomal delivery of cargo [20], which in this case, are single cytosolic proteins bearing in their amino acid sequence a distinctive pentapeptide (KFERQ-like) targeting motif [21]. Recognition of the CMA substrate proteins by the Hsc70 co-chaperone complex is followed by their binding to the CMA receptor at the lysosomal membrane, the lysosome-associated membrane protein type 2A (LAMP-2A)[22], and the subsequent unfolding [23] and translocation [24] of the substrate across the lysosomal membrane (Fig. 1). A luminal form of hsc70 assists in the translocation of substrates, which, upon reaching the lumen, undergo rapid degradation [24].
All autophagic processes share common functions in the cell and have proven to be essential for the maintenance of cellular homeostasis and in the adaptation of cells to the changing extracellular environment [25–27]. One of the first functions described for autophagy was its role in the maintenance of the cellular energetic balance through the recycling of the constitutive elements of the degraded components [28]. This function becomes especially important for cell survival during conditions of nutrient deprivation, but also contributes to the cellular adaptation to arrival of an excess of nutrients, such as lipids, which if accumulated in large amounts inside cells, can become toxic [28, 29]. During starvation, selective breakdown of non-essential proteins frees amino acids that can be used to sustain synthesis of proteins required for survival under these stress conditions. In addition, mobilization of intracellular energy stores, lipid droplets, and glycogen through autophagy has been recently shown to contribute to maintain a positive energetic balance during the starvation period [30]. A second important function of autophagy is to serve as a quality control mechanism that assures maintenance of the integrity of the proteome and the renewal of cellular organelles. Autophagy contributes to the elimination of damaged or misfolded proteins before or even after they organize into protein aggregates or other aberrant protein assemblies which can be toxic for the cell [31–33]. Out of the different types of autophagy-mediated degradation of organelles, mitophagy (degradation of mitochondria) and the autophagy of the endoplasmic reticulum during stress have received considerable attention for their important roles in the cellular defense against toxicity resulting from these organelles’ malfunctioning [34–37]. The large degradative capacity of the autophagic system makes autophagy an important mechanism in all those processes requiring major cellular remodeling or elimination of entire regions of cytosol. Thus, autophagy aids in the removal of cellular components that are no longer needed during specific stages of development, cellular differentiation and in tissue remodeling [38–41]. Moreover, the beneficial role of autophagy in eliminating unwanted cytosolic material further extends to pathogens that reach this compartment upon cell invasion and underlies the basis of the role of autophagy in innate and adaptive immunity, by direct degradation of the invading agents and antigen presentation [42].
This diverse array of physiological functions of the autophagic process justifies the growing number of links established between autophagy malfunctioning and human disease. In some pathologies, failure of autophagy is secondary to the main cause of the disease but still contributes further to its aggravation, whereas in other conditions, a primary defect in autophagy has been identified as the main cause of disease [43, 44]. Autophagy malfunctioning has been described in pathologies resulting from alterations in protein homeostasis such as common neurodegenerative disorders including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and several tauopathies. Disease presents when clearance of the pathogenic proteins by autophagy is no longer possible, although often times it is the pathogenic protein that interferes with proper autophagic functioning, thus contributing not only to its own poor degradation, but also to defective degradation of other proteins eliminated by this pathway [45]. Cancer was the first human disease in which a primary defect in an autophagy-related gene was described. Further studies have revealed the complexity of the interplay between autophagy and oncogenic processes where autophagy can act either as a tumor suppressor (i.e. by providing genome stability [46–48]), or as a pro-oncogenic pathway that favors survival of the cancer cells during malignant transformation by assuring better quality control and supporting the high energetic demands of their rapid division [49–51]. The contribution of autophagy to host cell immunity against invading pathogens justifies current efforts to enhance the autophagic response in infectious processes. However, in recent years it has become apparent that certain pathogens can hijack autophagic compartments and utilize them for their replication and survival in the host, thus highlighting the need for the development of a more customized anti-infectious approach to the manipulation of autophagy [42]. Readers interested in an in-depth review of the interplay of autophagy and disease are referred to some of the recent comprehensive reviews on this topic [39, 42–44].
3. Selectivity in autophagy
As mentioned in the previous section, the concept of selectivity in autophagy has been revised in recent years in light of the growing evidence supporting targeted removal of cytosolic components by autophagy. Efforts to dissect the molecular basis of autophagic selectivity have also contributed to unveiling a previously unknown cross-communication among the different autophagic pathways and of these processes with the ubiquitin/proteasome system.
According to the type of specific cargo degraded, new terms to describe this selective nature of macroautophagy have been coined, such as reticulophagy, for the degradation of ER [52], mitophagy for mitochondrial degradation [34], ribophagy for ribosomes [53], xenophagy for pathogens [42], pexophagy for peroxisomes [54], lipophagy for lipid droplets [30] and aggrephagy for aggregates [55]. The selectivity in these specialized types of macroautophagy is mediated by a family of cargo recognition molecules that includes proteins such as p62, NBR1, or NDP52 [56, 57]. These proteins contain a region for directly binding molecular tags in the cargo to be degraded, and at the same time, are capable of binding essential autophagy components such as LC3 through a dedicated region known as LIR (LC3 interacting region) [58]. Tags recognized by these proteins include specific proteins exposed on the surface and protein post-translational modifications, such as ubiquitination, which links macroautophagy and the proteasome system. Although initially identified in aggregates, these receptors can also mediate macroautophagy of other cellular components such as mitochondria, peroxisomes and even pathogens [56]. Recent studies have demonstrated that in addition to receptors and tagging, recognition of cargo by the macroautophagy machinery may also depend on intrinsic properties of the cargo itself. For example, reduced mobility of proteins in the surface of aggregates seems to facilitate their recognition by macroautophagy by permitting the assembly of the integral components of the autophagosome on top of the aggregate [59]. In other instances, selectivity seems to be mediated by molecular chaperones, as is the case of the hsc70-mediated degradation of aggregate proteins by macroautophagy known as chaperone-assisted selective autophagy (CASA) [60].
In the case of microautophagy in addition to the selective degradation of cytosolic proteins by selective endosomal microautophagy in mammals [19], selectivity has also been described in yeast for the degradation of the nuclear fractions (piecemeal microautophagy; micronucleophagy) [17], peroxisomes (micropexophagy) [61] and glycogen [62].
The characteristic manner in which substrates are selected and targeted to the lysosomal compartment by CMA makes this pathway solely dedicated to selective protein removal. This selectivity has been demonstrated to assure removal of, for example, damaged proteins, without disturbing normally functioning proteins nearby in the case of mild oxidative stress [63] or exposure to UV light [64] and toxic protein-denaturing compounds [65]. This selectivity is also utilized by mammalian cells to eliminate subsets of proteins no longer necessary during nutrient deprivation, such as, for example, glycolytic enzymes [66], for recycling of their constitutive amino acids. Close to 30 bona-fide CMA substrates have been identified and validated to date [67] but sequence analysis reveals that up to 30% of cytosolic proteins bear putative CMA targeting motifs and could be, in theory, amenable for degradation by this pathway.
4. Multi-level conversations among proteolytic systems
Although proteolytic systems have been traditionally studied and dissected as individual isolated pathways, growing evidence supports the existence of an intricate network of communications both among the different autophagic pathways and in-between these pathways and the ubiquitin/proteasome system.
Despite the fact that current studies have only begun to scratch the surface of the proteolytic cross-communication, it has already become evident that there are different levels of interaction among these systems. We comment here on the interactions for which experimental evidence has been provided and that can be, in a way, organized around three main types of observations: 1) those in which changes in one proteolytic pathway have an impact on the activity of the other; 2) those in which different proteolytic systems share components and substrates and 3) those instances in which essential components of one pathway become substrates of another proteolytic pathway that thus contribute to its regulation (Fig. 1).
4.1. Changes in one proteolytic pathway affect other proteolytic systems
The first evidence for communication among proteolytic pathways originated from the observation that cells often respond to chemical or genetic blockage of one proteolytic pathway by upregulating others. Later studies have revealed that these reactive changes also occur under physiological and pathological conditions, when activation of one pathway can compensate for the failure of another.
Pharmacological blockage of the proteasome often results in compensatory upregulation of macroautophagy as supported by studies in cardiomyocytes [68], different cancer cell lines [69, 70] and neuronal cell lines and neurons in culture [71, 72]. The same holds true in whole organisms such as flies upon genetic ablation of proteasome components [73] (Fig. 2). This response was also demonstrated in the context of aggregopathies where pathogenic proteins that accumulate in the affected tissues in these disorders, such as α-synuclein in Parkinson’s disease or mutant huntingtin in Huntington’s disease, often interfere with the normal functioning of the proteasome [74–77]. Activation of macroautophagy under these conditions serves to facilitate the removal of these proteins as they organize into multimeric irreversible complexes (oligomers or aggregates) no longer removable by other proteolytic systems and this often enhances cell survival [70–72, 74]. Whether the reactive activation of macroautophagy occurs in response to the presence of these protein inclusions or directly because of the defective proteasome activity needs further clarification. In some instances, proteins such as p53 [72] have been proposed to link both systems because p53, often degraded by the proteasome, has been shown to have an activating effect on macroautophagy [78–80]. Transcriptional upregulation of some of the Atgs or inhibition of negative regulators of macroautophagy such as mTOR in response to proteasome blockage have also been described in other instances [70, 81] but the mechanisms that modulate this upregulation remain, for the most part, unknown. Recent studies have also shown pronounced upregulation of CMA when the proteasome is blocked although the mechanisms behind these changes remain unknown [82].
Figure 2. Compensatory mechanisms in intracellular proteolysis.
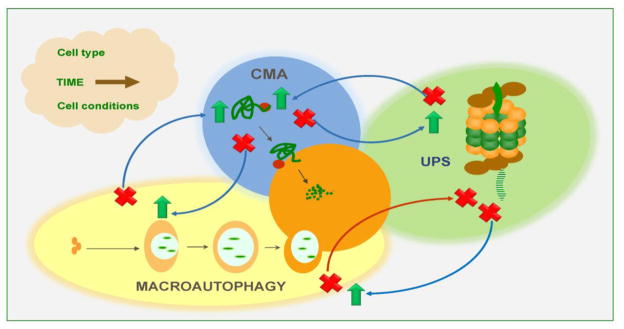
Changes in the activity of one proteolytic system impact the activity of the other systems. So far, most of the changes described are of compensatory nature whereby blockage of one pathway (marked with a red cross) leads to upregulation of another one (marked with a green arrow). However, there are also instances in which blockage of one pathway also interferes with the functionality of another. This proteolytic crosstalk may be influenced by many factors, as the ones shown in the cloud.
Most cells activate macroautophagy in response to blockage of CMA and vice versa [64, 83] (Fig. 2). However despite this bidirectional compensation, these pathways are not redundant and, although under basal conditions the upregulation of the functional pathway is enough to maintain cellular homeostasis, the affected cells fail to orchestrate a proper response to different stressors. For example, cells defective in CMA show constitutively high levels of macroautophagy and this upregulation is required for their normal homeostasis, as downregulation of this pathway leads to cellular failure [64]. However, upon exposure to oxidant or pro-oxidant agents, cells with compromised CMA display lower rates of survival despite macroautophagy still being fully active [64, 83]. Interestingly, the susceptibility to stress of these cells is clearly dependent on the type of stressor because for example, conditions such as heat shock, where macroautophagy may play a more important role due to the presence of protein aggregates, does not elicit a higher vulnerability in the CMA compromised cells. A plausible explanation could be that activation of CMA may be beneficial over macroautophagy only in those conditions in which damage mainly affects single proteins rather than organelles, especially given the capability of CMA for selective protein removal [63]. The cross-talk between macroautophagy and CMA, at least in most cells, works in both directions, as blockage of macroautophagy also leads to enhanced CMA activity [83]. In this case, it is less intuitive to understand what the beneficial effects of this upregulation of CMA could be when CMA can only degrade single soluble proteins but no toxic oligomeric protein complexes or organelles. It is possible that CMA may facilitate maintenance of the soluble proteome under these conditions while other pathways such as microautophagy or yet to be identified mechanisms may also be upregulated to take care of organelle turnover.
In contrast to this bi-directional relationship between CMA and macroautophagy, the cross-talk of the latter with the proteasome seems to compensate only in one direction. Thus although as described above, blockage of the proteasome leads to macroautophagy upregulation, blockage of macroautophagy often leads to reduced proteasome activity (Fig. 2) [84]. Different mechanisms may contribute to the compromise of proteasome activity. For example, in certain cells accumulation of p62 as result of the reduced activity of macroautophagy has been shown to compete with the deubiquitinating components of the 19S regulatory complex and ultimately being responsible for the decreased feeding of substrates into the proteasome catalytic core [84].
The reactive response of one proteolytic pathway to changes in another pathway is not universal, but rather could exhibit both cell type-specificity and also time-dependence (Fig. 2). Regarding the former, recent studies have demonstrated that the cross-talk between CMA and macroautophagy is no longer present in the case of cancer cells [50]. Experimental blockage of CMA in these cells does not elicit macroautophagy upregulation, but instead higher rates of protein degradation through the UPS have been detected under those conditions. Interestingly, this enhanced proteasome degradation seems to affect some subsets of proteins but not others although the mechanisms that determine this selectivity remain unknown [50]. Lastly, the duration of the functional compromise of one proteolytic pathway seems to also affect the response of the other pathways. For example although, as described above, cells often respond to chemical inhibition of the proteasome by upregulating macroautophagy, studies using long-term inhibitors of the proteasome have demonstrated the inability of the treated cells to properly upregulate macroautophagy in response to nutritional stress [85]. It is possible that the sustained constitutive upregulation of macroautophagy in response to proteasome blockage results in either a depletion of the macroautophagy effectors and/or reduced ability to modify the lysosomal compartment to accommodate any further increase in the amount of cargo delivered by macroautophagy.
Interestingly, in contrast to the growing number of studies demonstrating the impact that blockage of one proteolytic pathway has on others, there is still no supportive evidence that upregulation of one proteolytic pathway induces responsive changes in another proteolytic system. For example, chemical upregulation of macroautophagy does not seem to modify proteasome or CMA activities and genetic upregulation of CMA, at least in cultured cells, does not modify macroautophagy or UPS efficiency. These findings support that the cross-talk among pathways may preferentially have a compensatory function and goes along with the idea that these mechanisms of degradation are not redundant.
4.2. Different proteolytic pathways share components and substrates
Numerous examples support that the same protein can be degraded by different proteolytic systems depending on the cell type. Furthermore, even in the same cell, different pools of the same protein can undergo degradation by different proteolytic pathways. For example, in the case of IκBα, the inhibitor of the nuclear transcription factor kappa B, it has been shown to be degraded by the proteasome both in a ubiquitin dependent and ubiquitin-independent manner; it also contains motifs for calpain and caspase-mediated cleavage, and it bears in its amino acid sequence a targeting motif for CMA that is utilized under conditions of nutrient deprivation to initiate the activation of NF-κBα [86–89]. Metabolic labeling analysis for different periods of time permit identification of pools of IκBα with different half-lives that coexist in most cells. The very short half-life forms of IκBα are preferentially substrates for the proteasome system, whereas the lysosomal system degrades the long-lived pool through CMA [89]. This use of different proteolytic pathways is no longer the exception but it has become a common characteristic of a large fraction of the cellular proteome.
This multiplicity of pathways to degrade the same protein may confer cells with an extra level of post-translational regulation; thus in the case of IκBα, whereby reserving a fraction of this protein for lysosomal degradation, makes possible a transcriptional peak dependent on NFκB even when many other signaling pathways are repressed. In other instances the beneficial effect of the availability of different proteolytic pathways for a single substrate is used by the cells as a safety mechanism that guarantees the elimination of unwanted proteins or those which are prone to aggregate even when their normal degradation route is compromised. For example, wild-type alpha-synuclein is normally degraded in neurons by the ubiquitin/proteasome system and through CMA [90, 91], whereas macroautophagy has been shown to be capable of eliminating the mutant forms of this protein, in particular, when organized into oligomeric irreversible structures no longer amenable to degradation by the two above mentioned systems [92]. Having this “way out” guarantees cellular homeostasis, since blockage of macroautophagy in the affected neurons leads to severe proteotoxicity and often neuronal death.
The current challenge in the field is to identify the molecular determinants that decide the proteolytic pathway followed by a given protein. Some of these markers of degradation have been well characterized, such as the ubiquitin attachment for proteasome degradation [93] or the presence of a pentapeptide motif in the amino acid sequence that, when recognized by a chaperone, will deliver the protein for lysosomal degradation by CMA [67]. However, ubiquitin tagging for example can also be utilized for autophagy targeting. The first clue about the usage of ubiquitin by autophagy originated from studies in transgenic animals with compromised autophagy, where one of the most apparent modifications was the accumulation of polyubiquitinated proteins despite the fact that no major changes were observed in the proteasome system [94, 95]. Later, autophagy receptors were shown to recognize ubiquitin moieties on the surface of protein aggregates or the organelles such as peroxisomes before their degradation [58, 96]. However, the complexity of the ubiquitin code is larger than initially anticipated. For example the number of ubiquitin moieties conjugated to a protein and the lysine (K) in the ubiquitin used for the conjugation (linkage) have started to reveal a complex code in which some linkages (K48) are preferred by the proteasome whereas others (K63) are often identified in substrates undergoing degradation by macroautophagy [97, 98]. With more than 10 types of ubiquitin linkages already identified, the possibility of combined usage of lysines in the same chain and the occurrence of branching of chains indicates that the deciphering of this code is probably just in its beginning stages.
Although at a lower level of complexity, the CMA targeting motif also allows for some level of flexibility. For example, in addition to being functional in both directions and when placed in different regions of the same protein, the pentapetide that forms a CMA targeting motif can also be completed by post-translational modifications. Thus a functional motif can result from a tetrapeptide missing a positive or a negative charge through acetylation or phosphorylation of a fifth residue, respectively [99, 100]. This confers an important regulatory role to kinases, phosphatases, acetylating enzymes and deacetylases in the degradative fate of many proteins via CMA.
Chaperones have also been placed in the center of the degradative decision as they can recognize unfolded proteins and present their taggable regions to the pertinent enzymes [101]. Chaperones also mediate delivery of substrate proteins directly to the proteasome and to lysosomes, and in some cases such as CHIP (carboxyl terminus of Hsc70-interacting protein), they can even actively contribute to ubiquitin conjugation [102]. Although not in all cells, as described in previous sections, chaperones such as Bag1, Bag3 and hsp70 have also been involved in the targeting of aggregate proteins to degradation by macroautophagy through CASA [60]. However, aggregophagy can occur even in the absence of those chaperones with the assistance of the cargo-recognizing molecules [55]. In addition to the role of hps70 in macroautophagy, other members of the hsp70 family such as hsc70 participate in targeting of substrates for both CMA and endosomal microautophagy but the factors that determine delivery to one system or the other remain unknown. It is possible that because CMA can only handle proteins susceptible to unfolding, soluble single proteins are delivered by hsc70 to CMA but if proteins organize into irreversible oligomeric complexes, the same chaperone targets them to endosomal microautophagy where unfolding is no longer required prior to internalization and degradation [19].
Sharing of components among pathways is not limited to tagging but also effectors and regulators of one pathway can be utilized by another. One of the examples that has received much attention in recent days are E3 ubiquitin ligases such as Parkin, which, in addition to their role in proteasome substrate tagging, have been demonstrated to play an important function in the degradation of mitochondria through some forms of mitophagy [103–106]. Parkin is recruited to damaged mitochondria where it interacts with the mitochondria resident protein PTEN-induced kinase 1 (PINK1) and mediates ubiquitination of two mitochondrial fusion proteins MFN1 and MFN2, which in turn recruit other proteins to mitochondria to initiate mitophagy [107, 108]. Interestingly, further reinforcing the cooperativity between proteolytic systems, Parkin also promotes arrival to this mitochondria setting of other ubiquitin-proteasome system components that facilitate trimming and degradation of proteins from the outer membrane of the mitochondria before mitophagy can be initiated. The abundance and relative redundancy of E3 ligases in cells makes it unlikely that engagement of ligases such as Parkin in autophagy would diminish its availability for degradation of substrates through the proteasome system. However, once in a disease setting where the amount of pathogenic proteins that need to undergo degradation is high, and where some degree of compromise of quality control mechanisms can occur, it would not be surprising that effectors shared between two proteolytic systems could become limiting to one of them.
4.3. Components of one proteolytic pathway are degraded by another proteolytic system
A third elegant mechanism by which proteolytic systems interplay is by doing what proteolytic systems are intended to do, and that is degradation. There is a growing number of examples in which components of one proteolytic pathway become substrates for another proteolytic system. As with any other intracellular protein, those that form part of the ubiquitin-proteasome system or are engaged in autophagy also need to undergo continuous turnover and, in fact, changes in their degradation rates can exert an additional level of regulation over those proteolytic systems in which they participate.
The catalytic core of the proteasome is degraded as a whole complex by macroautophagy under conditions such as nutrient deprivation, thus contributing to the downregulation of proteasome-dependent degradation under those conditions [109]. However, the mechanisms that contribute to recognition of the proteasome by the macroautophagy machinery remain unknown. In contrast to this degradation triggered early in starvation, under basal conditions or when starvation is sustained beyond 12 hours, specific subunits of the proteasome seem to be selectively targeted for degradation by CMA [109]. In fact, a subset of subunits of the catalytic and regulatory cores of the proteasome bear in their sequence CMA targeting motifs and can be identified in the lumen of CMA active lysosomes if the activity of lysosomal proteases is inhibited [110]. It is possible that CMA plays a dual role over the proteasome system as a quality control enforcer, by eliminating excess subunits not used for assembly under basal conditions, and by exerting an additional regulatory role by limiting assembly of new proteasomes when nutrients are scarce.
Other components of the ubiquitin-proteasome system also utilize autophagy as their preferred method for degradation. For example ubiquilin, a protein that contributes to substrate presentation to the proteasome, has been detected inside autophagosomes, supporting its degradation through this pathway [111]. However, this protein becomes highly enriched in lysosomes active for CMA where it undergoes degradation during stress conditions [111].
Probably one of the most studied examples of a macroautophagy effector being degraded by the proteasome is p62. This cargo recognition protein required in many types of selective autophagy, can undergo degradation by the proteasome [84, 112]. Enhancing p62 degradation through the proteasome pathway could, at least in theory, limit the amount of this receptor available for macroautophagy.
In contrast to the vast number of substrates already identified for each proteolytic pathway, less is known regarding the degradation of the proteins involved in the degradation systems themselves. For example, except for the above mentioned cargo receptors and Atg8/LC3, the protein that serves as an essential constituent of the autophagosome whose degradation by autophagy is often used as a read out of autophagic flux [113], the degradation pathways followed by the other Atgs involved in macroautophagy are, for the most part, unknown. Likewise, in the case of CMA the degradation of the lysosomal receptor, LAMP-2A has been shown to contribute to modulate the activity of this pathway and has been extensively characterized, but the mechanisms that contribute to degrade the cytosolic chaperones that participate in this process are poorly characterized.
Future studies should provide a more complete view of this additional level of interplay that promises to have important regulatory functions.
5. Therapeutic implications of the proteolytic cross-talk and future challenges
Although much is still needed to learn about the rules that mediate the interplay among different proteolytic systems in cells, the growing number of disease conditions in which one proteolytic system compensates for failure of another highlights the potential value for molecular modulators of this cross-talk as attractive therapeutic targets in some human diseases associated to proteotoxicity.
The ability to upregulate one proteolytic pathway in response to failure of another may contribute to increasing the duration of the disease-free state, for instance, in different protein conformational disorders in which compromise of one of these pathways occurs. Although more detailed analysis is required, in conditions such as Parkinson’s disease where a direct blockage of the proteasome and CMA by α-synuclein, one of the main pathogenic proteins in this disorder, has been reported [90, 91], it is anticipated that the upregulation of macroautophagy observed in the affected neurons may help preserve cellular homeostasis even when the other two systems are compromised. Whether cellular toxicity and death are eventually precipitated either by a gradual increase in the level of blockage of these other two pathways, or due to the inability to sustain upregulated macroautophagy beyond some point or a combination of both will require future investigation.
Future studies should also address if the differences in the potency or even ability to activate these compensatory mechanisms account for cell and organ-specific differences in susceptibility to disease. For example, a gradual decrease with age in macroautophagy and CMA activity has been described in different organs [114] but whether this defect occurs concurrently or through the failure of one following the other after a period of compensatory upregulation remains unknown. A better understanding of both the sequence of events that end in the failure of each proteolytic system, as well as the molecular mechanisms that modulate the activation and maintain the compensatory activities should provide novel targets worth exploring for therapeutic purposes.
The rather limited information on the pathways that contribute to normal turnover of protein effectors for the different proteolytic pathways is probably the reason behind the lack of experimental examples of the applicability of this type of regulation. However, understanding the contribution that changes in the activity of one proteolytic system may have on the levels of effectors available for other proteolytic systems may be important from a therapeutic point of view. Thus, although blockage of one proteolytic pathway to increase effectors of another pathway may never be a good option, once the molecular determinants that trigger degradation of effectors of, for example, the proteasome by macroautophagy become elucidated, it is reasonable to think that interventions aimed at selectively preventing the targeting and degradation of these proteasome effectors by autophagy may be a realistic way to enhance proteasome activity when needed.
Overall, the cross-talk among proteolytic systems is still relatively unexplored but it is emerging as an important mechanism that cells adopt and use to their advantage during disease conditions. Consequently, understanding the principles and molecules that regulate this cross-talk could offer very interesting novel therapeutic approaches to modulate intracellular protein degradation and to preserve or restore protein homeostasis in disease settings.
References
- 1.De Duve C, Wattiaux R. Functions of lysosomes. Annual review of physiology. 1966;28:435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klionsky DJ, Codogno P, Cuervo AM, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Yoshimori T, Ohsumi Y. The Role of Atg Proteins in Autophagosome Formation. Annual Review of Cell and Developmental Biology. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 6.He C, Klionsky DJ. Regulation Mechanisms and Signaling Pathways of Autophagy. Annual Review of Genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki K, Ohsumi Y. Current knowledge of the pre-autophagosomal structure (PAS) FEBS Lett. 2010;584:1280–6. doi: 10.1016/j.febslet.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature cell biology. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–21. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–67. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell death and differentiation. 2011;18:571–80. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Kim YC, Fang C, et al. Differential Regulation of Distinct Vps34 Complexes by AMPK in Nutrient Stress and Autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vergne I, Deretic V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010;584:1313–8. doi: 10.1016/j.febslet.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dall’armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23:R33–45. doi: 10.1016/j.cub.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novak I, Dikic I. Autophagy receptors in developmental clearance of mitochondria. Autophagy. 2011;7:301–3. doi: 10.4161/auto.7.3.14509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakai Y, Koller A, Rangell L, Keller G, Subramani S. Peroxisome degradation by microautophagy in Pichia pastoris. Identification of specific steps and morphological intermediates. J Cell Biol. 1998;141:625–636. doi: 10.1083/jcb.141.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts P, Moshitch-Moshkovitz S, Kvam E, et al. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:129–141. doi: 10.1091/mbc.E02-08-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Developmental cell. 2011;20:131–9. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–5. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 21.Chiang HL, Dice JF. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. The Journal of biological chemistry. 1988;263:6797–805. [PubMed] [Google Scholar]
- 22.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–3. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 23.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. The Journal of biological chemistry. 2000;275:27447–56. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 24.Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. The Journal of cell biology. 1997;137:825–34. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 26.Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological reviews. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 27.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular Biology of the Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roth DM, Balch WE. Modeling general proteostasis: proteome balance in health and disease. Curr Opin Cell Biol. 2011;23:126–34. doi: 10.1016/j.ceb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar S, Ravikumar B, Rubinsztein DC. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol. 2009;453:83–110. doi: 10.1016/S0076-6879(08)04005-6. [DOI] [PubMed] [Google Scholar]
- 33.Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer. 2010;1:779–786. doi: 10.1177/1947601910383277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim I, Rodriguez-Enriquez S, Lemasters J. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawakami T, Inagi R, Takano H, et al. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol Dial Transplant. 2009;24:2665–72. doi: 10.1093/ndt/gfp215. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi-Nishino M, Fujita N, Noda T, et al. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy. 2010;6:301–3. doi: 10.4161/auto.6.2.11134. [DOI] [PubMed] [Google Scholar]
- 38.Ferdous A, Battiprolu PK, Ni YG, Rothermel BA, Hill JA. FoxO, autophagy, and cardiac remodeling. Journal of cardiovascular translational research. 2010;3:355–64. doi: 10.1007/s12265-010-9200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsukamoto S, Kuma A, Mizushima N. The role of autophagy during the oocyte-to-embryo transition. Autophagy. 2008;4:1076–8. doi: 10.4161/auto.7065. [DOI] [PubMed] [Google Scholar]
- 41.Singh R, Xiang Y, Wang Y, et al. Autophagy regulates adipose mass and differentiation in mice. The Journal of clinical investigation. 2009;119:3329–39. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. The Journal of Pathology. 2012;226:255–273. doi: 10.1002/path.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:806–11. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 47.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of clinical investigation. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes & Development. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Molecular biology of the cell. 2011;22:165–78. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kon M, Kiffin R, Koga H, et al. Chaperone-mediated autophagy is required for tumor growth. Science translational medicine. 2011;3:109ra117. doi: 10.1126/scitranslmed.3003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang RC, Levine B. Autophagy in cellular growth control. Febs Letters. 2010;584:1417–1426. doi: 10.1016/j.febslet.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hamasaki M, Noda T, Baba M, Ohsumi Y. Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic. 2005;6:56–65. doi: 10.1111/j.1600-0854.2004.00245.x. [DOI] [PubMed] [Google Scholar]
- 53.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nature cell biology. 2008;10:602–10. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 54.Dunn WA, Jr, Cregg JM, Kiel JA, et al. Pexophagy: the selective autophagy of peroxisomes. Autophagy. 2005;1:75–83. doi: 10.4161/auto.1.2.1737. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiology of disease. 2011;43:17–28. doi: 10.1016/j.nbd.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell cycle. 2009;8:1986–90. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 57.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nature immunology. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 58.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 59.Wong E, Bejarano E, Rakshit M, et al. Molecular determinants of selective clearance of protein inclusions by autophagy. Nat Commun. 2012;3:1240. doi: 10.1038/ncomms2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Current biology : CB. 2010;20:143–8. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 61.Veenhuis M, Salomons FA, Van Der Klei IJ. Peroxisome biogenesis and degradation in yeast: a structure/function analysis. Microscopy research and technique. 2000;51:584–600. doi: 10.1002/1097-0029(20001215)51:6<584::AID-JEMT8>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 62.Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy in glucose homeostasis. Pathology, research and practice. 2006;202:631–8. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proceedings of the National Academy of Sciences. 2006;103:5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cuervo AM, Hildebrand H, Bomhard EM, Dice JF. Direct lysosomal uptake of [agr]2-microglobulin contributes to chemically induced nephropathy. Kidney Int. 1999;55:529–545. doi: 10.1046/j.1523-1755.1999.00268.x. [DOI] [PubMed] [Google Scholar]
- 66.Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. American Journal of Physiology - Cell Physiology. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 67.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 68.Zheng Q, Su H, Tian Z, Wang X. Proteasome malfunction activates macroautophagy in the heart. Am J Cardiovasc Dis. 2011;1:214–26. [PMC free article] [PubMed] [Google Scholar]
- 69.Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. The American journal of pathology. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu K, Dunner K, Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29:451–62. doi: 10.1038/onc.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pan T, Kondo S, Zhu W, et al. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiology of disease. 2008;32:16–25. doi: 10.1016/j.nbd.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 72.Du Y, Yang D, Li L, et al. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy. 2009;5:663–75. doi: 10.4161/auto.5.5.8377. [DOI] [PubMed] [Google Scholar]
- 73.Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 74.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 75.Bennett E, Bence N, Jayakumar R, Kopito R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 76.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 77.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 78.Zhang XD, Wang Y, Zhang X, et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy. 2009;5 doi: 10.4161/auto.5.3.8174. [DOI] [PubMed] [Google Scholar]
- 79.Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy. 2008;4:870–3. doi: 10.4161/auto.6730. [DOI] [PubMed] [Google Scholar]
- 80.Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 81.Klappan AK, Hones S, Mylonas I, Bruning A. Proteasome inhibition by quercetin triggers macroautophagy and blocks mTOR activity. Histochemistry and cell biology. 2012;137:25–36. doi: 10.1007/s00418-011-0869-0. [DOI] [PubMed] [Google Scholar]
- 82.Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386. doi: 10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kaushik S, Massey A, Mizushima N, Cuervo AM. Constitutive Activation of Chaperone-mediated Autophagy in Cells with Impaired Macroautophagy. Mol Biol Cell. 2008;19:2179–92. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–27. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding Q, Dimayuga E, Martin S, et al. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003;86:489–497. doi: 10.1046/j.1471-4159.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 86.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–74. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 87.Milligan SA, Owens MW, Grisham MB. Inhibition of IkappaB-alpha and IkappaB-beta proteolysis by calpain inhibitor I blocks nitric oxide synthesis. Arch Biochem Biophys. 1996;335:388–95. doi: 10.1006/abbi.1996.9998. [DOI] [PubMed] [Google Scholar]
- 88.Miyamoto S, Seufzer BJ, Shumway SD. Novel IkappaB alpha proteolytic pathway in WEHI231 immature B cells. Mol Cell Biol. 1998;18:19–29. doi: 10.1128/mcb.18.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cuervo AM, Hu W, Lim B, Dice JF. IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol Biol Cell. 1998;9:1995–2010. doi: 10.1091/mbc.9.8.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 91.Stefanis L, Larsen K, Rideout H, Sulzer D, Greene L. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–60. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Webb J, Ravikumar B, Atkins J, Skepper J, Rubinsztein D. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 93.Walters KJ, Goh AM, Wang Q, Wagner G, Howley PM. Ubiquitin family proteins and their relationship to the proteasome: a structural perspective. Biochim Biophys Acta. 2004;1695:73–87. doi: 10.1016/j.bbamcr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 94.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 95.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 96.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–74. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–42. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–90. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lv L, Li D, Zhao D, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–30. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thompson LM, Aiken CT, Kaltenbach LS, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–99. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacol Res. 2012;66:484–93. doi: 10.1016/j.phrs.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–8. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tanaka A. Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Letters. 2010;584:1386–92. doi: 10.1016/j.febslet.2010.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tanaka A, Cleland MM, Xu S, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. The Journal of cell biology. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Van Humbeeck C, Cornelissen T, Hofkens H, et al. Parkin interacts with Ambra1 to induce mitophagy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:10249–61. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. Journal of Cell Science. 2012;125:795–9. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gegg ME, Cooper JM, Chau KY, et al. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Human molecular genetics. 2010;19:4861–70. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cuervo AM, Palmer A, Rivett AJ, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. 1995;227:792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x. [DOI] [PubMed] [Google Scholar]
- 110.Kaushik S, Bandyopadhyay U, Sridhar S, et al. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011;124:495–9. doi: 10.1242/jcs.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rothenberg C, Srinivasan D, Mah L, et al. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010;19:3219–32. doi: 10.1093/hmg/ddq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374–8. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 113.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal Turnover, but Not a Cellular Level, of Endogenous LC3 is a Marker for Autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 114.Cuervo AM. Autophagy and Aging: keeping that old broom working. Trends in Genetics. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]